

Abstract
The highly conserved enzyme arginyl-tRNA-protein transferase (Ate1) mediates arginylation, a posttranslational modification that is only incompletely understood at its molecular level. To investigate whether arginylation affects actin-dependent processes in a simple model organism, Dictyostelium discoideum, we knocked out the gene encoding Ate1 and characterized the phenotype of ate1-null cells. Visualization of actin cytoskeleton dynamics by live-cell microscopy indicated significant changes in comparison to wild-type cells. Ate1-null cells were almost completely lacking focal actin adhesion sites at the substrate-attached surface and were only weakly adhesive. In two-dimensional chemotaxis assays toward folate or cAMP, the motility of ate1-null cells was increased. However, in three-dimensional chemotaxis involving more confined conditions, the motility of ate1-null cells was significantly reduced. Live-cell imaging showed that GFP-tagged Ate1 rapidly relocates to sites of newly formed actin-rich protrusions. By mass spectrometric analysis, we identified four arginylation sites in the most abundant actin isoform of Dictyostelium, in addition to arginylation sites in other actin isoforms and several actin-binding proteins. In vitro polymerization assays with actin purified from ate1-null cells revealed a diminished polymerization capacity in comparison to wild-type actin. Our data indicate that arginylation plays a crucial role in the regulation of cytoskeletal activities.
INTRODUCTION
The posttranslational modifications (PTMs) that have been described for the cytoskeletal protein actin include phosphorylation, ubiquitination, methylation, acetylation, and carbonylation (Terman and Kashina, 2013) and were shown to be important for the integrity of a large number of different cells. In addition, it has been found that in cultured fibroblasts β-actin, but not γ-actin, undergoes arginylation, which was thought to be crucial for the organization of actin and the formation of lamellipodia in motile cells (Karakozova et al., 2006; Kashina, 2006). Arginylation is a widespread PTM that was discovered more than 50 yr ago and was linked to the ubiquitin-dependent N-end rule pathway of protein degradation (Kaji et al., 1963). More recent research revealed that arginylation is relevant for a variety of important biological processes such as embryogenesis, cardiovascular development, neuronal crest cell migration, and cancer (Kwon et al., 2002; Rai et al., 2008; Kurosaka et al., 2010).
Arginylation is triggered by an evolutionary conserved enzyme that transfers an arginine from tRNA onto the N-terminus of proteins, the arginyltransferase 1 (Ate1, EC 2.3.2.8.). Knockout of Ate1 in mice caused severe defects in cardiovascular development and angiogenesis and led to embryonic lethality (Kwon et al., 2002). N-terminal arginylation of β-actin facilitates cell migration (Karakozova et al., 2006) and actin polymerization in vivo (Saha et al., 2010), independently of proteasome-mediated degradation. More recently, it was reported that not only N-terminal residues but also actin side chains can be arginylated by Ate1 in vivo (Wang et al., 2014), and it was shown that the nucleotide coding sequence of actin regulates the rate of protein arginylation in vivo (Zhang et al., 2010).
Not only actin but also a number of other proteins have been shown to undergo arginylation, including important structural components and regulators of the cytoskeleton (Wong et al., 2007). These results underscore the complexity and relevance of arginylation and highlight its putative effects on important cellular processes such as cell adhesion and migration. For example, arginylation of a proteolytic fragment of the cell adhesion protein talin mediates an essential role in cell–cell adhesion (Zhang et al., 2012). Arginylation of myosin in platelets affects its regulation by phosphorylation and its normal function in contractility (Lian et al., 2014). Ate1 proteins have been described in many eukaryotic species (Manahan and App, 1973; Balzi et al., 1990; Kwon et al., 1999; Rai and Kashina, 2005), and despite the lack of strong overall sequence homology, they seem to share conserved enzyme activities.
In the present study, we have investigated the role of Ate1 in the social amoeba Dictyostelium discoideum. Dictyostelium is a molecular model organism that is increasingly being used to explore not only general cell biological functions of specific proteins but also to investigate their basic contributions to human diseases (Müller-Taubenberger et al., 2013). Similarly to human neutrophils, Dictyostelium cells are highly motile, and their repertoire of cytoskeletal proteins comprises the main representatives of higher eukaryotes. Here we present evidence that in D. discoideum Ate1 arginylates actin and most probably other regulators of actin cytoskeleton dynamics and show that Ate1 functions play an important role for the regulation of cell adhesion and migration.
RESULTS
Identification of Ate1 in Dictyostelium discoideum
The D. discoideum genome encodes only one Ate1 family member (DDB0238346). Dictyostelium discoideum Ate1 (DdAte1) was identified in a standard protein blast search (Shiryev et al., 2007) with the mouse Ate1 protein sequence (Q9Z2A5) as query, comprises 629 amino acids (74 kDa), and is encoded by a 1890-base-pair gene located on chromosome 1 (positions 2333199–2335088 on the Watson strand). Here we present the functional characterization of Ate1 in the social amoeba D. discoideum. Our results strongly suggest that DdAte1 represents an evolutionarily conserved precursor of metazoan Ate1.
Dictyostelium Ate1 is similar to other Ate1 proteins
Ate1 proteins have an enzymatically active site at the N-terminus (amino acid residues 23–99 in DdAte1) and a conserved C-terminal domain (amino acid residues 318–560 in DdAte1) with almost no effects on the enzymatic activity (Kwon et al., 1999) (Figure 1A, black boxes). Sequence analysis revealed an identity of 53% for the N-terminal and 60% for C-terminal Ate1 domains compared with the human Ate1, respectively (Figure 1A). In functional Ate1 proteins, four cysteines are critical for the enzymatic activity (Li and Pickart, 1995a,b; Kwon et al., 1999). DdAte1 contains these conserved cysteine residues at amino acid positions 26, 29, 79, and 80 (Supplemental Figure S1, black stars). The C-terminal region of Ate1 proteins is highly variable, both in sequence and in length (Kwon et al., 1999), but the C-terminus of DdAte1 includes a cysteine at position 516 that is conserved in other Ate1 proteins, although it is not required for enzymatic activity (Supplemental Figure S1, gray star). In addition, DdAte1 contains an unusual insert of 79 amino acids between positions 356 and 435 that interrupts the C-terminal Ate1 domain. This region has no homology to other known protein domains and is only present in DdAte1 but not in other amoebozoan Ate1 family proteins (Figure 1, A and B). Interestingly, in Drosophila sechellia and Arabidopsis thaliana, Ate1 proteins have amino acid insertions at various positions of the C-terminus (Supplemental Figure S1, cyan, red, and green boxes).
FIGURE 1:

Comparison of DdAte1 with Ate1 proteins from different organisms. (A) Schematic organization of D. discoideum Ate1 in comparison to Ate1 proteins from other organisms. The black boxes indicate the conserved N- (Nt-Ate1 domain) and C-terminal (Ct-Ate1 domain) arginyltransferase homology domains. The sequences of D. discoideum and human Ate1 share an overall identity of 54%. Numbers indicate the length of the proteins in amino acid residues. (B) Phylogenetic tree of Ate1 proteins that were identified by blast searches at NCBI. The tree was computed with the constraint-based multiple sequence alignment tool COBALT (neighbor joining) at NCBI (Papadopoulos and Agarwala, 2007). The sequences used to compile the tree originate from diverse taxa, including monocots (light green; Triticum uratum [EMS49035], Aegilops tauschii [EMT26921], Oryza sativa [NP001055690]), eudicots (dark green; Arabidospsis thaliana [BAD44222], Arabidospsis lyrata [XP002873220]), worms (light blue; Caenorhabditis elegans [P90914]), amoebozoa (red; Dictyostelium fasciculatum [XP004357377], Polyspondylium pallidum [EFA83779], D. discoideum [XP647040], D. purpureum [XP_003285818]), mammals (blue; Mus musculus Isoform 1 [NP_038827.2], M. musculus Isoform 2 [NP_001258272.1], M. musculus Isoform 3 [NP_001025066.1], M. musculus Isoform 4 [NP_001129526.1], Bos mutus [ELR60396.1], Homo sapiens Isoform 1 [NP_001001976], H. sapiens Isoform 2 [NP_008972], H. sapiens Isoform 3 [NP_001275663], H. sapiens Isoform 4 [NP_001275664], H. sapiens Isoform 5 [NP_001275665]), flies (orange; Drosophila simulans [XP_002082298], Drosophila sechellia [XP002034657], Drosophila melanogaster [AAL83965], Drosophila ananasae [XP_001960010]), and yeast (pink; Saccharomyces cerevisiae [P16639]). (C) Structural predictions for Ate1 proteins from mouse (M. musculus (Isoform 1) [NP_038827.2]), D. sechellia [XP002034657], and D. discoideum [XP647040]). The predicted extensions within the arginyltransferase domain are indicated in light blue. The unique C-terminal part of D. discoideum Ate1 (red, amino acid residues 548–629) most probably does not interfere with the exposed active site of the enzyme. (D) Active sites of Ate1 modeled proteins from mouse, D. sechellia, and D. discoideum are highlighted. The exposed active sites in the first globular domain are very well conserved. The four cysteine residues relevant for the enzymatic activity are exposed at the outer face of the protein.
