

Abstract
Neuronal mRNAs can be packaged in reversibly stalled polysome granules before their transport to distant synaptic locales. Stimulation of synaptic metabotropic glutamate receptors (mGluRs) reactivates translation of these particular mRNAs to produce plasticity-related protein; a phenomenon exhibited during mGluR-mediated LTD. This form of plasticity is deregulated in Fragile X Syndrome, a monogenic form of autism in humans, and understanding the stalling and reactivation mechanism could reveal new approaches to therapies. Here, we demonstrate that UPF1, known to stall peptide release during nonsense-mediated RNA decay, is critical for assembly of stalled polysomes in rat hippocampal neurons derived from embryos of either sex. Moreover, UPF1 and its interaction with the RNA binding protein STAU2 are necessary for proper transport and local translation from a prototypical RNA granule substrate and for mGluR-LTD in hippocampal neurons. These data highlight a new, neuronal role for UPF1, distinct from its RNA decay functions, in regulating transport and/or translation of mRNAs that are critical for synaptic plasticity.
SIGNIFICANCE STATEMENT The elongation and/or termination steps of mRNA translation are emerging as important control points in mGluR-LTD, a form of synaptic plasticity that is compromised in a severe monogenic form of autism, Fragile X Syndrome. Deciphering the molecular mechanisms controlling this type of plasticity may thus open new therapeutic opportunities. Here, we describe a new role for the ATP-dependent helicase UPF1 and its interaction with the RNA localization protein STAU2 in mediating mGluR-LTD through the regulation of mRNA translation complexes stalled at the level of elongation and/or termination.
Keywords: long-term depression, protein synthesis, RNA binding protein, RNA granule, RNA helicase, stalled polysome
Introduction
In neurons, mRNA transport and localized translation are crucial for synaptic plasticity (Jung et al., 2014). Proper spatiotemporal control of translation in dendrites or developing axons necessitates reliable suppression of protein synthesis during mRNA transport and a mechanism for subsequent reactivation at the appropriate time and destination. Most described repression mechanisms target translation initiation on particular mRNAs (Jung et al., 2014), and it would follow that their transport precedes polysome formation. In contrast, we and others have described a neuronal ribonucleoprotein complex (a neuronal RNA granule) consisting of stalled polysomes, where initiation of mRNAs has already occurred and repression is at the level of either elongation or termination (Elvira et al., 2006; Darnell et al., 2011; Graber et al., 2013b). The composition of the neuronal RNA granule seems to be particularly suited for the fast bursts of protein synthesis that have been observed in metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD). This type of localized synaptic plasticity requires protein synthesis yet paradoxically occurs even in the absence of translation initiation, presumably due to the reactivated translation of preexisting polysomes (Graber et al., 2013b).
One protein implicated in stalled polysomes is the fragile X mental retardation protein, FMRP (Darnell et al., 2011). A phosphorylated form of FMRP has been previously linked with stalled polysomes (Ceman et al., 2003). Indeed, FMRP is dephosphorylated upon mGluR stimulation, and this is required for the proper synthesis of proteins during mGluR-LTD (Niere et al., 2012). However, in the absence of FMRP, mRNA transport is largely unaffected (Steward et al., 1998). LTD proteins in this context are instead constitutively synthesized in dendrites independently of mGluR stimulation, leading to enhanced LTD upon mGluR stimulation (Hou et al., 2006; Nosyreva and Huber, 2006). Thus, FMRP appears to be necessary for stabilization of stalled polysomes but not their initial formation. In contrast, the mRNA binding protein STAU2 is required for mGluR-LTD and has been associated with the transport and translation of at least one FMRP and LTD target mRNA, Map1b, via its 3′ untranslated region (UTR) (Lebeau et al., 2011).
STAU2 interacts with the RNA decay factor UPF1 (Miki et al., 2011; Park et al., 2013), and these proteins were found in the RNA granule proteome (Elvira et al., 2006; El Fatimy et al., 2016). UPF1 is a critical ATPase/helicase in nonsense-mediated RNA decay (NMD) pathways, where it is recruited to the stop codon through association with eRF1 and is involved in recognition of improper stop codons that are often present in mis-spliced mRNAs (Chang et al., 2007). One aspect of UPF1 function is to stall translation through interactions with eIF3, another abundant component of RNA granules (Elvira et al., 2006; Isken et al., 2008, El Fatimy et al., 2016), and to stall peptide release through binding to eRF3 (Ivanov et al., 2008). These characteristics made UPF1 an attractive candidate to play a mechanistic role in stalling polysomes.
Our results highlight a new role for UPF1 in regulating mRNA targets at the level of transport and translation, distinct from its RNA decay functions, and provide mechanistic insights into how stalled polysomes can be formed before transport and reactivated locally at synapses during synaptic plasticity.
Materials and Methods
Animals and cell culture.
Sprague Dawley rats were obtained from Charles River Canada. All animal experiments were approved by the Animal Ethics Committees of the Montreal Neurological Institute and Université de Montréal, and abided by the guidelines of the Canadian Council on Animal Care. Rat primary hippocampal neurons were dissected from embryonic day 18 Sprague Dawley embryos of either sex and supplemented with new medium (neurobasal media supplemented with 1% (v/v) N2 and penicillin/streptomycin, 2% (v/v) B27 and 0.5 mm Gluta-MAX; Thermo Fisher Scientific) every 6–8 d as previously described (Lebeau et al., 2011). Electrophysiology experiments were performed with 18–25 DIV dissociated hippocampal neuron cultures, whereas all other experiments were performed with 8–10 DIV cultures. Neurons were plated on 12 mm poly-L-lysine-coated #1.5 German glass coverslips (Neuvitro) at a density of 100,000 cells per well of a 12-well tissue culture plate. For super-resolution experiments, neurons were plated on poly-L-lysine-coated LAB-TEK II chambered #1.5 coverglass slides (Thermo Fisher Scientific). For knockdown experiments with single RNAi transduction, cultures were transduced with a multiplicity of infection (MOI) of 3–10. For rescue experiments with double transduction, cultures were transduced with Stau2 RNAi with an MOI of 3 and with STAU2 constructs, each with an MOI of 5 or with Upf1 RNAi with an MOI of 3 and with UPF1 rescue at an MOI of 0.5. Although only a subset of neurons was transduced with this MOI, they could be detected by the mCHERRY expressed from a separate promoter on the lentivirus. A minimum of 3 d was allowed for transduction and knockdown. Transduction efficiency was assessed in each experiment by GFP or mCHERRY expression. HEK293T cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS, sodium pyruvate, penicillin, and streptomycin.
DNA constructs and lentiviruses.
The GFP.STAU2.flag plasmid (expressing the human 59 kDa isoform of STAU2) has been previously described (Graber et al., 2013b). C-terminally tagged UPF1.myc plasmid (expressing human UPF1) was a kind gift from Dr. Luc Desgroseillers, and RIP1.myc plasmid was a kind gift from Dr. Phil Barker. STAU2 C-terminal deletion and domain mutation constructs were created by PCR amplification of regions using GFP.STAU2.flag as template and inserting in-frame with GFP and the Flag tag. The F207A ribosome binding mutation was generated by site-directed mutagenesis. Plasmid sequences were verified by Sanger sequencing.
To efficiently knock down STAU2 or UPF1 in rat hippocampal neurons, lentiviruses were generated that express short hairpin RNA (shRNA) driven by a CMV promoter as previously described (Thomas et al., 2009). A nontargeting, “scrambled” shRNA sequence (AATTCTCCGAACGTGTCACGT), a sequence targeting all of the rat isoforms of STAU2 (ACTAGTGGACGCTTTATAGCC), a sequence targeting rodent UPF1 isoforms (AGCAGCTTGTGGTAAATATAC), or a sequence targeting rodent PNRC2 (TGGTGATGGTGGTTCACTAAA), designed using the Invitrogen Block-iT RNAi designer (thus minimizing the possibility of off-target designs), were cloned into a lentiviral expression vector downstream of an emerald GFP (emGFP) ORF (pRRL.emGFP.shRNA). For rescue experiments, the RNAi-resistant GFP.STAU2.flag or the GFP.C351.flag (UPF1 binding mutation) ORFs were excised by restriction digest from the pcDNA plasmids used in the coimmunoprecipitation experiments and subcloned into the lentiviral expression plasmid. The UPF1 rescue construct was purchased from Vectorbuilder and expressed mCHERRY from a viral promoter and human UPF1 from an EF1A promoter (pLV[Exp] − mCherry/Neo-EF1A > hUPF1[ORF003793]*/FLAG pLV[Exp]). All clones were verified by Sanger sequencing.
VSV G-pseudotyped virus was packaged by transient transfection of HEK293T helper cells with Lipofectamine 2000 (Thermo Fisher Scientific) pMD2.g, pRSV-Rev, and pMDLg/pRRE packaging plasmids together with the pRRL.emGFP.shRNA. Virus was collected over 48 h with cell debris removed by microfiltration. Virus was then concentrated by centrifugation, resuspended in PBS, pH 7.3, and stored at −80°C. Virus was titered based on GFP expression in HEK293T cells. Transduction efficiency was assessed in each experiment by GFP or mCHERRY expression.
Coimmunoprecipitation and immunoblotting.
For experiments to determine whether 3,5-dihydroxyphenylglycine (DHPG)-induced changes in phosphorylation of UPF1, hippocampal cultures (10 DIV) were incubated in the presence or absence of 50 μm (S)-3,5-DHPG (Tocris Bioscience) for 10 min and then harvested, lysed in Laemmli buffer, and the proteins separated by SDS-PAGE and immunoblotted first for phospho-UPF1 (1:500; anti-Ser1127; #0701016; Millipore) followed by anti-rabbit coupled to HRP, then stripped and reblotted with anti-UPF1 (1:10,000; #ab133564, Epitomics) followed by anti-rabbit coupled to HRP. To determine loading, the membrane was stripped one additional time and probed with anti-GAPDH (1:1000; #sc-25778, Santa Cruz Biotechnology) followed by anti-rabbit coupled to HRP.
