

Summary
Activation of Gαq-coupled receptors by inflammatory mediators inhibits cold-sensing TRPM8 channels, aggravating pain and inflammation. Both Gαq and the downstream hydrolysis of phosphatidylinositol 4, 5-bisphosphate (PIP2) inhibit TRPM8. Here, I demonstrate that direct Gαq gating is essential for both the basal cold sensitivity of TRPM8 and TRPM8 inhibition elicited by bradykinin in sensory neurons. The action of Gαq depends on binding to three arginine residues in the N terminus of TRPM8. Neutralization of these residues markedly increased sensitivity of the channel to agonist and membrane voltage and completely abolished TRPM8 inhibition by both Gαq and bradykinin while sparing the channel sensitivity to PIP2. Interestingly, the bradykinin receptor B2R also binds to TRPM8, rendering TRPM8 insensitive to PIP2 depletion. Furthermore, TRPM8-Gαq binding impaired Gαq coupling and signaling to PLCβ-PIP2. The crosstalk in the TRPM8-Gαq-B2R complex thus determines Gαq gating rather than PIP2 as a sole means of TRPM8 inhibition by bradykinin.
Keywords: TRPM8, cold, G protein, GPCR signaling, PIP2, protein-protein interaction, bradykinin, inflammatory mediator, pain
Graphical Abstract
Highlights
-
•
Gαq governs the cold sensitivity of TRPM8 and TRPM8 inhibition by bradykinin
-
•
Gαq gates TRPM8 through engaging three basic arginine sites on TRPM8
-
•
TRPM8 inhibition by bradykinin depends on Gαq gating sites but not PIP2 signaling
-
•
Bradykinin receptor prevents the PIP2 sensitivity of TRPM8 channels
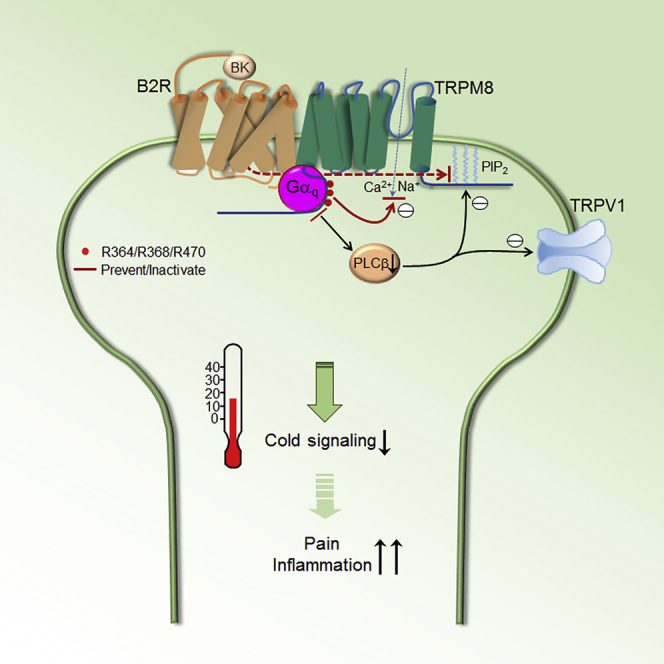
TRPM8 channels are inhibited by receptors coupled to Gαq, contributing to pain and inflammation. Zhang reveals Gαq gating sites on TRPM8 and shows that bradykinin receptor solely uses Gαq gating sites for TRPM8 inhibition upon activation, while depriving the channel of sensitivity to PIP2.
Introduction
TRPM8 channels detect a wide range of cold temperatures, spanning from innocuous cooling to noxious cold (Madrid et al., 2009, McKemy et al., 2002, Peier et al., 2002). It is also activated by cooling-mimetic compounds, such as menthol and its derivative, WS-12. The basal cold sensitivity of TRPM8 is critical to many physiological processes ranging from cold sensation to core body temperature regulation (Bautista et al., 2007, Colburn et al., 2007, Dhaka et al., 2007, Gavva et al., 2012), to basal tear secretion (Parra et al., 2010). Furthermore, activation of TRPM8 inhibits inflammatory and neuropathic pain, mediating an analgesic effect (Dhaka et al., 2007, Knowlton et al., 2013, Liu et al., 2013, Proudfoot et al., 2006), though it causes cold hypersensitivity in some cases (Colburn et al., 2007, De Caro et al., 2018, Knowlton et al., 2013). TRPM8 has also been implicated in the inhibition of tissue inflammatory responses and inflammatory cytokine release (Caceres et al., 2017, Ramachandran et al., 2013, Wang et al., 2017). Activation of TRPM8, therefore, inhibits both pain and inflammation, which may underlie cold therapy that has been used to relieve pain and inflammation for hundreds of years.
However, TRPM8 was inhibited during inflammation due to the actions of released inflammatory mediators such as bradykinin (BK) and histamine (Linte et al., 2007, Premkumar et al., 2005, Zhang et al., 2012). Inhibition of TRPM8, therefore, disinhibits the anti-pain and anti-inflammatory effects of TRPM8, contributing to aggravated pain, inflammation, and possibly dry eye diseases (Liu et al., 2013, Yang et al., 2018, Zhang et al., 2012).
BK and histamine act on Gαq-coupled receptors. Initially, TRPM8 inhibition induced by BK was attributed to dephosphorylation of TRPM8 by activated protein kinase C (PKC) (Premkumar et al., 2005). Another appealing possibility is that activated Gαq-PLCβ catalyzes hydrolysis of the membrane lipid phosphatidylinositol 4, 5-bisphosphate (PIP2), which is essential for TRPM8 activation (Liu and Qin, 2005, Rohács et al., 2005), leading to TRPM8 inhibition. Indeed, PIP2 hydrolysis-mediated TRPM8 inactivation has been implicated in TRPM8 desensitization and cold adaptation (Daniels et al., 2009, Rohács et al., 2005).
Adding to the complexity, we have found that activated Gαq directly inhibits TRPM8 in vitro (Zhang et al., 2012). However, it is not yet clear whether direct Gαq gating of TRPM8 also occurs in native sensory neurons and which mechanisms are mainly responsible for TRPM8 inhibition by inflammatory mediators in vivo. After all, activated Gαq will inevitably activate downstream PLCβ, triggering the concomitant breakdown of PIP2. It is also not known how Gαq directly gates TRPM8 and the relative role of Gαq gating and PIP2 signaling in TRPM8 modulation.
In this research, I have found that direct Gαq gating is crucial for regulating the basal cold sensitivity of TRPM8 and is the sole mechanism of TRPM8 inhibition elicited by BK in native sensory neurons without significant involvement of PLCβ-PIP2 signaling or Gα11 in this process. Importantly, I revealed the Gαq gating mechanism and identified three basic arginine residues on the N-terminal melastatin homology region (MHR) 1–3 in TRPM8 as Gαq gating sites. Mutation of these sites entirely abolished TRPM8 inhibition by activated Gαq and BK and also significantly reduced TRPM8 inhibition by histamine. I further elucidated the mechanism for the lack of a role of PIP2 in TRPM8 inhibition by BK, and I found that it is determined by a bidirectional crosstalk in the TRPM8-Gαq-B2R complex. Our data also suggest independent and cooperative modulation of TRPM8 by Gαq and PIP2.
Results
Gαq Is Crucial for the Cold Sensitivity of TRPM8 in Sensory Neurons
We have previously shown that activated Gαq directly inhibits TRPM8 channels in vitro (Zhang et al., 2012), but it remains unknown whether this mechanism also occurs to sensory neurons. To discriminate between Gαq gating and PIP2 signaling in TRPM8 modulation in sensory neurons, I took advantage of Gαq knockout (KO) mice, in which Gα11 remains intact and will take over the activation of downstream PLCβ-PIP2 signaling.
Cell-attached patch clamping was used to record TRPM8-mediated firing responses in small-to-medium sensory dorsal root ganglia (DRG) neurons, because the cell-attached mode preserves intracellular modulatory factors, minimizing artificial disruption of intracellular signaling (Madrid et al., 2006); furthermore, TRPM8 exhibits run-down in other patch configurations (Liu and Qin, 2005). Figure 1A shows that a ramp drop in bath temperatures elicited firing discharges in a DRG neuron at 26.6°C. A second cold ramp evoked similar firing responses with little desensitization. To verify that cold-elicited firing is mediated by TRPM8, DRG neurons were exposed to PBMC, a specific TRPM8 antagonist (Knowlton et al., 2011), during the second cold ramp. As shown in Figure 1B, PBMC completely blocked firing induced by the second cold challenge. Furthermore, the same neurons also responded to the specific TRPM8 agonist WS-12 (Figures 1A and 1B), validating that cold-induced firing is mediated by TRPM8. The effects of PBMC and WS-12 were, therefore, used to identify TRPM8-mediated cold responses in DRG neurons. With this approach, ∼30% of probed cells responded to cold, and ∼68% of these cold-sensitive neurons were TRPM8+. TRPM8-independent firing responses were excluded from further analysis.
Figure 1.

Gαq Is Crucial for the Cold Sensitivity of TRPM8+ DRG Neurons
(A–D) Firing responses evoked by two consecutive cold ramps and WS-12 (1 μM) in wild-type (WT) (A and B) and Gαq−/− (C and D) DRG neurons in the absence (A and C) or presence (B and D) of PBMC (50 nM). Arrows indicate cold activation threshold.
(E) Histogram distribution of cold activation threshold of DRG neurons from experiments similar to those in (A)–(D). WT, n = 25; Gαq−/−, n = 24.
(F) Bar summary of threshold for cold activation of DRG neurons.
(G and H) Summary of total number of firing (G) and average peak amplitude of firing (H) responses evoked during cold application period from the same experiments as described above.
(I) Summary of total number of firing responses evoked during capsaicin application (0.5 μM, 10 s).
Error bars in all figures represent mean ± SEM. ∗p < 0.05; ∗∗∗p < 0.001; NS, not significant. See also Figure S1.