The domain architecture of DdAte1 corresponds to that of Ate1 proteins from other amoebozoa, plants, and flies (Figure 1A, black boxes). Given the difference of DdAte1 to homologues from other species at the amino acid level, the tertiary structure could provide further evidence for the conservation of the protein. Currently, no crystal structure for any Ate1 protein is available. Therefore, selected Ate1 protein sequences were used in SWISS-MODEL (Guex and Peitsch, 1997; Schwede et al., 2003; Arnold et al., 2006) for three-dimensional-structure predictions. Surprisingly, the predicted tertiary structures of Mus musculus, Dictyostelium sechellia, and D. discoideum Ate1 are highly similar to each other (Figure 1C). In particular, the exposed active site is quite well conserved in the first globular domain of the modeled proteins (Figure 1D). The Ate1 protein of D. sechellia includes a short 48-amino-acid-residue-long stretch at amino acid positions 239–287 (Supplemental Figure S1, cyan box). The tertiary structure predictions are not affected despite the difference in size of both DdAte1 and D. sechellia Ate1. The very last C-terminal part (amino acid residues 548–629) of DdAte1 (Figure 1C, red color) could not be modeled into the C-terminal globular domain as it is predicted to contain a random-coil sequence with a long α-helix, and most probably this part does not have any effect at the exposed active site of the enzyme (Figure 1D).
Our findings suggest the high conservation of DdAte1 on the structural level compared with Ate1 proteins from higher organisms. COBALT alignment and phylogenetic analysis of the DdAte1 protein sequence with the nearest Ate1 proteins of other species highlights the close relationship and the ancestry of Ate1 proteins. The phylogenetic comparison (Figure 1B) indicates that amoebozoan Ate1 proteins are more ancient than their homologues in flies and mammals, and Polysphondylium pallidum and Dictyostelium fasciculatum have more diverged variants of Ate1 proteins compared with D. discoideum and Dictyostelium purpureum.
Subcellular localization of Ate1 in Dictyostelium
Saccharomyces cerevisiae Ate1p was shown to be located predominantly in the nuclei of yeast cells (Kwon et al., 1999). Two of the mouse Ate1 isoforms have been reported either to be completely cytosolic (Ate1-2p) or to localize in both cytosol and nucleus (Ate1-1p) (Kwon et al., 1999). To investigate the subcellular localization of DdAte1, a C-terminal GFP fusion construct was generated and transfected into Dictyostelium wild-type cells. DdAte1-GFP localizes to the cytosol and is enriched in the nucleus and pseudopodial protrusions (Figure 2, A and B). DdAte1-GFP localization in the cytosol and the nucleus is more persistent than in transient pseudopodial protrusions. Fluorescence intensities of DdAte1-GFP expressing cells were measured in nuclei, cytosol, and lamellipodia. DdAte1-GFP was slightly more prominent in cortical protrusions than in the nucleus (Figure 2C). A more detailed analysis of the intensity profiles of cells expressing both DdAte1-GFP and the filamentous actin marker RFP-LimEΔcoil, revealed that the peak of DdAte1-GFP fluorescence lags behind the RFP-LimEΔcoil signal (Figure 2, D and E). This indicates that DdAte1-GFP is not colocalizing at sites where actin is polymerized to actively expand the front but peaks ∼0.9 µm behind this zone (Figure 2E). The localization of DdAte1-GFP in protruding pseudopodia is similar to coronin and Aip1, two actin-binding proteins involved in the disassembly of filamentous actin (Ishikawa-Ankerhold et al., 2010). In fluorescence recovery after photobleaching (FRAP) experiments, DdAte1-GFP exhibited a considerably mobile behavior in lateral protrusions and reappeared in the bleached region with a half-time of recovery of ∼10 s (Figure 2, F and G, and Supplemental Movie 1), indicating a rather high exchange rate within the cytosol. The localization of DdAte1-GFP at cortical protrusions was not affected by bleaching of the cytosolic fraction. Since the localization of Ate1-GFP was to some extent similar to that of GFP, we conducted control experiments with cells expressing only GFP. In contrast to DdAte1-GFP, the localization of GFP at cortical protrusions was affected by bleaching of the cytosol.
FIGURE 2:

Subcellular localization of DdAte1. (A) DdAte1-GFP expressing cells were recorded by live-cell spinning disk confocal microscopy. DdAte1-GFP localizes to the cytoplasm and is enriched in nuclei and in the cell cortex during the formation of pseudopodia. (B) Live-cell spinning disk confocal microscopy of a DdAte1-GFP expressing cell (left). The intensity profile along the line is plotted (right). The fluorescent signal is stronger in the nucleus and cortical pseudopodia. (C) Fluorescence intensity of DdAte1-GFP in the cytosol, nucleus, and protrusions. Fluorescence intensities of the region of interest were measured with ImageJ within a 4 × 4 square pixel area (0.251 μm2) for n = 54 cells. Fluorescence intensity in protrusions and nuclei showed an enrichment compared with the cytosol. For the statistical analysis, GraphPad Prism Software employing one-way analysis of variance (ANOVA) was used. There is no significance difference between nucleus and protrusion (ns), whereas differences of cytosol versus nucleus and cytosol versus protrusions are significant (p < 0.0097) (**). Bars are ±SD. (D) Live-cell spinning disk confocal microscopy of a Dictyostelium cell expressing both DdAte1-GFP and RFP-LimEΔcoil. (E) The intensity profiles of both fluorescence channels along the lines in D are plotted. (F) Selected time points of a representative FRAP experiment at the protrusion of DdAte1-GFP expressing cells (Supplemental Movie 1). The fluorescence in a protrusion (within the white square at time point 0 s) was bleached with a point-focused 473-nm laser pulse. Spinning disk confocal microscopy images with a time-lapse acquisition rate of six stacks per minute and a maximum intensity projection of five slices per image stack are shown. (G) DdAte1-GFP in protrusions shows a high exchange rate. The graph shows recovery kinetics from three independent measurements performed using a spinning disk microscope (mean ± SD). Scale bars, 5 μm in A, D, and F; 10 µm in B.
Movie S1.
Representative FRAP experiment at a protrusion of a Dictyostelium cell expressing Ate1-GFP. The photo-bleaching event is stamped into the movie at 20 s. Scale bar, 5 μm.
To study the localization of DdAte1 during cytokinesis, DdAte1-GFP expressing cells undergoing mitosis were recorded by live-cell microscopy (Supplemental Figure S2A and Supplemental Movie 2). DdAte1-GFP did not colocalize with cortical actin, the contractile ring, or any other actin-rich protrusion formed during cell division but was cytosolic and slightly enriched at lateral protrusions on completion of cell division (Supplemental Figure S2A, arrows). Live-cell recordings of cells showing a more prominent nuclear localization revealed that DdAte1-GFP is exported from the nucleus at the onset of cytokinesis and remains exclusively cytosolic during cell division indicating a cell-cycle-specific localization of Ate1 (Supplemental Figure S2A). The enrichment of DdAte1-GFP in nuclei is consistent with a potential nuclear localization signal (NLS) in the 79-amino-acid-residue insert region (Supplemental Figure S1, dotted box). The stretch of amino acid residues 417–425 is similar to the basic consensus sequence of known nuclear localization signals predicted by ELM (Eukaryotic Linear Motif) (http://elm.eu.org) and NLS mapper (http://nls-mapper.iab.keio.ac.jp).
Movie S2.
Spinning disk confocal imaging of a Dictyostelium cell expressing Ate1-GFP during cell division.
The appearance of the DdAte1-GFP in different cellular compartments and its dynamic behavior indicates that Ate1 very likely has specific roles in these locations. These compartments include in particular the nucleus and sites of the actin turnover like pseudopods. Our primary goal in the present study was to investigate the effects of Ate1 with respect to cytoskeletal functions.
Ate1-null cells are impaired in substrate adhesion
Ate1 is nonessential in yeast, plants, and worms (Balzi et al., 1990; Yoshida et al., 2002), but knockout of Ate1 in mice and flies led to embryonic lethality (Kwon et al., 2002; Saha and Kashina, 2011). To investigate the relevance of Ate1 in Dictyostelium, we generated a knockout construct to disrupt the ate1 gene by homologous recombination (Supplemental Figure S3A). Ate1-knockout cells were tested by PCR for the integration of the blasticidin resistance gene, and four independent clones were identified (Supplemental Figure S3B). Ate1 is not essential in Dictyostelium as growth and development were not substantially affected after knockout of the ate1 gene. Nevertheless, ate1-null cells showed some remarkable features. They were significantly reduced in size and had a rounder appearance compared with wild-type cells (Figure 3A). To quantify the decrease in cell size, a volume measurement with hematocrit tubes was performed. Ate1-null cells had an ∼20% smaller volume than wild-type cells (Figure 3B), but the sizes of nuclei and the DNA content were not conspicuously altered compared with wild type.