For experiments to determine the effectiveness of knockdown and rescue, hippocampal neurons were transduced at 7 DIV and, 3–7 d later, were harvested and lysed in Laemmli buffer. Antibodies described above were used along with anti-STAU2 (kind gift of Dr. Michael Kiebler) and anti-PNRC2 (1:1000, #NBP1-74252, Novus Biologicals).
HEK293T cells were seeded in 6-well plates and transfected 24 h later with 1 μg each of the appropriate plasmid. Cells were lysed in nondenaturing coimmunoprecipitation buffer (150 mm NaCl, 1 mm EDTA, 1% (v/v) Triton X-100, 50 mm Tris-HCl, pH 7.4) supplemented with Roche Complete EDTA-free protease inhibitor mixture (Roche); 200 μg of lysate was incubated overnight with Flag antibody-conjugated beads (Sigma-Aldrich) followed by extensive washing with lysis buffer. Immunoprecipitated Flag-tagged species were eluted from beads with 2 μg/ml of Flag peptide (Sigma-Aldrich), resuspended in Laemmli buffer, resolved by SDS-PAGE, and transferred to PVDF membrane for Western blotting. For coimmunoprecipitations, membranes were probed with rabbit polyclonal anti-myc or mouse anti-Flag M2 monoclonal antibodies (Sigma-Aldrich) followed by anti-rabbit and anti-mouse secondary antibodies conjugated to infrared dyes CF680 and CF770 (Biotium), respectively. Secondary antibody detection was performed using the Licor Odyssey system (LI-COR Biosciences), or standard HRP detection with film was used.
Ribopuromycylation (RPM) and STAU2, UPF1 immunocytochemistry.
To enrich for stalled polysomes, rat hippocampal neurons at 8–10 DIV were incubated with 5 μm homoharringtonine (HHT; Tocris Bioscience) in 1 ml of supplemented neurobasal media for 10 min. RPM has been described previously (Graber et al., 2013b). Briefly, puromycin (100 μm) and emetine (200 μm) (both from Sigma-Aldrich) were then added, and the cells were incubated at 37°C for an additional 5 min. Free puromycin was removed with a digitonin wash on ice, and cells were fixed at room temperature for 15 min with 4% PFA/sucrose. Cells were permeabilized with 0.1% Triton X-100 and blocked with 5% BSA. Puromycin was detected with 1:1000 of mouse anti-puromycin for 1 h (clone 2A4) (David et al., 2012), UPF1 was detected with 1:500 rabbit monoclonal anti-RENT1/UPF1 (Epitomics), and STAU2 was detected with 1:500 rabbit anti-STAU2 (Dr. Michael Kiebler); 1:1000 of anti-mouse or anti-rabbit Alexa-568 secondary antibody was incubated with samples for 1 h (Thermo Fisher Scientific). For super-resolution imaging, primary antibodies were detected by incubation with 1:1000 or 1:10,000 of anti-mouse Alexa-647 and anti-rabbit Alexa-568 for 1 h. To reduce antibody drift during single-molecule localization, antibody-antigen pairs were postfixed for 10 min at room temperature with 4% PFA/sucrose.
Azidohomoalanine (AHA) labeling.
Neurons were first incubated with methionine-free HEPES-buffered solution (120 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 25 mm HEPES, 30 mm glucose) for 30 min at 37°C followed by addition of 5 μm HHT and 25 μm AHA (Thermo Fisher Scientific) for 10 min before induction with 50 μm (S)-3,5-DHPG (Tocris Bioscience) for a further 10 min. DHPG was removed from the cells with one wash of PBS and incubated for a final 10 min with fresh AHA and HHT to resolve any DHPG-mediated increases in protein synthesis. Cells were fixed as above. Subsequent detection of AHA has been described previously (Graber et al., 2013b). For quantification of AHA, AHA intensity was measured between 50 and 60 μm from the soma boundary. All values were normalized to the average intensity of the scrambled control from that experiment.
MAP1B induction.
Neurons were incubated with HHT for 10 min before coincubation with 50 μm DHPG for an additional 10 min. Cells were fixed as above, and standard immunocytochemistry was performed with 1:200 mouse anti-MAP1B clone AA6 (Santa Cruz Biotechnology) and 1:1000 anti-mouse Alexa-568, both for 1 h. For quantification of MAP1B, MAP1B intensity was measured between 50 and 60 μm from the soma boundary. All values were normalized to the average intensity of the scrambled control from that experiment.
Droplet digital RT-PCR.
Total RNA was isolated from 2 × 106 hippocampal neurons using RNAqueous Total RNA isolation kit following the manufacturer's protocol (Thermo Fisher Scientific). cDNA was synthesized from 1 μg of total RNA using Superscript II reverse transcriptase with dT18 primer (Thermo Fisher Scientific). Standard curves and the appropriate annealing temperatures were obtained by quantitative PCR (Eppendorf realplex) for the following rat-specific primer sets (listed as forward, reverse) that were designed to span an intron: Map1b: CACACGAGGGGAAGAGAAGG, TTAGTGGTTCCTGGTCCTGC; Upf1: GTCACAGACTCAAGATAACATCACG, GACTAAATCCTCATTACCAGAGTCA; Stau2: ACTCCCACCTCTTCCTGTGA, CATACTCGGGTCCAGCCTTT; Gapdh: TCATGACCACAGTCCATGCC, ATCACGCCACAGCTTTCCAG; Rgs4: CATCGGCTGGGATTTCTGCT, GCTCACCCTCTGGCAAGTTA; Cplx1: GGCATACGAGATAAGTATGGCATC, CTTCTTGGGTCGAGTCAGGC; Arc: GCCAGTCTTGGGCAGCATAG, ACTGGTATGAATCACTGCTGGG; Dexi: AGAGGCCCGCCTGCTC, GGATCTGGCAACTCCCCATC; Arf1: GGCGGCCTGAGGTCTCT, TTGCCAAAAAGGCCCTTGAA; Gap43: AGGAGGAGAAAGAAGCTGTAGAT, GTTCTTGGTCAGCCTCGGG. Droplet digital PCR was performed with varying amounts of cDNA using QX200 ddPCR EvaGreen Supermix (Bio-Rad) together with the QX200 Droplet Digital PCR system (Bio-Rad) according to the manufacturer's protocol. Following droplet generation, the PCR step was performed using a C1000 thermal cycler (Bio-Rad) with the following parameters: 1 cycle at 94°C for 1 min, 45 cycles at 94°C for 15 s followed by 58°C for 1 min with a ramp rate of 2°C/s, 1 cycle at 4°C for 5 min, and 1 cycle at 90°C for 5 min. Droplets were flagged as positive or negative for PCR product using the QX200 Droplet Reader, and Quantasoft software version 1.7.4 was used to determine absolute copy number per microliter.
Single-molecule RNA-FISH (smRNA-FISH).
Neurons were fixed on glass coverslips with formaldehyde as above, washed with PBS, and permeablized with 70% ethanol for at least 1 h. Neurons were rehydrated with 10% (v/v) formamide and 2× SSC (300 mm NaCl, 30 mm sodium citrate). Neurons were then incubated with 25 pmol of Map1b Stellaris FISH probes (Biosearch Technologies). This set of 48, 21-mer oligos, each of which is conjugated to Quasar 570 fluorophores was designed to target nucleotides 93-1726 of the 7386 nucleotide rat Map1b mRNA (GenBank accession NM_019217). The probe sequences are as follows: (5′-3′): GAGGAAGGCAATTTCTGCTG, GCAGCATGTTCAAAGTCTTC, CCAGTCTTTCTGATCTTTTG, CATGGGAGAGTCAAACTGCT, TGACTCTCTCATCTTTCACA, CTCTCTTTGGAAGTAGCTGA, GGTGTCGAAGTCTCTTTTTT, TGGTTCAGTTTCAGTGAGTG, GATGTCTCTTCAAGCTTTGT, AGTCTGAATTACACCTGCTT, AACTGCCTGTTCTAGACTTA, CTCTTCAAAAAGTCCCTGGA, CATCCCTGATTTATCTTCAT, CACTCAGCTCGTAGTAATCA, TTCATCTTGTTGGTTCTTGG, CTTGGTTCTTCAGGTAACTC, CGGCCAAAGTTAAACCCAAG, CCAGAGGGGAAAGATCATGG, CAGGTAATCATCTCCTTCAT, GAGGATGTTTCAACTTGGGT, AGAAGCTAATCTGGACCTGG, TTGACTGTCTGGTTTTACGG, TTGTTGAACACACAGTACCC, AAAGGTCAGTGGCCAAATCT, GAGGTTTGCTCATAGGTTAC, AGGTGCTGTTTCTTTAGTTG, TTTCTTGGCAGCAACATCTA, CTGTCCAAAGTCACTGACTT, CTGGGTGACATCAGAAGGTA, CTACTTCCTGGAACAAGCAG, ATCAGTCGTGGTTTGTACTA, AATCGTCGTAGGTTTCTGTT, ATCAATGGTGGTCTCATCTT, CTCTCTCCTCTTTAGGAATA, TTTTTCCTTCTCACTTCATC, TTTTCCGCTTAACACAGGAG, ATTCCATCAGTGACTTTGTC, TCTTGGGAGGGAAGAACGTT, GGAGCTGTTCAGAGAGAACG, GTGAAGAGTAGCTTGGAGGA, CTTTGGAGGAGTGCGAATGA, GGTTGGTTAATGAGCCGAAG, GTTGATCCGATTTTGGACTT, GATGTTCTTTAGAGAGCCAC, CATGGTGAGCATTGTCAAGT, TGTCAATCTTCACGTTACCA, TCTCGGAAGTTCAGCTTTTG. To detect rat Map2 mRNA (GenBank accession NM_013066.1), a probe-set was synthesized with the following Quasar 570-conjugated DNA sequences: GAGGAAGGCAATTTCTGCTG, GCAGCATGTTCAAAGTCTTC, CCAGTCTTTCTGATCTTTTG, CATGGGAGAGTCAAACTGCT, TGACTCTCTCATCTTTCACA, CTCTCTTTGGAAGTAGCTGA, GGTGTCGAAGTCTCTTTTTT, TGGTTCAGTTTCAGTGAGTG, GATGTCTCTTCAAGCTTTGT, AGTCTGAATTACACCTGCTT, AACTGCCTGTTCTAGACTTA, CTCTTCAAAAAGTCCCTGGA, CATCCCTGATTTATCTTCAT, CACTCAGCTCGTAGTAATCA, TTCATCTTGTTGGTTCTTGG, CTTGGTTCTTCAGGTAACTC, CGGCCAAAGTTAAACCCAAG, CCAGAGGGGAAAGATCATGG, CAGGTAATCATCTCCTTCAT, GAGGATGTTTCAACTTGGGT, AGAAGCTAATCTGGACCTGG, TTGACTGTCTGGTTTTACGG, TTGTTGAACACACAGTACCC, AAAGGTCAGTGGCCAAATCT, GAGGTTTGCTCATAGGTTAC, AGGTGCTGTTTCTTTAGTTG, TTTCTTGGCAGCAACATCTA, CTGTCCAAAGTCACTGACTT, CTGGGTGACATCAGAAGGTA, CTACTTCCTGGAACAAGCAG, ATCAGTCGTGGTTTGTACTA, AATCGTCGTAGGTTTCTGTT, ATCAATGGTGGTCTCATCTT, CTCTCTCCTCTTTAGGAATA, TTTTTCCTTCTCACTTCATC, TTTTCCGCTTAACACAGGAG, ATTCCATCAGTGACTTTGTC, TCTTGGGAGGGAAGAACGTT, GGAGCTGTTCAGAGAGAACG, GTGAAGAGTAGCTTGGAGGA, CTTTGGAGGAGTGCGAATGA, GGTTGGTTAATGAGCCGAAG, GTTGATCCGATTTTGGACTT, GATGTTCTTTAGAGAGCCAC, CATGGTGAGCATTGTCAAGT, TGTCAATCTTCACGTTACCA, TCTCGGAAGTTCAGCTTTTG, ATGCTTCCAGAAGAGGAGAC. The probes were incubated with the cells in hybridization buffer (1 mg/ml yeast tRNA, 200 μg/ml BSA, 10% (w/v) dextran sulfate, 2 mm vandal ribonucleoside complex, 10% (w/v) formamide, 2× SSC) overnight in a humidified chamber at 37°C. Neurons were washed twice with 10% (v/v) formamide, 2× SSC at 37°C. Fresh GLOX buffer (0.37 mg/ml glucose oxidase, 300 AU catalase, 0.4% (w/v) glucose, 2× SSC, 10 mm Tris-HCl, pH 8.0) was added before mounting the coverslips on glass slides for imaging.