I then performed similar experiments in DRG neurons isolated from Gαq-deficient mice. I first validated the ablation of Gαq, but not Gα11, in Gαq-lacking DRG neurons (Figure S1A). Gαq-deficient DRG neurons responded to two consecutive cold ramps similarly to wild-type (WT) neurons (Figure 1C), but they started to fire at a higher temperature threshold (Figures 1C and 1D). The cold responses were blocked by PBMC, and the same neurons also responded to WS-12, confirming that cold-evoked firing responses are TRPM8 dependent (Figures 1C and 1D). Overall, deleting Gαq markedly shifted the temperature threshold for TRPM8 activation in DRG neurons toward higher temperature ranges in both low- and high-threshold cold-sensing neurons (Figure 1E), leading to a significant increase of 3.2°C in the cold activation threshold (Figure 1F) (WT, 26.16°C ± 0.6°C; Gαq−/−, 29.37°C ± 0.61°C; p < 0.001), consistent with the finding of the basal inhibition of TRPM8 channels by endogenous Gαq (Zhang et al., 2012). Notably, cold temperatures also induced much more TRPM8-dependent firing events in Gαq KO neurons than in WT neurons (Figures 1A–1D and 1G), though there was no significant difference in firing amplitude between WT and KO neurons (Figure 1H). Enhanced TRPM8 responses are not caused by indirect upregulation of TRPM8, because TRPM8 expression was not altered in Gαq KO neurons (Figure S1B). In contrast to cold, firing events induced by capsaicin were not significantly different between WT and Gαq KO neurons (Figure 1I), suggesting that increased firing responses in Gαq-lacking neurons are specific to cold and unlikely to be mediated by indirect effects of Gαq deletion on other action potential transducing channels such as voltage-gated sodium channels.
These experiments demonstrate that Gαq tonically inhibits the basal cold sensitivity of TRPM8 channels in DRG neurons and suggest that Gαq is present in both low- and high-threshold cold-sensing DRG neurons.
Gαq Is Essential for Inhibiting Firing Responses Induced by Bradykinin
To investigate whether Gαq is also crucial for TRPM8 inhibition induced by BK in DRG neurons, cells were stimulated by WS-12, and induced firing activity was monitored with cell-attached recording. Under control conditions, no significant changes in firing were observed in either WT or Gαq KO neurons after vehicle perfusion (1 min) (Figures 2A, 2D, and 2G). The same procedure was then used to investigate the effect of BK on TRPM8. Of note, the functional BK receptor B2R is co-expressed in 33.3% TRPM8+ DRG neurons (Zhang et al., 2012) but in 83.9% TRPV1+ DRG neurons, indicated by the proportion of neurons in which TRPV1 is sensitized by BK (Zhang et al., 2008) (also discussed later). In fact, BK-induced inhibition of TRPM8 was only observed in TRPV1+ neurons (Zhang et al., 2012). Therefore, to ensure that the recorded neurons express B2R and to preclude the neurons lacking B2R from confounding analysis, I selected TRPM8+ DRG neurons exhibiting TRPV1 sensitization and/or TRPM8 inhibition in response to BK as an index of B2R expression. As expected, WS-12-elicited firing responses were significantly inhibited and even completely eliminated by BK in some WT DRG neurons (Figures 2B and 2G). Furthermore, the response to capsaicin in this neuron was also sensitized by BK (Figure 2C), confirming functional B2R expression. In contrast, BK did not inhibit TRPM8-triggered firing in Gαq-lacking DRG neurons and, in fact, slightly increased firing elicited by WS-12, though it was not statistically significant compared to control (Figures 2E and 2G). The lack of inhibitory effect was not due to lack of expression of B2R or Gα11 or impaired PLCβ signaling in Gαq-deficient DRG neurons, because firing responses elicited by capsaicin in the same neurons were sensitized by BK (Figures 2F and 2H), an event depending on the Gαq/11-PLCβ signaling pathway (Lukacs et al., 2007, Zhang et al., 2008); furthermore, Gα11 is expressed in every DRG neuron (Han et al., 2006). As complementary evidence, BK also sensitized TRPV1 in Gαq null neurons to a degree similar to that in WT neurons in whole-cell patch clamping (Figures 2I, 2J, and 7J); moreover, the sensitization was completely prevented by the phospholipase C (PLC) inhibitor U73122, validating the involvement of PLCβ in this process. These data suggest that PLCβ signaling is not impaired in DRG neurons lacking Gαq, consistent with the finding that Gαq, rather than Gα11, plays a major role in TRPM8 inhibition (Li and Zhang, 2013, Zhang et al., 2012).
Figure 2.

Gαq Is Essential for BK-Induced Inhibition of TRPM8-Dependent Firing Responses in DRG Neurons
(A–C) Representative firing responses elicited by WS-12 (1 μM) or capsaicin (Cap, 0.5 μM) (C) in WT DRG neurons before and after vehicle (A) or BK (1 μM, 1 min) (B and C). (B) and (C) indicate the same neuron.
(D–F) Example firing responses evoked by WS-12 or capsaicin (F) in DRG neurons lacking Gαq before and after vehicle (D) or BK (E and F). (E) and (F) indicate the same neuron.
(G) Scatterplot of ratio of action potentials (APs) induced by WS-12 after and before BK in experiments similar to those in (A)–(D). ∗∗p = 0.00588; NS, p = 0.107.
(H) Summary of ratio of firing responses evoked by capsaicin after and before BK from experiments similar to those in (C) and (F).
(I) Example currents evoked by capsaicin (100 nM, 5 s) in a Gαq null DRG neuron. The gap indicates BK treatment (1 μM, 1 min).
(J) Collective results of sensitization fold caused by BK from experiments similar to those in (I). The sensitization was abolished by U73122 (2.5 μM). The number of experiments is denoted above each bar.
All data indicate mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant.
Figure 7.

TRPM8 Is Insensitive to PIP2 Depletion in the Presence of B2R
(A) Translocation of Tubby-R332H-cYFP induced by rapamycin (1 μM) in HEK293 cells co-expressing mRFP-FKBP-5-ptase domain and PM-FRB-CFP. Scale bars, 10 μM.
(B) Real-time quantification of membrane fluorescence (FPM) relative to cytosol fluorescence (FCytosol) in the top left cell from (A).
(C) Example of whole-cell inward and outward currents elicited by menthol in HEK293 cells co-expressing mRFP-FKBP-5-ptase and PM-FRB-CFP together with either TRPM8 and B2R or TRPM8-TM and B2R. Red traces indicate cells pretreated with rapamycin (Rapa, 1 μM, 1 min).
(D) Collective results from experiments similar to those in (C).
(E) Binding of TRPM8 to B2R, but not to H1R, in a nickel-beads (Ni-NTA) pull-down assay performed on HEK293 cell lysate expressing the proteins as indicated.
(F) Binding between PLCβ-his (6×) and Gαq Q209L is reduced in the presence of TRPM8 in a nickel beads pull-down assay. Lane 1 indicates total cell lysate (TCL).
(G) Translocation of Tubby-R332H-cYFP at different time points after the addition of BK (1 μM) in HEK293 cells co-expressing B2R or together with TRPM8 or C-terminal deleted (TRPM8-CD). Scale bars, 10 μM. On the right, real-time quantification of membrane Tubby fluorescence is indicated relative to cytosol fluorescence from the cells in (G).
(H) Summary of results similar to those in (G).
(I) Example inward currents evoked by capsaicin (Cap, 100 nM, 5 s) in HEK293 cells expressing TRPV1 and B2R or together with TRPM8 before and after BK treatment (1 μM, red). TRPM8 co-expression is indicated by currents elicited by menthol (Men, 200 μM).
(J) Summary of TRPV1 sensitization fold induced by BK in experiments similar to those in (I). BK-induced sensitization of TRPV1 was also significantly different between TRPM8+ and TRPM8− DRG neurons indicated by responses to menthol.
All error bars represent mean ± SEM. ∗∗p < 0.01; ∗∗p < 0.05; ∗∗∗p < 0.001; NS, not significant. See also Figure S4.
Altogether, these experiments demonstrate that Gαq is crucial for both the basal and BK-induced TRPM8 inhibition in DRG neurons. Neither Gα11 nor downstream PLCβ signaling is involved in these processes.
Identification of Gαq Effector Sites on TRPM8 Channels
Activated Gαq rapidly inhibits TRPM8 through binding to TRPM8 (Zhang et al., 2012). Therefore, Gαq binding sites on TRPM8 should constitute critical channel gating sites. To ascertain which terminus of TRPM8 contains functional Gαq binding sites, the GST (glutathione S-transferase)-coupled N or C terminus of TRPM8 was used to pull down different chimeric Gαq proteins in which different functional regions in the distal C terminus of Gαq are replaced by Gαi (Figure 3A) (Zhang et al., 2012). In general, the binding of Gαq to the N terminus is stronger than that to the C terminus of TRPM8 (Figures 3B and 3C), suggesting that the N terminus of TRPM8 is more critical to Gαq gating. The most potent binding to the N terminus of TRPM8 was found with Gαqi, 2Gαqi, and 3Gαqi (Figure 3B), which also exert a potent inhibitory effect on TRPM8 (Zhang et al., 2012). Reduced binding to the N terminus was observed with 4Gαqi and 5Gαqi (Figure 3B), which exhibit a diminished inhibitory effect on TRPM8 due to the replacement of Switch III in Gαq by the equivalent part from Gαi in these chimeras (Figure 3A) (Zhang et al., 2012). The good correlation between the binding of Gαq chimeras to the N terminus and the functional effect on TRPM8 further suggests that the N terminus of TRPM8 contains Gαq gating sites. The binding experiment also supports that the N terminus mainly binds to Switch III in Gαq (Figure 3A), a major region also responsible for the gating of TRPM8 (Zhang et al., 2012), consistent with the suggestion of a critical role of the N-terminal TRPM8 in Gαq gating. In contrast, reduced binding to the C terminus of TRPM8 was observed in 3Gαqi (Figure 3C), in which the α3 helix downstream of Switch III in Gαq is replaced (Figure 3A), suggesting that the C terminus of TRPM8 primarily binds to the α3 helix, a region also responsible for binding to PLCβ (Figure 3A) (Venkatakrishnan and Exton, 1996, Waldo et al., 2010).
Figure 3.

Delineation of GαqBinding Regions in TRPM8
(A) Schematic diagram shows different Gαqi chimeras and regions binding to the N- and C-terminus of TRPM8, and PLCβ.
(B and C) GST-coupled N terminus (B) and C terminus (C) of TRPM8 bind to different Gαq chimeras in pull-down assays. IP, immunoprecipitation.
(D) Schematic diagram shows different N-terminal fragments coupled to GST.
(E) The binding of Gαq Q209L to different GST-coupled N-terminal fragments of TRPM8. Total cell lysate (TCL) denotes Gαq expression.
(F) The binding of Gαq Q209L to different C-terminal fragments coupled to GST (top) in a GST pull-down assay (bottom).
(G) Schematic diagram depicts the relationship of Gαq binding regions (N7, N8, and TRP) and other domains.
To further delineate Gαq binding regions in the N terminus, a series of different N-terminal fragments coupled to the GST tag were constructed for Gαq pull-down assay (Figure 3D). With this approach, two small regions, N7 (F245-E398) and N8 (R451-N606), were found to be responsible for the binding of Gαq to the N terminus (Figures 3E and 3G). These two short regions, therefore, likely contain the functional Gαq effector sites. Using a similar approach, I have also found that the main Gαq binding region in the C terminus is located in the proximal C terminus of TRPM8 (Figure 3F), a region also containing the TRP domain critical to TRPM8 gating and modulation (Figure 3G). Taken together, Gαq binds to N7 and N8 in the N terminus and also to the proximal C terminus of TRPM8, although N7 and N8 are more likely to be the functional Gαq gating regions (Figure 3G). Reciprocally, the N and C termini of TRPM8 mainly bind to Switch III and the α3 helix in Gαq, respectively (Figure 3A).