FIGURE 3:

The analysis of ate1-null cells reveals that substrate contact areas of mutant cells are much smaller and lack actin-containing adhesion points. (A) Phase-contrast microscopy images of live cells showing that ate1-null cells are smaller in comparison to wild-type cells (compare also to Supplemental Figure S3C). Scale bar, 10 μm. (B) Determination of the average cell volume of wild-type and ate1-null cells as described under Materials and Methods revealed that the cell volume of ate1-null cells is ∼ 20% smaller than the volume of the wild type. The experiment was repeated three times. Each value corresponds to the average of 5 × 106 cells. (C) RICM revealed that ate1-null cells are much less adhesive to glass surfaces. Wild-type and mutant cells were plated on glass coverslips and tracked for 100 frames in 10-s intervals. Scale bars, 5 µm. (D) The contact area for every cell was measured using ImageJ. From these data, the maximal contact area was calculated for wild-type (n = 27), ate1-null (n = 57), and rescue (n = 39) cells. The area of adhesion to the substrate of ate1-null cells is 40% smaller than the area of wild-type cells. (E) Confocal microscopy of GFP-LimEΔcoil expressing wild-type and ate1-null cells. Scale bars, 5 µm. (F) TIRF microscopy of wild-type and ate1-null cells expressing GFP-LimEΔcoil. Scale bars, 5 µm. (G) Quantification of adhesion points in wild-type (n = 12) and ate1-null (n = 12) cells. The quantification revealed a significant decrease of adhesion points in ate1-null compared with wild-type cells. For the statistical analysis, one-way ANOVA-Tukey’s multiple comparison test was used for D, unpaired t test for B and G. ** and *** p < 0.0001, p < 0.05 was considered significant. The red lines indicate mean values.
Although ate1-null cells stained with TRITC-phalloidin showed no significant differences in the localization or the overall amount of F-actin (Supplemental Figure S3C; also see Figure 7A later in this article), the smaller and more rounded appearance of the cells was an indication for a possible impact of DdAte1 on cortical actin cytoskeleton functions. Since cells lacking Ate1 were characterized by a more rounded shape, reflection interference contrast microscopy (RICM) was used to investigate the adhesiveness of ate1-null cells to the substrate (Figure 3C and Supplemental Movies 3 and 4). Ate1-null cells showed a substantial decrease (40%) of the mean contact area (Figure 3D). This reduced adhesion was rescued by the expression of Ate1-GFP in ate1-null cells to almost wild-type levels (Figure 3, C and D, and Supplemental Movie 5).
FIGURE 7:

Actin isolated from ate1-null cells reveals different isoelectric properties and lower polymerization capacity in comparison to actin from wild type. (A) Triton-insoluble cytoskeletons were extracted from wild-type and ate1-null cells and subjected to Western blot analysis using anti-actin antibodies. The amount of F-actin in the Triton-insoluble cytoskeleton of ate1-null cells is comparable to wild type. (B) Protein transfer shown in A was controlled by Ponceau S staining. (C) Lysates prepared from wild-type and ate1-null cells were separated by two-dimensional PAGE, and proteins were stained with Coomassie Brilliant blue. The overall appearance of actin isoforms is nearly the same in wild-type and ate1-null cells, but ate1-null cells lack the long basic tail of the major actin isoforms. (D) Actin polymerization assays using actin isolated from wild-type or ate1-null cells and 10% pyrenylated actin.
Movie S3.
RICM of a Dictyostelium wild-type cell. Dark areas indicate close contact to the substrate surface. Note the constant spreading and de-attachment to the substratum. Left: phase contrast, right: RICM. Bar, 5 μm.
Movie S4.
RICM of a Dictyostelium ate1-null cell. Dark areas indicate a close contact to the substrate surface. Ate1-null cells never spread on the substratum as closely as do wild-type cells. Left: phase contrast, right: RICM. Bar, 5 μm.
Movie S5.
RICM of a Dictyostelium Ate1-rescued cell (ate1-null cell expressing Ate1-GFP). Dark areas indicate close contact to the substrate surface. Note the constant spreading and de-attachment to the substratum as observed in wild type (Movie 3). Left: phase contrast, right: RICM. Bar, 10 μm.
In Dictyostelium, actin has a critical role in cell adhesion by forming transient adhesive foci at the contact areas to the substrate (Bretschneider et al., 2004). These short-lived patches can be visualized in live cells by the expression of the F-actin marker GFP-LimEΔcoil (Figure 3, E and F, and Supplemental Movie 6). Whereas the cortical actin cytoskeleton appeared unaffected in ate1-null cells, F-actin–containing adhesion sites at the substrate were almost completely absent in ate1-null cells (Figure 3, E and F, and Supplemental Movies 7, 8, and 9). We quantified adhesion points in wild-type and ate1-null cells (Figure 3G). ate1-null cells lacked actin adhesive foci almost completely. This observation suggests that either a subfraction of actin or a cytoskeletal component that is required for cell–substrate adhesion is affected by the absence of functional Ate1 in Dictyostelium. Expression of RFP-LimEΔcoil in ate1-rescued cells showed that the formation of actin foci at the substrate adhesion sites was almost completely restored (Figure 4B). When ate1-null cells expressing GFP-LimEΔcoil were flattened by compression under agar, they formed actin-containing structures at the substrate facing area, but these structures showed a different dynamicity compared with transient actin foci. The phenotypes of ate1-null and rescued cells indicate a crucial requirement of Ate1-mediated arginylation for the formation of actin adhesions mediating the transient contacts to the substratum.
FIGURE 4:
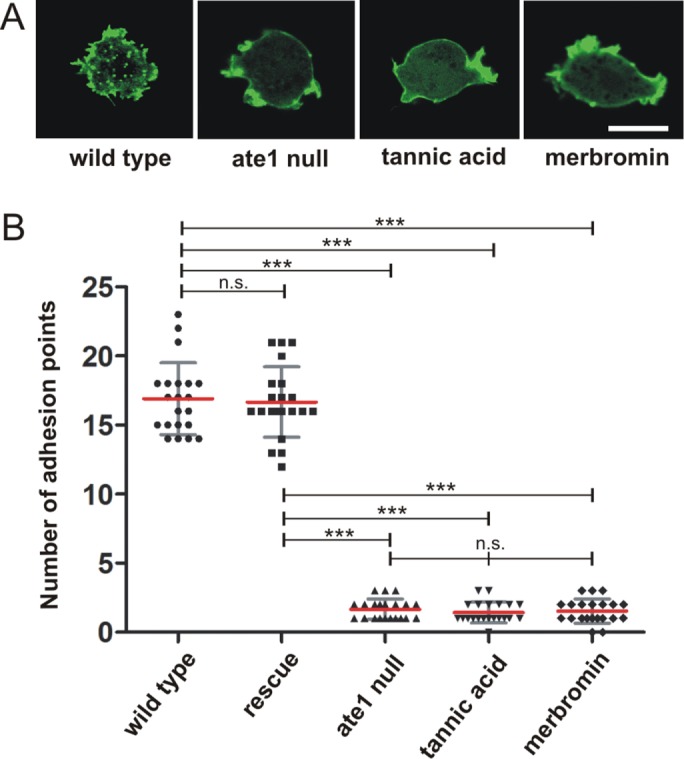
Small molecule inhibitors mimic the ate1-null phenotype. (A) Wild-type cells expressing GFP-LimEΔcoil were treated with 30 µM tannic acid or 50 µM merbromin for 24 h. Untreated wild-type, ate1-null, and inhibitor-treated wild-type cells (tannic acidic or merbromin) expressing GFP-LimEΔcoil were recorded by confocal microscopy, and the actin focal adhesion dots were counted and quantified as displayed in B. Scale bar, 10 µm. (B) Adhesion point quantification was conducted manually using the LSM Image Examiner. Ate1-null and inhibitor-treated cells showed a significant decrease of adhesion points compared with wild-type cells (n = 21 for all groups). One-way ANOVA-Tukey’s multiple comparison test revealed significant differences of ate1-null and inhibitor-treated cells versus wild-type and Ate1-GFP rescue cells as shown in the graph. ***p < 0.0001. Differences between wild-type and rescue cells were not significant (ns); p < 0.05 was considered significant. Error bars are ±SD.
Movie S6.