Confocal imaging and quantitation of Map1b mRNA and puromycylated puncta.
Neurons were imaged using a Zeiss LSM-710 confocal microscope with a 63× oil-immersion objective (NA = 1.4). ImageJ was used for image postprocessing (e.g., neurite straightening using the “Straighten” plugin), including quantitation. For quantitation of puromycylated puncta, straightened images of neurites were thresholded so that only high-intensity puncta were visible, and their numbers counted and normalized to neurite length. For smRNA-FISH, a Laplacian of Gaussian transformation was applied to confocal images using the ImageJ plugin “LoG3D” to enhance edges of individual puncta. Images were then thresholded, and outline masks of puncta were generated and quantitated in ImageJ using the “Analyze particles” tool.
Super-resolution imaging.
Single-molecule localization was performed using a Vutara SR-350 Stochastic Optical Reconstruction Microscope (STORM; Vutara) microscope in the presence of a photo-switching buffer (20 mm MEA, 144 mm 2-mercaptoethanol, 2 mm cyclo-octatetraene, 170 AU glucose oxidase, 2000 AU catalase; all sourced from Sigma-Aldrich). At least 5000 exposure frames were taken, and localizations were calculated using Vutara's proprietary algorithms. Point clouds with ball size diameter reflecting localization accuracy were plotted using Vutara visualization software. A stringent confidence cutoff of 0.9 was used together with post hoc denoising by Delaunay Tessellation Field Interpolation to remove diffuse background localizations. Localization data points (x, y, z coordinates) were exported, and individual RPM/UPF1 puncta were plotted with the scatterplot3d R package. Euclidean distance between localization points in three-space were calculated using a custom MATLAB script with the formula, , and kernel density estimates were plotted using the geom_density function of the ggplot2 R package.
Electrophysiology and mGluR-LTD.
Coverslips with cultured hippocampal neurons were placed in a recording chamber mounted on an inverted microscope equipped with epifluorescence (Nikon Eclipse Ti-S). Cultures were perfused at 1 ml/min with Tyrode's solution at room temperature containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2, 0.1 picrotoxin, and 0.001 tetrodotoxin, pH 7.37–7.41, adjusted to 295–305 mOsmol with KOH. Whole-cell recordings were obtained from hippocampal neurons with borosilicate micropipettes (3–7 mΩ) filled with intracellular solution containing the following (in mm): 125 K-gluconate, 2.6 KCl, 1.3 NaCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, 0.1 EGTA, and 14 phosphocreatine-Tris, pH 7.2–7.3, 275–280 mOsmol. Neurons were selected for recordings by visual identification in phase contrast in experiments without transduction and by GFP expression in transduction experiments. Recordings were made in voltage-clamp mode using a Multiclamp 700B amplifier (Molecular Devices). The cell holding potential was maintained at −60 mV, and series resistance was routinely monitored. Recorded signals were low-pass filtered at 2 kHz, digitized at 20 kHz, and stored on a PC using Digidata 1440A acquisition board and pClamp10 software (Molecular Devices). Data were only included if the holding current (<200 pA) was stable and series resistance (<32 mΩ) varied <25% of initial value.
LTD was induced by bath application of the Group I mGluR agonist (S)-3,5-DHPG (100 μm; Abcam) for 5 min. The translation inhibitors emetine (40 μm) and HHT (20 μm) were bath-applied 10 min before, during, and 30 min after DHPG application. Miniature EPSCs (mEPSCs) were recorded in 5 min episodes throughout the experiments. For analysis, mEPSCs were detected and measured over two 5 min time periods, before (−5 to 0 min) and after (25–30 min) DHPG application using MiniAnalysis program (Synaptosoft) after lowpass Gaussian filtering of traces at 1 kHz. Detection threshold was set at 7 pA, and all detected events were verified by visual inspection. Cells with mEPSC frequency ≤0.5 Hz were excluded from analysis.
Experimental design and statistical analysis.
In general, all experiments were planned to have at least three replicates from separate hippocampal cultures. Because, for most of the experiments, the amount of error was not known a priori, it was difficult to accomplish effective power analysis to preplan the number of experiments. Unless otherwise stated in figure legends, statistical p values were determined using a one-tailed (where appropriate) or two-tailed, unpaired Student's t test for two group comparisons and one-way ANOVA followed by Šidák post hoc multiple-comparison correction for >2 groups using Graphpad Prism version 6. For electrophysiology experiments, one-tailed, paired t tests on mEPSC parameters before versus after DHPG application were performed using Graphpad Prism. Any distinct analysis is described in the figure legends. Bar plots are presented as the mean ± SEM, and in-text values are stated as the mean ± SD. The experimenter was blinded for the analysis of neurites labeled for AHA, RPM, MAP1B protein, and Map1b mRNA.
Results
UPF1 is required for the presence of stalled polysomes in neurites
To examine the role of UPF1 in the generation of stalled polysomes, we constructed a lentivirus encoding GFP and short-hairpin RNAi targeted to Upf1. Three days after infection of hippocampal neuron cultures, levels of UPF1 were significantly decreased (50%) compared with neurons transduced with a lentivirus expressing a nontargeting (scrambled) RNAi as measured by immunofluorescence (Fig. 1A,B). Western blotting indicated a similar decrease in UPF1 expression in total cell lysates (Fig. 1C). Next, we assessed the number of stalled polysomes in neurites with lowered UPF1 expression using an in situ run-off assay in hippocampal cultures. Briefly, translating polysomes are allowed to complete translation in the presence of an inhibitor (HHT) that prevents formation of new polysomes (Graber et al., 2013b). The remaining stalled polysomes are detected by RPM, where puromycin is covalently attached to polysome-bound nascent peptide chains that are fixed in position with the elongation inhibitor emetine (David et al., 2012). Reducing the expression of UPF1 was sufficient to decrease the number of stalled polysomes seen in distal dendrites (>50 μm from soma) by ∼50% (Fig. 1D). Stimulation of mGlu receptors with DHPG can reactivate translation of stalled polysomes encoding proteins required for mGluR-LTD (Graber et al., 2013b). This burst of protein synthesis can be measured after first blocking initiation-dependent protein synthesis with HHT, allowing ribosome run-off and then measuring new, initiation-independent protein synthesis by pulsing with the methionine analog AHA (Graber et al., 2013b). A fluorophore is then chemically attached to AHA and detected by fluorescence microscopy. Using this assay, we found that decreasing expression of UPF1 blocked induction of initiation-independent translation by DHPG (Fig. 1E). To rule out off-target effects of our Upf1 RNAi, we rescued DHPG-induced initiation-independent protein synthesis with a lentivirus expressing human UPF1, which lacks the sequence targeted by the RNAi. This vector also expresses mCHERRY on a separate cistron. Expression of this lentivirus rescues the knockdown of UPF1 (Fig. 1F) and the activation of initiation-independent protein synthesis by DHPG (Fig. 1G), demonstrating that the effect of UPF1 on blocking initiation-independent protein synthesis was not an off-target effect of the RNAi.
Figure 1.