Activated Gαq gates TRPM8 through Switch III (Zhang et al., 2012), a region undergoing major conformational changes upon exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on Gαq during Gαq activation (Lambright et al., 1996, Waldo et al., 2010), consistent with the structural requirement for effector interaction. Notably, Switch III contains a cluster of acidic residues. Surface charge modeling of Gαq also revealed that Switch III is negatively charged (Figure 4A). Therefore, I hypothesized that the Gαq effector sites on TRPM8 are positive, allowing for electrostatic interaction with negative Switch III in Gαq. Interestingly, a cluster of basic arginine and lysine residues were found on N7, with a distribution pattern similar to those of acidic residues on Switch III in Gαq (Figure 4A). These basic residues were then neutralized by mutation to glutamine.
Figure 4.

Identification of Gαq Effector Sites on TRPM8
(A) Molecular surface charge representation of Gαq. Key domains and distance are shown. Underneath is the alignment of charged residues (in red) in Switch III of Gαq and N7-TRPM8.
(B) Example inward and outward whole-cell currents elicited by menthol (200 μM, 5 s) at −60 mV and 60 mV, respectively, in HEK293 cells expressing TRPM8 and mutants as indicated.
(C) Summary of TRPM8 peak currents from similar experiments to those in B (n = 19–28).
(D) Summary of the effect of 3Gαqi on the peak currents of TRPM8 and R470Q elicited by menthol (n = 22–35).
(E) Averaged current-voltage (I-V) traces of TRPM8 and R470Q with or without 3Gαqi co-expression (n = 16–27).
(F) Binding of Gαq Q209L to GST-N7 and R470Q mutant in a GST pull-down assay.
(G) On the left is a diagram showing different truncations of N8-TRPM8. On the right, the binding of truncated N8 fragments to Gαq Q209L is indicated.
(H) Summary of inward and outward currents of different TRPM8 mutants elicited by menthol.
(I) GST pull-down assay shows the binding of Gαq Q209L to GST-N8 and mutants as indicated.
(J) Summary of currents of R364Q-R368Q mutant with or without co-expression of 3Gαqi. The number of experiments is indicated above each bar.
(K) Average I-V relationship of R364Q-R368Q mutant with (n = 27) or without (n = 20) the co-presence of 3Gαqi.
All error bars represent mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant.
In principle, the mutation of Gαq effector sites on TRPM8 should disrupt Gαq binding, abolish the inhibitory effect of Gαq, and potentiate the function of TRPM8 channels. Indeed, I found that neutralizing R470 markedly enhanced TRPM8 inward and outward currents evoked by menthol, while mutation of other nearby basic residues either reduced TRPM8 currents or had no significant effect (Figures 4B and 4C). Moreover, R470Q mutant abolished the inhibition of outward current and reduced the inhibition of inward current caused by 3Gαqi, an activated Gαq chimera containing Q209L mutation, while downstream signaling to PLCβ was selectively ablated (Figure 4D) (Zhang et al., 2012), although 3Gαqi still significantly inhibited the inward current of R470Q (Figure 4D). Depolarization-induced outward currents in R470Q were also much larger than those in WT TRPM8 but were still inhibited by 3Gαqi, as seen in the WT channel (Figure 4E). Notably, mutating R470 completely eliminated the binding of activated Gαq to the N7 fragment (Figure 4F), demonstrating that R470 is crucial to the binding between Gαq and N7. Taken together, these results suggest that R470 is a critical Gαq effector site on TRPM8. However, the incomplete abrogation of Gαq-elicited inhibition of TRPM8 by R470Q mutation suggests the presence of other unidentified Gαq effector sites, likely on N8 (discussed later).
There are no obvious clusters of positive residues on N8. To narrow down the Gαq binding region in N8, a progressive truncation of N8 coupled to GST was generated and used to pull down Gαq (Figure 4G). A prominent reduction in Gαq binding was observed when the distal 36 amino acids were truncated from the C terminus of N8 (Figure 4G), suggesting that these 36 amino acids contain Gαq binding sites. Meanwhile, basic residues in N8 were individually neutralized to glutamine. Currents mediated by these mutated channels were then monitored using whole-cell patch clamping. I found that neutralization of two individual arginine residues (R364Q and R368Q) on N8 both significantly enhanced TRPM8 currents (Figure 4H). TRPM8 currents were further increased after mutating these two sites in combination (R364Q-R368Q) (Figure 4H). However, all other mutants were without significant effect. Coincidently, R364 and R368 fall within the distal 36 amino acids in N8 that are also critical to Gαq binding (Figure 4G). As anticipated, neutralization of these two sites in N8 was also sufficient to disrupt the binding between N8 and Gαq (Figure 4I), suggesting that these two sites are also Gαq effector sites. Consistent with this idea, the inhibitory effects of 3Gαqi on the inward and outward currents of TRPM8 evoked by menthol were abolished in the double mutant (R364Q-R368Q) (Figure 4J). However, voltage gating of R364Q-R368Q was still inhibited by 3Gαqi (Figure 4K).
Altogether, Gαq binds to two small regions in the N terminus of TRPM8 (N7 and N8), and three positively charged arginine residues (R364, R368, and R470) within these regions function as Gαq effector sites. These experiments also support the notion that gating of TRPM8 by menthol and voltage is separable through independent mechanisms, though they are allosteric during TRPM8 gating.
Gαq Effector Sites Are Responsible for Gαq Gating of TRPM8
I next mutated all three basic arginine residues on TRPM8 to glutamine and named the mutant TRPM8-TM (triple mutant). As expected, TRPM8-TM exhibited much larger currents than the WT channel and completely abolished the inhibitory effects of 3Gαqi on TRPM8 currents elicited by menthol (Figures 5A and 5B). The sensitivity of TRPM8-TM to menthol was also dramatically increased, leading to a marked leftward shift in dose-response curve compared to that of WT TRPM8 (WT, EC50 = 148.8 μM ± 8.1 μM; TM, EC50 = 40.6 μM ± 5.3 μM) (Figure 5C). However, this marked leftward shift was not observed in mouse embryonic fibroblast (MEF) cells lacking endogenous Gαq/11 (TRPM8 in Gαq/11−/−, EC50 = 75.1 μM ± 2.8 μM; TM in Gαq/11−/−, EC50 = 61.4 μM ± 5.0 μM), supporting the idea that increased sensitivity of TRPM8-TM is due to relief of basal inhibition from endogenous Gαq/11 and not caused by indirect alterations to the intrinsic biophysical properties of the channel. These data are also consistent with the basal inhibition of TRPM8 by endogenous Gαq in DRG neurons (Figures 1C–1G). Notably, TRPM8-TM also entirely abolished the inhibition of 3Gαqi on TRPM8 outward currents evoked by a depolarization voltage ramp (Figure 5D). A further detailed analysis of voltage gating of TRPM8 through applying a series of depolarization voltage steps revealed that TRPM8-TM shifted the voltage gating of TRPM8 toward negative potentials close to resting membrane potential, resulting in a markedly reduced V1/2 (WT, V1/2 = 116.0 mV ± 11.0 mV; TM, V1/2 = 66.5 mV ± 5.9 mV) (Figures 5E–5G). It also shows that TRPM8-TM abolished the inhibitory effect of activated 3Gαqi on V1/2 observed in the WT channels (Figure 5G) (Zhang et al., 2012).
Figure 5.

Gαq Gates TRPM8 through Three Arginine Residues on TRPM8
(A) Representative inward and outward currents of TRPM8 and TRPM8-TM activated by menthol (200 μM, 5 s).
(B) Summary of currents of TRPM8 and TRPM8-TM with or without 3Gαqi from experiments similar to those in (A). Data are mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001.
(C) Dose-response relationship of TRPM8 and TRPM8-TM expressed in HEK293 and Gαq/11−/− MEF cells in response to menthol. Curves are fitted with the Hill equation. TRPM8 (♦), EC50 = 148.8 μM ± 8.1 μM; TRPM8-TM (▲), EC50 = 40.6 μM ± 5.3 μM; TRPM8 in Gαq/11−/− MEF (□), EC50 = 75.1 μM ± 2.8 μM; TM in Gαq/11−/− (△), EC50 = 61.4 μM ± 5.0 μM). n = 7–15.
(D) Average I-V relationship of TRPM8-TM with (pink, n = 13) or without (black, n = 14) 3Gαqi.
(E) Example currents of TRPM8 and TRPM8-TM evoked by voltage steps from −120 mV up to 200 mV in 20-mV increments. Maximal current was evoked at 140 mV for TRPM8-TM.
(F) Normalized conductance (G)-voltage relationship from the cells in (E) fitted with the Boltzmann equation, giving rise to V1/2 and slope factor as follows: TRPM8, 103.5 mV and 36.2 mV; TRPM8-TM, 62.0 mV and 36.3 mV.
(G) Collective results of V1/2 from experiments similar to those in (E) and (F). The number of experiments is shown above each bar.
(H) Side stereoview of a ribbon representation of the TRPM8 structure (6BPQ).
(I) Side stereoview of central cavity formed by the N terminus of two opposite TRPM8 subunits. Distance measurements between domains and residues are indicated.
Error bars represent mean ± SEM. ∗∗p < 0.01; NS, not significant. See also Figure S2.
Altogether, TRPM8-TM completely abolished the inhibitory effects of 3Gαqi on TRPM8 activation triggered by both menthol and voltage, demonstrating that three arginine residues (R364, R368, and R470) in the N terminus of TRPM8 are Gαq effector sites responsible for the Gαq gating of TRPM8.
In accordance with the recently revealed TRPM8 structure (Yin et al., 2018), R364 and R368 are located near the bottom stack layer of the cytoplasmic domain (CD) of TRPM8, sitting at a turning loop between two helixes in MHR1-2 (Figure 5H). R470 also sits at a loop between two helixes (HTH3a and HTH3b) in the MHR3 domain. Three arginine residues constitute a contiguous plane lining a cytoplasmic cavity formed by the CD of TRPM8 (Figure 5I). Of note, the C-terminal Gαq binding TRP domain and the bottom S6 from the pore domain form the dome of the cytoplasmic cavity. The height of the cavity measured from the bottom of S6 to R368 is 83 Å. The distance of R470 from two opposite subunits is 68 Å, and the distance of two opposite R368 residues is 82 Å (Figure 5I). Therefore, the size of the cavity is, in principle, sufficient to accommodate Gαq with a dimension of 43 Å × 61 Å (Figure 4A). In this case, the top of Gαq would be close to the TRP domain and the intracellular mouth of the channel pore. Conformational changes in Gαq upon activation may be transduced to the TRP domain and the pore domain, resulting in the allosteric gating of TRPM8.