Actin network dynamics visualized by expression of LimEΔcoil-GFP in Dictyostelium discoideum wild-type cells. Recording was by confocal microscopy. The focal plane was moved in z-axis towards the substrate adhesion site. Scale bar, 10 μm.
Movie S7.
Actin network dynamics visualized by expression of LimEΔcoil-GFP in Dictyostelium discoideum ate1-null cells (clone #1-21). Recording was by confocal microscopy. The focal plane was moved in z-direction. Scale bar, 10 μm.
Movie S8.
Actin network dynamics visualized by expression of LimEΔcoil-GFP in Dictyostelium discoideum ate1-null cells (clone #2-17). Recording was by confocal microscopy. The focal plane was moved in z-direction. Scale bar, 10 μm.
Movie S9.
Actin network dynamics visualized by expression of LimEΔcoil-GFP in Dictyostelium discoideum wild-type (left) and ate1-null (right) cells. Recording was by TIRF microscopy. The focal plane was moved in z-direction. Scale bar, 5 μm.
Small molecule inhibitors mimic the Ate1-mutant phenotype
To test whether the phenotype of ate1-null cells depends on arginylation mediated by the DdAte1, we used two small molecule inhibitors, tannic acid and merbromin. Both inhibitors have been reported previously to inhibit Ate1 activity in higher cells and thereby regulating arginylation-dependent protein degradation, cell motility, and angiogenesis (Saha et al., 2012). Wild-type cells expressing GFP-LimEΔcoil were treated with 30 μM tannic acid or 50 μM merbromin for 12, 24, or 48 h. Cell growth was not affected by the addition of the inhibitors (Supplemental Figure S4). However, whereas the untreated control cells showed actin adhesion foci at the substrate, these were hardly detectable in inhibitor-treated cells, resulting in a phenotype similar to ate1-null cells (Figure 4A). We quantified the effect of the small molecule inhibitors by determining the number of actin spots in treated and nontreated cells (Figure 4B). The treated cells had a significantly reduced number of actin foci, similar to ate1-null cells, indicating that the observed phenotype depends on arginylation carried out by Ate1 in Dictyostelium. In cells treated for longer periods (>72 h) with tannic acid or merbromin, pseudopod formation was virtually abolished, and the cortical actin network was reduced similarly to the effect described for higher eukaryotic cells (Saha et al., 2012).
Deficiency of Ate1 causes increased directed migration in two dimensions
To monitor whether the observed small contact area of ate1-null cells to the substrate (Figure 3, C and D) has an impact to the ability of the cells to migrate, we analyzed random motility, and chemotactic movement in a gradient of folic acid under agar, or a gradient of cAMP in two dimensions. In comparison to wild-type cells, random motility of vegetative ate1-nulls cells was not significantly different (Figure 5A). However, in under-agar folic acid chemotaxis assays, growth-phase ate1-null cells showed a significant increase in cell speed as well as a reduction of straightness and directionality (Figure 5B). Velocity of aggregation-competent ate1-null cells migrating in a gradient of cAMP was also slightly increased compared with wild type (Figure 6A and Supplemental Movie 10). These results suggest that the reduced contact area to the substrate in ate1-null cells compared with wild type results in an increase in the velocity of directional migration in two dimensions both in folate and in cAMP chemotaxis.
FIGURE 5:

Increased migration speed of ate1-null cells in under-agar folate chemotaxis. (A) Random motility was determined for growth-phase wild-type and ate1-nulls attached to a glass surface without a chemotactic stimulus. (B) Under-agarose assays of growth-phase wild-type and ate1-null cells were performed in low 35-mm standard-bottom µ-dishes (Ibidi) as described in Materials and Methods. Ate1-null cells move faster than wild-type cells showing a significant increase in cell speed. Straightness and directionality are decreased in cells lacking Ate1. ***p < 0.0001 and *p = 0.0227, respectively; p < 0.05 was considered significant. The red lines indicate the mean of speed, straightness, or directionality.
FIGURE 6:

Chemotaxis of aggregation-competent ate1-null cells is impaired in three-dimensional compared with two-dimensional environments. Ate1-null cells expressing GFP-LimΔcoil were starved together with wild-type cells expressing Cherry-LimEΔcoil. (A) The migratory behavior of wild-type cells expressing Cherry-LimΔcoil (n = 54), and ate1-null expressing GFP-LimΔcoil cells (n = 74) was analyzed in a two-dimensional chemotaxis micropipette assay toward cAMP. The average speed of ate1-null cells was significantly higher than the average speed of wild-type cells. *p = 0.0045. The data from four individual experiments were averaged. (B) Migration speed analyzed during three-dimensional chemotaxis conditions using µ-Slide Chemotaxis3D chambers (Ibidi). In three-dimensional environments, ate1-null cells (n = 342) migrated significantly slower than wild-type cells (n = 342) toward cAMP. **p < 0.0001. The speed was analyzed using the Bitmap Imaris Software. The red lines indicate the mean speed; p < 0.05 was considered significant.
Movie S10.
Chemotaxis of ate1-null cells expressing GFP-LimEΔcoil (green), and wild-type cells expressing RFP-LimEΔcoil (red). Fluorescently labeled LimEΔcoil visualizes filamentous actin, and was employed here to distinguish wild-type and ate1-null cells. Aggregation-competent cells were exposed to chemoattractant gradients by moving a micropipette filled with cAMP. Cells were recorded by confocal microscopy. Note that mutant cells (green) migrate faster than wild type (red). Scale bar, 20 μm.
Directed migration of Ate1-mutant cells in three dimensions is impaired
The migratory behavior of Dictyostelium cells in three-dimensional environments can be significantly different from migration under two-dimensional conditions on a solid substrate (Zhao et al., 2013). To test whether motility in three dimensions is affected in ate1-null cells, we established a setup to measure the speed of chemotaxing cells toward cAMP in a three-dimensional collagen environment (Roth et al., 2015). Interestingly, the mean speed of ate1-null cells was considerably reduced in three-dimensional cell migration. Ate1-null cells migrated with an average speed of 9.3 µm/min, whereas wild-type cells showed an average speed of 13.9 µm/min (Figure 6B). These results indicate that arginylation is crucial for the migratory behavior of Dictyostelium cells in three-dimensional environments.
Arginylaton of Dictyostelium actin
To examine differences in the total amount of actin of ate1-null versus wild-type cells, Western blots of total cell lysates and Triton X-100-insoluble cytoskeleton extracts were performed (Figure 7A). The total amount of actin and the amount of F-actin are very similar in wild-type and ate1-null cells (Figure 7B). To examine the isoelectric properties of actin from ate1-null cells in comparison to wild type, two-dimensional PAGE of total cellular lysates was performed (Figure 7C). The pattern formed by the different actin isoforms in two-dimensional PAGE revealed that lysates of ate1-null cells lack the characteristic comet tail of actin spots that are visible in wild type, suggesting that these isoforms are less positively charged. The total amount of actin appeared nearly identical in wild-type and ate1-null cells (Figure 7C). To analyze the polymerization properties of nonarginylated actin in comparison to wild-type actin, we isolated actin from wild-type and ate1-null cells in parallel and performed polymerization assays (Figure 7D). Actin purified from ate1-null cells showed a lower polymerization capacity compared with actin isolated from wild-type cells.
To further analyze whether actin or a fraction of actin in Dictyostelium is arginylated, we prepared actomyosin precipitates highly enriched in actin from wild-type cells and analyzed the samples by Orbitrap mass spectrometry. The results are summarized in Table 1. Dictyostelium actins can be arginylated on glutamic acid (E). According to the identified peptides, two actins (Act8 and Act22) were identified to be arginylated in their N-terminal regions and with high confidence side chains of six actins (Act3, Act8, Act10, Act17, Act22, and Act23) can be arginylated. These actin isoforms include the most abundant actin isoform Act8 that constitutes more than 95% of the total actin expressed in Dictyostelium (Joseph et al., 2008). Other variant actins were not identified as potential targets of Ate1.
TABLE 1:
Arginylation sites of D. discoideum actin isoforms.
 |
By mass spectrometric analysis of actin isolated from D. discoideum, the listed amino acid residues were identified as potential arginylation sites on side chains of glutamic acid (E) indicated in red in the identified peptide sequence. Act8 is the most abundant actin isoform and representative of a group of 17 identical actins encoded by different actin genes (Joseph et al., 2008).
Actin was the main constituent of the actomyosin precipitate that was used for the mass spectrometric analysis. In addition to actin, myosin-II and actin-binding proteins were present in the precipitate (Konzok et al., 1999). Orbitrap mass spectrometry identified potential arginylation sites of several actin-binding proteins. These include the ß-subunit of F-actin capping protein, coronin, the actin cross-linking proteins α-actinin and filamin (Abp120), and myosin-II (Table 2). Interestingly, a putative calcium-dependent cell adhesion protein (Cad3) was also identified in the actomyosin fraction as an arginylated protein.
