

Abstract
Acetylcholine is released in the prefrontal cortex (PFC) and is a key modulator of cognitive performance in primates. Cholinergic stimulation has been shown to have beneficial effects on performance of cognitive tasks, and cholinergic receptors are being actively explored as promising targets for ameliorating cognitive deficits in Alzheimer's disease. We hypothesized that cholinergic stimulation of PFC during performance of a cognitive task would augment neuronal activity and neuronal coding of task attributes. We iontophoretically applied the general cholinergic receptor agonist carbachol onto neurons in dorsolateral PFC (DLPFC) of male rhesus macaques performing rule-guided prosaccades and antisaccades, a well established oculomotor task for testing cognitive control. Carbachol application had heterogeneous effects on neuronal excitability, with both excitation and suppression observed in significant proportions. Contrary to our prediction, neurons with rule-selective activity exhibited a reduction in selectivity during carbachol application. Cholinergic stimulation disrupted rule selectivity regardless of whether it had suppressive or excitatory effects on these neurons. In addition, cholinergic stimulation excited putative pyramidal neurons, whereas the activity of putative interneurons remained unchanged. Moreover, cholinergic stimulation attenuated saccade direction selectivity in putative pyramidal neurons due to nonspecific increases in activity. Our results suggest excessive cholinergic stimulation has detrimental effects on DLPFC representations of task attributes. These findings delineate the complexity and heterogeneity of neuromodulation of cerebral cortex by cholinergic stimulation, an area of active exploration with respect to the development of cognitive enhancers.
SIGNIFICANCE STATEMENT The neurotransmitter acetylcholine is known to be important for cognitive processes in the prefrontal cortex. Removal of acetylcholine from prefrontal cortex can disrupt short-term memory performance and is reminiscent of Alzheimer's disease, which is characterized by degeneration of acetylcholine-producing neurons. Stimulation of cholinergic receptors is being explored to create cognitive enhancers for the treatment of Alzheimer's disease and other psychiatric diseases. Here, we stimulated cholinergic receptors in prefrontal cortex and examined its effects on neurons that are engaged in cognitive behavior. Surprisingly, cholinergic stimulation decreased neurons' ability to discriminate between rules. This work suggests that overstimulation of acetylcholine receptors could disrupt neuronal processing during cognition and is relevant to the design of cognitive enhancers based on stimulating the cholinergic system.
Keywords: acetylcholine, carbachol, DLPFC, iontophoresis, macaque, rule
Introduction
Acetylcholine (ACh) is central to optimal cognitive performance in primates (Ballinger et al., 2016). Lesions of the basal forebrain nuclei, which provide corticopetal cholinergic innervation in primates (Mesulam et al., 1983), cause deficits in a variety of contexts, including acquisition of visual discriminations (Ridley et al., 1984), shifting of spatial attention (Voytko et al., 1994), and mnemonic tasks such as delayed match-to-sample (Aigner et al., 1991). The importance of prefrontal cholinergic tone was delineated by Croxson et al. (2011), who tested macaques on a variety of tasks after cholinergic deafferentation of prefrontal cortex (PFC). Ablation of cholinergic neurons revealed selective, delay-dependent deficits in spatial working memory performance, but not in other PFC-dependent behavioral tasks.
Extensive degeneration of cholinergic neurons is also a hallmark of Alzheimer's disease (Whitehouse et al., 1982). Cholinesterase inhibitors are the mainstay of pharmacological treatment, but their efficacy has been questioned (Amenta et al., 2001). Alternative measures of enhancing cholinergic stimulation are being investigated as treatment strategies (Caccamo et al., 2009; Foucault-Fruchard and Antier, 2017). For example, recent evidence indicates that intermittent electrical stimulation of the basal forebrain cholinergic nuclei is beneficial to working memory performance (Liu et al., 2017). In addition, advancements in the design of cholinergic agonists allow researchers to dissociate the actions of specific receptor subtypes (Bubser et al., 2012; Ferreira-Vieira et al., 2016). Although cholinergic stimulation has shown some promise in improving cognitive performance in macaques (Buccafusco and Terry, 2004; Tsukada et al., 2004) and Alzheimer's disease patients (Bodick et al., 1997), its influence on local PFC neurophysiology has been less examined in primates (Inoue et al., 1983; Sawaguchi and Matsumura, 1985).
Previously, we examined the role of muscarinic receptors in macaque dorsolateral PFC (DLPFC) by iontophoretically applying the general muscarinic receptor antagonist scopolamine during performance of a conditional saccade task, in which subjects made a saccade toward a peripheral stimulus (prosaccade) or made an antisaccade away from a stimulus, depending on a previous instruction cue (Major et al., 2015). This cognitive control task, which incorporates working memory for task rules, prepotent response inhibition, and flexible stimulus–response associations, is sensitive to DLPFC integrity (Pierrot-Deseilligny et al., 2003; Condy et al., 2007; Hussein et al., 2014). Moreover, deficits in antisaccade performance are a reliable indicator of prefrontal dysfunction in neuropsychiatric disorders, including Alzheimer's disease and schizophrenia (Fukushima et al., 1994; Kaufman et al., 2010). Iontophoretic blockade of muscarinic receptors resulted in general inhibition of prefrontal neuron activity and decreased selectivity related to rule, stimulus, and response encoding (Major et al., 2015), exemplifying the cortical basis of cognitive deficits following pharmacological insults to the cholinergic system (Klinkenberg and Blokland, 2010). Here, we explored the neurophysiological consequences of locally stimulating cholinergic receptors in macaque DLPFC using the general cholinergic agonist carbachol. Carbachol is a cholinomimetic, more resistant to cholinesterase-mediated breakdown than ACh, and therefore hypothesized to have longer-lasting effects than ACh (Rosenberry et al., 2008). Extensively explored for its effects on neuronal physiology, carbachol results in the generation of graded persistent activity in rodent entorhinal cortical neurons (Egorov et al., 2002) and augments gamma oscillations in the mouse medial PFC, which are associated with working memory performance (Pafundo et al., 2013). Based on our previous results (Major et al., 2015), and other studies which found that stimulation of DLPFC nicotinic receptor subtypes enhanced spatial working memory representations (Yang et al., 2013; Sun et al., 2017), we hypothesized that iontophoretic application of carbachol in macaque DLPFC would increase neuronal discharge rates and augment neuronal representation of task attributes during performance of prosaccades and antisaccades.
We found that carbachol had complex effects on DLPFC neuronal excitability, increasing discharge rates in putative pyramidal neurons. Contrary to our hypothesis, cholinergic stimulation of DLPFC disrupted neuronal representation of task rules in working memory and diminished saccade direction selectivity in putative pyramidal neurons. Our findings indicate that continuous pharmacological stimulation of the DLPFC cholinergic system is detrimental to cognitive performance.
Materials and Methods
Experimental procedures were performed on two adult male macaque monkeys (Macaca mulatta; age 8–11 years, weight 9–12 kg) in accordance with the Canadian Council of Animal Care policy and a protocol approved by the Animal Care Committee of the University of Western Ontario Council on Animal Care. These two monkeys were previously the subjects of other published studies, including iontophoretic investigations of muscarinic blockade (Major et al., 2015), dopaminergic receptors (Vijayraghavan et al., 2016), and a multielectrode investigation of the systemic effects of ketamine on prefrontal cortex (Skoblenick and Everling, 2012, 2014; Ma et al., 2015; Skoblenick et al., 2016). Both animals had a plastic head restraint and plastic recording chambers implanted above their right lateral PFC as described previously (Skoblenick and Everling, 2012).
Behavioral task.
The behavioral task and physiological techniques are similar to those described in previous reports (Major et al., 2015; Vijayraghavan et al., 2016). Briefly, animals performed a variant of the prosaccade and antisaccade task (Everling et al., 1998), in which the task rule had to be maintained in working memory (Fig. 1A). After the monkey fixates on a central white spot (0.2°, 300 ms, fixation window 4° × 4°), this fixation spot briefly changed color to red or green (100 ms), indicating the task rule, before reverting to white. The rule cue had to be maintained in working memory, while the subject maintained fixation over the delay period (800–1300 ms). Subsequently, the fixation spot disappeared and after a brief gap period (150–300 ms) a peripheral stimulus was presented pseudorandomly to the left or right of the fixation spot (17° eccentricity). Subjects had to make the appropriate saccade, specified by the current rule, either toward (prosaccade) or away from (antisaccade) the stimulus within 500 ms to receive liquid reward. Task, behavior monitoring, and reward delivery were controlled using CORTEX (National Institutes of Mental Health). The gap period was used to increase task difficulty (Everling et al., 1998). Rule colors were counterbalanced between subjects and rule/stimulus combinations were presented in pseudorandom order.
Figure 1.
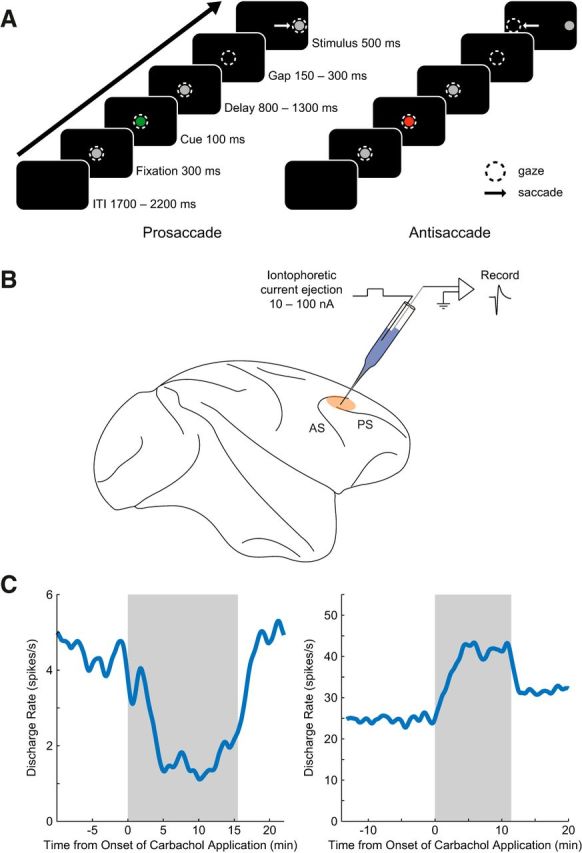
Experimental paradigm and single neuron recording with concurrent iontophoresis. A, After central fixation, a green or red cue was flashed, signifying a prosaccade or antisaccade trial, respectively. This task rule was maintained over an 800–1300 ms delay and a short gap. Monkeys then performed a saccade toward (prosaccade) or away from (antisaccade) the peripheral stimulus to receive liquid reward. Dashed circles represent gaze of the animal and white arrows represent saccade direction. ITI, Intertrial interval. B, Single neuron extracellular recordings were performed in the right dorsolateral prefrontal cortices of two rhesus macaques. Custom-made glass iontophoretic electrodes were used to eject general cholinergic receptor agonist carbachol onto neurons. Beige area represents recording locus. AC, Arcuate sulcus; PS, principal sulcus. C, Effect of carbachol on discharge rate over experiment time course. Left, Discharge rate of an example neuron is shown over the course of the recording. This neuron was inhibited during application of carbachol (shaded gray). Right, Carbachol application excited neuronal discharge rate of a different example neuron. Discharge rates were derived from 1 s bins and smoothed with a 200 s width.