Gαq Gating Is Crucial for TRPM8 Inhibition by Inflammatory Mediators
The inflammatory mediators BK and histamine inhibit TRPM8 through binding, respectively, to B2R and H1R, two Gαq-coupled G-protein-coupled receptors (GPCRs). Both activated Gαq and concomitant PIP2 hydrolysis cause TRPM8 inhibition. The identified TRPM8-TM selectively deficient for Gαq gating but without impairment in PLCβ signaling and PIP2 sensitivity (discussed later) will be an excellent tool for determination of the relative role of direct Gαq gating and PIP2 signaling in TRPM8 inhibition during inflammatory signaling.
TRPM8 undergoes run-down in the whole-cell configuration, even under Ca2+-free conditions (Figure S3), likely due to the breakdown of intracellular PIP2 and/or polyphosphate (Liu and Qin, 2005, Zakharian et al., 2009). Furthermore, intracellular dialysis under the whole-cell configuration may disrupt Gαq coupling and GPCR signaling. To avoid these artificial effects and to minimize the intervention of whole-cell dialysis in intracellular signaling, cells were briefly pretreated with inflammatory mediators (1 min), allowing for intact completion of intracellular signaling before entering the whole-cell configuration for monitoring TRPM8 currents. In HEK293 cells co-expressing TRPM8 and B2R, a short pretreatment with BK elicited a robust inhibition of the inward and outward currents of TRPM8 (Figures 6A and 6B), consistent with the previous findings (Zhang et al., 2012). However, the potent inhibition was completely abolished in TRPM8-TM (Figures 6A and 6B). Similarly, TRPM8-TM also completely abrogated BK-elicited inhibition of voltage gating of TRPM8 (Figure 6C). These results suggest that BK-induced TRPM8 inhibition is solely due to direct Gαq gating, consistent with the aforementioned finding in sensory DRG neurons lacking Gαq (Figures 2D–2G). This conclusion is also supported by the previous pharmacological evidence showing that inhibition of PLCβ by U73122 had no effect on BK-induced TRPM8 inhibition (Zhang et al., 2012), arguing against a possible role for PIP2 hydrolysis in the functional coupling between B2R and TRPM8.
Figure 6.

Gαq Gating Sites Are Essential for TRPM8 Inhibition by Inflammatory Mediators
(A) Example inward and outward currents elicited by menthol (200 μM, 5 s) in HEK293 cells co-expressing B2R and TRPM8 or TRPM8-TM without or with BK pretreatment (1 μM, 1 min, red).
(B) Summary of results similar to those in (A).
(C) Average I-V relationship of TRPM8 and TRPM8-TM with or without BK treatment (n = 7–13) from experiments similar to those in (A).
(D) Findings from experiments similar to those in (A) but from cells co-expressing H1R. Some cells were pretreated with histamine (His, 10 μM, 1 min, red) or together with U73122 (2.5 μM, blue).
(E) Summary of results similar to those in (D).
(F) Average I-V relationship of TRPM8 and TRPM8-TM with or without histamine and U73122 treatment (n = 6–16) from experiments similar to those in (D).
All data indicate mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant. See also Figure S3.
Potent inhibition of TRPM8 was also observed in cells co-expressing TRPM8 and H1R upon stimulation with histamine (Figure 6D). In this case, 93.5% of the peak inward current and 41% of the outward current of TRPM8 were inhibited by histamine, in agreement with the previous results (Zhang et al., 2012). Similar to BK treatment, TRPM8-TM abolished inhibition of outward current caused by histamine and significantly reduced histamine-induced inhibition of inward current, though histamine still inhibited 43% of the inward current of TRPM8-TM (Figures 6D and 6E). The remaining inhibition is likely contributed by PIP2 hydrolysis due to actions of activated PLCβ. Indeed, the previous pharmacological experiments showed that inhibition of PLCβ by U73122 partially alleviated TRPM8 inhibition by histamine, though it had no effect on BK-induced TRPM8 inhibition (Zhang et al., 2012). To further corroborate this idea, cells expressing TRPM8-TM lacking Gαq gating were also pretreated with U73122 for simultaneous prevention of PIP2 signaling. As expected, the inhibitory effect of histamine was completely abolished under this condition (Figures 6D and 6E).
Similar findings were also observed when TRPM8 was activated by membrane voltage. Figure 6F shows that inhibition of depolarization-induced outward current by histamine was completely abolished only by TRPM8-TM together with U73122 pretreatment, not by either alone. These data conclusively demonstrate that both direct Gαq gating and downstream PIP2 hydrolysis contribute to histamine-evoked TRPM8 inhibition, with Gαq gating playing a primary role (∼57%), and the involvement of other mechanisms is unlikely.
Taken together, direct Gαq gating of TRPM8 channels is the sole mechanism for BK-induced inhibition of TRPM8, without a significant role for PIP2 hydrolysis. However, both Gαq gating and PIP2 signaling contribute to histamine-triggered TRPM8 inhibition.
TRPM8 Is Insensitive to PIP2 Depletion in the Presence of B2R
Both B2R and H1R are Gαq-coupled GCPRs. How can PIP2 signaling play a role in the modulation of TRPM8 initiated by H1R, but not by B2R, though both receptors use Gαq gating for modulation of TRPM8? To elucidate this question, I used the inducible PIP2 depletion system in which 5-phosphatase (5-ptase) coupled to FK506-binding protein 12 (FKBP12) is rapidly recruited to the membrane through dimerization with a membrane-targeted fragment of mammalian target of rapamycin (mTOR) by rapamycin, resulting in the depletion of membrane PIP2 (Varnai et al., 2006). I first confirmed that the addition of rapamycin elicited a rapid reduction of membrane PIP2 with this system, as indicated by a prompt translocation of co-expressed Tubby-R332H-YFP, a reporter of membrane PIP2 level, to the cytoplasm (Figures 7A and 7B). I then investigated the effect of PIP2 depletion on TRPM8 currents in HEK293 cells expressing the inducible PIP2-depleting system. As anticipated, TRPM8 currents were markedly inhibited by rapamycin (Figures 7C and 7D). A similar extent of inhibition by rapamycin was also observed with TRPM8-TM, suggesting that mutating Gαq gating sites does not affect the sensitivity of TRPM8 to PIP2. However, strikingly, depletion of PIP2 with rapamycin ceased to inhibit either TRPM8 or TRPM8-TM when B2R was co-expressed (Figures 7C and 7D). Co-expressed B2R also significantly inhibited the basal currents of TRPM8 and TRPM8-TM, likely due to reduced sensitivity of the channels to basal membrane PIP2. In contrast, co-expression of H1R did not affect the responses of TRPM8 to PIP2 depletion (Figure S4), suggesting that B2R, but not H1R, alters the sensitivity of TRPM8 channels to PIP2. Consistent with this idea, TRPM8 binds to B2R, but not to H1R (Figure 7E). These results suggest that the binding of B2R to TRPM8 influences the PIP2 sensitivity of TRPM8 channels, leading to lack of response of TRPM8 to PIP2 depletion. These data also confirm the finding that direct Gαq gating is the sole mechanism of TRPM8 inhibition by BK, while histamine-induced TRPM8 inhibition involves both Gαq gating and PIP2 signaling.
Gαq engages and activates PLCβ through extensive contacts with the catalytic domain in PLCβ via Switch regions and the α3 helix in Gαq (Waldo et al., 2010), the same regions also binding to the N and C termini of TRPM8 (Figure 3A). Notably, a cluster of acidic residues in Switch III of Gαq crucial for TRPM8 gating was also found critical to PLCβ binding and activation (Venkatakrishnan and Exton, 1996), raising the possibility that TRPM8 competes with PLCβ for Gαq binding, thereby interfering with Gαq-PLCβ interaction and coupling. Indeed, TRPM8 prevented the binding of Gαq to PLCβ in a nickel-beads pull-down assay (Figure 7F). Consistently, co-expression of TRPM8 also significantly reduced Gαq-PLCβ signaling activated by BK, as indicated by a significantly smaller reduction of membrane Tubby-R332H-YFP fluorescence and, hence, less PIP2 hydrolysis (Figures 7G and 7H). The reduced PIP2 hydrolysis was, however, not observed in C-terminus-deleted TRPM8 (Figures 7G and 7H), supporting a role for the C terminus of TRPM8 in the intervention of Gαq-PLCβ signaling. Functionally, co-expression of TRPM8 significantly reduced TRPV1 sensitization in both transfected HEK293 cells and DRG neurons induced by BK (Figures 7I and 7J), an event depending on Gαq-PLCβ-PKC signaling (Zhang et al., 2008), further demonstrating that TRPM8 prevents Gαq-PLC signaling.
In summary, B2R-Gαq and TRPM8 crosstalk, forming a bidirectional signaling complex. During Gαq gating of TRPM8, TRPM8 binding also reciprocally interferes with Gαq binding and signaling to PLCβ and PIP2. Concomitant B2R binding further renders TRPM8 insensitive to PIP2. The crosstalk in the TRPM8-Gαq-B2R signaling complex therefore fosters an outcome that direct Gαq gating—rather than PIP2 signaling—is a sole mechanism for TRPM8 inhibition triggered by BK during inflammation.
Discussion
In this study, I have investigated the gating mechanisms of TRPM8 by Gαq and its role in the modulation of the cold sensitivity of TRPM8 during inflammatory signaling. I found that Gαq constitutively binds to and gates TRPM8, inhibiting the basal cold sensitivity of TRPM8 in sensory DRG neurons under resting conditions. Direct Gαq gating is also responsible for TRPM8 inhibition elicited by BK and histamine under inflammatory conditions.
I further elucidated the mechanism of lack of a role for PIP2 in the modulation of TRPM8 by BK. Surprisingly, B2R binds to TRPM8, rendering the channel insensitive to PIP2 depletion. Meanwhile, TRPM8-Gαq binding interferes with Gαq-PLCβ binding and signaling, resulting in diminished sensitization of TRPV1. B2R-Gαq and TRPM8, therefore, form a bidirectional signaling complex: on the one hand, Gαq and B2R engage TRPM8, leading to direct Gαq gating and diminished PIP2 sensitivity of TRPM8, respectively; on the other hand, TRPM8 binding reciprocally prevents Gαq-PLCβ binding and signaling, leading to reduced PIP2 hydrolysis. The bidirectional signaling crosstalk in the B2R-Gαq-TRPM8 complex, therefore, collectively determines direct Gαq gating as a sole means to inhibit TRPM8 by BK. Furthermore, inhibition of sensitization of TRPV1 by co-expressed TRPM8 suggests that a non-channel-function-dependent mechanism also contributes to the analgesic effect of TRPM8. In contrast to B2R, H1R neither binds to TRPM8 nor influences the PIP2 sensitivity of TRPM8, consistent with the finding that PLCβ-PIP2 signaling plays a role in histamine-evoked TRPM8 inhibition. These results suggest that the PIP2 sensitivity of TRPM8 varies across DRG neurons influenced by co-expressed B2R.
