

Abstract
Cold allodynia is a common symptom of neuropathic and inflammatory pain following peripheral nerve injury. The mechanisms underlying this disabling sensory alteration are not entirely understood. In primary somatosensory neurons, cold sensitivity is mainly determined by a functional counterbalance between cold-activated TRPM8 channels and Shaker-like Kv1.1–1.2 channels underlying the excitability brake current IKD. Here we studied the role of IKD in damage-triggered painful hypersensitivity to innocuous cold. We found that cold allodynia induced by chronic constriction injury (CCI) of the sciatic nerve in mice, was related to both an increase in the proportion of cold-sensitive neurons (CSNs) in DRGs contributing to the sciatic nerve, and a decrease in their cold temperature threshold. IKD density was reduced in high-threshold CSNs from CCI mice compared with sham animals, with no differences in cold-induced TRPM8-dependent current density. The electrophysiological properties and neurochemical profile of CSNs revealed an increase of nociceptive-like phenotype among neurons from CCI animals compared with sham mice. These results were validated using a mathematical model of CSNs, including IKD and TRPM8, showing that a reduction in IKD current density shifts the thermal threshold to higher temperatures and that the reduction of this current induces cold sensitivity in former cold-insensitive neurons expressing low levels of TRPM8-like current. Together, our results suggest that cold allodynia is largely due to a functional downregulation of IKD in both high-threshold CSNs and in a subpopulation of polymodal nociceptors expressing TRPM8, providing a general molecular and neural mechanism for this sensory alteration.
SIGNIFICANCE STATEMENT This paper unveils the critical role of the brake potassium current IKD in damage-triggered cold allodynia. Using a well-known form of nerve injury and combining behavioral analysis, calcium imaging, patch clamping, and pharmacological tools, validated by mathematical modeling, we determined that the functional expression of IKD is reduced in sensory neurons in response to peripheral nerve damage. This downregulation not only enhances cold sensitivity of high-threshold cold thermoreceptors signaling cold discomfort, but it also transforms a subpopulation of polymodal nociceptors signaling pain into neurons activated by mild temperature drops. Our results suggest that cold allodynia is linked to a reduction of IKD in both high-threshold cold thermoreceptors and nociceptors expressing TRPM8, providing a general model for this form of cold-induced pain.
Keywords: α-DTx, 4-AP, Kv1 channels, PBMC, thermotransduction, TRPM8
Introduction
Painful hypersensitivity to innocuous cold, or cold allodynia, is a prevalent symptom of neuropathic and inflammatory pain induced by peripheral nerve damage. Despite major advances in our understanding of the mechanisms underlying the diverse forms of cold-induced pain in response to axonal damage, including cold allodynia, the molecular and neural bases of this invalidating sensory alteration have not been entirely elucidated (Yin et al., 2015). In trigeminal and DRG neurons, the concerted operation of several transduction and voltage-gated ion channels shape their net excitability in response to temperature changes (Vriens et al., 2014; González et al., 2015). In low- and high-threshold cold thermoreceptors, cold sensitivity is largely dependent on the counterbalance between the functional expression of TRPM8 channels, the main molecular entity responsible for the cold-activated excitatory current (Icold) (McKemy et al., 2002; Peier et al., 2002; Latorre et al., 2011; McCoy et al., 2011; Almaraz et al., 2014; Madrid and Pertusa, 2014) and Shaker-like Kv1.1 and Kv1.2 channels, the molecular counterpart underlying the inhibitory potassium current IKD (Viana et al., 2002; Madrid et al., 2009; McKemy, 2013; González et al., 2015). In these cold-sensitive neurons (CSNs), IKD current works as an excitability brake, contributing to set their thermal threshold under physiological conditions (Viana et al., 2002; Madrid et al., 2009). This fast-activating slow-inactivating voltage-dependent K+ current dampens the effect of the cold-induced depolarizing TRPM8-dependent current, shifting the temperature threshold of the neuron to higher values (i.e., lower temperatures) and reducing the net response of CSNs to temperature drops. IKD exerts its action at membrane potentials subthreshold to the action potential firing (Storm, 1988; Viana et al., 2002), reducing the excitability of the neuron and preventing an unspecific activation by temperature reductions of normally cold-insensitive neurons of other somatosensory modalities (Viana et al., 2002; Belmonte et al., 2009; Madrid et al., 2009).
Changes in the functional expression pattern of voltage-gated K+ channels in response to axonal damage in murine models have been reported (Rasband et al., 2001; Kim et al., 2002; Yang et al., 2004; Cao et al., 2010; Duan et al., 2012; Zhao et al., 2013; Fan et al., 2014; Calvo et al., 2016; Wang et al., 2016). Nevertheless, despite the relevance of IKD current in cold sensitivity, the possibility that axonal damage may induce a modification of the functional expression of this current, thereby inducing exacerbated responses to innocuous cold in CSNs, and the appearance of mild-cold sensitivity in nociceptors in response to chronic nerve injury still has not been explored, and its relative contribution to this sensory alteration is still unknown.