Neuronal recordings and pharmacology.
Carbachol was iontophoretically administered using custom-made seven-barreled glass iontophoretic electrodes, which were modified from the design of Millar and Williams (1989) and fabricated as described previously (Major et al., 2015). Briefly, a 50 μm diameter tungsten wire (Midwest Tungsten Service), which served as the recording electrode, was electrochemically etched and inserted into the central capillary of a seven-barreled glass pipette (Friedrich and Dimmock). The glass was then pulled over the wire resulting in a multi-barrelled electrode with a fine tip (PMP107L-e Multipipette Puller, MicroData Instrument). Typical recording electrode impedances ranged from 0.5 to 1 MΩ (measured at 1 kHz). Carbachol (carbamoylcholine chloride; Tocris Bioscience; 100 mm in pH 3 deionized water) was top-filled into the peripheral capillaries of the multi-barrelled glass and pushed pneumatically to the tip of the iontophoretic electrode. Drugs were ejected using a Neuro Phore BH-2 iontophoretic ejection system (Harvard Apparatus). Constant ejection currents (5–100 nA, median: 30 nA) were manually set and a retention current of −8 nA was used to prevent leakage of drug from the barrel when ejection currents were not applied. Current balancing was not used as drug ejection with such low current strengths typically does not stimulate neurons or create electrophysiological noise with the electrode design used here (Vijayraghavan et al., 2007). Previous iontophoretic studies have shown there is no effect of pH of the drug solution on neuronal discharge rate (Disney et al., 2007; Vijayraghavan et al., 2007). Carbachol was used as a proxy for ACh due to its greater resistance to acetylcholinesterase-mediated breakdown (Rosenberry et al., 2008), which we hypothesized would result in more reliable stimulation of cholinergic receptors. Carbachol has been shown to have slightly larger magnitudes and duration of effect than ACh in cat (Crawford et al., 1966) and rat cortex (Bassant et al., 1990). The electrode was mounted on a hydraulic micromanipulator (MO-95, Narishige Group) and lowered into DLPFC (Fig. 1B) through a 23-gauge dura-penetrating stainless steel guide tube.
Neuronal signals were amplified, digitized, and filtered (300 Hz–6 kHz, four-pole Bessel) with an OmniPlex Neural Data Acquisition System (Plexon). Neuron waveforms were sorted offline in principal component space (Offline Sorter, Plexon) and analyzed offline (MATLAB, MathWorks). Spike density functions were created by convolving spike trains with a 50 ms Gaussian activation function (Richmond et al., 1987).
We collected data from blocks of trials with no drug application (control condition), followed by drug delivery (drug condition). For some sessions, an additional recovery condition was collected after cessation of drug application. Conditions typically lasted longer than 10 min (Fig. 1C) and neurons with <8 correct trials for each rule (prosaccade/antisaccade) and saccade direction (leftward/rightward) combination in any of the conditions were discarded from further analysis (n = 5). Behavioral effects are not usually expected with microiontophoretic drug application because the small amount of drug released does not spread to a large enough volume of cortical neuropil to affect behavior, especially in areas of broad specialization like PFC (Vijayraghavan et al., 2007).
Data analysis.
Discharge rate analyses were performed in several epochs over the course of the trial: “entire” trial epoch (1500 ms before to 1000 ms after stimulus onset), fixation epoch (0–200 ms after fixation onset), cue epoch (0–200 ms after colored cue onset), and delay epoch (600 ms before to 70 ms after peripheral stimulus onset). Based on prior studies, this delay epoch is when PFC neurons are found to display maximal rule discriminability (Everling and DeSouza, 2005; Bongard and Nieder, 2010). We also analyzed the stimulus epoch (0–400 ms after peripheral stimulus onset), post-saccade epoch (0–400 ms after saccade onset), and intertrial interval (0–1000 ms after reward onset). We excluded neurons with very low discharge rates (<1 spike/s in both control and drug conditions) from the analysis as the low firing rates precluded reliable analysis of physiological effects of the drug. The task-selectivity profile of each included neuron was determined by performing an ANOVA on the trial discharge rates in the cue and delay epochs with two factors: drug condition and rule. Neurons with a significant main effect of rule or an interaction of rule and drug (p < 0.05) were classified as rule-selective neurons (“rule neurons”). Magnitude of rule selectivity was further quantified using area under the receiver operating characteristic curve (AUROC; 1000 steps; Green and Swets, 1966). AUROCs were computed from the mean discharge rates during the delay epoch for prosaccades and antisaccades. AUROC values range from 0 to 1. By convention, neurons showing higher activity (preference) for the prosaccade rule were deemed to possess AUROC values >0.5. The AUROC values for neurons with greater activity for the antisaccade rule would thus be <0.5 and were subtracted from 1, therefore reported AUROC values were for preferred versus nonpreferred rule. An AUROC of 1 signified a completely selective neuron with nonoverlapping distributions of preferred and nonpreferred rule discharge rates. An AUROC of 0.5 signified a lack of rule discriminability, wherein preferred and nonpreferred rule discharge rate distributions completely overlapped. Analysis of task selectivity was also performed on the stimulus epoch with three-way ANOVA (factors: drug condition, rule, and peripheral stimulus direction), where neurons with a significant main effect of stimulus direction or a significant interaction between stimulus direction and condition were classified as “visual neurons”. These neurons significantly discriminated between peripheral stimuli on the left versus right side of the screen, regardless of trial rule. Similarly, discharge rates in the post-saccade epoch were explored with three-way ANOVA (factors: drug condition, rule, and saccade direction) to classify “saccade neurons”, with a significant main effect of saccade direction or significant interaction between saccade direction and drug condition. Activity of these neurons discriminated between leftward and rightward saccade directions. Selectivities of visual and saccade neurons were also quantified with AUROC between the contralateral and ipsilateral stimulus directions or saccade directions, respectively. AUROC data are reported for the preferred versus nonpreferred direction. Since a change in AUROC can be explained by changes in either the mean discharge rates or trial-to-trial discharge rate variances between trial types (e.g., prosaccade vs antisaccade trials), neuronal reliability was measured with Fano factor: trialwise discharge rate variance divided by the mean. Fano factors for preferred and nonpreferred rule trails were determined separately then averaged together, as these two trials types intrinsically vary between each other in rule-selective neurons.
To compare discharge rate changes across task-selective neurons, mean discharge rates for preferred and nonpreferred trial types in the control and drug conditions were normalized as follows:
 |
The preferred and nonpreferred trial type would be the preferred rule and nonpreferred rule in the case of rule selectivity, and would be preferred and nonpreferred saccade direction in the case of saccade direction selectivity. xt is the mean discharge rate for a given trial type in control or drug (e.g., activity in control preferred rule trials); xmin and xmax are the minimum and maximum discharge rates among the four values: control preferred, control nonpreferred, drug condition preferred, and drug condition nonpreferred mean discharge rate; and xnorm is the normalized mean discharge rate for that given trial type (e.g., preferred rule trials in control condition). Relative contributions of changes in mean and variability of discharge rate on changes in neuronal selectivity were estimated for the population of rule-selective neurons with the following multiple linear regression model:
 |
Here, ΔFRpref represents the carbachol-induced change in normalized discharge rate for the preferred rule between the control and carbachol conditions. β coefficients represent the slope between the respective predictor and change in AUROC when all other predictors are held constant.
We also performed an analysis of action potential waveforms to classify neuronal types (“broad-spiking” putative pyramidal neurons and “narrow-spiking” putative nonpyramidal neurons), using methodology derived from previous studies (Mountcastle et al., 1969; Mitchell et al., 2007; Johnston et al., 2009). Action potential waveforms were extracted from the delay epoch during the control condition at a temporal resolution of 25 μs (40 kHz sampling frequency) and increased to 1 μs resolution with spline interpolation. After aligning to voltage trough and averaging all waveforms, duration from waveform trough (negative deflection) to peak (positive deflection) was measured. Neurons that did not show the typical shape of a downward trough, followed by a positive peak in voltage, were removed from further waveform analysis (n = 15; Jacob et al., 2013). A neuron was defined as a broad-spiking neuron (putative pyramidal neuron) if trough to peak duration was >270 μs (Johnston et al., 2009; Ma et al., 2015) and defined as a narrow-spiking neuron (putative nonpyramidal neuron) if trough to peak duration was <270 μs.
Statistical analysis.
Wilcoxon rank sum test was used for determining significance of changes in discharge rate among individual neurons. Significance of changes in population discharge rate, Fano factor, and selectivity (AUROC) were determined using the Wilcoxon signed rank test. Fisher's exact test was used to determine whether excitability or suppression of discharge rate of task-selective neurons was contingent on trial preference (e.g., preference for prosaccades or antisaccades, or ipsilateral or contralateral saccade directions). To determine unimodality of the distribution of trough to peak durations of narrow- and broad-spiking neurons, we performed Hartigan's Dip Test (Hartigan and Hartigan, 1985).