I also revealed the molecular details of TRPM8-Gαq interaction and the functions of different binding regions. The present results support the hypothesis that the N terminus of TRPM8 is crucial to Gαq gating of TRPM8 through binding to Switch III in Gαq, while the proximal C terminus interferes with Gαq-PLCβ-PIP2 signaling through binding to the α3 helix in Gαq (Figure 3A). The proximal C terminus contains the TRP domain that is critical to the binding and sensitivity of TRPM8 to PIP2 (Rohács et al., 2005). Gαq binding to the TRP domain may, thus, also influence TRPM8-PIP2 binding and the resultant TRPM8 sensitivity to PIP2, in addition to diminished PLCβ-PIP2 signaling. It is, therefore, likely that Gαq gating and PIP2 signaling also crosstalk and cooperate to modulate TRPM8, in addition to their separate and independent roles in the modulation of TRPM8.
Apart from BK and histamine, my previous research suggests that activation of two other Gαq-coupled GPCRs— the muscarinic receptor M1R and the chloroquine receptor MrgprA3—also inhibit TRPM8 through direct Gαq gating (Li and Zhang, 2013, Than et al., 2013). Interestingly, pharmacological inhibition of PLCβ with U73122 did not exert any effect on TRPM8 inhibition induced by the activation of either of these two GPCRs in a manner similar to that of B2R, suggesting that direct Gαq gating, but not PLCβ-PIP2 signaling, is involved in the functional coupling of TRPM8 to both M1R and MrgprA3. Direct Gαq gating may be a primary mechanism underlying TRPM8 modulation by the broad family of Gαq-coupled GPCRs.
This study also provided important insights into the mechanisms of Gαq gating of TRPM8 channels. I found that Gαq gates TRPM8 through electrostatic interactions with three basic arginine residues on the N-terminal MHR1–3 of TRPM8. Neutralization of these residues abrogated Gαq binding and its inhibitory effect on TRPM8. The mutation also dramatically enhanced TRPM8 sensitivity and shifted voltage gating close to resting membrane potential, explaining the enhanced cold sensitivity and cold-evoked firing response in DRG neurons lacking Gαq. These unveiled Gαq gating sites provide insights into the general gating mechanisms of TRPM8 channels and will advance our understanding of TRPM8 modulation in physiology and diseases. They also suggest a critical role for the MHR domain in the gating of TRPM8 and, potentially, other members of the TRPM family.
How does Gαq gate TRPM8 channels? Our results suggest that R470 and the TRP domain in TRPM8 contact with Switch III and the α3 helix in Gαq, respectively (Figure 3A). This notion is further supported by structural measurements showing that the distance between R470 and the TRP domain (∼36 Å) matches the length of the α3 helix of Gαq (∼30 Å) (Figures 4A and 5I). Structural modeling of TRPM8-Gαq interaction also suggests that R470 is close to E245 on Gαq and that the top of the α3 helix is close to the bottom of the TRP domain (<10 Å) (Figure S2). However, R364 and R368 near the bottom of the cavity are far from R470 (∼47 Å) (Figure 5I). Thus, it seems impracticable that Switch III binds simultaneously to both R470 and R364-R368. However, structural modeling suggests that R364-R368 are close to the helical domain of Gαq (Figure S2), which also exhibits rich negative charges (Figure 4A), allowing for electrostatic interaction. This interaction may be responsible for the residual binding between the N terminus of TRPM8 and 5Gαqi (Figure 3B). Overall, Gαq is likely tethered to the cytoplasmic cavity of TRPM8 through interactions with the top TRP domain, lateral R470, and bottom R364-R368 in the cytoplasmic cavity via the α3 helix, Switch III, and the helical domain, respectively (Figure S2). These interactions likely impede structural coupling between different TRPM8 domains, leading to allosteric inhibition of TRPM8 gating. Another prominent feature of the model is that the top of Gαq is close to S6 in addition to the TRP domain of TRPM8. Conformation changes during Gαq activation may, thus, allow Gαq to interact with both the TRP and pore domains, which are critical for integrating allosteric gating signals from cold, cooling agonists, voltage, and PIP2 (Yin et al., 2019), mediating the allosteric gating effects of Gαq. As Switch III in Gαq is one of the regions undergoing major conformational changes upon Gαq activation and accounts for the majority of binding to TRPM8, while little structural changes occurs to the α3 helix and the helical domain, it is likely that R470 acts as a major Gαq gating site, while R364-R368 function as ancillary tethering sites assisting in Gαq gating. However, the exact mechanisms of TRPM8 gating by Gαq await future determination of the structure of the TRPM8-Gαq complex.
TRPM8 has been implicated in a plethora of pathological conditions and diseases, including pain (Colburn et al., 2007, De Caro et al., 2018, Dhaka et al., 2007, Knowlton et al., 2013, Liu et al., 2013, Proudfoot et al., 2006), inflammation (Caceres et al., 2017, Ramachandran et al., 2013, Wang et al., 2017), itch (Palkar et al., 2018), thermoregulation and energy metabolism (Li et al., 2018, Reimúndez et al., 2018), cancer (Yee, 2015), dry eye diseases (Yang et al., 2018), and airway diseases (Liu et al., 2018). TRPM8 has thus become an attractive therapeutic target for treating these conditions and diseases. Potent inhibition of TRPM8 by Gαq-coupled GPCRs provides additional therapeutic possibilities for alleviating these diseases and conditions, for example, through existing GPCR agonists or antagonists for modulating TRPM8 activities in affected tissues and organs. Furthermore, in terms of the marked impact of the Gαq effector sites on TRPM8 gating, Gαq gating sites will constitute excellent targets for high-fidelity drug discovery for effective manipulation of TRPM8 function.
Altogether, the discovery of a prominent gating mechanism of TRPM8 by Gαq and its application to inflammation in this research will advance the understanding of the role of TRPM8 in physiology and diseases and provide the basis and guidance for developing intervention strategies for combating TRPM8-associated diseases.
Star★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HA.11 | Covance | Cat# MMS-101R-1000; RRID:AB_291262 |
| Mouse monoclonal anti-β-Tubulin | Sigma-Aldrich | Cat# T4026; RRID:AB_477577 |
| Rabbit polyclonal anti-Gaq | Santa Cruz | Cat# sc-393; RRID:AB_631536 |
| Rabbit polyclonal anti-Ga11 | Santa Cruz | Cat# sc-394; RRID:AB_2111195 |
| Rabbit polyclonal anti-PLCβ | Santa Cruz | Cat# sc-9050; RRID:AB_2165496 |
| Rabbit polyclonal anti-TRPM8 | Alomone labs | Cat# ACC-049; RRID:AB_2040254 |
| Anti-GST HRP conjugated | GE healthcare | Cat# RPN1236; RRID:AB_771429 |
| Mouse monoclonal anti-V5 tag | Thermo Fisher | Cat# R960-25; RRID:AB_2556564 |
| ECL HRP-linked rabbit whole Ab | GE healthcare | Cat# NA934; RRID:AB_772206 |
| ECL HRP-linked mouse whole Ab | GE healthcare | Cat# NA931; RRID:AB_772210 |
| Biological Samples | ||
| Mice dorsal root ganglia | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM media | Thermo Fisher | Cat# 11564446 |
| Protease inhibitors | Roche Diagnostics | Cat# 11836170001 |
| FBS | Thermo Fisher | Cat# 11550356 |
| TurboFect reagent | Thermo Fisher | Cat# 15325016 |
| Mouse Laminin | Becton Dickinson | Cat# 354232 |
| NGF | Promega | Cat# G5141 |
| Poly-L-lysine | Sigma-Aldrich | Cat# P9155 |
| Cytosine β-D-arabinofuranoside | Sigma-Aldrich | Cat# C1768 |
| Collagenase type IV | Worthington | Cat# LS004186 |
| Bradykinin | Tocris Bioscience | Car# 3004 |
| Rapamycin | Tocris Bioscience | Cat# 1292 |
| Quick-change mutagenesis kit | Agilent Technologies | Cat# 200521 |
| PBMC | Focus Biomolecules | Cat# 10-1413 |
| Glutathione-agarose | Sigma-Aldrich | Cat# G4510 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL11268 |
| Gαq−/− MEF cells | Provided by Prof. Stefan Offermanns | Zywietz et al., 2001 |
| Experimental Models: Organisms/Strains | ||
| Gαq knockout mice | Provided by Prof. Stefan Offermanns | Offermanns et al., 1997b |
| Recombinant DNA | ||
| Rat TRPM8-V5-His | Zhang et al., 2012 | N/A |
| G protein alpha q | cDNA resource center | Cat# GNA0Q00000 |
| Bradykinin receptor B2R | cDNA resource center | Cat# BDKB20TN00 |
| Histamine receptor H1R | cDNA resource center | Cat# HRH010TN00 |
| 6xHis-tag PLCβ1 | Provided by Prof. Elliott M. Ross (UTSMC) | N/A |
| mRFB-FKBP-5-ptase | Provided by Dr. Gerry Hammond (University of Pittsburgh) | Varnai et al., 2006 |
| PM-FRB-CFP | Provided by Dr. Gerry Hammond | Varnai et al., 2006 |
| Tubby-R332H-cYFP | Provided by Dr. Gerry Hammond | Quinn et al., 2008 |
| GST-pcDNA3 | Hasan et al., 2017 | N/A |
| Software and Algorithms | ||
| Sigmaplot | Systat Software Inc. | https://systatsoftware.com/products/sigmaplot/ |
| pClamp11 | Molecular Device | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite |
| Discovery studio 4.5 | BIOVIA | https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| Axopatch 200B amplifier | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/amplifiers/axon-instruments-patch-clamp-amplifiers#gref |
| Confocal microscope | Leica | https://www.leica-microsystems.com/products/confocal-microscopes/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xuming Zhang (x.zhang39@aston.ac.uk).
Experimental Model and Subject Details
Animals
Gαq−/− mice were kindly provided by Prof. Stefan Offermanns (Max-Planck-Institute, Bad Nauheim, Germany). The male and female mice and their littermates were maintained on a C57BL/6 background housed in a 12h light/dark cycle with food and water ad libitum. The experiments on mice were approved by the ethical review committee of Aston University and UK home office and carried out in accordance with the Animal Scientific Procedures Act 1986 in the UK. The knockout mice are viable but exhibit impaired motor coordination and defective platelet activation (Offermanns et al., 1997a, Offermanns et al., 1997b).