TABLE 2:
Potential arginylation sites identified in cytoskeletal proteins of D. discoideum contracted actomyosin precipitates.
 |
By mass spectrometric analysis of contracted actomyosin precipitates prepared from D. discoideum as described under Materials and Methods, potential arginylation sites in cytoskeletal and cell adhesion proteins were identified. The data were analyzed using DTASelect v2.1.3 software (Wong et al., 2007; Xu et al., 2009).
DISCUSSION
Our phenotypic analysis of mutant cells provides strong evidence that DdAte1, the arginyltransferase expressed in D. discoideum, is involved in the regulation of cell adhesion and cytoskeletal activities, and thus affects cell migration. Although it is very likely that DdAte1 has other targets in addition to actin and actin-binding proteins, a regulatory effect on actin cytoskeleton dynamics is indicated both by the results of the mass spectrometric analysis of contracted actomyosin samples, and the localization studies with GFP-tagged DdAte1.
The actinome of Dictyostelium was thoroughly characterized in an earlier study, and a systematic comparison of the 33 actins has shown that the main differences in the individual amino acid sequences are located in the N-terminal parts (Joseph et al., 2008). Our study shows that as in other organisms, Dictyostelium actins can be arginylated at their N-termini as well as at internal amino acid side chains. In Dictyostelium, the most abundant actin isoform is encoded by 17 distinct genes (Act8 group), whereas the protein sequences of the other 16 actin variants can be almost identical with the conventional actin or differ rather drastically (Joseph et al., 2008). Our results indicate that in Dictyostelium the most predominant actin isoform Act8 (representing the Act8 group), as well as the closely related variants Act3, Act10, Act17, Act22, and Act23 are arginylated. The detailed nature of the modification in the variant actin isoforms will be difficult to analyze unless their specific functions are known.
The ratio of total actin versus F-actin was not altered in ate1-null cells, but the analysis of actin polymerization properties showed a lower polymerization capacity for actin isolated from ate1-null cells compared with wild-type actin. This result is in agreement with data obtained for nonarginylated actin isolated from Ate1-knockout embryonic fibroblasts (Saha et al., 2010) and supports the assumption that arginylation is important for the regulation of actin polymerization.
Our mass spectrometric analysis of contracted actomyosin precipitates not only identified distinct arginylated actin isoforms but also indicated that the actin-binding proteins coronin, α-actinin, filamin (Abp120), and capping protein as well as myosin-II are arginylated. These proteins are crucial regulators of cytoskeletal functions, and recent work already has described arginylation of myosin-II and α-actinin in muscle cells (Kurosaka et al., 2012; Cornachione et al., 2014). The findings underscore the significance of arginylation, and an important task will be to dissect the regulatory functions of Ate1 specifically in muscle and nonmuscle cells.
The role of DdAte1 in regulating cytoskeletal activities became evident by the dominant effect of knocking out the gene encoding DdAte1. Knockout of ate1 causes a reduction of cell size, as well as a lack of cell adhesion points, an increased speed of directed migration in two dimensions, and reduced cell migration in three dimensions. The phenotype of ate1-null cells could be rescued by expression of a functional DdAte1-GFP in the knockout strains. The question of how Ate1 alters cell adhesion and cell migration still remains enigmatic. It has been postulated that arginylation is a general mechanism that regulates actin isoform segregation in vivo and participates in the formation of loose actin networks at the leading edge of the cell (Kashina, 2006; Saha et al., 2010). Loss of arginylation has already been reported to cause impaired cell migration in different cell types, for instance, in neuronal cells (Kurosaka et al., 2010; Pavlyk et al., 2018). The results presented here extend these studies and show that Ate1 is of importance for the migratory ability of cells. We provide evidence that Dictyostelium ate1-knockout cells, when exposed to chemotactic gradients, move more rapidly on solid substrates probably due to reduced substrate adhesion, suggesting that the cells become more slippery. However, when cells lacking Ate1 move in three-dimensional environments, cell migration is considerably impaired. The findings indicate that adhesive properties are important for chemotactic movement under more confined conditions. Obviously, the focal adhesions are important for migration in more challenging environments in three-dimensions when the cells move primarily in a bleb-driven mode (Zatulovskiy et al., 2014). It will be of high interest to investigate whether other cells that move in a similar manner like Dictyostelium, for instance, human primary neutrophils, are equally affected by a down-regulation or an impairment of Ate1 functions.
Our results indicate a more specialized function of arginylated actin or other proteins, in particular for the establishment of adhesion points to the substratum. A similar mutant phenotype, reduced attachment to the substratum combined with increased cell speed during chemotactic movement in a gradient of cAMP has been described earlier in mutants lacking the cytoskeletal protein talin A (Niewöhner et al., 1997). This study implied that cell motility can be uncoupled from traction forces. For the protein-kinase Phg2 it has been shown that Phg2 functions are important for the modeling of actin-rich focal sites and cell motility (Gebbie et al., 2004). Phg2, like talin A or myosin VII, plays a role in cellular adhesion and organization of the cytoskeleton. Phg2-mutant cells showed reduced motility, and on adhesion to a glass surface, were characterized by unusally massive actin assemblies present in the central part of the cell contact zone but, in contrast to ate1-null cells, were able to form actin foci (Gebbie et al., 2004). Thus, from these results and data in the present study, it has to be concluded that the dynamic establishment of actin foci to the substratum is a very complex process that is still not completely understood in its details. One possible link could be provided by Cad3, a putative cell adhesion molecule that was detected as arginylated protein by our mass spectrometric analysis of the actomyosin fraction. Functions of Cad3 have not been investigated so far, but it is a putative Ca2+-dependent cell adhesion protein with high homology to the well-explored Dictyostelium cell adhesion protein Cad1 (CadA, gp24) (Wong et al., 1996).
DdAte1-GFP is present in the cytosol and shows an enrichment in the nucleoplasm. In contrast to F-actin, DdAte1-GFP is not evenly distributed in the cell cortex. Live-cell imaging revealed that DdAte1-GFP is recruited to newly extended cellular protrusions, and a detailed analysis showed that the localization of DdAte1-GFP peaked behind the polymerization zone of actin. This localization is very similar to that of coronin, a protein involved in the disassembly of actin (Bretschneider et al., 2009) Coronin might be one of the cytoskeletal proteins directly regulated by arginylation as our mass spectrometric data indicated an arginylation site in coronin.
Taken together, Dictyostelium provides an interesting system for future studies to investigate the importance of Ate1 for the regulation of cytoskeletal proteins that are involved in adhesion or migration. Another unexplored field is the role of arginylation during development. An earlier study of arginylation in Dictyostelium already provided evidence for distinct substrates that were differentially arginylated during development (Bohley et al., 1991). The fact that Ate1 is found in different cellular compartments indicates diverse functions for the enzyme. In Dictyostelium, Ate1 localizes similarly to Ate1p-1 in mice, but in contrast to mice, the knockout of Ate1 in Dictyostelium cells is not lethal. This suggests that unlike higher eukaryotes, in Dictyostelium, plants, and worms the activity of Ate1 is dispensable for the survival of the organisms. Among the unicellular model organisms, Dictyostelium is one of the few examples, in which an orthologue of Ate1 was found, and thus the organism may closely reflect the last eukaryotic common ancestor. This makes Dictyostelium a very promising system to investigate the minimal requirements of arginylation and its basic roles for the interplay and regulation of actin and actin-binding proteins in the future.
MATERIALS AND METHODS
Sequence analysis
BLAST analyses were performed using the D. discoideum Ate1 protein sequence with the setting all nonredundant GenBank CDS translations+PDB+SwissProt+PIR+PRF excluding environmental samples from WGS projects. Only known proteins were used to generate the phylogenetic tree. For this purpose, protein sequences were aligned with COBALT (National Center for Biotechnology Information [NCBI]), and the phylogenetic tree was generated with the fast minimum evolution method. After export in Newick tree format, the phylogenetic tree was edited with FigTree v1.3.1 software. Structure modeling was performed with I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). Because no suitable template is available for Ate1 proteins so far, no sample input was performed. The resulted models were viewed and edited using SWISS-MODEL and Swiss-PdbViewer 4.1.0 software (Guex and Peitsch, 1997; Schwede et al., 2003; Arnold et al., 2006). The most similar modeled structures are shown.
Cell culture, gene replacement, and transformation of Dictyostelium
Cells of the axenic D. discoideum strain AX2-214, here designated as wild type, or mutant cells derived from it, were cultivated in nutrient HL5 medium (Formedium) at 21°C, either in shaken suspension at 150 rpm or in polystyrene culture dishes. To induce starvation, cells were washed twice in 17 mM Soerensen’s phosphate buffer (PB), pH 6.0, and shaken at a density of 107 cells/ml in the buffer.