UPF1 is required for DHPG-mediated reactivation of translation from stalled polysomes in neurites. A, Schema of the lentivirus transduction protocol to efficiently express shRNA-targeting Upf1 or a nontargeting control (scrambled RNAi) in primary neurons. Lentivirus expression was monitored with an emGFP ORF fused upstream of the shRNA cassette. Neurons were cultured for an additional 72 h before fixation. B, Representative confocal images (left) and quantitation (right) of UPF1 protein in neurons at day 10 expressing indicated RNAi. Data are from 12 individual neurons from three independent experiments. For explanatory notes regarding statistics used throughout the article, see Materials and Methods. C, Immunoblot (IB) showing steady-state levels of UPF1 in neurons expressing RNAi. GAPDH was used as a loading control. Apparent molecular weight is indicated on left in kDas. Data are representative of three independent experiments. D, Reduced UPF1 expression decreases the number of stalled polysomes in neurites. Representative confocal images of straightened neurites (left) and quantitation (right) of stalled polysomes >50 μm from the cell soma detected by RPM in the presence of HHT. Data are from 15–24 neurons from three different cultures. E, UPF1 is required for mGluR-mediated, initiation-independent protein synthesis. AHA incorporation (Click-AHA) into nascent peptides was measured in distal neurites in the presence of HHT following activation of mGluR with 50 μm DHPG in neurons expressing indicated RNAi. Data are from 30–49 neurons from four different cultures (for quantification, see Materials and Methods). F, Rescue of UPF1 levels in hippocampal cultures. Neurons were transduced with scrambled, Upf1 RNAi, or Upf1 RNAi plus a lentivirus expressing mCHERRY and human UPF1 ORFs (resistant to rat Upf1 RNAi) from separate promoters. Neurons from three independent cultures were fixed and stained with anti-UPF1 and imaged for emGFP (reporting RNAi cassette) and mCHERRY (reporting UPF1 rescue cassette). Average levels of UPF1 from 10 cell bodies transduced with Upf1 RNAi or Upf1 RNAi + UPF1 rescue lentiviruses were standardized to average UPF1 levels in 10 scrambled RNAi-transduced neurons from cultures transduced at the same time. Results are from three independent neuron cultures. G, Same as E, but with expression of both Upf1 RNAi plus UPF1 rescue plasmid. Expression of UPF1 rescued the ability of DHPG to induce increased incorporation of AHA. Data are from 15–26 neurons from three different cultures. *p < 0.05; **p < 0.01; ***p < 0.001.
Knocking-down UPF1 should increase levels of mRNAs normally regulated by NMD. Previously, the protein ARC, whose mRNA is a constitutive target for NMD due to an intron in its 3′ UTR, was shown to be upregulated when NMD is disrupted (Giorgi et al., 2007). Indeed, droplet digital RT-PCR results from our UPF1 knockdowns are consistent with this upregulation, with Arc mRNA increasing to 191 ± 8.85% over scrambled controls, n = 2. Could the effects we observe in UPF1 knockdowns be due to upregulation of NMD targets? To assess this, we knocked down expression of Proline-rich Nuclear Receptor Coregulatory protein 2 (PNRC2), a protein that interacts with UPF1 and that is required for NMD (Cho et al., 2009). Although we observed an efficient knockdown of PNRC2 in hippocampal neuronal cultures (Fig. 2A), we saw no significant change in the induction of AHA incorporation in neurites following DHPG treatment in neurons lacking PNRC2 (Fig. 2B). These data indicate that the observed decrease in translation from reactivated stalled polysomes in UPF1 knockdowns appears to be independent of its primary function in NMD.
Figure 2.

DHPG-mediated translation occurs independently of NMD and requires UPF1 for proper localization of a target mRNA. A, Diagram depicting function of PNRC2, acting as a bridge between the decapping and NMD (or SMD) machineries on an mRNA substrate (top). Representative immunoblot of PNRC2 in neurons expressing indicated RNAi (bottom). B, PNRC2-dependent NMD is not required for mGluR-mediated, initiation-independent protein synthesis. The AHA incorporation assay was performed as in Figure 1E on neurons expressing scrambled or Pnrc2 RNAi. Data are from 19–29 neurons from three independent cultures. C, Droplet digital RT-PCR of Map1b (left) or Upf1 (right) mRNAs normalized to Gapdh mRNA from total lysates of hippocampal neurons expressing RNAi. Data are from three independent experiments. A one-tailed t test was used to determine significance of Upf1 knockdown. D, Reduced UPF1 expression decreases the number of Map1b mRNA copies in neurites determined using smRNA-FISH. Representative confocal images of straightened neurites after application of a Laplacian of Gaussian transformation to highlight mRNA puncta edges are shown. Segmentation and thresholding of individual puncta (“particle mask”) allowed quantitation of mRNA in the soma, proximal, and distal neurites (bottom). Data are from 20–39 neurons obtained from three different cultures. E, Immunoblot of total neuronal lysates showing effect of DHPG on total and phospho-UPF1 (P-UPF1) levels (top) and quantitation from four independent experiments showing change in phospho:total-UPF1 and UPF1:GAPDH expression following 50 μm DHPG treatment for 10 min (bottom). A one-sample, two-tailed t test was used to determine significance. *p < 0.05; **p < 0.01; ***p < 0.001.
One explanation for the paucity of stalled polysomes in distal dendrites lacking UPF1 is that their mRNA cargo might be degraded in the soma. We therefore chose to assess Map1b mRNA expression in this context as it encodes an essential protein for the expression of mGluR-LTD and whose synthesis occurs in a rapid and highly localized manner (Davidkova and Carroll, 2007; Lebeau et al., 2011; Graber et al., 2013b). We have previously shown that Map1b mRNA is present in ribosome-containing RNA granules (Lebeau et al., 2011) and undergoes initiation-independent translation upon stimulation of mGlu receptors (Graber et al., 2013b), all features that are consistent with stalled polysomes. Knockdown of UPF1 had no effect, however, on total Map1b mRNA levels in neurons measured using droplet digital RT-PCR (Fig. 2C), demonstrating that UPF1 does not directly regulate levels of Map1b mRNA through NMD. This left the possibility that Map1b-stalled polysomes do not form in the context of decreased UPF1 expression, or form but become mislocalized. Alternatively, these mRNAs may still be properly localized to the dendrite but undergo leaky, dispersed translation that could explain the decrease in the number of stalled polysome puncta. To test whether Map1b mRNA is still localized to dendrites, we looked at the subcellular distribution of endogenous Map1b mRNA using smRNA-FISH (Raj et al., 2008). In neurons expressing nontargeting RNAi, we were able to detect many copies of Map1b throughout the cell soma, with some also visible in neurites (Fig. 2D). Strikingly, in neurons expressing Upf1 RNAi, there was a significant decrease in the number of Map1b messages per micron of neurite length in both proximal (<50 μm) and distal (>50 μm from the soma boundary) neurites, whereas somatic Map1b did not significantly change (Fig. 2D).
Thus, we conclude that, in the absence of UPF1, reactivation of stalled polysomes in neuronal dendrites is inhibited, most likely due to the lack of formation and/or transport of these structures into dendrites.
If UPF1 is critical for the stalling process, one might expect that post-translational modification of UPF1 could play a role in release of the stall. Indeed, we found that DHPG induced a significant dephosphorylation of UPF1 (Fig. 2E), consistent with the known activation of phosphatases by DHPG (Niere et al., 2012). Thus, dephoshorylation of UPF1 could play a role in the release of stalled polysomes following DHPG treatment.
UPF1 is colocalized with stalled polysomes in distal neurites
UPF1 has been associated with RNA transport granules in previous studies (Barbee et al., 2006; Giorgi et al., 2007) and is present in the proteomics of RNA granules (Elvira et al., 2006; El Fatimy et al., 2016). We took advantage of our ability to identify stalled polysomes using RPM and then assayed their association with UPF1 in super-resolution using Dual Color 3D-STORM (Fig. 3) (Juette et al., 2008). HHT-ribopuromycylated neurons were fixed and probed for puromycin and UPF1 using appropriate primary antibodies, incubated with standard secondary antibodies and followed by an additional fixation step to minimize movement of antibody complexes. Ribopuromycylated puncta in hippocampal neurons were first located by low-resolution wide-field immunofluorescence (Fig. 3A) before surveying individual fluorophore-conjugated secondary antibody localizations at super-resolution (Fig. 3B,C). We collected super-resolution localizations of both Alexa-568 (labeling puromycin) and Alexa-647 (labeling UPF1) puncta in samples saturated with secondary antibodies (high density probes at 1:1000 dilution; Fig. 3B) and samples labeled with limited secondary antibody (low density probes at 1:10,000 dilution; Fig. 3C) to minimize the chances of detecting nonspecific antibody complexes. Intriguingly, we found that the topography of localizations, when plotted with Gaussian blur (point-splatting), highlights structures that are suggestive of multiple UPF1 molecules (or a single molecule complexed with multiple secondary antibodies) in complex with the apex of a stalled polysome (Fig. 3B,C). Supporting this supposition, the sizes of the densely puromycylated structures were found to be generally uniform and consistent with the expected diameter of a puromycylated ribosome with associated primary and secondary antibodies (∼25–50 nm). We used a commercially available monoclonal antibody against UPF1 whose epitope remains proprietary; therefore, it is difficult to determine whether the granules we observed contained one or more UPF1 molecules. However, we did observe, in general, fewer UPF1 relative to puromycin localizations (individual localizations for a typical granule are plotted in Fig. 3D). While the number of puromycin localizations appears too large for a single polysome, uncertainties concerning the number of secondary antibody fluorophores and the possible recounting of fluorophores due to movement during the lengthy imaging procedure preclude definitive conclusions on this point. If multiple polysomes are present in a granule, it would suggest some tertiary organizing feature in RNA granules as has recently been suggested by EM micrographs of purified RNA granules (El Fatimy et al., 2016). Further, we found that puromycin and UPF1 localizations resided in close proximity to each other. We extended this qualitative analysis by calculating the point-to-point distances for each localization species (UPF1-UPF1, puromycin-puromycin, and UPF1-puromycin) and plotting their densities as a function of distance in nanometers. Figure 3E describes the distribution of the localizations found in the single puncta represented in Figure 3D. By surveying the maxima of these densities for each species across multiple RPM puncta from independent experiments, we observed that peak heteromolecular (UPF1-puromycin) distances were significantly different from peak homomolecular (UPF1-UPF1 or puromycin-puromycin) distances, consistent with a specific molecular complex rather than nonspecific association of secondary antibodies (Fig. 3F). Thus, by using a super-resolution approach, we were able to sufficiently resolve submicron, anisotropic structures that are consistent with helical polysomes bound to UPF1.