Results
Extracellular recordings and microiontophoretic carbachol application were performed in 100 DLPFC neurons. After exclusion of neurons due to cutoff criteria (see Materials and Methods) 83 neurons (50 from Monkey T, 33 from Monkey O) over the course of 79 recording sessions remained for further analysis (37 from Monkey T, 42 from Monkey O). Throughout control and drug conditions, monkeys performed the prosaccade and antisaccade task (described in Materials and Methods). Recovery from drug effects was tested in 28 recording sessions. Although minute decreases in performance were observed for both prosaccade (correct performance; mean ± SEM; control: 95.1 ± 0.01%, carbachol: 93.4 ± 0.01%) and antisaccade trials (control: 90.7 ± 0.02%, carbachol: 88.0 ± 0.02%), this declining trend continued into the recovery condition (recovery prosaccade: 92.7 ± 0.01%, recovery antisaccade: 86.4 ± 0.02%), and therefore was more likely a consequence of non-drug-related factors, such as waning motivation over the course of the experiment. Similarly, reaction times were longer in the carbachol condition (control prosaccade: 138 ± 1 ms, carbachol prosaccade: 142 ± 1 ms; control antisaccade: 192 ± 3 ms, carbachol antisaccade: 194 ± 3 ms), but continued to increase in recovery conditions (recovery prosaccade: 144 ± 1 ms, recovery antisaccade: 196 ± 3 ms).
Effect of carbachol on discharge rates
We examined the effects of microiontophoretic application of carbachol at various current doses on discharge rates of 83 prefrontal neurons. We found that carbachol had heterogeneous effects on population activity. Figure 2A shows the mean discharge rate of each neuron in control (abscissa) and after drug application (ordinate). Carbachol application did not have a significant overall effect on population neuronal activity (p = 0.212, Wilcoxon signed rank test; median change in discharge rate of +0.4 spikes/s), although individual neurons were excited or suppressed. Of the 83 neurons, discharge rates of 41 neurons (49%) were significantly increased (entire epoch, p < 0.05; Wilcoxon rank sum test), 32 neurons (39%) were inhibited, and discharge rates of 10 neurons (12%) were unaffected after carbachol application (Fig. 2A). Further, change in population discharge rate was nonsignificant in every task epoch (fixation epoch: p = 0.241, cue: p = 0.124, delay: p = 0.238, stimulus: p = 0.256, post-saccade: p = 0.364, intertrial interval: p = 0.316; Wilcoxon signed rank test), indicating that the drug did not differentially affect discharge rate during a specific component of the task.
Figure 2.

Effects of carbachol on prefrontal neuron discharge rate. A, Discharge rates during control (abscissa) and carbachol application (ordinate) are plotted (filled circles: neurons significantly excited or inhibited by carbachol application; open circles: no significant effect of carbachol application; entire epoch, p < 0.05, Wilcoxon rank sum test). Dashed equality line is shown. Median change in discharge rate was +0.4 spikes/s, although discharge rates were not significantly affected by carbachol as a population (p = 0.212, Wilcoxon signed rank test). Inset, Pie chart with proportions of neurons that were significantly excited (red; n = 41, 49%), inhibited (blue; n = 32, 39%), or not affected (white; n = 10, 12%) by carbachol application. Discharge rates are calculated from 1500 ms before stimulus onset to 1000 ms after stimulus onset. B, Bar charts depicting the mean trial discharge rates for two example neurons. Top, Increasing dose of carbachol progressively increased discharge rate of this neuron. Cessation of drug ejection resulted in a significant, partial recovery of discharge rate. All changes between sequential doses or recovery were significant. Bottom, In another neuron, increasing doses of carbachol resulted in gradually stronger inhibition of neuronal excitability. Partial recovery was observed. Statistical significances were determined by Wilcoxon ranked sum test with Holm-Bonferroni correction for multiple comparisons. C, Population effects of carbachol dose are shown. Mean discharge rates during control, low (≤30 nA), and high (>30 nA) doses of carbachol (dose ranges based on median split of all applied doses) are shown for neurons that were significantly excited (top) or inhibited (bottom) by carbachol. In excited neurons, higher doses resulted in a further significant increase to discharge rate. Among suppressed neurons, higher doses of carbachol did not result in further suppression of discharge rate. Significance determined by Wilcoxon signed rank test with Holm–Bonferroni correction for multiple comparisons. D, Recovery of population discharge rates after cessation of carbachol application. Top, Neurons significantly excited by carbachol (p < 0.05, Wilcoxon rank sum test) that were tested for recovery exhibited partial, albeit nonsignificant (p = 0.169) recovery after cessation of carbachol application. Bottom, Suppressed neurons did not recover discharge rates during our observed recovery condition. Error bars indicate SEM. In all panels, asterisks indicate significant (p < 0.05) comparisons with Holm–Bonferroni correction for multiple comparisons, where applicable.
To further explore the heterogeneous effects of carbachol application on prefrontal neuronal activity, we examined 23 neurons on which successive doses of carbachol were applied, and wherein drug application resulted in a significant change (increase or decrease) in the discharge rate (entire epoch, p < 0.05; Wilcoxon rank sum test). Figure 2B demonstrates the effects of progressively increasing doses of carbachol on two of these DLPFC neurons that were excited and suppressed by cholinergic stimulation (top and bottom, respectively). Increasing doses of carbachol application on the neuron depicted in the top progressively and significantly increased discharge rate (control: 5.3 ± 0.1 spikes/s; highest dose of carbachol: 14.2 ± 0.3 spikes/s). After cessation of drug application, there was significant partial recovery from carbachol-induced excitation, with neuronal discharge rate declining to 12.2 ± 0.2 spikes/s. However, carbachol application did not have an excitatory effect on all recorded neurons. This trend is illustrated by the neuron depicted in Figure 2B (bottom). This neuron was gradually suppressed after successive doses of carbachol (control: 9.1 ± 0.2 spikes/s; highest dose of carbachol: 3.2 ± 0.1 spikes/s), followed by partial recovery of discharge rate (control: 8.0 ± 0.2 spikes/s).
We further analyzed effects of dose on discharge rates in the 23 aforementioned neurons with multiple carbachol dose applications (13 excited, 10 inhibited) by grouping doses into “low” and “high”, based on a median split of the applied dose range (median carbachol dose = 30 nA). Figure 2C shows the effects of low and high doses of carbachol on neurons that were excited (top) and inhibited (bottom) by drug application. Discharge rates of carbachol-excited neurons increased monotonically with carbachol dose. Low-dose application caused significant increase in excitability compared with control, and high-dose application resulted in further increases to neuronal discharge rate (Fig. 2C, top; entire epoch). Carbachol-suppressed neurons, however, did not show monotonicity of suppression with drug dose (Fig. 2C, bottom). High-dose carbachol application (>30 nA) could not further suppress neuronal discharge rates. It is noteworthy that this was not due to a floor effect, as neurons still had substantial activity after high-dose application.
We tested 27 neurons (15 excited, 12 inhibited) for recovery from physiological effects after cessation of drug application. Figure 2D shows these data for neurons that were significantly excited (top) or inhibited (bottom) by carbachol application. Neurons that were excited by carbachol had reduction in discharge rates after drug cessation, though not reaching significance, thus indicating partial recovery from drug effects (entire epoch, p = 0.169; Wilcoxon signed rank test). However, in neurons that were significantly suppressed, recoveries did not occur during our recording sessions of ∼10 min (p = 1). Thus, differential effects of carbachol on neuronal excitability were accompanied by different post-drug physiological effects.
Effect of carbachol on task-selective neurons
Next, we examined the effects of cholinergic stimulation with carbachol on task selectivity of neurons in the prosaccade and antisaccade task. Our task involved retaining the specified rule in working memory, and we explored the effects of carbachol on working memory representation of the rule in prefrontal neuronal activity (Everling and DeSouza, 2005). We identified task selectivity in prefrontal neurons using a two-way ANOVA with factors: rule (prosaccade or antisaccade trial) and drug condition (control or carbachol), using discharge rates during the delay epoch of the task. Based on the ANOVA, we classified 24 neurons as possessing rule selectivity (rule neurons; significant main effect of rule or interaction of rule and drug). We assessed the magnitude of rule selectivity in this population with the AUROC metric (see Materials and Methods). Figure 3A displays an example neuron that had a greater delay epoch discharge rate in the control condition during antisaccade trials (17.4 ± 1.4 spikes/s) compared with prosaccade trials (9.0 ± 1.3 spikes/s). Upon carbachol application, the baseline activity of this neuron was excited (fixation epoch control vs carbachol discharge rate: p < 0.0001; Wilcoxon rank sum test) and selectivity for the task rule was decreased (carbachol antisaccade: 16.3 ± 0.8 spikes/s, carbachol prosaccade: 11.5 ± 0.8 spikes/s; control AUROC: 0.74, carbachol AUROC: 0.67). Another prefrontal neuron (Fig. 3B) displayed a preference for antisaccades during the control condition delay epoch (antisaccade: 11.2 ± 0.4 spikes/s, prosaccade: 8.3 ± 0.3 spikes/s). Similar to the first example, this neuron was excited in the fixation (p < 0.0001) and delay epochs (control: 9.8 ± 0.2 spikes/s, carbachol: 12.5 ± 0.1 spikes/s, p < 0.0001) after carbachol application and rule selectivity was diminished (control AUROC: 0.76, carbachol AUROC: 0.59).
Figure 3.