DRG neurons and cell lines
DRG neurons were isolated from male and female wild-type or Gαq−/− mice of 2-7 days after birth following sacrifice by cervical dislocation and cultured as described previously (Than et al., 2013, Zhang et al., 2008). Briefly, isolated DRGs were treated with type IV collagenase (Worthington, LS004186) followed by trituration. The dissociated DRG neurons were then seeded onto coverslips coated with poly-L-lysine (Sigma, P9155) and laminin (BD, 354232) and maintained in DMEM media (Thermo fisher, 11564446) containing 10% fetal bovine serum (Thermo Fisher, 11550356), 2mM L-glutamine, 100IU/ml penicillin, 100 μg/ml streptomycin supplemented with 5 μM cytosine β-D-arabinofuranoside (Sigma, C1768) and 50ng/ml nerve growth factor (Promega, G5141). The cultured DRG neurons were used within 24 hours after isolation.
HEK293 cell line was purchased from ATCC, and MEF cells derived from Gαq/11−/− mice were obtained from Prof. Stefan Offermanns (Max-Planck-Institute, Germany) as described previously (Zhang et al., 2012). The cells lines were maintained in high glucose DMEM media containing 10% fetal bovine serum, 100IU/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified incubator (5% CO2). Cells were used between 15 and 30 passage numbers. The cell lines were derived from embryos and the sex of these cell lines is thus not available.
Method Details
Cell transfection
HEK293 and MEF cells were transfected with plasmid cDNAs using TurboFect transfection reagent (Thermo Fisher Scientific, 15325016) as described previously (Hasan et al., 2017, Zhang et al., 2012). Briefly, 4 μg cDNA in 400 μL serum-free media was mixed with 8 μL TurboFect reagent followed by incubation for 20 min at RT. The transfection complexes were then added to the cells and incubated for 24h. For single cell electrophysiology, cells were co-transfected with GFP for identification of successfully transfected cells.
Molecular Biology
GST-coupled TRPM8 cytoplasmic fragments were constructed by amplifying relevant N- and C-terminal fragments from rTRPM8 with PCR followed by cloning into the GST-pCDNA3 vector via BamHI and EcoRI (Hasan et al., 2017). Truncation of GST-coupled protein fragments was produced by introducing a stop codon at targeted sites using Quick-Change mutagenesis kit (Agilent Technologies, 200521). The kit was also used to generate all the TRPM8 mutants. cNDAs for HA-B2R (BDKB20TN00) and HA-H1R (HRH010TN00) and Gαq (GNA0Q00000) were from the Missouri cDNA resource center. PLCβ-His (6x) cNDA is a kind gift from Dr. Elliott Ross (University of Texas Southwestern Medical Center at Dallas). The membrane lipid PIP2 depletion was induced using mRFB-FKBP-5-ptase catalytic domain together with membrane targeted PM-FRB-CFP (kindly provide by Dr. Gerry Hammond, University of Pittsburgh), as described (Varnai et al., 2006). All other cDNA constructs including TRPM8-V5-His, Tubby-R332H-YFP and Gαqi chimeras have been described in the previous paper (Zhang et al., 2012).
Pull down assay and western blot
GST pull down assay was performed as described previously (Hasan et al., 2017, Zhang et al., 2012). Briefly, GST-coupled TRPM8 cytoplasmic fragments expressed from HEK293 cells were isolated using GST-agarose (Sigma, G4510), and then incubated with protein lysate containing Gαq Q209L in a binding buffer (20mM Tris-HCl, pH 7.4, 150mM NaCl, 1% NP-40, 1mM EDTA, 1mM EGTA and protease inhibitor cocktails (Roche, 11836170001)) at 4°C overnight. Bound Gαq proteins were eluted by boiling in sample buffer followed by separation in 10% SDS-PAGE gel and detection with primary anti-Gαq (Santa Cruz, sc-393) and secondary HRP-linked rabbit antibody (GE healthcare, NA934). The blots were stripped and redetected with HRP-conjugated anti-GST (GE healthcare, RPN1236).
Nickel beads pull down assay was conducted as described previously with slight modifications (Zhang et al., 2012). Briefly, HEK293 cells expressing TRPM8-V5-His with HA-B2R or with HA-H1R were solubilized in lysis buffer (20mM Tris-HCl, pH7.4, 300mM NaCl, 1% NP-40, 10% glycerol, 0.2mM EDTA, 20mM imidazole plus protease inhibitor cocktail). Protein lysates were then incubated with nickel beads (QIAGEN) at 4°C overnight. After thorough washing in lysis buffer, nickel beads were boiled in sample buffer. Eluted proteins were resolved on 7.5% SDS-PAGE gel followed by western blot detection with anti-HA (Covance, 101R-1000) and anti-V5 (Thermo Fisher, R960-25) together with secondary HRP-linked mouse antibody (GE healthcare, NA931). Similar nickel pull down assay was also performed for determination of the effect of TRPM8 on Gαq binding to hexahistidine-tagged PLCβ (Figure 7E). In this case, HEK293 cell lysate expressing TRPM8 was added to the cell lysate containing PLCβ-his and Gαq Q209L. After incubation at 4°C overnight, the cell lysate mixture was thoroughly washed and bound Gαq protein was then detected with anti-Gαq followed by stripping and redetection with anti-PLCβ (Santa Cruz, sc-9050). All the blots shown are representative of at least three independent experiments.
The expression level of TRPM8, Gαq and Gα11 in Gαq−/− mice was detected using western blot with anti-TRPM8 (Alomone labs, ACC-049), anti-Gαq (Santa Cruz, sc-393) and anti-Gα11 (Santa Cruz, sc-394), respectively. The blots were also stripped and redetected with anti-β-tubulin (Sigma, T4026) for verifying equal loading across samples.
Electrophysiology
All electrophysiology recordings were obtained using an Axopatch 200B patch amplifier (Molecular Devices) as described previously (Hasan et al., 2017, Zhang et al., 2012). Briefly, patch pipettes were fabricated from thin-walled glass capillary using a pipette puller (Sutter instrument) with a resistance between 2.5 ∼4.0 MΩ and filled with internal solution (in mM): 140 KCl, 2.0 MgCl2, 5 EGTA. 10 HEPES, pH 7.4 with KOH. Ca2+-free extracellular solution was used and contained (in mM): 140 NaCl, 4 KCl, 10 HEPES, 1 MgCl2, 5 EGTA, 5 Glucose, pH7.4 with NaOH. Ca2+-containing Hanks solution was largely similar to Ca2+-free solution except that 5mM EGTA in Ca2+-free solution was replaced with 2mM CaCl2. For studying the effect of GPCR and PIP2 signaling on TRPM8 in Figure 6 and 7, cells were pretreated with BK (Tocris, 3004) or histamine or rapamycin (Tocris, 1292) for 1min to allow intact completion of signaling before entering whole-cell configuration. After establishment of whole-cell, series resistance was 80% compensated. Signals were analog filtered at 1 KHz using a low-pass Bessel filter of the amplifier and digitized with Digidata 1440A (Molecular Devices). Recordings were made at RT with a holding potential at −60mV or + 60mV.
The current-voltage (I-V) relationship of TRPM8 was determined using a ramp depolarization from −120mV to +120mV in 650 msec. Voltage-dependent activation of TRPM8 was further investigated from currents evoked by depolarization voltage steps from −120mV up to +200mV in 20mV increments with every step lasting for 100msec.The half-maximal activation voltage (V1/2) was calculated using the Boltzmann equation as described previously (Zhang et al., 2012).
Cell-attached recordings were performed as described previously (Madrid et al., 2006, Perkins, 2006, Zhang et al., 2012). Patch pipettes were pulled from thick-walled glass tubing with a resistance between 8 ∼12 MΩ and a tight seal with seal resistance over 1GΩ was typical obtained. Cell-attached voltage-clamp mode was then used to measure action potential currents in DRG neurons evoked by different stimuli under normal Ca2+-containing Hanks solution (see above). Cell-attached patch clamping maintains the intracellular contents of cells allowing for reliable and long lasting recording of action potentials. As TRPM8 is mainly expressed in small-to-medium sized DRG neurons (Dhaka et al., 2008), we probed at random the responses of small-to-medium DRG neurons to cold, WS-12 and PBMC (Focus Biomolecules, 10-1413) for identification of TRPM8+ neurons. Normal Hanks solution was used for both bath and pipette solutions.
Fluorescence imaging
Imaging of Tubby-R332H-YFP in live cells were performed using a Leica confocal microscope (Leica) as described previously (Li and Zhang, 2013, Than et al., 2013, Zhang et al., 2012). Fluorescence signals were acquired every 0.75 s. Bradykinin (1 μM) or rapamycin (1 μM) was added to the bath solution during imaging. Membrane PIP2 depletion was estimated by quantifying the ratio of fluorescence signals on the membrane to those in the cytosol using ImageJ (NIH).
Protein structure modeling
The structure of TRPM8 (PDB: 6BPQ) and Gαq (PDB: 3OHM) was analyzed and modeled using Discovery studio 4.5 (BIOVIA).
Quantification and Statistical Analysis
Firing events of DRG neurons and TRPM8 currents were quantified and analyzed using the Clampfit 10.2 software (Molecular Devices). The number of cells was denoted in parentheses above the bars of each figure and derived from at least three different days of preparations. All the data passed the normality test and are shown as mean ± SEM. Significance between groups was determined using Student’s t test or one-way ANOVA, and considered significant at p < 0.05.
Acknowledgments
I would like to thank Dr. Dan Rathbone for assistance with protein structure modeling and Dr. Gerry Hammond for help with live imaging and analysis. This work was supported by an MRC grant (G0801387) and intramural funding from Aston University. Original data for this work are available through Aston data Explorer at https://doi.org/10.17036/researchdata.aston.ac.uk.00000427.
Author Contributions
X.Z. designed and performed experiments, analyzed data, and wrote the paper.
Declaration of Interests
The author declares no competing interests.
Published: June 18, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.080.