Figure 3.
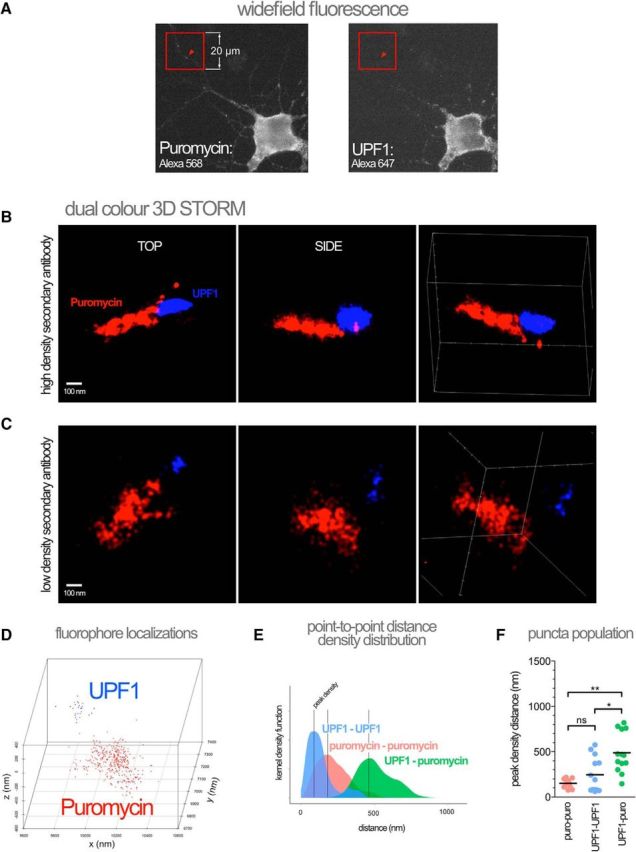
Super-resolution microscopy reveals stalled polysomes colocalized with UPF1 in distal neurites. A, Stalled polysome puncta were first detected in widefield fluorescence mode on the STORM using RPM in the presence of HHT (the location of an RPM puncta is indicated by arrowheads in each channel) before acquisition of the super-resolution image in both channels. The UPF1 signal was below detection during widefield acquisition due to the short exposure times needed to minimize bleaching. B, Reconstructed image of an RPM puncta based on two-color localizations in three dimensions (Dual Color 3D STORM) using high-density secondary antibodies to detect anti-puromycin and anti-UPF1 primary antibodies. Individual fluorophore localizations on conjugated secondary antibodies plotted using the point-splatting feature of the Vutara software to highlight structure. Top, side, and angled views are shown. C, Same as in B, but using 1:10,000 instead of 1:1000 secondary antibody dilution. D, Individual fluorophore localizations of the polysome-UPF1 complex in C. E, Kernel density plot summarizing point-to-point distances of localizations shown in D. Vertical lines indicate density maxima at distance x (“peak density distance”). F, Peak density distances for a population of polysome-UPF1 complexes (n = 13). Error bar indicates the mean. A Kruskal–Wallis nonparametric ANOVA was performed with Dunn's multiple-comparisons post hoc test. Data were collected from three different neuronal cultures. *p < 0.05; **p < 0.01.
Characterization of a STAU2 mutation that fails to interact with UPF1
We have previously shown that STAU2, but not its paralog STAU1, is required for production of MAP1B during mGluR-LTD in rat primary hippocampal neurons and slices (Lebeau et al., 2011). Consistent with this, we also found that STAU2 colocalizes with ribosomes and stalled polysomes (Lebeau et al., 2011; Graber et al., 2013b). We now find that, similar to our results with Upf1 RNAi, reduction of STAU2 expression using RNAi targeting all major rat isoforms (Fig. 4A,B), reduces the number of stalled polysomes (Fig. 4C) and decreases the amount of Map1b mRNA found in distal neurites (Fig. 4D). Importantly, no effect on localization was seen for Map2 mRNA in either Stau2 or Upf1 RNAi-expressing neurons, indicating that the mislocalization effect for Map1b cannot be generalized to all mRNAs (Fig. 4E). Similar to UPF1 (Fig. 2C), there was no effect of knocking down STAU2 on the total level of Map1b mRNA despite a strong reduction (>80%) in Stau2 mRNA (Fig. 4F).
Figure 4.
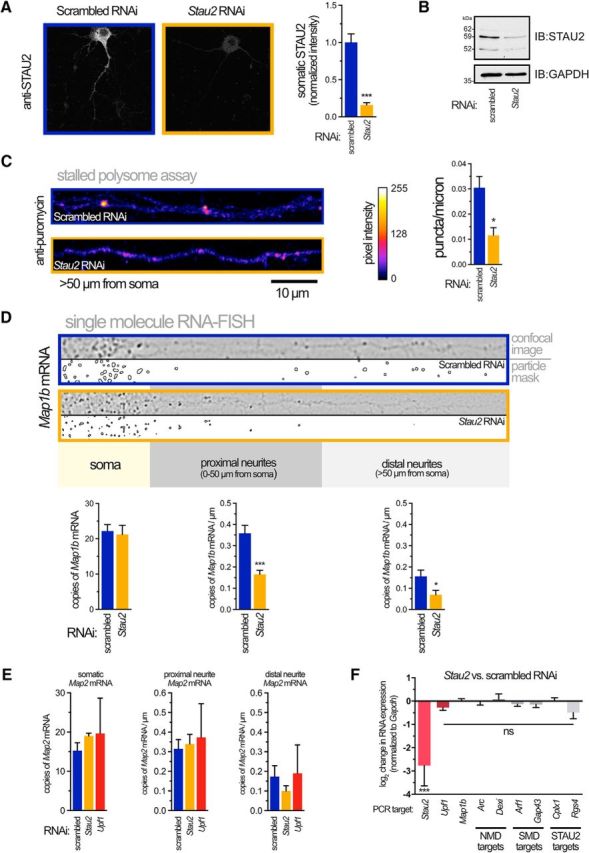
STAU2 is required for the presence of stalled polysomes in neurites. A, STAU2 protein was knocked down with the same lentivirus RNAi strategy as outlined in Figure 1A. Representative confocal images (left) and quantitation (right) of STAU2 protein in neurons expressing indicated RNAi. B, Representative immunoblot showing steady-state levels of STAU2 isoforms in neurons expressing RNAi. GAPDH was used as a loading control. C, Reduced STAU2 expression decreases the number of stalled polysomes in neurites. Representative confocal images of straightened neurites (left) and quantitation (right) of stalled polysomes >50 μm from the cell soma detected by RPM in the presence of HHT. Data were obtained from 15–24 neurons from three different cultures. D, Reduced STAU2 expression decreases the number of Map1b mRNA copies in neurites determined using smRNA-FISH. Representative confocal images of straightened neurites after application of a Laplacian of Gaussian transformation to highlight mRNA puncta edges. Segmentation and thresholding of individual puncta (“particle mask”) allowed quantitation of mRNA in the soma, proximal, and distal neurites (bottom). Data were obtained from 20–39 neurons from three different cultures. E, Quantitation of Map2 smRNA-FISH puncta in neurons expressing indicated RNAi and analyzed as in D. Data are from 3 or 4 neurons from two different cultures. F, Expression of target mRNAs in total hippocampal neuron lysates expressing Stau2 versus scrambled RNAi measured by droplet digital RT-PCR. Expression of each indicated PCR target was normalized to Gapdh. A one-sample, two-tailed t test was used to determine significance; n = 3 or 4 independent cultures. *p < 0.05; ***p < 0.001.
Together, these results suggest a functional relationship between STAU2 and UPF1 in the context of RNA transport, translation, or both. Interestingly, both STAU1 and STAU2 have been previously implicated in RNA decay together with UPF1, where their interactions were found to enhance exon-junction complex-independent RNA decay (so-called Staufen-mediated RNA decay [SMD]) (Park et al., 2013). This interaction triggers UPF1 phosphorylation that facilitates the recruitment of RNA-decapping factors, although the exact mechanisms and proteins involved are still not well understood (Kim et al., 2005). However, the loss of Staufen-mediated decay is unlikely to explain our results as (1) in our cultures, we do not observe upregulation of mRNAs known to be targets of SMD after STAU2 knockdown (Fig. 4F); and (2) knockdown of PNRC2 does not affect the DHPG-induced upregulation of initiation-independent RNA translation (Fig. 2B), even though PNRC2 is also required for SMD (Cho et al., 2012). It should also be noted that neither Upf1 mRNA nor NMD targets (Arc, Dexi) are upregulated after knockdown of STAU2, although one mRNA previously shown to be regulated by STAU2, Rgs4 (Heraud-Farlow et al., 2013), was slightly downregulated, albeit nonsignificantly (Fig. 4F). UPF1 knockdown also did not alter Stau2 mRNA levels (93.6 ± 8.7% of scrambled controls).
Using transiently expressed epitope-tagged versions of human UPF1 and the 59 kDa isoform of STAU2 (an RNA granule-associated isoform that is highly expressed in hippocampal neurons) (Duchaîne et al., 2002), we were able to confirm their coimmunopurification in HEK293T (Fig. 5A,B). Importantly, we were unable to pull down another myc-tagged protein, RIP-myc, with FLAG-tagged STAU2, indicating that the STAU2-UPF1 interaction is specific (Fig. 5B). Miki et al. (2011) have previously shown that STAU2 and UPF1 directly interact with each other in a cell-free system. However, this interaction could depend on indirect binding through RNA or RNA-ribosome complexes in HEK293T cells. To address this, we treated lysates with RNase A before immunoprecipitation and found that, despite the loss of the ribosomal protein S6 (RPS6; indicative of ribosome-RNA association) in the nuclease-treated IP, STAU2 complexes still contained UPF1, although we cannot say that RNA-ribosome complexes do not facilitate this interaction (Fig. 5C).
Figure 5.