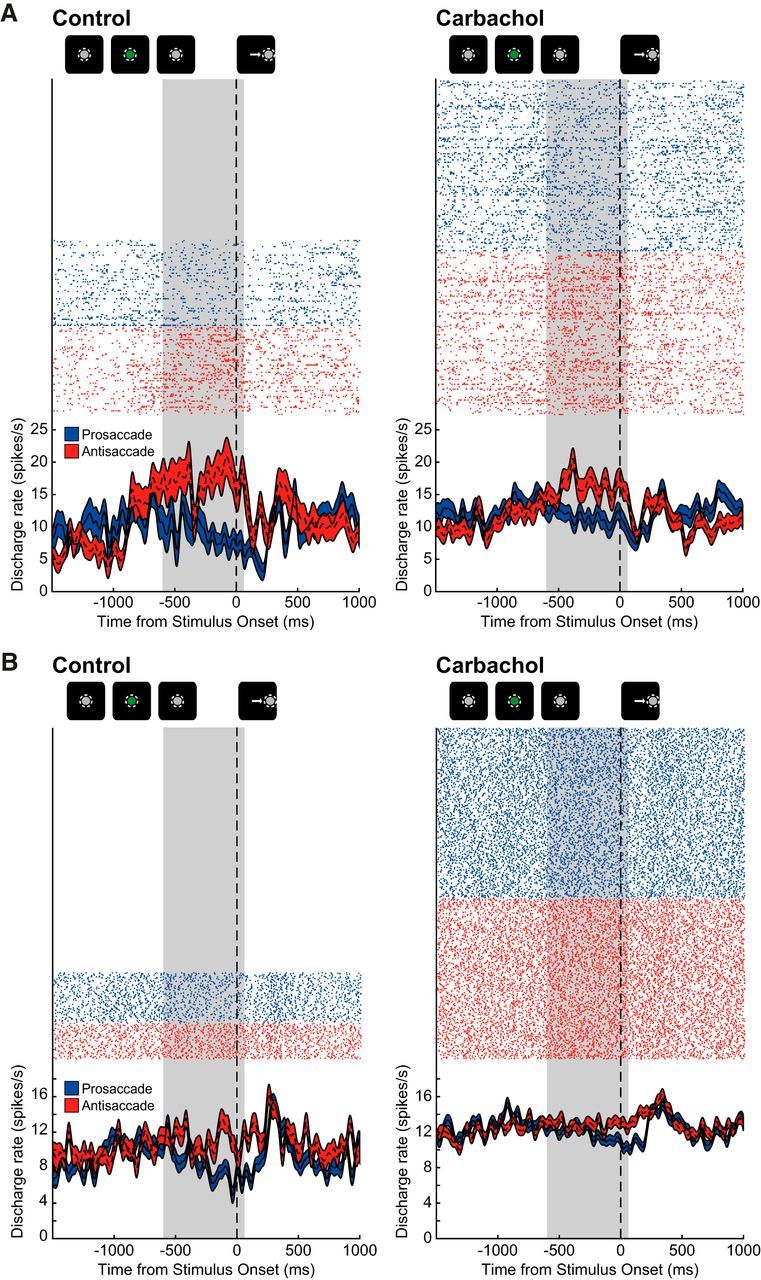
Effect of carbachol application on two representative rule-selective prefrontal neurons. A, Rasters (top; each row of dots is from a single trial) and spike density functions (bottom; mean ± SEM) are shown for a neuron with preferentially higher discharge rate during antisaccade trials (red) compared with prosaccade trials (blue) during the delay epoch, before (left) and after (right) iontophoretic application of carbachol. Delay epoch is shaded in gray (see Materials and Methods). Qualitative schematic of main trial events is shown above. After carbachol application, this rule neuron increases delay epoch discharge rate and preference for antisaccade trials is reduced. Rasters and spike density functions are aligned to onset of peripheral stimulus. B, A neuron with selectivity for antisaccades during the delay epoch is shown. All colors and conventions like A. After carbachol application, discharge rate is increased, especially during prosaccade trials, and rule preference is diminished.
For the 24 rule neurons, we examined changes in population rule selectivity due to carbachol application (15 from Monkey T, 9 from Monkey O; 14 prosaccade-preferring, 10 antisaccade-preferring). Figure 4A shows the population normalized spike density functions (mean ± SEM) of rule neurons before (left; blue: preferred rule, green: nonpreferred rule) and after carbachol application (right). Change in rule selectivity was quantified using AUROC. Figure 4B shows the AUROCs for each rule neuron during control (abscissa) and carbachol conditions (ordinate). Although drug effects on selectivity of individual neurons varied, carbachol significantly decreased population rule selectivity (p = 0.0397, Wilcoxon signed rank test; blue: prosaccade-rule-preferring, red: antisaccade-rule-preferring). We also examined Fano factor as a measure of trial-to-trial variability of neuronal spike count. Fano factor was not significantly affected during application of carbachol (control: 2.7 ± 0.4, carbachol: 2.4 ± 0.3, p = 0.0675). As reduced rule AUROC can be due to either reduced difference in prosaccade versus antisaccade discharge rates or increased neuronal variability, we explored the relative contributions of changes in neuronal discharge rate and variability using multiple linear regression with four predictors: change in normalized preferred rule discharge rate, change in normalized nonpreferred rule discharge rate, change in preferred rule variance, and change in nonpreferred rule variance (see Materials and Methods). This multiple regression model was significant (F(4,19) = 25.56, p < 0.0001) with R2 = 0.843. As seen in Table 1, carbachol-induced change in normalized preferred and nonpreferred rule discharge rates and change in preferred rule variance were significant predictors of change in AUROC. Of these three factors, change in preferred and nonpreferred rule discharge rate contributed the most to altered selectivity, as the magnitude of their β coefficients were much larger than that of preferred rule variance.
Figure 4.
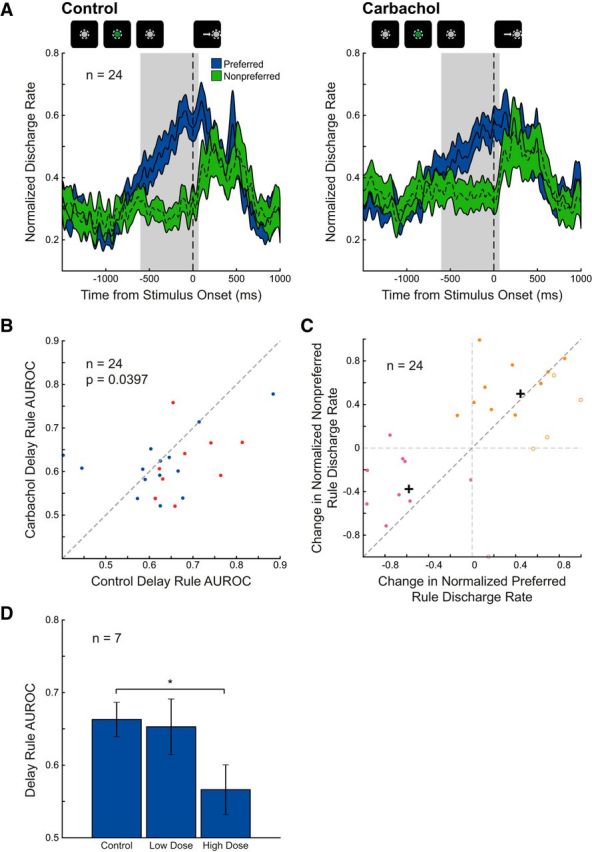
Effects of carbachol on population rule selectivity. A, Mean normalized spike density functions of preferred (blue) and nonpreferred (green) rule trials for 24 DLPFC rule-selective neurons during control (left) and carbachol conditions (right) are shown. Neurons were identified as rule-selective by ANOVA (see Materials and Methods). Overall difference between discharge rates for preferred and nonpreferred rules diminished. B, Rule selectivity (measured by AUROC) is shown for each rule neuron during control (abscissa) and carbachol application (ordinate). Drug application significantly decreased selectivity of rule neurons, as quantified by AUROC (p = 0.0397, Wilcoxon signed rank test). Prosaccade-preferring and antisaccade-preferring rule neurons are represented in blue and red, respectively. Dashed equality line is shown. C, Change in normalized discharge rate during preferred rule trials for 24 rule neurons (abscissa) is compared with change in normalized discharge rate of nonpreferred rule trials (ordinate). Based on k-means cluster analysis (k = 2), two clusters of neurons were identified (labeled pink and orange; filled circles: neurons with decreased AUROC; open circles: neurons with increased AUROC) with centroids shown as black crosshairs. D, Dose-dependent effects on rule selectivity by carbachol. Mean delay epoch rule AUROCs are shown for seven rule neurons that were subject to both low (≤30 nA) and high doses (>30 nA) of carbachol (split by median of applied dose range). High doses of carbachol resulted in significant diminishment of delay epoch rule selectivity in this subset of rule neurons whereas low-dose carbachol did not. Significance determined by Wilcoxon signed rank test (p < 0.05; asterisk indicates significant comparison) with Holm–Bonferroni correction for multiple comparisons.
Table 1.
Summary of multiple linear regression analysis for rule neurons
| Predictor of ΔAUROC | β Coefficient | p |
|---|---|---|
| ΔFRpref | 0.231 | 1.04e-7 |
| ΔFRnonpref | −0.193 | 9.32e-6 |
| ΔVarpref | −0.0494 | 0.0162 |
| ΔVarnonpref | 0.000602 | 0.983 |
β coefficients and p values from the multiple regression analysis (see Materials and Methods) are shown to asses the impact of four predictors on change in rule AUROC: ΔFRpref, change in mean normalized discharge rate during preferred rule trials; ΔFRnonpref, change in mean normalized discharge rate during nonpreferred rule trials; ΔVarpref, change in normalized variance during preferred rule trials; ΔVarnonpref, change in normalized variance during nonpreferred rule trials.
We further explored the relative changes in preferred and nonpreferred discharge rate in Figure 4C. Change in normalized preferred discharge rate is plotted (abscissa) against change in normalized nonpreferred discharge rate (ordinate). Neurons in which carbachol application reduced or increased the AUROC are labeled with filled and unfilled circles, respectively. Because change in AUROC is largely determined by the change in preferred and nonpreferred discharge rate, neurons above the equality line (dark gray dashed line) generally experienced decreased AUROC and neurons below the line increased AUROC. We observed two distinct clusters of neurons (k-means clustering, k = 2; centroids shown as crosshairs), one of suppressed neurons (n = 10, 42%; labeled in purple), and one of excited neurons (n = 14, 58%; orange). Carbachol reduced selectivity of almost all suppressed neurons, although the change in selectivity did not reach significance (n = 10, p = 0.0840). This decreased selectivity was due to greater inhibition during preferred rule trails compared with nonpreferred rule trials. Carbachol had more equivocal effects on the AUROC of excited rule neurons (n = 14, p = 0.296). Some neurons decreased selectivity due to relatively greater excitation of nonpreferred rule (above equality line), and others increased selectivity due to greater excitation of preferred rule. Thus, carbachol both excited and suppressed discharge rate of rule neurons, often resulting in decreased selectivity, which can be attributed to either a relatively greater suppression of activity for preferred rules or a relatively greater increase in nonpreferred rule discharge rate.
Selectivity of prosaccade-preferring rule neurons (Fig. 4B, blue) were not significantly affected by carbachol (n = 14, p = 0.463), but selectivity of antisaccade-preferring rule neurons (red) was significantly decreased (n = 10, p = 0.0371). The preference of rule neurons for prosaccade or antisaccade did not have any bearing on whether rule neurons were excited or inhibited by carbachol (p = 0.421, Fisher's exact test).
In some rule neurons (n = 7), multiple doses of carbachol were applied (Fig. 4C). In these rule neurons, low doses (<30 nA) did not significantly change AUROC values (p = 0.688), but high doses (>30 nA) resulted in significant diminishment of rule selectivity (p = 0.0156). Thus, disruption of rule selectivity in the delay period by cholinergic stimulation is dose-dependent.