Here we show that cold allodynia in response to peripheral nerve damage by chronic constriction is linked to a reduction in the IKD current in both high-threshold CSNs and in a subpopulation of nociceptive neurons. These nociceptors, which are normally insensitive to moderate cooling, became sensitive to mild cold temperatures by the functional reduction of IKD. Although no major differences in TRPM8-dependent currents were found, TRPM8 channels appear to be critical to cold sensitivity of CSNs from both control and injured animals because the specific TRPM8 blocker 1-phenylethyl-4-(benzyloxy)-3-methoxybenzyl(2-aminoethyl) carbamate (PBMC) suppressed cold-induced responses in both groups of neurons. Consistent with these results, our mathematical model of CSNs shows that a reduction in IKD density, as the one observed in injured mice, shifts the thermal threshold to higher temperatures in neurons where the cold-induced excitatory current depends on TRPM8-like channels. Simulations using this model support the idea that this brake potassium current is a critical factor to determine the broad thermal threshold of CSNs. Moreover, in this model, the reduction of IKD induces cold sensitivity in former cold-insensitive (nociceptive) neurons expressing low levels of TRPM8-like current. Thus, our results suggest that the transformation of high-threshold CSNs and nociceptors into neurons sensitive to innocuous and mild cold respectively, due to a reduction of IKD density induced by chronic constriction injury in TRPM8-positive neurons, would be part of the main neural and molecular mechanisms that underpin painful hypersensitivity to cold induced by this form of chronic nerve damage.
Materials and Methods
Animals.
This study was performed using male young adult (P21-P40) BALBC mice. Animals were housed maximum four per cage in a 12 h light/dark cycle with food and water ad libitum and killed with CO2. All experiments were conducted according to bioethical guidelines of the Comisión Nacional de Investigación Científica y Tecnológica de Chile (CONICYT) (reference number 018/241) and have been approved by the Bioethical Committee of the University of Santiago de Chile (reference number 927/76).
Model of cold allodynia induced by axonal damage.
We used peripheral axonal injury by chronic constriction of the sciatic nerve as a model of nerve damage manifesting cold allodynia in mice hindpaw (Bennett and Xie, 1988). In brief, under ketamine and xylazine anesthesia, the right sciatic nerve was exposed after an incision to the skin and the separation of the heads of the biceps femoris muscle. At 5 mm (proximal) of the peroneal-tibial bifurcation, two loose silk ligatures with 8-0 chromic gut separated by 3 mm were tied around the exposed nerve, until an ∼80% of its original diameter, and the incision was closed. For sham controls, the sciatic nerve was exposed but not ligated.
Acetone evaporative cooling assay.
Cold sensitivity was assessed with the acetone evaporation assay. The sample size was calculated using a power of 80% and an error (α) of 0.05%; assuming a change of three times in nociceptive score, and an estimated SD of 0.4 and 0.6 in sham and CCI group, the sample size was 6 animals per group. Mice were habituated to the behavior room for a minimum of 3 h before testing and to the experimental chambers for at least 20 min before testing. Experiments were performed during the light period. The same investigators performed the scoring in all of the behavioral tests, which were performed blind with regard to the type of operation. For acetone-evoked evaporative cooling, mice were placed in round plastic chambers on a metal mesh platform. A drop (∼20 μl) of acetone was applied to the plantar surface of the hindpaws, and the number of the following nocifensive events were monitored over the following 60 s and an arbitrary score was assigned to the behavior: 0 indicated no response, 0.5 licking response, 1 flinching and brushing of the hindpaw, 2 strong flinching, and 3 strong flinching, licking, and paw guarding. Nocifensive responses were observed during the first minute after acetone application and measurements were repeated 3 times with a 10 min interval to obtain a mean value. The events for the contralateral paw measured under the same conditions were subtracted from the ipsilateral one to give the final net acetone score. A value of 0 indicates no difference between cold-evoked sensations of the ipsilateral and contralateral hindpaws, whereas a positive value indicates cold allodynia. Solutions, either drug or vehicle (saline solution) was injected into the plantar surface of the hindpaw, in a volume of 5 μl using a 30 gauge needle coupled to a Hamilton syringe. For the experiments using 4-aminopyridine (4-AP), acetone test was performed 10 min after injection of the drug or vehicle.
Cell culture.