UPF1 interacts with the TBD of STAU2. A, Schematic of DNA constructs used for coimmunoprecipitation assays. NES, Nuclear Export Signal. B, Coimmunoprecipitation of Flag-tagged STAU2 and Myc-tagged UPF1 in HEK293T cells. Myc-tagged RIP1, a protein not involved in this complex, was not immunoprecipitated with Flag-tagged STAU2 (right), indicating that the interaction seen with UPF1 and STAU2 was not due to promiscuous binding or specific to the Myc and Flag peptide tags. C, Flag coimmunoprecipitation as in B, but incubated with 100 μg/ml RNase A. There is loss of endogenous RPS6 from complex with RNase A treatment and retention of UPF1. D, Coimmunoprecipitation demonstrating that removal of the STAU2 C terminus along with the TBD (C351) abrogates binding of UPF1 but has no effect on binding to the ribosome (RPS6). In contrast, a mutation in dsRBD3 of STAU2 (F207A), but not the full-length or C351 mutation, disrupts ribosomal interactions as indicated by reduced IP of endogenous RPS6. E, Coimmunoprecipitation of STAU2 proteins harboring C-terminal deletion mutations. F, Putative UPF1 binding region in STAU2 based on the coimmunoprecipitation experiments. The homology of this region compared with STAU1 is low (ClustalW alignment), indicating that the interaction with UPF1 might be different between these two proteins. G, Immunoblot of STAU2 and GFP expression in hippocampal neurons expressing scrambled or Stau2 RNAi (expressing GFP on a separate cistron; “RNAi virus GFP”) or RNAi plus a rescue lentivirus expressing the GFP-STAU2 or the C351 mutation constructs that are both resistant to the rat-specific RNAi. The STAU2 antibody used does not detect the STAU2 C351 species. GAPDH was used as loading control. H, mGluR-mediated induction of MAP1B protein synthesis requires the UPF1 binding region. MAP1B protein levels were assessed by immunofluorescence in distal neurites of hippocampal neurons after 10 min following stimulation with 50 μm DHPG or with vehicle. Neurites from 25–36 neurons and four independent cultures were analyzed. A one-tailed, one-way ANOVA with Šidák multiple hypothesis correction was used. *p < 0.05.
To begin to understand how STAU2-UPF1 interactions might affect mRNA translation, we attempted to define the specific region(s) in the 59 kDa isoform of human STAU2 that is necessary for binding to UPF1. We created several Flag-tagged constructs that expressed STAU2 domains (incorporating the STAU2 nuclear export signal to ensure cytoplasmic expression; Fig. 5A). We found that, on their own, none of the defined double-stranded RNA binding domains (dsRBDs) or the C-terminal tubulin binding domain (TBD) were sufficient for interaction with UPF1 (data not shown). However, a C-terminal deletion mutation that lacked the TBD (C351), but retained dsRBDs 1–4, was sufficient to abrogate binding to UPF1 (Fig. 5D).
We next asked whether the loss of UPF1 binding with the STAU2 C351 mutation could be indirect, resulting from impaired STAU2 interaction with ribosome/RNA complexes that are presumably pulled down in our immunoprecipitations lacking RNase A treatment. We detected the presence of RPS6 in our immunoprecipitated STAU2-UPF1 complexes, suggesting the presence of monosome/polysome complexes. To confirm this, we introduced a point mutation in dsRBD3 of STAU2 (F207A; numbering based on Uniprot ID: Q9NUL3-2) that is analogous to the F135A mutation in its paralog STAU1 that has previously been reported to ablate RNA binding and curtail binding to the ribosome, presumably by altering the secondary structure of the former (Luo et al., 2002). We observed decreased affinity of this mutation construct for RPS6 relative to wild-type (Fig. 5D), likely due to a decrease in associated monosomes/polysomes. Importantly, however, the ability to bring down RPS6 is unaffected in immunoprecipitants from cells expressing the C351 mutation versus wild-type STAU2 (Fig. 5D). These data suggest that the C351 mutation does not impair STAU2 interactions with monosomes/polysomes but does affect the interaction with UPF1, consistent with an independent interaction between STAU2 and UPF1. It should be noted that the C351 mutation could still affect STAU2 function(s) other than binding UPF1, such as heterodimerization (Gleghorn et al., 2013).
To better define the region in the C terminus that is necessary for UPF1 binding, we created several additional C-terminal deletion mutations. Full binding, indistinguishable from the wild-type, was observed in the C430 mutation, which contained the entire TBD but lacked the remaining C-terminal residues (Fig. 5E). Critically, we found that the middle portion of the TBD was necessary, but not sufficient for efficient binding to UPF1. Interestingly, this binding region has very low amino acid homology to Staufen 1 (Fig. 5F), perhaps suggesting that Staufen 1 and 2 interact with UPF1 in different ways, although the functional significance of this, if any, remains unclear.
STAU2-UPF1 interactions are necessary for DHPG induction of MAP1B protein in distal neurites
STAU2 is required for DHPG induction of MAP1B protein (Lebeau et al., 2011). This occurs independently of translation initiation (Graber et al., 2013b) and is presumably the result of peptide release from polysomes that have been reactivated. To determine whether the STAU2-UPF1 interaction is important for this functional release of stalled polysomes, we used a rescue paradigm where STAU2 levels were reduced by our lentiviral RNAi in hippocampal rat neurons and then rescued using a separate lentivirus expressing either human full-length STAU2 or human STAU2 lacking the UPF1 binding domain (C351), which are both resistant to rat-specific RNAi. Both constructs expressed at equivalent levels (reported by GFP immunodetection in Fig. 5G) and the full-length STAU2 construct expressed at similar levels to the endogenous 59 kDa STAU2 (Fig. 5G, top; the STAU2 antibody does not detect C351 expression). As previously shown using transient siRNA transfection by Lebeau et al. (2011), knockdown of STAU2 in this context blocked the DHPG-mediated induction of MAP1B protein in distal neurites (Fig. 5H). Demonstrating that this is not an off-target effect, we were able to rescue induction of MAP1B with coexpression of RNAi-resistant human STAU2, but not the STAU2 harboring the C351 mutation (Fig. 5H). These results suggest that DHPG induction of MAP1B expression requires the STAU2-UPF1 interaction, although we cannot rule out the possibility that the loss of the DHPG-induced increase was due to a separate function of STAU2 that was also lost with the C351 mutation.
mGluR-LTD in dissociated hippocampal neurons does not require translation initiation
We next examined the role of UPF1 and STAU2 in regulating synaptic plasticity mediated by reactivated translation from stalled polysomes. We first determined whether mGluR-LTD recorded at the single-cell level in cultured hippocampal neurons was dependent on reactivation of stalled polysomes using voltage-clamp recordings of mEPSCs. In cultured hippocampal neurons, mGluR-LTD is associated with an internalization of GluA receptors, resulting in a reduction of mEPSC frequency with no change in amplitude (Waung et al., 2008; Sanderson et al., 2011). Consistent with these previous reports, DHPG-induced LTD in long-term hippocampal neuron cultures was associated with a decrease in mEPSC frequency, but not amplitude, following bath application of DHPG (Fig. 6A,B).
Figure 6.

mGluR-LTD in cultured neurons is protein synthesis-dependent yet translation initiation-independent. A, Representative traces of mEPSCs (dots above traces) over time during whole-cell recording from cultured (18–25 DIV) rat hippocampal neurons. B, Summary bar graphs for all cells, showing LTD of mEPSC frequency, but not amplitude, 30 min after application of DHPG. LTD was prevented by the translation elongation inhibitor emetine (40 μm) but not by the translation initiation inhibitor HHT (20 μm). Data are accumulated from 4–9 neurons from 4 or 5 independent experiments. *p < 0.05.
We recently showed that mGluR-LTD at Schaffer collateral synapses is insensitive to HHT, an inhibitor of translation initiation or, more precisely, of the first round of elongation. Furthermore, mGluR-LTD was prevented by emetine, an inhibitor of translation elongation (Graber et al., 2013b). Thus, we first verified whether mGluR-LTD was similarly independent of translation initiation in dissociated cultured neurons. Application of the elongation inhibitor emetine blocked the decrease in mEPSC frequency induced by DHPG, whereas incubation with the initiation inhibitor HHT did not (Fig. 6A,B). Mean amplitude of mEPSCs was not significantly changed by DHPG in these experiments. These results, showing a block of mGluR-LTD by the translation elongation inhibitor emetine confirm that long-term synaptic plasticity in cultured hippocampal neurons is dependent on de novo protein synthesis. Moreover, the lack of inhibition of mGluR-LTD by the translation initiation inhibitor HHT suggests that this plasticity is dependent on regulation downstream of initiation, through reactivated translation from stalled polysomes.
UPF1 and its interaction with STAU2 are required for mGluR-LTD in hippocampal neurons
Having established that cultured neurons show mGluR-LTD independent of translation initiation, but dependent on translation elongation in response to DHPG, we next wanted to determine whether UPF1 and its interaction with STAU2 on stalled polysomes were required for mGluR-LTD. We assessed DHPG-induced LTD of mEPSCs in neurons expressing Upf1 or scrambled RNAi lentivirus and found that the depression of mEPSC frequency, without change in amplitude, was prevented in neurons with reduced UPF1 expression relative to those expressing scrambled RNAi (Fig. 7A,B).
Figure 7.

mGluR-LTD is impaired by STAU2 and UPF1 knockdown, and its rescue requires UPF1-STAU2 interaction. A, Representative traces of mEPSCs during whole-cell recording from cultured rat hippocampal neurons transduced with indicated lentiviruses (expressing GFP and/or mCHERRY). B, Summary bar graphs for all cells, showing that LTD of mEPSC frequency, but not amplitude, induced by DHPG (scrambled RNAi-treated cells) is impaired in cells with knockdown of UPF1 or STAU2, and rescued in cells with RNAi-resistant STAU2 overexpression but not in cells with overexpression of STAU2 lacking the UPF1 binding region (C351 mutation). Data are accumulated from 5–9 neurons from 3–6 independent experiments. *p < 0.05; **p < 0.01.