Unlike the disruptive effects on rule representation in the delay epoch, population rule selectivities were unaffected in the cue epoch (n = 11, p = 0.0830, Wilcoxon signed rank test; 4 prosaccade-preferring, 7 antisaccade-preferring), stimulus epoch (n = 25, p = 0.757; 11 prosaccade-preferring, 14 antisaccade-preferring), and post-saccade epoch (n = 24, p = 0.265; 6 prosaccade-preferring, 18 antisaccade-preferring).
We examined the effects of cholinergic stimulation on neurons with selectivity for other attributes observed in the task. We identified 19 prefrontal neurons that were selective for peripheral stimulus location during the stimulus epoch (8 contralateral-stimulus-preferring, 11 ipsilateral-stimulus-preferring). Additionally, 40 neurons were selective for the direction of saccade in the post-saccade epoch (32 contralateral-saccade-preferring, 8 ipsilateral-saccade-preferring). Carbachol application had equivocal effects on population selectivity of both peripheral stimulus direction (control vs carbachol stimulus direction AUROC: p = 0.968, Wilcoxon signed rank test) and for the saccade direction (control vs carbachol saccade direction AUROC: p = 0.122).
Effects of carbachol on putative pyramidal and nonpyramidal neurons
Past studies have reported that drug-induced modulation of discharge characteristics, signal-to-noise ratio, and coding of task attributes can be different between cortical neuronal classes (Mountcastle et al., 1969; Jacob et al., 2013; Ma et al., 2015; Thiele et al., 2016). To ascertain whether cholinergic stimulation had differential effects on putative pyramidal neurons and interneurons defined by electrophysiological characteristics, we performed a similar analysis, whereby we classified prefrontal neurons (n = 68; see Materials and Methods) as broad-spiking (putative pyramidal neurons) or narrow-spiking (putative nonpyramidal neurons; see Materials and Methods; Johnston et al., 2009). The distribution of waveform trough to peak durations was not unimodal (Fig. 5A; p = 0.00910, Hartigans' Dip Test; Hartigan and Hartigan, 1985). Based on a previous study in our laboratory, neurons with a waveform trough to peak duration <270 μs were classified as narrow-spiking and neurons with a trough to peak duration >270 μs were classified as broad-spiking (Fig. 5B; green: mean narrow-spiking waveforms, pink: broad-spiking waveforms), yielding 21 narrow waveform neurons (31%) and 47 broad waveform neurons (69%).
Figure 5.

Effects of carbachol on putative pyramidal and nonpyramidal neurons. Waveforms were extracted and trough to peak duration was measured in 68 neurons (see Materials and Methods). A, Histogram of trough to peak durations. Trough to peak duration distribution was not unimodal as determined by Hartigans' Dip Test (p = 0.00910). Similar to previous reports, a threshold of 270 μs (dashed line) was used to classify neuronal waveforms as narrow-spiking (green, putative nonpyramidal neurons) or broad-spiking (pink, putative pyramidal neurons). 30 μs bin width. B, Normalized waveforms aligned to initial trough (negative inflection) are shown with narrow-spiking (green) or broad-spiking (pink) label. C, Discharge rates among narrow-spiking neurons were not significantly affected by carbachol (entire epoch, p = 0.876, Wilcoxon signed rank test). Shown as filled circles, discharge rates of 8 neurons (38%) were significantly excited (p < 0.05, Wilcoxon rank sum test) and 10 (48%) were significantly suppressed. D, Broad-spiking neurons were significantly excited by carbachol (p = 0.0444, Wilcoxon signed rank test). Discharge rates of 27 neurons (57%) were significantly excited (p < 0.05, Wilcoxon rank sum test) and 16 (34%) were significantly suppressed.
Discharge rates of narrow-spiking neurons were not significantly affected by carbachol application at the population level (Fig. 5C; entire epoch, p = 0.876, Wilcoxon signed rank test). Population discharge rate of narrow-spiking neurons was not significantly excited or suppressed in any task epoch (fixation epoch: p = 0.639, cue: p = 0.664, delay: p = 0.931, stimulus: p = 0.986, post-saccade: p = 0.848, intertrial interval: p = 0.903). In addition to the lack of significant effect on discharge rates, carbachol also did not affect the selectivity of task-selective narrow-spiking neurons for any task attributes, including rule selectivity during the delay and cue epochs, stimulus direction selectivity in the stimulus epoch, and saccade direction selectivity in the post-saccade epoch.
In contrast, we found that broad-spiking neurons were significantly excited by carbachol application at the population level (Fig. 5D; p = 0.0444), notwithstanding some individual neurons that were suppressed. Broad-spiking neurons were also significantly excited in the fixation, cue, delay, and stimulus epochs (p = 0.0235, p = 0.0163, p = 0.0490, p = 0.0455, respectively), but not in the post-saccade (p = 0.0865) or intertrial interval epochs (p = 0.0904).
Carbachol did not significantly affect selectivity of broad-spiking rule-selective neurons in the cue or delay epochs, or broad-spiking visual neurons in the stimulus epoch. However, although we found that saccade direction selectivity in the population of 40 saccade neurons was not significantly changed by carbachol, we found that the subset of this population comprised of broad-spiking neurons did show changes in population saccade selectivity. Figure 6A shows a prefrontal neuron with higher discharge rate for contralateral saccades (2.9 ± 0.4 spikes/s) than for ipsilateral saccades (1.8 ± 0.3 spikes/s) during the post-saccade epoch, regardless of trial rule. Although the baseline activity of this neuron was not significantly excited (fixation epoch control vs carbachol, p = 0.06, Wilcoxon rank sum test) this neuron was excited in the post-saccade epoch (control: 2.4 ± 0.3 spikes/s, carbachol: 3.8 ± 0.2 spikes/s, p = 0.000415) and selectivity for saccade direction was reduced (control AUROC: 0.61, carbachol AUROC: 0.53). Population normalized spike density functions of broad-spiking saccade neurons show this augmentation of population discharge rates (Fig. 6B). Analysis of saccade direction selectivity by AUROC (Fig. 6C; red: contralateral, n = 14; blue: ipsilateral, n = 4) revealed that carbachol application resulted in a small, but significant, decrease in population selectivity (mean control AUROC: 0.66 ± 0.02, mean carbachol AUROC: 0.62 ± 0.01, p = 0.0429; Wilcoxon signed rank test). We also examined Fano factor as a measure of trial-to-trial variability of neuronal spike count. Fano factor was not significantly affected during application of carbachol (control: 1.5 ± 0.2, carbachol: 1.6 ± 0.2, p = 0.286). We explored the relative contributions of changes in neuronal discharge rate and variability in decreasing selectivity in broad-spiking saccade neurons using a multiple linear regression model similar to that used for rule neurons. This multiple regression model was significant (F(4,13) = 14.43, p = 0.000104) with R2 = 0.816. Carbachol-induced change in normalized preferred and nonpreferred saccade direction discharge rates and change in preferred rule variance were significant predictors of change in AUROC (Table 2). Change in preferred and nonpreferred direction discharge rate contributed more to altered selectivity than change in preferred direction variance, as measured by their β coefficients.
Figure 6.
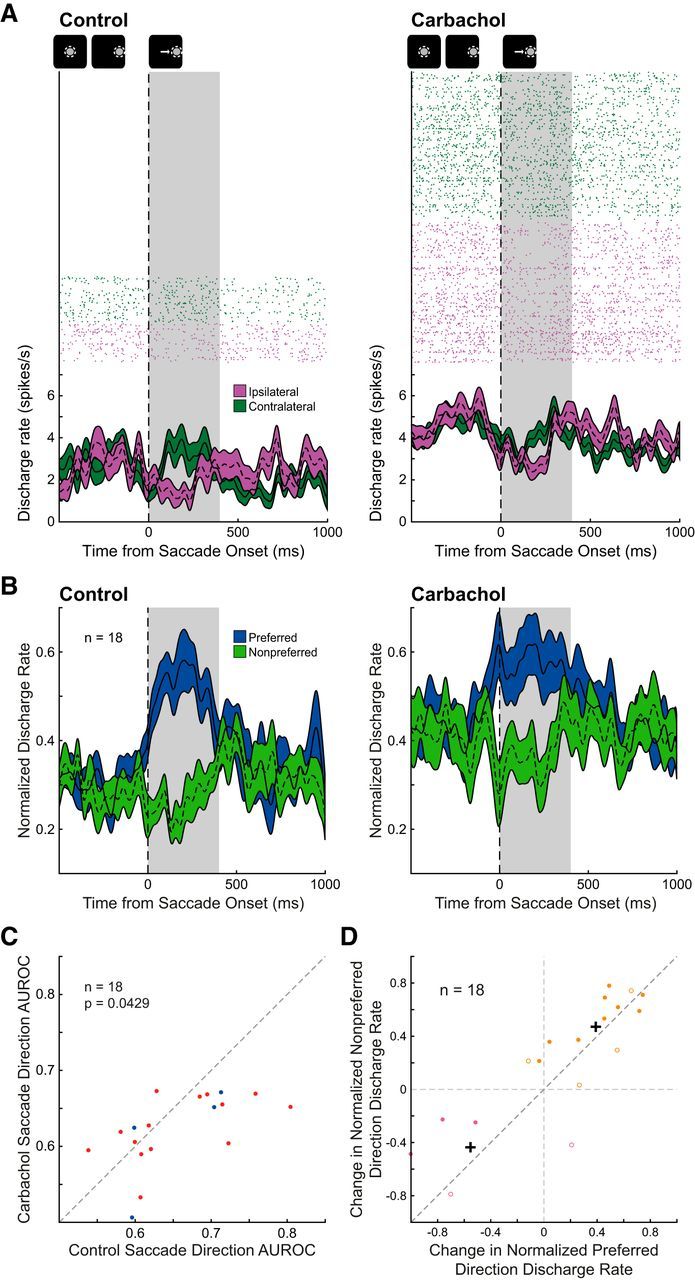
Effect of carbachol on saccade direction selectivity of broad-spiking, putative pyramidal neurons. A, Rasters (above) and spike density functions (bottom) aligned on saccade onset are shown for a neuron with preferentially higher discharge rate during contralateral saccade trials (green) compared with ipsilateral saccade trials (purple) during the post-saccade epoch is shown before (left) and after (right) iontophoretic application of carbachol. The post-saccade epoch is shaded in gray (see Materials and Methods). Qualitative schematic of main trial events is shown above. After carbachol application, this saccade neuron increased discharge rate in the post-saccade epoch, with a greater increase for ipsilateral trials, thereby reducing saccade direction selectivity. B, Mean normalized spike-density functions of preferred (blue) and nonpreferred (green) saccade trials for 18 DLPFC saccade-direction-selective neurons during control (left) and carbachol conditions (right) are shown. C, Carbachol significantly decreased selectivity of broad-spiking saccade neurons, as quantified by AUROC (abscissa: control AUROC values, ordinate: drug AUROC values; n = 18, p = 0.0429, Wilcoxon signed rank test). Ipsilateral-saccade-preferring and contralateral-saccade-preferring neurons are represented in blue and red, respectively. Dashed equality line is shown. D, Change in normalized discharge rate during preferred saccade direction trials of the 18 saccade neurons is compared with change in normalized discharge rate during nonpreferred saccade direction trials. Based on k-means cluster analysis (k = 2), two clusters of neurons are shown (pink and orange; filled circles: neurons with decreased AUROC, open circles: neurons with increased AUROC) with centroids shown as black crosshairs.