For expression of fluorescently tagged Ate1, AX2 wild-type and ate1-null cells were transformed by electroporation with pDEX-derived plasmids enabling the expression of Ate1-GFP or GFP-Ate1. Transformants were selected by addition of either 10 µg/ml G418 (Sigma-Aldrich) or 7.5 µg/ml Blasticidin-S (ICN Biomedicals) and cloned on lawns of bacteria.
For the Ate1-GFP expression construct, the full-length Ate1 (DDB_G0269024) sequence was amplified by PCR using genomic D. discoideum DNA as template and oligonucleotides AAAATGGAAAGATTTTTAATGCAATCATC and TTTAAAATAGAAAATTAATTCTTTTGTTAATTC. The nucleotide sequence of Ate1 was cloned into the EcoRI-site of the expression vector pDEX27 (Müller-Taubenberger, 2006).
For the ate1-knockout construct, a 5′-UTR and a 3′-UTR of the Ate1 (DDB_G0269024) sequence were amplified by PCR using genomic Dictyostelium DNA as template. For the 5′-UTR oligonucleotides GATCAGGGTTGGCGCAGATCAGG and TCATCAGGATGAATGATTGGTATTAAAG and for the 3′-UTR GGTGAAATTAAAACACCAAAACCAGG and TAACACTAACATCAGAAAATGAAAGG were used. Both fragments were cloned sequentially into the vector pLPBLP (Faix et al., 2004). Wild-type AX2 cells were transformed with the gene replacement construct (Supplemental Figure S3A) by electroporation using a Bio-Rad gene pulser at 0.8–0.9 kV and 3 μF in 4-mm cuvettes. Transformants were selected by addition of 10 μg blasticidin-S (ICN Biomedicals) per milliliter and cloned by spreader dilutions on lawns of nonpathogenic Klebsiella aerogenes on SM-agar plates. Independent ate1-knockout clones originated from different plates and were identified by testing genomic DNA for insertion of the resistance cassette into the ate1 gene by PCR.
For expression of Ate1-GFP, GFP-LimEΔcoil, and/or RFP-LimEΔcoil, Dictyostelium AX2 wild-type or ate1-null cells were transformed with the expression vector(s) and selected by either addition of 10–20 μg/ml geneticin (Sigma-Aldrich) and/or hygromycine (Calbiochem). The expression of the constructs was tested by Western blot analysis and fluorescence microscopy.
Cell biological analyses
To determine the packed cell volume of wild-type and ate1-null cells, a modified hematocrit assay was used. Cells were washed twice with phosphate buffer, adjusted to 1 × 107 per ml, and 0.5 ml were filled into hematocrit tubes (Wintrobe) and centrifuged at 1000 rpm in a Rotanta centrifuge (Hettich) at 4°C for 5 min.
Preparation of Triton-insoluble cytoskeleton and actin
Triton X-100–insoluble cytoskeleton was prepared as described (Brink et al., 1990). Cells (7.5 × 106) were resuspended in 150 µl cold TEDA-BP buffer (10 mM Tris, 1 mM ethylene glycol-bis(aminoethyl ether)-N,N, N′,N′-tetraacetic acid [EGTA], 1 mM dithiothreitol [DTT], 0.02% NaN3, 1 mM benzamidin, 1 mM phenylmethylsulfonyl fluoride, pH 7.6) with 1% Triton X-100 and kept on ice for 15 min. The sample was warmed to room temperature (RT) for 15 min and centrifuged for 5 min at 10,000 × g. The pellet was resuspended in SDS–PAGE sample buffer and immediately analyzed.
Dictyostelium discoideum actin was prepared in parallel from 5-l cultures of wild-type and ate1-null cells as described (Eichinger et al., 1991). N-(1-pyrenyl) iodoacetamide-actin (pyrenyl-actin) was used in polymerization assays essentially as described (Eichinger et al., 1991).
Two-dimensional PAGE
Protein samples were adjusted by the addition of 2 ml of a solution containing 7.0 M urea, 2.0 M thio urea, 4% CHAPS (Sigma-Aldrich), 4% Servalyt, 0.4 g/ml ampholytes (Pharmacia Biotec), 0.2 M DTT, and protease inhibitors. Samples were then applied to the top of the wide-range immobilized pH gradient (IPG) strips and incubated overnight at RT covered with mineral oil. The swollen strips were subjected to electrophoresis for 1 h at 300 V and 16 h at 3500 V isoelectric focusing. Following electrophoresis, the IPG strips were extruded and equilibrated for 15 min into equilibration buffer 1 (50 mM Tris–HCl [pH 8.8], 6 M urea, 30% glycerol, 2% SDS, and 0.1 M DTT), and 15 min in equilibration buffer 2 (50 mM Tris–HCl [pH 8.8], 6 M urea, 30% glycerol, 2% SDS, and 0.15 M IAA). Second-dimension 12%-polyacrylamide gels with a thickness of 1 mm were freshly prepared in an Ettan DALTsix gel caster. The first dimension IPG strips were secured on top of the slab gels. The proteins were electrophoretically separated overnight at 10 mA (gel running buffer 0.1% SDS, 25 mM Tris–HCl, 185 mM glycine). Following electrophoresis, gels were stained with Coomassie Brilliant blue R-250.
Immunofluorescence microscopy
For immunolabeling, AX2 wild-type or ate1-null cells, settled onto glass coverslips, were either fixed with picric acid/formaldehyde for 20 min and postfixed with 70% ethanol for 10 min or fixed with 0.5% glutaraldehyde, 0.5% Triton X-100 in 1× PHEM-buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 4 mM MgSO4 × 7 H2O) for 5 min. For visualization of filamentous actin, cells were stained with Alexa Fluor 488-phalloidin (Molecular Probes). DNA was visualized by staining with 4′,6-diamidino-2-phenylindole (DAPI).
Light microscopy and image processing of fixed samples was conducted as described recently using a Cell Observer HS system (Zeiss) equipped with a PlanApo 100×/NA 1.4 objective, an Axiocam MRm Rev. 3 charge-coupled device (CCD) camera, a piezo stage, and the Axiovision 4.7 fast iterative deconvolution software package (Schulz et al., 2009).
Live-cell microscopy
Cells were seeded in glass-bottom petri dishes (Fluorodish; WPI) and flattened by agar overlay (Fukui et al., 1987). Confocal images were taken using an inverted LSM 510 Meta confocal microscope (Zeiss) equipped with a Neofluar 63×/NA 1.4 or a Neofluar 100×/NA 1.3 oil immersion objective, or a Cell Observer (Zeiss) with Yokogawa CSU-X1 spinning disk technology, and equipped with a PlanApo 100×/NA 1.4 oil objective and two Evolve 512 electron multiplying charge-coupled device (EMCCD) cameras (Photometrics). For excitation, the 488-nm argon ion laser line and the 543-nm helium-neon laser line were used, and emission was collected using 505- to 530-nm or 585- to 615-nm bandpass filters.
RICM imaging (Verschueren, 1985) was performed with an inverted LSM 510 laser scanning confocal microscope (Zeiss). Images were obtained using the 633-nm line of a He-Ne laser, a reflection mirror in place of the dichroic beam-splitter, a fully open pinhole, and an oil-immersion Neofluar 100×/NA 1.4 lens. The second dichroic beam-splitter was removed from the light path. The cells were either in PB or in nutrient rich media. The parameters of the laser scanning system were adjusted so that focal contacts appeared black. For time-lapse movies, ∼100 images were collected at 10-s intervals. The contact area was selected manually and measured using the ImageJ or Imaris (Bitplane) software.
Total internal reflection fluorescence (TIRF) microscopy (Axelrod, 2003) was performed as described (Riedl et al., 2008).
FRAP experiments were performed under agar overlay (Fukui et al., 1987). Live-cell imaging was performed on a spinning disk confocal microscope (Cell Observer SD; Zeiss) equipped with a PlanApo 100×/NA1.4 objective, two Evolve EMCCD cameras (Photometrics), and a Rapp UGA-40-2L Galvo Scanner (Rapp Optoelectronics) for laser manipulations. For FRAP experiments a square region of interest covering less than 10% of the area of the cell was bleached with the 473-nm laser line of the Rapp UGA-40-2L system at maximum laser intensity. Fluorescence intensities during recovery of the moving region of interest were measured using ImageJ within a 4 × 4 square pixel area (0.251 μm2). FRAP experiments were evaluated as described (Samereier et al., 2010).
Inhibition of Ate1
Growth-phase Dictyostelium cells expressing GFP-LimEΔcoil were treated with 50 µM merbromin, or 30 µM tannic acid in HL-5 medium (Formedium) for 12–24 h. Inhibitor treated and control cells were recorded using a confocal LSM 510 Meta microscope (Zeiss), and the actin adhesion foci were counted and quantified. The counting was performed manually using the LSM Image Examiner software.