We previously reported that mGluR-LTD induced by DHPG was impaired in cultured hippocampal slices transiently transfected with Stau2 siRNA (Lebeau et al., 2011). Next, we verified the importance of STAU2 in mGluR-LTD in dissociated neurons using Stau2 RNAi lentivirus. Neurons expressing Stau2 RNAi did not show DHPG-induced depression of mEPSC frequency, consistent with our previous observations in slices (Fig. 7A,B). There was, however, a decrease in mEPSC amplitudes when Stau2 was knocked down, that was not seen with UPF1. This may be due to regulation of additional messages by STAU2 independent of its role in stalled polysomes (Heraud-Farlow et al., 2013). Indeed, our droplet digital RT-PCR results revealed that one of the previously identified STAU2 mRNA targets was slightly downregulated in STAU2 knockdowns (Rgs4; Fig. 4F). Decreases in this and presumably other mRNAs stabilized by STAU2 may underlie the changes in amplitude seen with its loss, but not with that of UPF1.
Next, we attempted to rescue the deficit in mGluR-LTD in neurons expressing Stau2 RNAi with lentiviruses that express RNAi-resistant human STAU2. Infection with STAU2-expressing virus restored the DHPG-induced LTD of mEPSC frequency, indicating that the Stau2 RNAi phenotype was not due to off-target effects (Fig. 7A,B). Finally, we sought to determine whether the UPF1-STAU2 interaction was necessary for the STAU2 rescue of mGluR-LTD by expressing the STAU2 C351 mutation, which cannot interact with UPF1 into the Stau2 RNAi background. Expression of the mutated STAU2 failed to rescue the DHPG-induced LTD of mEPSC frequency (Fig. 7A,B). Interestingly, the mutated STAU2 did rescue the change in mEPSC amplitude, consistent with this effect of STAU2 being mediated through a UPF1-independent role of STAU2 and also demonstrating that the C351 mutation does not perturb all STAU2 functions.
Together, these data demonstrate that UPF1, as well as STAU2, are required for mGluR-LTD in hippocampal neurons and, moreover, highlight the importance of their direct interaction in the transport and/or translation of RNA granules that participate in protein synthesis-dependent synaptic plasticity.
Discussion
A new role for UPF1 in the formation of neuronal granules comprising stalled polysomes
We have characterized a novel function for the RNA helicase UPF1 in the regulation of transport RNA granules consisting of stalled polysomes in neurons. UPF1 is present in stalled polysomes by proteomic analysis (Elvira et al., 2006; El Fatimy et al., 2016), and we demonstrate that it occupies a polarized position in neuronal RNA granules determined using super-resolution microscopy (Fig. 3). Knockdown of UPF1 in hippocampal neurons results in the following: (1) fewer stalled polysomes in neurites; (2) reduced transport of Map1b, an mRNA stored in stalled polysomes; (3) ablation of DHPG-induced, initiation-independent protein synthesis (Fig. 1); and (4) a block in mGluR-LTD expression, which is known to involve stalled polysomes (Fig. 7). Together, these data strongly argue for the importance of UPF1 in this form of regulated transport and translation in neurons.
There are a number of possible functions for UPF1 in regulating stalled polysomes: UPF1 may be important in their formation, stability, ability to be translationally activated, or some combination thereof. One attractive model would be that, when a ribosome translating an mRNA reaches the stop codon, UPF1 is recruited together with the peptide release factors eRF1 and eRF3, and instead of signaling decay or stimulating release as in non-neuronal cells, a process initiating the formation of a stalled polysome would ensue. Such a mechanism would also require STAU2, mediating either UPF1 recruitment or stabilization and may require additional, neuron-specific trans-acting proteins or regulatory RNAs, or cis-acting sequence factors, such as alternative 3′ UTRs.
It is possible that the effect of knocking down UPF1 indirectly regulates stalled polysomes; mRNAs normally targeted for NMD through the presence of introns in their 3′ UTRs may be longer-lived, thus exhibiting enhanced translation in this context (Kurosaki and Maquat, 2013). However, the best-known LTD-related protein that is also an NMD target, ARC (Giorgi et al., 2007), would have been expected to facilitate mGluR-LTD by its increased levels in neurons lacking sufficient UPF1, not lead to a loss of mGluR-LTD, as we observed. Moreover, decreases in Map1b mRNA localization to dendrites would be difficult to explain by a loss of NMD-mediated Map1b degradation. Finally, in our effort to dissect disparate UPF1 functions, knockdown of PNRC2, a protein that mediates decapping as part of the NMD/SMD function of UPF1 had no effect on translation from reactivated stalled polysomes (new; Fig. 2). These data strongly support the concept that the canonical NMD/SMD functions of UPF1 do not play a role in DHPG-mediated changes in translation of plasticity-related proteins. It remains to be seen whether these roles can be dissected in the context of synaptic plasticity and in particular mGluR-LTD.
Perhaps the most parsimonious conclusion drawn from the observed reduction in Map1b mRNA copy number and translation in neurites with reduced UPF1 expression is that the latter functions in Map1b RNA transport rather than translation per se. This could occur in a number of ways. First, UPF1 interactions might be a prerequisite for proper transport of a Map1b stalled polysome. Second, UPF1 interactions might be required for stalled polysome formation, without which Map1b becomes mislocalized. CLIP analysis has shown that UPF1 interacts with mRNA in both EJC-dependent and -independent manners such that many copies of UPF1 proteins are scattered throughout the message (Hurt et al., 2013; Zünd et al., 2013). Importantly, Zünd et al. (2013) showed that UPF1 is preferentially displaced from coding regions on mRNAs with associated ribosomes while binding in 3′ UTRs is retained. This observation is consistent with our super-resolution data showing a large number of UPF1 localizations at the apical end of a polysomal structure. UPF1 does indeed specifically interact with the Map1b 3′ UTR (Hurt et al., 2013), so it is certainly possible that downregulation of the former could affect the transport of Map1b stalled polysomes, possibly by disturbing Staufen binding sites. Importantly, however, UPF1 (or STAU2) knockdown does not appear to adversely affect steady-state levels of Map1b in neurons (Figs. 2C, 4F). These data suggest that the NMD functions of UPF1, whether direct or indirect, are not involved in this context and highlight a tantalizing new function for this protein.
Implications of a role for STAU2-UPF1 interactions in RNA granules
The interaction between STAU2 and UPF1 has now been seen in a number of independent studies (Miki et al., 2011; Park et al., 2013; Flury et al., 2014). A previous study, however, identified the dsRBD2 and dsRBD3 domains as sufficient to bind to UPF1 and did not require C-terminal domains (Miki et al., 2011). In contrast, we found that deletion of the C terminus up to and including the TBD almost completely removed UPF1 binding without affecting STAU2 interactions with the ribosome. These results are not necessarily contradictory and suggest the possibility of two interacting domains, one including dsRBD2 and dsRBD3 and the second involving the TBD, both of which are required for full binding. For the purposes of our study, the loss of interaction with the removal of the C-terminal (C351 mutation) allowed us to more specifically examine the role of STAU2-UPF1 interactions because disrupting dsRBD3 would confound interpretation by removing most dsRNA binding for STAU2, including association with the ribosome (Luo et al., 2002). The TBD and dsRBD4 were sufficient for binding of STAU1 to UPF1 (Kim et al., 2005), although we show that removing the TBD was sufficient to prevent binding even in the presence of dsRBD4. Previous reports indicate that STAU2 binds to UPF1 more effectively than STAU1 (Park et al., 2013; Flury et al., 2014), and this may be explained either by the additional binding site in dsRBD2 or dsRBD3 or differences in the TBD involved in UPF1 binding (for a clustalW alignment of STAU2 vs STAU1 that shows the low percentage identity of residues in this region, see Fig. 5F).
Implications for the release of stalled polysomes
mGluR-LTD requires fast, local translation (Huber et al., 2000). We have previously shown that this translation is independent of translation initiation (Graber et al., 2013b), suggesting that LTD proteins are generated by finishing translation of previously initiated mRNAs. Based on our data and previous studies, we propose that at least three interlocked mechanisms could be implicated in releasing this form of stalled translation. Phosphorylated FMRP is necessary for stalling or stabilization of stalled polysomes, and FMRP dephosphorylation is required for mGluR-LTD, presumably by releasing the FMRP-mediated “brake” on stalled polysomes (Richter and Coller, 2015). Second, eEF2 phosphorylation is required for mGluR-LTD; and although the underlying reasons are still unclear, it is possible that this is due to a requirement for release of stalled polysomes (Graber et al., 2013a). Indeed, other forms of plasticity that require eEF2 phosphorylation also appear to be due to a need for reactivation of stalled polysomes (McCamphill et al., 2015). If release of mRNAs is stalled through UPF1-STAU2 interactions (Fig. 8), then this would also be subject to signal transduction mechanisms induced by mGluR-LTD. Interestingly, we also observed dephosphorylation of UPF1 after induction of mGluR-LTD (Fig. 2). Alternatively, once initially stalled through UPF1-STAU2 interactions, FMRP- and eEF2-mediated controls may predominate in the transported RNA granule. The role of UPF1-STAU2 interactions in release of a stalled polysome will be difficult to dissect if, as we suggest in this study, the latter do not form in the absence of this interaction.
Figure 8.

Model illustrating how UPF1-STAU2 interactions might contribute to local protein synthesis from stalled polyribosomes in synaptic plasticity. Target mRNAs, such as Map1b, harbor STAU2-UPF1 complexes in their 3′ UTRs that may prevent proper translation termination (indicated by eRF1 at the stop codon) and/or stall elongation, thus creating a stalled polysome complex. In the absence of either STAU2 or UPF1, Map1b mRNA fails to be transported into neurites. After transport of the stalled polysome complex to synapses, its reanimation is dependent on UPF1-STAU2 interactions, as mGluR-LTD fails to be expressed in the presence of HHT and a STAU2 mutant protein that still interacts with ribosomes but cannot form a complex with UPF1. This interaction depends on the TBD present in STAU2, which likely mediates interaction with the cysteine-histidine-rich (CH) domain of UPF1.