Table 2.
Summary of multiple linear regression analysis for broad-spiking saccade neurons
| Predictor of ΔAUROC | β Coefficient | p |
|---|---|---|
| ΔFRpref | 0.194 | 1.69e-5 |
| ΔFRnonpref | −0.144 | 0.00520 |
| ΔVarpref | −0.0599 | 0.00140 |
| ΔVarnonpref | 0.0195 | 0.414 |
β coefficients and p values from the multiple regression analysis (see Materials and Methods) are shown to asses the impact of four predictors on change in saccade direction AUROC: ΔFRpref, change in mean normalized discharge rate during preferred saccade direction trials; ΔFRnonpref, change in mean normalized discharge rate during nonpreferred saccade direction trials; ΔVarpref, change in normalized variance during preferred saccade direction trials; ΔVarnonpref, change in normalized variance during nonpreferred saccade direction trials.
We further explored the relative changes in preferred and nonpreferred discharge rate in Figure 6D. Neurons are labeled with filled and unfilled circles, indicating carbachol condition AUROC was less than or greater than control AUROC, respectively. Because change in AUROC is largely determined by these two variables, change in preferred and nonpreferred discharge rate, neurons above the equality line (dark gray dashed line) often had decreased AUROCs and neurons below the line, increased selectivity. Similar to the overall subset of broad-spiking neurons, saccade-direction-selective broad-spiking neurons were more often excited (n = 13, 72%; orange) by carbachol than suppressed (n = 5, 28%; purple). Carbachol reduced selectivity of almost all excited neurons (n = 13, p = 0.0479). There was no relationship between whether a neuron was excited or suppressed by carbachol and whether it preferred ipsilateral or contralateral saccade (p = 1, Fisher's exact test).
Thus, carbachol-induced effects on saccade selectivity were found in broad-spiking, putative pyramidal neurons, but not in narrow-spiking, putative nonpyramidal neurons. Carbachol mostly excited discharge rate of broad-spiking saccade neurons, resulting in decreased selectivity, which can be attributed to relatively greater increase in nonpreferred saccade direction discharge rate.
Discussion
In this study we examined the effects of localized cholinergic stimulation on primate DLPFC neurons engaged in a clinically relevant oculomotor task, which involved using a rule maintained in working memory to produce the appropriate saccadic responses to visual stimuli. Local carbachol application both excited and suppressed DLPFC neurons. Surprisingly, we also found carbachol disrupted neuronal rule representation in working memory, due to either suppression of preferred rule activity or excitation of nonpreferred rule activity. Moreover, broad-spiking putative pyramidal neurons were excited after cholinergic agonist application, and postsaccadic directional selectivity in these neurons was attenuated largely due to preferentially increased activity for the nonpreferred saccade direction.
Effect of carbachol on neuronal discharge rate in DLPFC
We found that local cholinergic stimulation excited a greater proportion of DLPFC neurons than those that were inhibited. However, our observation of both facilitation and inhibition with carbachol is consistent with previous iontophoretic applications of ACh in macaque DLPFC (Inoue et al., 1983; Sawaguchi and Matsumura, 1985), orbitofrontal (Aou et al., 1983), premotor (Nelson et al., 1973), motor (Matsumura et al., 1990), and primary visual cortex (V1; Soma et al., 2012). Mixed effects of ACh on neuronal activity have also been observed in marmoset V1 (Roberts et al., 2005; Zinke et al., 2006), cat V1 (Sato et al., 1987), rat medial PFC (Pirch et al., 1992; Nagy et al., 2014), and in guinea pig cortical slices (McCormick and Prince, 1985). Intriguingly, Sawaguchi and Matsumura (1985) found that ACh-excited and ACh-inhibited DLPFC neurons were found in separate layers.
We also found that carbachol increased activity in putative pyramidal neurons, while having equivocal effects in putative interneurons. Heterogeneity in responses to carbachol on interneurons has also been reported previously in rodent medial PFC (Pafundo et al., 2013), hippocampal slices (Zheng et al., 2011), and insular cortical slices (Yamamoto et al., 2010).
Effects of carbachol on task selectivity of DLPFC neurons
Systemic blockade of muscarinic receptors has detrimental effects on cognitive performance in a variety of tasks (Klinkenberg and Blokland, 2010), including spatial working memory. In macaque DLPFC, systemic injections of muscarinic antagonist scopolamine disrupted spatial working memory (Zhou et al., 2011), whereas local iontophoretic application strongly attenuated selectivity for all task attributes, including rule selectivity (Major et al., 2015). We therefore hypothesized that stimulation of DLPFC neuronal cholinergic receptors would augment the selectivity of rule representation in working memory. Contrary to our prediction, we found that carbachol reduced selectivity of rule neurons and broad-spiking neurons with saccade direction selectivity. Selectivity of rule neurons was reduced by two different mechanisms: in carbachol-inhibited neurons, preferred rule activity was suppressed more than nonpreferred rule activity, whereas in carbachol-excited neurons, activity for the nonpreferred rule increased more than preferred. Additionally, carbachol decreased postsaccadic selectivity in putative pyramidal saccade-direction-selective neurons, due to greater increase in nonpreferred saccade direction activity.
Our results suggest that cholinergic stimulation can weaken DLPFC task representations. Herrero et al. (2008) examined cholinergic modulation and attentional enhancement in macaque V1. They found that lower dose ACh application increased neuronal activity and enhanced attentional modulation, whereas at higher doses, attentional modulation was unaffected or even disrupted due to nonspecific increase in neuronal activity. Although we did not find systematic enhancement in rule selectivity at lower doses (Fig. 4D), disruption of rule at higher doses in carbachol-excited neurons agree with Herrero et al. (2008), wherein ceiling effects in increased excitability impaired task representation. Similarly, Zinke et al. (2006) found that ACh iontophoresis broadened orientation tuning of most ACh-excited neurons in marmoset V1, possibly due to ceiling effects in optimal stimulus-induced responses and increased activity to stimuli with nonpreferred orientations. Regardless of stimulus orientation, Sato et al. (1987) found that ACh increased stimulus-evoked responses in cat V1, resulting in no systematic effects on orientation selectivity. These findings from several groups suggest that cholinergic stimulation of cortical regions resulted in “inverted-U” dose-dependency of physiological effects on cortical neurons, whereby too little or too much ACh is detrimental to task performance and neuronal selectivity. This phenomenon has also been demonstrated in the dopaminergic system (Vijayraghavan et al., 2007). Our carbachol results may reflect the rightmost portion of a cholinergic inverted-U, whereby excessive cholinergic stimulation is disruptive to cognitive processing. Unlike the aforementioned investigations, we did not observe population enhancement of task-related selectivity during low-dose cholinergic stimulation. However, we cannot discount that the relative potency of carbachol compared with ACh may have precluded the potential observation of improvements in neuronal task-selectivity. Future experiments contrasting ACh and carbachol in the same paradigm may clarify this possibility.
We found trial-to-trial variance was not significantly affected after carbachol. Moreover, regression analysis suggested that effects on trial discharge rate variability had less impact on rule selectivity relative to changes in activity. Similarly, Herrero et al. (2008) found that ACh-induced changes in Fano factor did not contribute to attentional modulation of macaque V1 neurons.
Recently, Liu et al. (2017) examined the effects of electrical stimulation of the macaque nucleus basalis during performance of a delayed match-to-sample task. Intriguingly, they found that continuous stimulation was detrimental to task performance, but intermittent stimulation resulted in significant improvement. Our results showing reduced task selectivity during continuous iontophoretic carbachol application in DLPFC suggest overstimulation of the cholinergic system can be detrimental to cognitive performance.
Because carbachol is a general cholinergic agonist, both nicotinic and muscarinic receptors could potentially mediate these effects on physiology and task selectivity. Previously it was reported that muscarinic receptor antagonism blocked the effects of ACh iontophoresis on orbitofrontal cortex (Aou et al., 1983) and DLPFC (Inoue et al., 1983). We found that carbachol inhibited a significant population of DLPFC neurons and disrupted rule selectivity in some neurons with activity suppression. Because nicotinic actions in macaque DLPFC reported heretofore were excitatory (Yang et al., 2013; Sun et al., 2017), the inhibitory actions of carbachol may be muscarinic. Indeed, muscarinic receptors can directly suppress prefrontal neurons via activation of G-protein coupled inward-rectifying potassium or SK channels (Gulledge and Stuart, 2005). Although activation of nearby interneurons is another potential mechanism to inhibit neuronal activity (Disney and Aoki, 2008; Disney et al., 2014), we believe this is less likely as we did not observe significant excitation in narrow-spiking neurons, similar to previous reports (Gulledge et al., 2007; Pafundo et al., 2013). Further supporting a role of muscarinic receptors, and akin to previous studies applying ACh to DLPFC (Nelson et al., 1973; Sawaguchi and Matsumura, 1985), we generally found that carbachol effects had longer latencies of onset and partial recovery (seconds to minutes; Fig. 1C). This is inconsistent with fast ionotropic actions mediated by nicotinic receptors, since recoveries from nicotinic agonist stimulation are rapid (Disney et al., 2007).