Two-dimensional migration assays
Under-agarose assays were performed in low 35-mm standard-bottom µ-dishes (Ibidi). 0.7% (wt/vol) Biozym LE agarose (Biozym Scientific GmbH) in SM-medium (10 g/l Bacto Peptone, 1 g/l yeast extract, 1.9 g KH2PO4, 0.6 g/l K2HPO4, 0.43 g MgSO4, 10 g/l glucose, pH 6.5) was melted, and 2.5 ml of solution were immediately poured into µ-dishes. After hardening of the agarose, three troughs of 10 × 2 mm located 5 mm apart from each other were cut in the center of the dish. For assays, dishes were prepared freshly. Wild-type and ate1-null cells were harvested, washed with Soerensen buffer (14.6 mM KH2PO4, 2 mM Na2HPO4, pH 6.0) once, and resuspended in MB medium (14 g/l Bacto Peptone, 7 g/l yeast extract, 4.26 g MES buffer, pH 6.9). Cells (5 × 104) in equal volumes were loaded into the two outer troughs, and 1 mM folic acid in MB medium was loaded into the central trough. Directed cell migration was observed after 3–4 h of incubation at 22°C inside a moistened chamber.
Cell migration was imaged using an Axiovert 200M (Zeiss) equipped with an A-Plan 10×/NA 0.25 Ph1 lens and an AxioCamMR camera. Recorded time series consisted of 1500 phase contrast images taken every 10 s. Owing to technical limitations, for data analysis time series were split to 500 frames each, from which 20 cells were randomly selected for tracking. Cell trajectories were manually rendered using ImageJ (http://imagej.nih.gov/ij/) and the manual tracking plug-in (http://rsbweb.nih.gov/ij/plugins/track/track.html). Readout of velocity and straightness was achieved using the Ibidi Chemotaxis tool software, and directionality was calculated as the ratio of cell displacement along the x-axis to Euclidean distance.
Two-dimensional chemotaxis micropipette assay
Chemotactic stimulation with a micropipette filled with cAMP was performed as previously described (Gerisch, 2009; Müller-Taubenberger and Ishikawa-Ankerhold, 2013). To measure directed cell movement, ate1-null expressing GFP-LimEΔcoil and wild-type cells expressing DdCherry-LimEΔcoil were mixed and starved for 7 h at 22°C. Cells were stimulated with a micropipette filled with 10–4 M cAMP. Images were taken at intervals of 8 s using a LSM 510 Meta confocal microscope (Zeiss) with a 63×/NA 1.3 Neofluar oil-immersion objective. Images were analyzed with Fiji win 32, an enhanced and extended version of the ImageJ software package using the plug-in Mtrack J. Results of four different experiments were pooled.
Three-dimensional migration assays
For analysis of three-dimensional migration, starved cells were mixed with 1.5 mg/ml rat-tail collagen type I (Ibidi), 0.2% NaHCO3, 6.7 mM NaOH, 1.2 mM CaCl2 in Soerensen phosphate buffer. The cell–collagen mix was then filled into a µ-Slide Chemotaxis3D (Ibidi), and after polymerization of the collagen for 30 min at RT, a gradient of 100 nM cAMP was established across the observation channel. Migration was recorded for 1 h and 40 min at intervals of 20 s using an Axiovert 200M microscope equipped with a A-Plan 10×/NA 0.25 Ph1 lens and AxioVision4 software (Zeiss). Speed and directionality of migrating cells were analyzed using the Imaris software (Bitplane).
Mass spectrometry
Actomyosin was purified from Dictyostelium cells as described (Konzok et al., 1999), and the proteins of the actin-enriched fraction were precipitated with trichloroacetic acid as described (Xu et al., 2009). The proteins were dissolved with 5× Invitrisol, 100 mM Tris (pH 8.5) with 0.1% RapiGest, and then sonicated for 2 h. After reduction and alkylation, proteins were digested sequentially by LysC for 4 h and trypsin at 37°C overnight. Samples were desalted using a Pierce C18 spin column (Thermo Fisher). All peptides were separated by a home-packed Aqua C18 column (75 µm ×15 cm, 3 µm, 100 Å; Phenomenex) with a proxeon EASY-nLC liquid chromatography system. Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Peptides eluted from the LC column were directly electrosprayed into the mass spectrometer with the application of a distal 1.8 kV spray voltage. Data-dependent tandem spectrometry (MS/MS) analysis was performed on an Orbitrap Elite-ETD mass spectrometer (Thermo Fisher, San Jose, CA). A cycle of one full-scan MS spectrum (m/z 300–1800) was acquired followed by 20 MS/MS events, half of which were CID and half were ETD events, sequentially generated on the 1st to the 10th abundant precursors for CID and ETD fragmentation. The number of microscans was one for both MS and MS/MS scans, and the maximum ion injection times were 10 and 50 ms, respectively. The dynamic exclusion settings used were as follows: repeat count, 1; repeat duration, 30 s; exclusion list size, 500; and exclusion duration, 60 s.
DTASelect filtering was performed using DTASelect v2.1.3 followed by manual data validation by comparing theoretical and experimental spectra as described (Wong et al., 2007; Xu et al., 2009). Manually validated peptides of actin arginylation sites listed in Table 1 are highlighted in yellow in the DTASelect file (Supplemental Table S1). MS1 precursors with intensity and time information are shown in Supplemental Table S2.
Dynamic modification was set on glutamic acid, aspartic acid, glutamine, and asparagine for arginylation (+156.1011 Da).
Mass spectrometry data were searched against UniProt whole D. discoideum protein database using Integrated Proteomics Pipeline (Xu et al., 2006).
Statistics
For the statistical analysis, GraphPad Prism software was used.
Supplementary Material
Acknowledgments
We thank Marlis Fürbringer, Daniela Rieger, Stephanie Lindholz, and Gudrun Trommler for technical assistance, Matthias Samereier (LMU Munich) for help with migration assays, and Anna Kashina (University of Pennsylvania) for advice during initial phases of the study. We thank Ralph Gräf (University of Potsdam) for his support and the possibility to use microscopes of his laboratory. We thank Min Huang (NCPS Shanghai) and Robin Park (Scripps Research Institute, La Jolla) for help during analysis of mass spectrometry data. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 914/A07 to A.M.-T.). We are grateful for support of the Imaging facility of the SFB 914/Z01 to H.I.-A. and Steffen Massberg (LMU Munich).
Abbreviations used:
- DdAte1
Dictyostelium discoideum Arg-transfer RNA protein transferase 1
- RT
room temperature.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-02-0132) on December 26, 2018.