Supplemental Information
References
- Bautista D.M., Siemens J., Glazer J.M., Tsuruda P.R., Basbaum A.I., Stucky C.L., Jordt S.E., Julius D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204–208. doi: 10.1038/nature05910. [DOI] [PubMed] [Google Scholar]; Bautista, D.M., Siemens, J., Glazer, J.M., Tsuruda, P.R., Basbaum, A.I., Stucky, C.L., Jordt, S.E., and Julius, D. (2007). The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204-208. [DOI] [PubMed]
- Caceres A.I., Liu B., Jabba S.V., Achanta S., Morris J.B., Jordt S.E. Transient receptor potential cation channel subfamily M member 8 channels mediate the anti-inflammatory effects of eucalyptol. Br. J. Pharmacol. 2017;174:867–879. doi: 10.1111/bph.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]; Caceres, A.I., Liu, B., Jabba, S.V., Achanta, S., Morris, J.B., and Jordt, S.E. (2017). Transient receptor potential cation channel subfamily M member 8 channels mediate the anti-inflammatory effects of eucalyptol. Br. J. Pharmacol. 174, 867-879. [DOI] [PMC free article] [PubMed]
- Colburn R.W., Lubin M.L., Stone D.J., Jr., Wang Y., Lawrence D., D’Andrea M.R., Brandt M.R., Liu Y., Flores C.M., Qin N. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54:379–386. doi: 10.1016/j.neuron.2007.04.017. [DOI] [PubMed] [Google Scholar]; Colburn, R.W., Lubin, M.L., Stone, D.J., Jr., Wang, Y., Lawrence, D., D’Andrea, M.R., Brandt, M.R., Liu, Y., Flores, C.M., and Qin, N. (2007). Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379-386. [DOI] [PubMed]
- Daniels R.L., Takashima Y., McKemy D.D. Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 2009;284:1570–1582. doi: 10.1074/jbc.M807270200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Daniels, R.L., Takashima, Y., and McKemy, D.D. (2009). Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 284, 1570-1582. [DOI] [PMC free article] [PubMed]
- De Caro C., Russo R., Avagliano C., Cristiano C., Calignano A., Aramini A., Bianchini G., Allegretti M., Brandolini L. Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Br. J. Pharmacol. 2018;175:1691–1706. doi: 10.1111/bph.14177. [DOI] [PMC free article] [PubMed] [Google Scholar]; De Caro, C., Russo, R., Avagliano, C., Cristiano, C., Calignano, A., Aramini, A., Bianchini, G., Allegretti, M., and Brandolini, L. (2018). Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Br. J. Pharmacol. 175, 1691-1706. [DOI] [PMC free article] [PubMed]
- Dhaka A., Murray A.N., Mathur J., Earley T.J., Petrus M.J., Patapoutian A. TRPM8 is required for cold sensation in mice. Neuron. 2007;54:371–378. doi: 10.1016/j.neuron.2007.02.024. [DOI] [PubMed] [Google Scholar]; Dhaka, A., Murray, A.N., Mathur, J., Earley, T.J., Petrus, M.J., and Patapoutian, A. (2007). TRPM8 is required for cold sensation in mice. Neuron 54, 371-378. [DOI] [PubMed]
- Dhaka A., Earley T.J., Watson J., Patapoutian A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 2008;28:566–575. doi: 10.1523/JNEUROSCI.3976-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dhaka, A., Earley, T.J., Watson, J., and Patapoutian, A. (2008). Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 28, 566-575. [DOI] [PMC free article] [PubMed]
- Gavva N.R., Davis C., Lehto S.G., Rao S., Wang W., Zhu D.X. Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain. 2012;8:36. doi: 10.1186/1744-8069-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gavva, N.R., Davis, C., Lehto, S.G., Rao, S., Wang, W., and Zhu, D.X. (2012). Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain 8, 36. [DOI] [PMC free article] [PubMed]
- Han S.K., Mancino V., Simon M.I. Phospholipase Cbeta 3 mediates the scratching response activated by the histamine H1 receptor on C-fiber nociceptive neurons. Neuron. 2006;52:691–703. doi: 10.1016/j.neuron.2006.09.036. [DOI] [PubMed] [Google Scholar]; Han, S.K., Mancino, V., and Simon, M.I. (2006). Phospholipase Cbeta 3 mediates the scratching response activated by the histamine H1 receptor on C-fiber nociceptive neurons. Neuron 52, 691-703. [DOI] [PubMed]
- Hasan R., Leeson-Payne A.T., Jaggar J.H., Zhang X. Calmodulin is responsible for Ca2+-dependent regulation of TRPA1 Channels. Sci. Rep. 2017;7:45098. doi: 10.1038/srep45098. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hasan, R., Leeson-Payne, A.T., Jaggar, J.H., and Zhang, X. (2017). Calmodulin is responsible for Ca2+-dependent regulation of TRPA1 Channels. Sci. Rep. 7, 45098. [DOI] [PMC free article] [PubMed]
- Knowlton W.M., Daniels R.L., Palkar R., McCoy D.D., McKemy D.D. Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS ONE. 2011;6:e25894. doi: 10.1371/journal.pone.0025894. [DOI] [PMC free article] [PubMed] [Google Scholar]; Knowlton, W.M., Daniels, R.L., Palkar, R., McCoy, D.D., and McKemy, D.D. (2011). Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS ONE 6, e25894. [DOI] [PMC free article] [PubMed]
- Knowlton W.M., Palkar R., Lippoldt E.K., McCoy D.D., Baluch F., Chen J., McKemy D.D. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J. Neurosci. 2013;33:2837–2848. doi: 10.1523/JNEUROSCI.1943-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]; Knowlton, W.M., Palkar, R., Lippoldt, E.K., McCoy, D.D., Baluch, F., Chen, J., and McKemy, D.D. (2013). A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J. Neurosci. 33, 2837-2848. [DOI] [PMC free article] [PubMed]
- Lambright D.G., Sondek J., Bohm A., Skiba N.P., Hamm H.E., Sigler P.B. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]; Lambright, D.G., Sondek, J., Bohm, A., Skiba, N.P., Hamm, H.E., and Sigler, P.B. (1996). The 2.0 A crystal structure of a heterotrimeric G protein. Nature 379, 311-319. [DOI] [PubMed]
- Li L., Zhang X. Differential inhibition of the TRPM8 ion channel by Gαq and Gα 11. Channels (Austin) 2013;7:115–118. doi: 10.4161/chan.23466. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li, L., and Zhang, X. (2013). Differential inhibition of the TRPM8 ion channel by Gαq and Gα 11. Channels (Austin) 7, 115-118. [DOI] [PMC free article] [PubMed]
- Li C., Li J., Xiong X., Liu Y., Lv Y., Qin S., Liu D., Wei R., Ruan X., Zhang J. TRPM8 activation improves energy expenditure in skeletal muscle and exercise endurance in mice. Gene. 2018;641:111–116. doi: 10.1016/j.gene.2017.10.045. [DOI] [PubMed] [Google Scholar]; Li, C., Li, J., Xiong, X., Liu, Y., Lv, Y., Qin, S., Liu, D., Wei, R., Ruan, X., Zhang, J., et al. (2018). TRPM8 activation improves energy expenditure in skeletal muscle and exercise endurance in mice. Gene 641, 111-116. [DOI] [PubMed]
- Linte R.M., Ciobanu C., Reid G., Babes A. Desensitization of cold- and menthol-sensitive rat dorsal root ganglion neurones by inflammatory mediators. Exp. Brain Res. 2007;178:89–98. doi: 10.1007/s00221-006-0712-3. [DOI] [PubMed] [Google Scholar]; Linte, R.M., Ciobanu, C., Reid, G., and Babes, A. (2007). Desensitization of cold- and menthol-sensitive rat dorsal root ganglion neurones by inflammatory mediators. Exp. Brain Res. 178, 89-98. [DOI] [PubMed]
- Liu B., Qin F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005;25:1674–1681. doi: 10.1523/JNEUROSCI.3632-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, B., and Qin, F. (2005). Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 1674-1681. [DOI] [PMC free article] [PubMed]
- Liu B., Fan L., Balakrishna S., Sui A., Morris J.B., Jordt S.E. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain. 2013;154:2169–2177. doi: 10.1016/j.pain.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, B., Fan, L., Balakrishna, S., Sui, A., Morris, J.B., and Jordt, S.E. (2013). TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 154, 2169-2177. [DOI] [PMC free article] [PubMed]
- Liu H., Liu Q., Hua L., Pan J. Inhibition of transient receptor potential melastatin 8 alleviates airway inflammation and remodeling in a murine model of asthma with cold air stimulus. Acta Biochim. Biophys. Sin. (Shanghai) 2018;50:499–506. doi: 10.1093/abbs/gmy033. [DOI] [PubMed] [Google Scholar]; Liu, H., Liu, Q., Hua, L., and Pan, J. (2018). Inhibition of transient receptor potential melastatin 8 alleviates airway inflammation and remodeling in a murine model of asthma with cold air stimulus. Acta Biochim. Biophys. Sin. (Shanghai) 50, 499-506. [DOI] [PubMed]
- Lukacs V., Thyagarajan B., Varnai P., Balla A., Balla T., Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 2007;27:7070–7080. doi: 10.1523/JNEUROSCI.1866-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lukacs, V., Thyagarajan, B., Varnai, P., Balla, A., Balla, T., and Rohacs, T. (2007). Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 27, 7070-7080. [DOI] [PMC free article] [PubMed]
- Madrid R., Donovan-Rodríguez T., Meseguer V., Acosta M.C., Belmonte C., Viana F. Contribution of TRPM8 channels to cold transduction in primary sensory neurons and peripheral nerve terminals. J. Neurosci. 2006;26:12512–12525. doi: 10.1523/JNEUROSCI.3752-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Madrid, R., Donovan-Rodriguez, T., Meseguer, V., Acosta, M.C., Belmonte, C., and Viana, F. (2006). Contribution of TRPM8 channels to cold transduction in primary sensory neurons and peripheral nerve terminals. J. Neurosci. 26, 12512-12525. [DOI] [PMC free article] [PubMed]
- Madrid R., de la Peña E., Donovan-Rodriguez T., Belmonte C., Viana F. Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J. Neurosci. 2009;29:3120–3131. doi: 10.1523/JNEUROSCI.4778-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; Madrid, R., de la Peña, E., Donovan-Rodriguez, T., Belmonte, C., and Viana, F. (2009). Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J. Neurosci. 29, 3120-3131. [DOI] [PMC free article] [PubMed]
- McKemy D.D., Neuhausser W.M., Julius D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature. 2002;416:52–58. doi: 10.1038/nature719. [DOI] [PubMed] [Google Scholar]; McKemy, D.D., Neuhausser, W.M., and Julius, D. (2002). Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52-58. [DOI] [PubMed]
- Offermanns S., Hashimoto K., Watanabe M., Sun W., Kurihara H., Thompson R.F., Inoue Y., Kano M., Simon M.I. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc. Natl. Acad. Sci. USA. 1997;94:14089–14094. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]; Offermanns, S., Hashimoto, K., Watanabe, M., Sun, W., Kurihara, H., Thompson, R.F., Inoue, Y., Kano, M., and Simon, M.I. (1997a). Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc. Natl. Acad. Sci. USA 94, 14089-14094. [DOI] [PMC free article] [PubMed]