A new role for UPF1 in long-term synaptic plasticity
It has been suggested that specific transport complexes are involved in distinct forms of plasticity. We have previously shown that STAU2 is required for mGluR-LTD (Lebeau et al., 2011), whereas the paralogous STAU1 is necessary for the late phase of NMDA receptor-dependent LTP (Lebeau et al., 2008). Interestingly, although late-LTP and mGluR-LTD both require protein synthesis, late-LTP is blocked by either translation initiation or elongation inhibitors in contrast to mGluR-LTD, which is blocked only by elongation inhibitors (Graber et al., 2013b). Because both STAU1 and STAU2 can bind to UPF1 (Park et al., 2013; Flury et al., 2014), our finding that mGluR-LTD requires the UPF1-STAU2 interaction raises the intriguing possibility that UPF1-STAU1 complexes might function in late-LTP. However, it remains to be determined how such a differential interaction by UPF1 may regulate the translation of different subsets of mRNAs in mGluR-LTD and late-LTP. Thus, it would be of interest to explore how a differential interaction of UPF1 with either STAU1 or STAU2 may regulate the synthesis of distinct plasticity related proteins through stalled polysomes, and direct the switch between LTD and LTP.
Footnotes
This work was supported by Canadian Institutes of Health Research and Fonds de Recherche du Québec-Santé (FRQS) to J.-C.L. and W.S.S. T.E.G. was supported by FRQS and Canadian Institutes of Health Research Postdoctoral Fellowships. E.F. was supported by FRQS Postdoctoral Fellowship. S.H.-S. was supported by Université de Montréal and FRQS Graduate Studentships. J.-C.L. is the recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology. W.S.S. is a James McGill Professor. We thank Drs. Thomas Stroh and Liliana Pedraza for helpful discussions and expert advice in regard to super-resolution imaging; Congyao Zha for immunoblotting; and Ryan Sanford and Dr. Louis Collins for providing expertise regarding the measurement of super-resolution localization distances in 3D space and for the MATLAB script required to calculate point-to-point distances presented in Figure 3.
The authors declare no competing financial interests.
References
- Barbee SA, Estes PS, Cziko AM, Hillebrand J, Luedeman RA, Coller JM, Johnson N, Howlett IC, Geng C, Ueda R, Brand AH, Newbury SF, Wilhelm JE, Levine RB, Nakamura A, Parker R, Ramaswami M (2006) Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 52:997–1009. 10.1016/j.neuron.2006.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST (2003) Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet 12:3295–3305. 10.1093/hmg/ddg350 [DOI] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF (2007) The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76:51–74. 10.1146/annurev.biochem.76.050106.093909 [DOI] [PubMed] [Google Scholar]
- Cho H, Kim KM, Kim YK (2009) Human proline-rich nuclear receptor coregulatory protein 2 mediates an interaction between mRNA surveillance machinery and decapping complex. Mol Cell 33:75–86. 10.1016/j.molcel.2008.11.022 [DOI] [PubMed] [Google Scholar]
- Cho H, Kim KM, Han S, Choe J, Park SG, Choi SS, Kim YK (2012) Staufen1-mediated mRNA decay functions in adipogenesis. Mol Cell 46:495–506. 10.1016/j.molcel.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261. 10.1016/j.cell.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW (2012) Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 197:45–57. 10.1083/jcb.201112145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidkova G, Carroll RC (2007) Characterization of the role of microtubule-associated protein 1B in metabotropic glutamate receptor-mediated endocytosis of AMPA receptors in hippocampus. J Neurosci 27:13273–13278. 10.1523/JNEUROSCI.3334-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaîne TF, Hemraj I, Furic L, Deitinghoff A, Kiebler MA, DesGroseillers L (2002) Staufen2 isoforms localize to the somatodendritic domain of neurons and interact with different organelles. J Cell Sci 115:3285–3295. [DOI] [PubMed] [Google Scholar]
- El Fatimy R, Davidovic L, Tremblay S, Jaglin X, Dury A, Robert C, De Koninck P, Khandjian EW (2016) Tracking the Fragile X Mental Retardation Protein in a highly ordered neuronal RiboNucleoParticles population: a link between stalled polyribosomes and RNA granules. PLoS Genet 12:e1006192. 10.1371/journal.pgen.1006192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvira G, Wasiak S, Blandford V, Tong XK, Serrano A, Fan X, del Rayo Sánchez-Carbente M, Servant F, Bell AW, Boismenu D, Lacaille JC, McPherson PS, DesGroseillers L, Sossin WS (2006) Characterization of an RNA granule from developing brain. Mol Cell Proteomics 5:635–651. 10.1074/mcp.M500255-MCP200 [DOI] [PubMed] [Google Scholar]
- Flury V, Restuccia U, Bachi A, Mühlemann O (2014) Characterization of phosphorylation- and RNA-dependent UPF1 interactors by quantitative proteomics. J Proteome Res 13:3038–3053. 10.1021/pr5002143 [DOI] [PubMed] [Google Scholar]
- Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ (2007) The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130:179–191. 10.1016/j.cell.2007.05.028 [DOI] [PubMed] [Google Scholar]
- Gleghorn ML, Gong C, Kielkopf CL, Maquat LE (2013) Staufen1 dimerizes through a conserved motif and a degenerate dsRNA-binding domain to promote mRNA decay. Nat Struct Mol Biol 20:515–524. 10.1038/nsmb.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TE, McCamphill PK, Sossin WS (2013a) A recollection of mTOR signaling in learning and memory. Learn Mem 20:518–530. 10.1101/lm.027664.112 [DOI] [PubMed] [Google Scholar]
- Graber TE, Hébert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille JC, Sossin WS (2013b) Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci U S A 110:16205–16210. 10.1073/pnas.1307747110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Sharangdhar T, Li X, Pfeifer P, Tauber S, Orozco D, Hörmann A, Thomas S, Bakosova A, Farlow AR, Edbauer D, Lipshitz HD, Morris QD, Bilban M, Doyle M, Kiebler MA (2013) Staufen2 regulates neuronal target RNAs. Cell Rep 5:1511–1518. 10.1016/j.celrep.2013.11.039 [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E (2006) Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51:441–454. 10.1016/j.neuron.2006.07.005 [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288:1254–1257. 10.1126/science.288.5469.1254 [DOI] [PubMed] [Google Scholar]
- Hurt JA, Robertson AD, Burge CB (2013) Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res 23:1636–1650. 10.1101/gr.157354.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE (2008) Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133:314–327. 10.1016/j.cell.2008.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE (2008) Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J 27:736–747. 10.1038/emboj.2008.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J (2008) Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods 5:527–529. 10.1038/nmeth.1211 [DOI] [PubMed] [Google Scholar]
- Jung H, Gkogkas CG, Sonenberg N, Holt CE (2014) Remote control of gene function by local translation. Cell 157:26–40. 10.1016/j.cell.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Furic L, Desgroseillers L, Maquat LE (2005) Mammalian Staufen1 recruits Upf1 to specific mRNA 3′UTRs so as to elicit mRNA decay. Cell 120:195–208. 10.1016/j.cell.2004.11.050 [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Maquat LE (2013) Rules that govern UPF1 binding to mRNA 3′ UTRs. Proc Natl Acad Sci U S A 110:3357–3362. 10.1073/pnas.1219908110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau G, Maher-Laporte M, Topolnik L, Laurent CE, Sossin W, Desgroseillers L, Lacaille JC (2008) Staufen1 regulation of protein synthesis-dependent long-term potentiation and synaptic function in hippocampal pyramidal cells. Mol Cell Biol 28:2896–2907. 10.1128/MCB.01844-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau G, Miller LC, Tartas M, McAdam R, Laplante I, Badeaux F, DesGroseillers L, Sossin WS, Lacaille JC (2011) Staufen 2 regulates mGluR long-term depression and Map1b mRNA distribution in hippocampal neurons. Learn Mem 18:314–326. 10.1101/lm.2100611 [DOI] [PubMed] [Google Scholar]
- Luo M, Duchaîne TF, DesGroseillers L (2002) Molecular mapping of the determinants involved in human Staufen-ribosome association. Biochem J 365:817–824. 10.1042/bj20020263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCamphill PK, Farah CA, Anadolu MN, Hoque S, Sossin WS (2015) Bidirectional regulation of eEF2 phosphorylation controls synaptic plasticity by decoding neuronal activity patterns. J Neurosci 35:4403–4417. 10.1523/JNEUROSCI.2376-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Kamikawa Y, Kurono S, Kaneko Y, Katahira J, Yoneda Y (2011) Cell type-dependent gene regulation by Staufen2 in conjunction with Upf1. BMC Mol Biol 12:48. 10.1186/1471-2199-12-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niere F, Wilkerson JR, Huber KM (2012) Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci 32:5924–5936. 10.1523/JNEUROSCI.4650-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM (2006) Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95:3291–3295. 10.1152/jn.01316.2005 [DOI] [PubMed] [Google Scholar]
- Park E, Gleghorn ML, Maquat LE (2013) Staufen2 functions in Staufen1-mediated mRNA decay by binding to itself and its paralog and promoting UPF1 helicase but not ATPase activity. Proc Natl Acad Sci U S A 110:405–412. 10.1073/pnas.1213508110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5:877–879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Coller J (2015) Pausing on polyribosomes: make way for elongation in translational control. Cell 163:292–300. 10.1016/j.cell.2015.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TM, Collingridge GL, Fitzjohn SM (2011) Differential trafficking of AMPA receptors following activation of NMDA receptors and mGluRs. Mol Brain 4:30. 10.1186/1756-6606-4-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Bakker CE, Willems PJ, Oostra BA (1998) No evidence for disruption of normal patterns of mRNA localization in dendrites or dendritic transport of recently synthesized mRNA in FMR1 knockout mice, a model for human fragile-X mental retardation syndrome. Neuroreport 9:477–481. 10.1097/00001756-199802160-00022 [DOI] [PubMed] [Google Scholar]
- Thomas S, Ritter B, Verbich D, Sanson C, Bourbonnière L, McKinney RA, McPherson PS (2009) Intersectin regulates dendritic spine development and somatodendritic endocytosis but not synaptic vesicle recycling in hippocampal neurons. J Biol Chem 284:12410–12419. 10.1074/jbc.M809746200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM (2008) Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59:84–97. 10.1016/j.neuron.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zünd D, Gruber AR, Zavolan M, Mühlemann O (2013) Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol 20:936–943. 10.1038/nsmb.2635 [DOI] [PubMed] [Google Scholar]