However, we cannot discount nicotinic involvement in carbachol's actions reported here. Although nicotinic receptor stimulation can augment working memory activity in PFC (Yang et al., 2013; Sun et al., 2017) and can improve cognitive performance (Terry et al., 2015), other studies have shown that low doses of nicotinic antagonist enhanced attentional performance in rodents (Hahn et al., 2011) and improved delayed match-to-sample performance in monkeys (Terry et al., 1999). In the physiological context, Yang et al. (2013) showed that low-dose iontophoretic stimulation of α7 nicotinic receptors during oculomotor delayed responses increased macaque DLPFC neuronal excitability and improved memory period spatial tuning, whereas α7 receptor antagonist reduced delay period activity and spatial tuning. Moreover, high-dose α7 receptor stimulation eroded tuning due to general activity increase for nonpreferred spatial directions, similar to the effects on DLPFC task selectivity reported here. Sun et al. (2017), in the same paradigm, found that nicotinic α4β2 receptor stimulation strengthened delay period activity for preferred spatial locations, while, interestingly, having no effect on neurons with saccade direction selectivity (Wang et al., 2004). Because we found that carbachol disrupted saccade direction selectivity of putative pyramidal cells, this suggests muscarinic receptors mediate carbachol's effects on postsaccadic activity. Notably, carbachol has a lower affinity and channel opening rate constant for nicotinic receptors than ACh (Akk and Auerbach, 1999). Thus, both muscarinic and nicotinic mechanisms may have contributed to carbachol's actions. Future experiments examining these receptor families with subtype-specific compounds will be necessary to delineate the signaling mechanisms that mediate the actions of carbachol on PFC task-related activity reported here.
The data reported here and previously (Major et al., 2015; Liu et al., 2017), suggest that continuous cholinergic receptor stimulation or blockade can be detrimental to prefrontal neuronal representations in cognitive tasks. Endogenous ACh is transiently released with high temporal precision (Parikh et al., 2007; Sarter et al., 2009) and continuous stimulation of cholinergic receptors may not be beneficial to cognitive performance (Bentley et al., 2011). Subtype-selective cholinergic agonists are being actively investigated to ameliorate cognitive dysfunction in neuropsychiatric disorders, including Alzheimer's disease and schizophrenia (Bodick et al., 1997; Wienrich et al., 2002; Shekhar et al., 2008). Our findings offer a cautionary note and suggest that general cholinergic stimulation using pharmacology may in fact be detrimental to cognitive functions. Future work with more selective agonists may shed light on which downstream signaling mechanisms are beneficial in treatment of cognitive dysfunction.
Footnotes
This work was supported by CIHR Grant FRN148365 to S.E.; the Canada First Research Excellence Fund to BrainsCAN; and the Ontario Graduate Scholarship, Natural Sciences and Engineering Research Council scholarship, and Alzheimer's Society London and Middlesex scholarship to A.J.M.
The authors declare no competing financial interests.
References
- Aigner TG, Mitchell SJ, Aggleton JP, DeLong MR, Struble RG, Price DL, Wenk GL, Pettigrew KD, Mishkin M (1991) Transient impairment of recognition memory following ibotenic-acid lesions of the basal forebrain in macaques. Exp Brain Res 86:18–26. [DOI] [PubMed] [Google Scholar]
- Akk G, Auerbach A (1999) Activation of muscle nicotinic acetylcholine receptor channels by nicotinic and muscarinic agonists. Br J Pharmacol 128:1467–1476. 10.1038/sj.bjp.0702941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta F, Parnetti L, Gallai V, Wallin A (2001) Treatment of cognitive dysfunction associated with Alzheimer's disease with cholinergic precursors: ineffective treatments or inappropriate approaches? Mech Ageing Dev 122:2025–2040. 10.1016/S0047-6374(01)00310-4 [DOI] [PubMed] [Google Scholar]
- Aou S, Oomura Y, Nishino H (1983) Influence of acetylcholine on neuronal activity in monkey orbitofrontal cortex during bar press feeding task. Brain Res 275:178–182. 10.1016/0006-8993(83)90433-X [DOI] [PubMed] [Google Scholar]
- Ballinger EC, Ananth M, Talmage DA, Role LW (2016) Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91:1199–1218. 10.1016/j.neuron.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassant MH, Baleyte JM, Lamour Y (1990) Effects of acetylcholine on single cortical somatosensory neurons in the unanesthetized rat. Neuroscience 39:189–197. 10.1016/0306-4522(90)90232-S [DOI] [PubMed] [Google Scholar]
- Bentley P, Driver J, Dolan RJ (2011) Cholinergic modulation of cognition: insights from human pharmacological functional neuroimaging. Prog Neurobiol 94:360–388. 10.1016/j.pneurobio.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM (1997) Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol 54:465–473. 10.1001/archneur.1997.00550160091022 [DOI] [PubMed] [Google Scholar]
- Bongard S, Nieder A (2010) Basic mathematical rules are encoded by primate prefrontal cortex neurons. Proc Natl Acad Sci U S A 107:2277–2282. 10.1073/pnas.0909180107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubser M, Byun N, Wood MR, Jones CK (2012) Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb Exp Pharmacol 208:121–166. 10.1007/978-3-642-23274-9_7 [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV (2004) Donepezil-induced improvement in delayed matching accuracy by young and old rhesus monkeys. J Mol Neurosci 24:85–91. 10.1385/JMN:24:1:085 [DOI] [PubMed] [Google Scholar]
- Caccamo A, Fisher A, LaFerla FM (2009) M1 agonists as a potential disease-modifying therapy for Alzheimer's disease. Curr Alzheimer Res 6:112–117. 10.2174/156720509787602915 [DOI] [PubMed] [Google Scholar]
- Condy C, Wattiez N, Rivaud-Péchoux S, Tremblay L, Gaymard B (2007) Antisaccade deficit after inactivation of the principal sulcus in monkeys. Cereb Cortex 17:221–229. 10.1093/cercor/bhj140 [DOI] [PubMed] [Google Scholar]
- Crawford JM, Curtis DR, Voorhoeve PE, Wilson VJ (1966) Acetylcholine sensitivity of cerebellar neurones in the cat. J Physiol 186:139–165. 10.1113/jphysiol.1966.sp008025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxson PL, Kyriazis DA, Baxter MG (2011) Cholinergic modulation of a specific memory function of prefrontal cortex. Nat Neurosci 14:1510–1512. 10.1038/nn.2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney AA, Aoki C (2008) Muscarinic acetylcholine receptors in macaque V1 are most frequently expressed by parvalbumin-immunoreactive neurons. J Comp Neurol 507:1748–1762. 10.1002/cne.21616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney AA, Aoki C, Hawken MJ (2007) Gain modulation by nicotine in macaque v1. Neuron 56:701–713. 10.1016/j.neuron.2007.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney AA, Alasady HA, Reynolds JH (2014) Muscarinic acetylcholine receptors are expressed by most parvalbumin-immunoreactive neurons in area MT of the macaque. Brain Behav 4:431–445. 10.1002/brb3.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA (2002) Graded persistent activity in entorhinal cortex neurons. Nature 420:173–178. 10.1038/nature01171 [DOI] [PubMed] [Google Scholar]
- Everling S, DeSouza JF (2005) Rule-dependent activity for prosaccades and antisaccades in the primate prefrontal cortex. J Cogn Neurosci 17:1483–1496. 10.1162/0898929054985455 [DOI] [PubMed] [Google Scholar]
- Everling S, Dorris MC, Munoz DP (1998) Reflex suppression in the anti-saccade task is dependent on prestimulus neural processes. J Neurophysiol 80:1584–1589. 10.1152/jn.1998.80.3.1584 [DOI] [PubMed] [Google Scholar]
- Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM (2016) Alzheimer's disease: targeting the cholinergic system. Curr Neuropharmacol 14:101–115. 10.2174/1570159X13666150716165726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucault-Fruchard L, Antier D (2017) Therapeutic potential of alpha7 nicotinic receptor agonists to regulate neuroinflammation in neurodegenerative diseases. Neural Regen Res 12:1418–1421. 10.4103/1673-5374.215244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima J, Fukushima K, Miyasaka K, Yamashita I (1994) Voluntary control of saccadic eye movement in patients with frontal cortical lesions and parkinsonian patients in comparison with that in schizophrenics. Biol Psychiatry 36:21–30. 10.1016/0006-3223(94)90058-2 [DOI] [PubMed] [Google Scholar]
- Green DM, Swets JA (1966) Signal detection theory and psychophysics. New York: Wiley. [Google Scholar]
- Gulledge AT, Stuart GJ (2005) Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci 25:10308–10320. 10.1523/JNEUROSCI.2697-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ (2007) Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol 97:2215–2229. 10.1152/jn.00493.2006 [DOI] [PubMed] [Google Scholar]
- Hahn B, Shoaib M, Stolerman IP (2011) Selective nicotinic receptor antagonists: effects on attention and nicotine-induced attentional enhancement. Psychopharmacology 217:75–82. 10.1007/s00213-011-2258-8 [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Hartigan PM (1985) The dip test of unimodality. Ann Stat 13:70–84. 10.1214/aos/1176346577 [DOI] [Google Scholar]
- Herrero JL, Roberts MJ, Delicato LS, Gieselmann MA, Dayan P, Thiele A (2008) Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454:1110–1114. 10.1038/nature07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein S, Johnston K, Belbeck B, Lomber SG, Everling S (2014) Functional specialization within macaque dorsolateral prefrontal cortex for the maintenance of task rules and cognitive control. J Cogn Neurosci 26:1918–1927. 10.1162/jocn_a_00608 [DOI] [PubMed] [Google Scholar]
- Inoue M, Oomura Y, Nishino H, Aou S, Sikdar SK, Hynes M, Mizuno Y, Katabuchi T (1983) Cholinergic role in monkey dorsolateral prefrontal cortex during bar-press feeding behavior. Brain Res 278:185–194. 10.1016/0006-8993(83)90237-8 [DOI] [PubMed] [Google Scholar]
- Jacob SN, Ott T, Nieder A (2013) Dopamine regulates two classes of primate prefrontal neurons that represent sensory signals. J Neurosci 33:13724–13734. 10.1523/JNEUROSCI.0210-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K, DeSouza JF, Everling S (2009) Monkey prefrontal cortical pyramidal and putative interneurons exhibit differential patterns of activity between prosaccade and antisaccade tasks. J Neurosci 29:5516–5524. 10.1523/JNEUROSCI.5953-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman LD, Pratt J, Levine B, Black SE (2010) Antisaccades: a probe into the dorsolateral prefrontal cortex in Alzheimer's disease: a critical review. J Alzheimers Dis 19:781–793. 10.3233/JAD-2010-1275 [DOI] [PubMed] [Google Scholar]