- Offermanns S., Toombs C.F., Hu Y.H., Simon M.I. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]; Offermanns, S., Toombs, C.F., Hu, Y.H., and Simon, M.I. (1997b). Defective platelet activation in G alpha(q)-deficient mice. Nature 389, 183-186. [DOI] [PubMed]
- Palkar R., Ongun S., Catich E., Li N., Borad N., Sarkisian A., McKemy D.D. Cooling relief of acute and chronic itch requires TRPM8 channels and neurons. J. Invest. Dermatol. 2018;138:1391–1399. doi: 10.1016/j.jid.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; Palkar, R., Ongun, S., Catich, E., Li, N., Borad, N., Sarkisian, A., and McKemy, D.D. (2018). Cooling relief of acute and chronic itch requires TRPM8 channels and neurons. J. Invest. Dermatol. 138, 1391-1399. [DOI] [PMC free article] [PubMed]
- Parra A., Madrid R., Echevarria D., del Olmo S., Morenilla-Palao C., Acosta M.C., Gallar J., Dhaka A., Viana F., Belmonte C. Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat. Med. 2010;16:1396–1399. doi: 10.1038/nm.2264. [DOI] [PubMed] [Google Scholar]; Parra, A., Madrid, R., Echevarria, D., del Olmo, S., Morenilla-Palao, C., Acosta, M.C., Gallar, J., Dhaka, A., Viana, F., and Belmonte, C. (2010). Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat. Med. 16, 1396-1399. [DOI] [PubMed]
- Peier A.M., Moqrich A., Hergarden A.C., Reeve A.J., Andersson D.A., Story G.M., Earley T.J., Dragoni I., McIntyre P., Bevan S., Patapoutian A. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108:705–715. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]; Peier, A.M., Moqrich, A., Hergarden, A.C., Reeve, A.J., Andersson, D.A., Story, G.M., Earley, T.J., Dragoni, I., McIntyre, P., Bevan, S., and Patapoutian, A. (2002). A TRP channel that senses cold stimuli and menthol. Cell 108, 705-715. [DOI] [PubMed]
- Perkins K.L. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J. Neurosci. Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; Perkins, K.L. (2006). Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J. Neurosci. Methods 154, 1-18. [DOI] [PMC free article] [PubMed]
- Premkumar L.S., Raisinghani M., Pingle S.C., Long C., Pimentel F. Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J. Neurosci. 2005;25:11322–11329. doi: 10.1523/JNEUROSCI.3006-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Premkumar, L.S., Raisinghani, M., Pingle, S.C., Long, C., and Pimentel, F. (2005). Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J. Neurosci. 25, 11322-11329. [DOI] [PMC free article] [PubMed]
- Proudfoot C.J., Garry E.M., Cottrell D.F., Rosie R., Anderson H., Robertson D.C., Fleetwood-Walker S.M., Mitchell R. Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr. Biol. 2006;16:1591–1605. doi: 10.1016/j.cub.2006.07.061. [DOI] [PubMed] [Google Scholar]; Proudfoot, C.J., Garry, E.M., Cottrell, D.F., Rosie, R., Anderson, H., Robertson, D.C., Fleetwood-Walker, S.M., and Mitchell, R. (2006). Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr. Biol. 16, 1591-1605. [DOI] [PubMed]
- Quinn K.V., Behe P., Tinker A. Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol. 2008;586:2855–2871. doi: 10.1113/jphysiol.2008.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]; Quinn, K.V., Behe, P., and Tinker, A. (2008). Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol 586, 2855-2871. [DOI] [PMC free article] [PubMed]
- Ramachandran R., Hyun E., Zhao L., Lapointe T.K., Chapman K., Hirota C.L., Ghosh S., McKemy D.D., Vergnolle N., Beck P.L. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl. Acad. Sci. USA. 2013;110:7476–7481. doi: 10.1073/pnas.1217431110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ramachandran, R., Hyun, E., Zhao, L., Lapointe, T.K., Chapman, K., Hirota, C.L., Ghosh, S., McKemy, D.D., Vergnolle, N., Beck, P.L., et al. (2013). TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl. Acad. Sci. USA 110, 7476-7481. [DOI] [PMC free article] [PubMed]
- Reimúndez A., Fernández-Peña C., García G., Fernández R., Ordás P., Gallego R., Pardo-Vazquez J.L., Arce V., Viana F., Señarís R. Deletion of the cold thermoreceptor TRPM8 increases heat loss and food intake leading to reduced body temperature and obesity in mice. J. Neurosci. 2018;38:3643–3656. doi: 10.1523/JNEUROSCI.3002-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reimundez, A., Fernandez-Peña, C., Garcia, G., Fernandez, R., Ordas, P., Gallego, R., Pardo-Vazquez, J.L., Arce, V., Viana, F., and Señaris, R. (2018). Deletion of the cold thermoreceptor TRPM8 increases heat loss and food intake leading to reduced body temperature and obesity in mice. J. Neurosci. 38, 3643-3656. [DOI] [PMC free article] [PubMed]
- Rohács T., Lopes C.M., Michailidis I., Logothetis D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005;8:626–634. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]; Rohacs, T., Lopes, C.M., Michailidis, I., and Logothetis, D.E. (2005). PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8, 626-634. [DOI] [PubMed]
- Than J.Y., Li L., Hasan R., Zhang X. Excitation and modulation of TRPA1, TRPV1, and TRPM8 channel-expressing sensory neurons by the pruritogen chloroquine. J. Biol. Chem. 2013;288:12818–12827. doi: 10.1074/jbc.M113.450072. [DOI] [PMC free article] [PubMed] [Google Scholar]; Than, J.Y., Li, L., Hasan, R., and Zhang, X. (2013). Excitation and modulation of TRPA1, TRPV1, and TRPM8 channel-expressing sensory neurons by the pruritogen chloroquine. J. Biol. Chem. 288, 12818-12827. [DOI] [PMC free article] [PubMed]
- Varnai P., Thyagarajan B., Rohacs T., Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 2006;175:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]; Varnai, P., Thyagarajan, B., Rohacs, T., and Balla, T. (2006). Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 175, 377-382. [DOI] [PMC free article] [PubMed]
- Venkatakrishnan G., Exton J.H. Identification of determinants in the alpha-subunit of Gq required for phospholipase C activation. J. Biol. Chem. 1996;271:5066–5072. doi: 10.1074/jbc.271.9.5066. [DOI] [PubMed] [Google Scholar]; Venkatakrishnan, G., and Exton, J.H. (1996). Identification of determinants in the alpha-subunit of Gq required for phospholipase C activation. J. Biol. Chem. 271, 5066-5072. [DOI] [PubMed]
- Waldo G.L., Ricks T.K., Hicks S.N., Cheever M.L., Kawano T., Tsuboi K., Wang X., Montell C., Kozasa T., Sondek J., Harden T.K. Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science. 2010;330:974–980. doi: 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]; Waldo, G.L., Ricks, T.K., Hicks, S.N., Cheever, M.L., Kawano, T., Tsuboi, K., Wang, X., Montell, C., Kozasa, T., Sondek, J., and Harden, T.K. (2010). Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330, 974-980. [DOI] [PMC free article] [PubMed]
- Wang X.P., Yu X., Yan X.J., Lei F., Chai Y.S., Jiang J.F., Yuan Z.Y., Xing D.M., Du L.J. TRPM8 in the negative regulation of TNFα expression during cold stress. Sci. Rep. 2017;7:45155. doi: 10.1038/srep45155. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, X.P., Yu, X., Yan, X.J., Lei, F., Chai, Y.S., Jiang, J.F., Yuan, Z.Y., Xing, D.M., and Du, L.J. (2017). TRPM8 in the negative regulation of TNFα expression during cold stress. Sci. Rep. 7, 45155. [DOI] [PMC free article] [PubMed]
- Yang J.M., Wei E.T., Kim S.J., Yoon K.C. TRPM8 channels and dry eye. Pharmaceuticals (Basel) 2018;11:E125. doi: 10.3390/ph11040125. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang, J.M., Wei, E.T., Kim, S.J., and Yoon, K.C. (2018). TRPM8 channels and dry eye. Pharmaceuticals (Basel) 11, E125. [DOI] [PMC free article] [PubMed]
- Yee N.S. Roles of TRPM8 ion channels in cancer: proliferation, survival, and invasion. Cancers (Basel) 2015;7:2134–2146. doi: 10.3390/cancers7040882. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yee, N.S. (2015). Roles of TRPM8 ion channels in cancer: proliferation, survival, and invasion. Cancers (Basel) 7, 2134-2146. [DOI] [PMC free article] [PubMed]
- Yin Y., Wu M., Zubcevic L., Borschel W.F., Lander G.C., Lee S.Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science. 2018;359:237–241. doi: 10.1126/science.aan4325. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yin, Y., Wu, M., Zubcevic, L., Borschel, W.F., Lander, G.C., and Lee, S.Y. (2018). Structure of the cold- and menthol-sensing ion channel TRPM8. Science 359, 237-241. [DOI] [PMC free article] [PubMed]
- Yin Y., Le S.C., Hsu A.L., Borgnia M.J., Yang H., Lee S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science. 2019;363:eaav9334. doi: 10.1126/science.aav9334. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yin, Y., Le, S.C., Hsu, A.L., Borgnia, M.J., Yang, H., and Lee, S.Y. (2019). Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 363, eaav9334. [DOI] [PMC free article] [PubMed]
- Zakharian E., Thyagarajan B., French R.J., Pavlov E., Rohacs T. Inorganic polyphosphate modulates TRPM8 channels. PLoS ONE. 2009;4:e5404. doi: 10.1371/journal.pone.0005404. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zakharian, E., Thyagarajan, B., French, R.J., Pavlov, E., and Rohacs, T. (2009). Inorganic polyphosphate modulates TRPM8 channels. PLoS ONE 4, e5404. [DOI] [PMC free article] [PubMed]
- Zhang X., Li L., McNaughton P.A. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59:450–461. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]; Zhang, X., Li, L., and McNaughton, P.A. (2008). Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59, 450-461. [DOI] [PubMed]
- Zhang X., Mak S., Li L., Parra A., Denlinger B., Belmonte C., McNaughton P.A. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat. Cell Biol. 2012;14:851–858. doi: 10.1038/ncb2529. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang, X., Mak, S., Li, L., Parra, A., Denlinger, B., Belmonte, C., and McNaughton, P.A. (2012). Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat. Cell Biol. 14, 851-858. [DOI] [PMC free article] [PubMed]
- Zywietz A., Gohla A., Schmelz M., Schultz G., Offermanns S. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. involvement of Gq but not G11. J. Biol. Chem. 2001;276:3840–3845. doi: 10.1074/jbc.M007819200. [DOI] [PubMed] [Google Scholar]; Zywietz, A., Gohla, A., Schmelz, M., Schultz, G., and Offermanns, S. (2001). Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. involvement of Gq but not G11. J. Biol. Chem. 276, 3840-3845. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.