REFERENCES
- Arnold K, Bordoli L, Kopp J, Schwede T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics , 195–201. [DOI] [PubMed] [Google Scholar]
- Axelrod D. (2003). Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol , 1–33. [DOI] [PubMed] [Google Scholar]
- Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. (1990). Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J Biol Chem , 7464–7471. [PubMed] [Google Scholar]
- Bohley P, Kopitz J, Adam G, Rist B, von Appen F, Urban S. (1991). Post-translational arginylation and intracellular proteolysis. Biomed Biochim Acta , 343–346. [PubMed] [Google Scholar]
- Bretschneider T, Anderson K, Ecke M, Müller-Taubenberger A, Schroth-Diez B, Ishikawa-Ankerhold HC, Gerisch G. (2009). The three-dimensional dynamics of actin waves, a model of cytoskeletal self-organization. Biophys J , 2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretschneider T, Diez S, Anderson K, Heuser J, Clarke M, Müller-Taubenberger A, Köhler J, Gerisch G. (2004). Dynamic actin patterns and Arp2/3 assembly at the substrate-attached surface of motile cells. Curr Biol , 1–10. [DOI] [PubMed] [Google Scholar]
- Brink M, Gerisch G, Isenberg G, Noegel AA, Segall JE, Wallraff E, Schleicher M. (1990). A Dictyostelium mutant lacking an F-actin cross-linking protein, the 120-kD gelation factor. J Cell Biol , 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornachione AS, Leite FS, Wang J, Leu NA, Kalganov A, Volgin D, Han X, Xu T, Cheng YS, Yates JR, 3rd, et al. (2014). Arginylation of myosin heavy chain regulates skeletal muscle strength. Cell Rep , 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger L, Noegel AA, Schleicher M. (1991). Domain structure in actin-binding proteins: expression and functional characterization of truncated severin. J Cell Biol , 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faix J, Kreppel L, Shaulsky G, Schleicher M, Kimmel AR. (2004). A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system. Nucleic Acids Res , e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, Yumura S, Yumura TK. (1987). Agar-overlay immunofluorescence: high-resolution studies of cytoskeletal components and their changes during chemotaxis. Methods Cell Biol , 347–356. [DOI] [PubMed] [Google Scholar]
- Gebbie L, Benghezal M, Cornillon S, Froquet R, Cherix N, Malbouyres M, Lefkir Y, Grangeasse C, Fache S, Dalous J, et al. (2004). Phg2, a kinase involved in adhesion and focal site modeling in Dictyostelium. Mol Biol Cell , 3915–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerisch G. (2009). Imaging actin cytoskeleton dynamics in Dictyostelium chemotaxis. Methods Mol Biol , 385–400. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis , 2714–2723. [DOI] [PubMed] [Google Scholar]
- Ishikawa-Ankerhold HC, Gerisch G, Müller-Taubenberger A. (2010). Genetic evidence for concerted control of actin dynamics in cytokinesis, endocytic traffic, and cell motility by coronin and Aip1. Cytoskeleton (Hoboken) , 442–455. [DOI] [PubMed] [Google Scholar]
- Joseph JM, Fey P, Ramalingam N, Liu XI, Rohlfs M, Noegel AA, Müller-Taubenberger A, Glöckner G, Schleicher M. (2008). The actinome of Dictyostelium discoideum in comparison to actins and actin-related proteins from other organisms. PLoS One , e2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji A, Kaji H, Novelli GD. (1963). A soluble amino acid incorporating system. Biochem Biophys Res Commun , 406–409. [DOI] [PubMed] [Google Scholar]
- Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR, 3rd, Mogilner A, Zebroski H, Kashina A. (2006). Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science , 192–196. [DOI] [PubMed] [Google Scholar]
- Kashina AS. (2006). Differential arginylation of actin isoforms: the mystery of the actin N-terminus. Trends Cell Biol , 610–615. [DOI] [PubMed] [Google Scholar]
- Konzok A, Weber I, Simmeth E, Hacker U, Maniak M, Müller-Taubenberger A. (1999). DAip1, a Dictyostelium homologue of the yeast actin-interacting protein 1, is involved in endocytosis, cytokinesis, and motility. J Cell Biol , 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S, Leu NA, Pavlov I, Han X, Ribeiro PA, Xu T, Bunte R, Saha S, Wang J, Cornachione A, et al. (2012). Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J Mol Cell Cardiol , 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S, Leu NA, Zhang F, Bunte R, Saha S, Wang J, Guo C, He W, Kashina A. (2010). Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet , e1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, Varshavsky A. (2002). An essential role of N-terminal arginylation in cardiovascular development. Science , 96–99. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Varshavsky A. (1999). Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol , 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pickart CM. (1995a). Binding of phenylarsenoxide to Arg-tRNA protein transferase is independent of vicinal thiols. Biochemistry , 15829–15837. [DOI] [PubMed] [Google Scholar]
- Li J, Pickart CM. (1995b). Inactivation of arginyl-tRNA protein transferase by a bifunctional arsenoxide: identification of residues proximal to the arsenoxide site. Biochemistry , 139–147. [DOI] [PubMed] [Google Scholar]
- Lian L, Suzuki A, Hayes V, Saha S, Han X, Xu T, Yates JR, 3rd, Poncz M, Kashina A., Abrams CS. (2014). Loss of ATE1-mediated arginylation leads to impaired platelet myosin phosphorylation, clot retraction, and in vivo thrombosis formation. Haematologica , 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manahan CO, App AA. (1973). An arginyl-transfer ribonucleic acid protein transferase from cereal embryos. Plant Physiol , 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Taubenberger A. (2006). Application of fluorescent protein tags as reporters in live-cell imaging studies. Methods Mol Biol , 229–246. [DOI] [PubMed] [Google Scholar]
- Müller-Taubenberger A, Ishikawa-Ankerhold HC. (2013). Fluorescent reporters and methods to analyze fluorescent signals. Methods Mol Biol , 93–112. [DOI] [PubMed] [Google Scholar]
- Müller-Taubenberger A, Kortholt A, Eichinger L. (2013). Simple system–substantial share: the use of Dictyostelium in cell biology and molecular medicine. Eur J Cell Biol , 45–53. [DOI] [PubMed] [Google Scholar]
- Niewöhner J, Weber I, Maniak M, Müller-Taubenberger A, Gerisch G. (1997). Talin-null cells of Dictyostelium are strongly defective in adhesion to particle and substrate surfaces and slightly impaired in cytokinesis. J Cell Biol , 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. (2007). COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics , 1073–1079. [DOI] [PubMed] [Google Scholar]
- Pavlyk I, Leu NA, Vedula P, Kurosaka S, Kashina A. (2018). Rapid and dynamic arginylation of the leading edge beta-actin is required for cell migration. Traffic , 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Kashina A. (2005). Identification of mammalian arginyltransferases that modify a specific subset of protein substrates. Proc Natl Acad Sci USA , 10123–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Wong CC, Xu T, Leu NA, Dong DW, Guo C, McLaughlin KJ, Yates JR, 3rd, Kashina A. (2008). Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development , 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat Methods , 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth H, Samereier M, Trommler G, Noegel AA, Schleicher M, Müller-Taubenberger A. (2015). Balanced cortical stiffness is important for efficient migration of Dictyostelium cells in confined environments. Biochem Biophys Res Commun , 730–735. [DOI] [PubMed] [Google Scholar]
- Saha S, Kashina A. (2011). Posttranslational arginylation as a global biological regulator. Dev Biol , 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Mundia MM, Zhang F, Demers RW, Korobova F, Svitkina T, Perieteanu AA, Dawson JF, Kashina A. (2010). Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol Biol Cell , 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Wang J, Buckley B, Wang Q, Lilly B, Chernov M, Kashina A. (2012). Small molecule inhibitors of arginyltransferase regulate arginylation-dependent protein degradation, cell motility, and angiogenesis. Biochem Pharmacol , 866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samereier M, Meyer I, Koonce MP, Gräf R. (2010). Live cell-imaging techniques for analyses of microtubules in Dictyostelium. Methods Cell Biol , 341–357. [DOI] [PubMed] [Google Scholar]
- Schulz I, Baumann O, Samereier M, Zoglmeier C, Gräf R. (2009). Dictyostelium Sun1 is a dynamic membrane protein of both nuclear membranes and required for centrosomal association with clustered centromeres. Eur J Cell Biol , 621–638. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. (2003). SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res , 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryev SA, Papadopoulos JS, Schaffer AA, Agarwala R. (2007). Improved BLAST searches using longer words for protein seeding. Bioinformatics , 2949–2951. [DOI] [PubMed] [Google Scholar]
- Terman JR, Kashina A. (2013). Post-translational modification and regulation of actin. Curr Opin Cell Biol , 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren H. (1985). Interference reflection microscopy in cell biology: methodology and applications. J Cell Sci , 279–301. [DOI] [PubMed] [Google Scholar]
- Wang J, Han X, Wong CC, Cheng H, Aslanian A, Xu T, Leavis P, Roder H, Hedstrom L, Yates JR, 3rd, Kashina A. (2014). Arginyltransferase ATE1 catalyzes midchain arginylation of proteins at side chain carboxylates in vivo. Chem Biol , 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Xu T, Rai R, Bailey AO, Yates JR, 3rd, Wolf YI, Zebroski H., Kashina A. (2007). Global analysis of posttranslational protein arginylation. PLoS Biol , e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EF, Brar SK, Sesaki H, Yang C, Siu CH. (1996). Molecular cloning and characterization of DdCAD-1, a Ca2+-dependent cell-cell adhesion molecule, in Dictyostelium discoideum. J Biol Chem , 16399–16408. [DOI] [PubMed] [Google Scholar]
- Xu T, Wong CC, Kashina A, Yates JR., 3rd (2009). Identification of N-terminally arginylated proteins and peptides by mass spectrometry. Nat Protoc , 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TV, Venable JD, Park SK, Cociorva D, Lu B, Liao L, Wohlschlegel J, Hewel J, Yates JR. (2006). ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol Cell Proteomics , S174. [Google Scholar]
- Yoshida S, Ito M, Callis J, Nishida I, Watanabe A. (2002). A delayed leaf senescence mutant is defective in arginyl-tRNA:protein arginyltransferase, a component of the N-end rule pathway in Arabidopsis. Plant J , 129–137. [DOI] [PubMed] [Google Scholar]
- Zatulovskiy E, Tyson R, Bretschneider T, Kay RR. (2014). Bleb-driven chemotaxis of Dictyostelium cells. J Cell Biol , 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Saha S, Kashina A. (2012). Arginylation-dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. J Cell Biol , 819–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Saha S, Shabalina SA, Kashina A. (2010). Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science , 1534–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gao R, Devreotes PN, Mogilner A, Zhao M. (2013). 3D arrays for high throughput assay of cell migration and electrotaxis. Cell Biol Int , 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.