- Klinkenberg I, Blokland A (2010) The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev 34:1307–1350. 10.1016/j.neubiorev.2010.04.001 [DOI] [PubMed] [Google Scholar]
- Liu R, Crawford J, Callahan PM, Terry AV Jr, Constantinidis C, Blake DT (2017) Intermittent stimulation of the nucleus basalis of meynert improves working memory in adult monkeys. Curr Biol 27:2640–2646.e4. 10.1016/j.cub.2017.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Skoblenick K, Seamans JK, Everling S (2015) Ketamine-induced changes in the signal and noise of rule representation in working memory by lateral prefrontal neurons. J Neurosci 35:11612–11622. 10.1523/JNEUROSCI.1839-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major AJ, Vijayraghavan S, Everling S (2015) Muscarinic attenuation of mnemonic rule representation in macaque dorsolateral prefrontal cortex during a pro- and anti-saccade task. J Neurosci 35:16064–16076. 10.1523/JNEUROSCI.2454-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura M, Sawaguchi T, Kubota K (1990) Modulation of neuronal activities by iontophoretically applied catecholamines and acetylcholine in the primate motor cortex during a visual reaction-time task. Neurosci Res 8:138–145. 10.1016/0168-0102(90)90066-N [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA (1985) Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci U S A 82:6344–6348. 10.1073/pnas.82.18.6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH (1983) Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 214:170–197. 10.1002/cne.902140206 [DOI] [PubMed] [Google Scholar]
- Millar J, Williams GV (1989) Effects of iontophoresis of noradrenaline and stimulation of the periaqueductal gray on single-unit activity in the rat superficial dorsal horn. J Comp Neurol 287:119–133. 10.1002/cne.902870109 [DOI] [PubMed] [Google Scholar]
- Mitchell JF, Sundberg KA, Reynolds JH (2007) Differential attention-dependent response modulation across cell classes in macaque visual area V4. Neuron 55:131–141. 10.1016/j.neuron.2007.06.018 [DOI] [PubMed] [Google Scholar]
- Mountcastle VB, Talbot WH, Sakata H, Hyvärinen J (1969) Cortical neuronal mechanisms in flutter-vibration studied in unanesthetized monkeys: neuronal periodicity and frequency discrimination. J Neurophysiol 32:452–484. [DOI] [PubMed] [Google Scholar]
- Nagy B, Szabó I, Csetényi B, Hormay E, Papp S, Keresztes D, Karádi Z (2014) Noradrenaline and acetylcholine responsiveness of glucose-monitoring and glucose-insensitive neurons in the mediodorsal prefrontal cortex. Brain Res 1543:159–164. 10.1016/j.brainres.2013.11.014 [DOI] [PubMed] [Google Scholar]
- Nelson CN, Hoffer BJ, Chu NS, Bloom FE (1973) Cytochemical and pharmacological studies on polysensory neurons in the primate frontal cortex. Brain Res 62:115–133. 10.1016/0006-8993(73)90623-9 [DOI] [PubMed] [Google Scholar]
- Pafundo DE, Miyamae T, Lewis DA, Gonzalez-Burgos G (2013) Cholinergic modulation of neuronal excitability and recurrent excitation-inhibition in prefrontal cortex circuits: implications for gamma oscillations. J Physiol 591:4725–4748. 10.1113/jphysiol.2013.253823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M (2007) Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron 56:141–154. 10.1016/j.neuron.2007.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrot-Deseilligny C, Müri RM, Ploner CJ, Gaymard B, Demeret S, Rivaud-Pechoux S (2003) Decisional role of the dorsolateral prefrontal cortex in ocular motor behaviour. Brain 126:1460–1473. 10.1093/brain/awg148 [DOI] [PubMed] [Google Scholar]
- Pirch JH, Turco K, Rucker HK (1992) A role for acetylcholine in conditioning-related responses of rat frontal cortex neurons: microiontophoretic evidence. Brain Res 586:19–26. 10.1016/0006-8993(92)91366-M [DOI] [PubMed] [Google Scholar]
- Richmond BJ, Optican LM, Podell M, Spitzer H (1987) Temporal encoding of two-dimensional patterns by single units in primate inferior temporal cortex: I. Response characteristics. J Neurophysiol 57:132–146. [DOI] [PubMed] [Google Scholar]
- Ridley RM, Bowes PM, Baker HF, Crow TJ (1984) An involvement of acetylcholine in object discrimination learning and memory in the marmoset. Neuropsychologia 22:253–263. 10.1016/0028-3932(84)90073-3 [DOI] [PubMed] [Google Scholar]
- Roberts MJ, Zinke W, Guo K, Robertson R, McDonald JS, Thiele A (2005) Acetylcholine dynamically controls spatial integration in marmoset primary visual cortex. J Neurophysiol 93:2062–2072. 10.1152/jn.00911.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberry TL, Sonoda LK, Dekat SE, Cusack B, Johnson JL (2008) Analysis of the reaction of carbachol with acetylcholinesterase using thioflavin T as a coupled fluorescence reporter. Biochemistry 47:13056–13063. 10.1021/bi8015197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM (2009) Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci 10:383–390. 10.1038/nrn2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Hata Y, Masui H, Tsumoto T (1987) A functional role of cholinergic innervation to neurons in the cat visual cortex. J Neurophysiol 58:765–780. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M (1985) Laminar distributions of neurons sensitive to acetylcholine, noradrenaline and dopamine in the dorsolateral prefrontal cortex of the monkey. Neurosci Res 2:255–273. 10.1016/0168-0102(85)90004-5 [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC (2008) Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry 165:1033–1039. 10.1176/appi.ajp.2008.06091591 [DOI] [PubMed] [Google Scholar]
- Skoblenick K, Everling S (2012) NMDA antagonist ketamine reduces task selectivity in macaque dorsolateral prefrontal neurons and impairs performance of randomly interleaved prosaccades and antisaccades. J Neurosci 32:12018–12027. 10.1523/JNEUROSCI.1510-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoblenick K, Everling S (2014) N-methyl-d-aspartate receptor antagonist ketamine impairs action-monitoring activity in the prefrontal cortex. J Cogn Neurosci 26:577–592. 10.1162/jocn_a_00519 [DOI] [PubMed] [Google Scholar]
- Skoblenick KJ, Womelsdorf T, Everling S (2016) Ketamine alters outcome-related local field potentials in monkey prefrontal cortex. Cereb Cortex 26:2743–2752. 10.1093/cercor/bhv128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma S, Shimegi S, Osaki H, Sato H (2012) Cholinergic modulation of response gain in the primary visual cortex of the macaque. J Neurophysiol 107:283–291. 10.1152/jn.00330.2011 [DOI] [PubMed] [Google Scholar]
- Sun Y, Yang Y, Galvin VC, Yang S, Arnsten AF, Wang M (2017) Nicotinic α4β2 cholinergic receptor influences on dorsolateral prefrontal cortical neuronal firing during a working memory task. J Neurosci 37:5366–5377. 10.1523/JNEUROSCI.0364-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Buccafusco JJ, Prendergast MA (1999) Dose-specific improvements in memory-related task performance by rats and aged monkeys administered the nicotinic-cholinergic antagonist mecamylamine. Drug Dev Res 47:127–136. [DOI] [Google Scholar]
- Terry AV Jr, Callahan PM, Hernandez CM (2015) Nicotinic ligands as multifunctional agents for the treatment of neuropsychiatric disorders. Biochem Pharmacol 97:388–398. 10.1016/j.bcp.2015.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele A, Brandt C, Dasilva M, Gotthardt S, Chicharro D, Panzeri S, Distler C (2016) Attention induced gain stabilization in broad and narrow-spiking cells in the frontal eye-field of macaque monkeys. J Neurosci 36:7601–7612. 10.1523/JNEUROSCI.0872-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada H, Nishiyama S, Fukumoto D, Ohba H, Sato K, Kakiuchi T (2004) Effects of acute acetylcholinesterase inhibition on the cerebral cholinergic neuronal system and cognitive function: functional imaging of the conscious monkey brain using animal PET in combination with microdialysis. Synapse 52:1–10. 10.1002/syn.10310 [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF (2007) Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10:376–384. 10.1038/nn1846 [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Major AJ, Everling S (2016) Dopamine D1 and D2 receptors make dissociable contributions to dorsolateral prefrontal cortical regulation of rule-guided oculomotor behavior. Cell Rep 16:805–816. 10.1016/j.celrep.2016.06.031 [DOI] [PubMed] [Google Scholar]
- Voytko ML, Olton DS, Richardson RT, Gorman LK, Tobin JR, Price DL (1994) Basal forebrain lesions in monkeys disrupt attention but not learning and memory. J Neurosci 14:167–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Vijayraghavan S, Goldman-Rakic PS (2004) Selective D2 receptor actions on the functional circuitry of working memory. Science 303:853–856. 10.1126/science.1091162 [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 215:1237–1239. 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]
- Wienrich M, Ceci A, Ensinger HA, Gaida W, Mendla KD, Osugi T, Raschig A, Weiser T (2002) Talsaclidine (WAL 2014 FU), a muscarinic M1 receptor agonist for the treatment of Alzheimer's disease. Drug Dev Res 56:321–334. 10.1002/ddr.10085 [DOI] [Google Scholar]
- Yamamoto K, Koyanagi Y, Koshikawa N, Kobayashi M (2010) Postsynaptic cell type-dependent cholinergic regulation of GABAergic synaptic transmission in rat insular cortex. J Neurophysiol 104:1933–1945. 10.1152/jn.00438.2010 [DOI] [PubMed] [Google Scholar]
- Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AF, Wang M (2013) Nicotinic alpha7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc Natl Acad Sci U S A 110:12078–12083. 10.1073/pnas.1307849110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Seeger T, Nixdorf-Bergweiler BE, Alzheimer C (2011) Layer-specific processing of excitatory signals in CA1 interneurons depends on postsynaptic M2 muscarinic receptors. Neurosci Lett 494:217–221. 10.1016/j.neulet.2011.03.016 [DOI] [PubMed] [Google Scholar]
- Zhou X, Qi XL, Douglas K, Palaninathan K, Kang HS, Buccafusco JJ, Blake DT, Constantinidis C (2011) Cholinergic modulation of working memory activity in primate prefrontal cortex. J Neurophysiol 106:2180–2188. 10.1152/jn.00148.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke W, Roberts MJ, Guo K, McDonald JS, Robertson R, Thiele A (2006) Cholinergic modulation of response properties and orientation tuning of neurons in primary visual cortex of anaesthetized Marmoset monkeys. Eur J Neurosci 24:314–328. 10.1111/j.1460-9568.2006.04882.x [DOI] [PMC free article] [PubMed] [Google Scholar]