Pairs of sham and CCI mice were killed by CO2 inhalation. Following decapitation, the L3-L5 DRGs ipsilateral to the nerve injury were removed and incubated in an enzymatic mixture, including collagenase Type XI (650 UI/ml; C7657, Sigma-Aldrich) and dispase (5 UI/ml; 17105-041 GIBCO, Thermo Fisher Scientific), in INC-mix solution (in mm as follows: 155 NaCl, 1.5 K2HPO4, 10 HEPES, 5 glucose, pH 7.4), during 40 min at 37°C. Sensory ganglia were mechanically dissociated using a polished Pasteur pipette and neurons were plated on poly-L-lysine-coated 6 mm #0 glass coverslips (Menzel-Gläser), maintained in MEM (Earle's salts, 111095080, GIBCO, Thermo Fisher Scientific) supplemented with MEM-vit (11120052, GIBCO, Thermo Fisher Scientific, 10% FBS (SH30910.03, Hyclone, GE Healthcare), 200 μg/ml streptomycin, 125 μg/ml penicillin (15140-122, GIBCO, Thermo Fisher Scientific), and used 6–12 h after plating for [Ca2+]i imaging experiments and electrophysiological recordings. Calcium imaging and patch-clamp experiments were performed in both conditions (sham and CCI), 7 d after surgery.
Heterologous expression of TRPM8.
HEK-293 cells (CRL-1573, RRID: CVCL_0045) stably expressing mTRPM8 (HEK-293-TRPM8-myc cells) were used to evaluate the blocking effect of PBMC (Pfizer) on TRPM8 channels. HEK-293-TRPM8-myc cells were cultured in DMEM containing 10% of FBS and antibiotics. Cells were trypsinized and replated on poly-L-lysine-coated 6 mm #0 glass coverslips (Menzel-Gläser) 24 h before calcium imaging and patch-clamp experiments.
Ca2+ imaging.
For ratiometric calcium imaging experiments, DRG neurons and HEK-293-TRPM8-myc cells were incubated with 5 μm fura-2 AM (F1221, Thermo Fisher Scientific) dissolved in standard extracellular solution supplemented with 0.02% Pluronic (P6867, Thermo Fisher Scientific) for 50 min at 37°C in darkness. Fluorescence measurements were made with an inverted Nikon Ti microscope fitted with a 12-bit cooled ORCA C8484-03G02 CCD camera (Hamamatsu). Fura-2 was excited at 340 and 380 nm at 0.5 Hz with a Polychrome V monochromator (Till Photonics), with exposure time no longer than 40 ms, and the emitted fluorescence was filtered with a 510 nm long-pass filter. Calibrated ratios were displayed online with HCImage version 2 software (Hamamatsu). Bath temperature (see below for details) was sampled simultaneously using a BAT-12 microprobe thermometer (Physitemp Instruments) connected to an IT-18 T-thermocouple, using Clampex 10 software (Molecular Devices), and digitized with an Axon Digidata 1440A AD converter (Molecular Devices).
Threshold temperature values for [Ca2+]i elevation were estimated as described by Madrid et al. (2009), by linearly interpolating the temperature at the midpoint between the last baseline point and the first point at which a rise in [Ca2+]i deviated by at least 4 times the SD of the baseline. Intracellular [Ca2+] increases observed in cultured CSNs are mainly due to Ca2+ entry through voltage-gated Ca2+ channels, which are activated during action potential firing. As have been previously shown, there is a tight correlation between threshold temperature detected on the [Ca2+]i signal and the threshold of action potential firing detected in the cell-attached mode (Viana et al., 2002; Madrid et al., 2006; González et al., 2015). This noninvasive method allowed us to determine the thermal threshold of several CSNs simultaneously. To quantify the percentage of CSNs in both sets of mice, a solution containing elevated K+ (30 mm KCl) was perfused at the end of the protocol to determine the viability of the neurons in the entire field; usually between 60% and 70% of the neurons respond to this stimulus. Only cells showing a [Ca2+]i increase in response to a 30 mm elevation in extracellular K+ were included in the analysis.
Electrophysiology.
Whole-cell voltage- or current-clamp recordings were performed simultaneously with temperature recordings. The extracellular standard solution for both sensory neurons and HEK-293 cells contained the following (in mm): 140 NaCl, 3 KCl, 1.3 MgCl2, 2.4 CaCl2, 10 HEPES, 10 glucose (298 mOsm/kg, pH 7.4, adjusted with NaOH). Standard patch-clamp pipettes (4–5 mΩ resistance) were made with GC150F-7.5 glass capillaries (Harvard Apparatus) and filled with intracellular solution containing the following (in mm): 105 K gluconate, 35 KCl, 8.8 NaCl, 10 HEPES, 0.5 EGTA, 4 MgATP, 0.4 NaGTP (300 mOsm/kg, pH 7.4, adjusted with KOH) (sensory neurons) and 140 CsCl, 0.6 MgCl2, 1 EGTA, and 10 HEPES, pH 7.4, adjusted with CsOH (276 mOsm/kg) (HEK-293 cells) (data not shown). Current and voltage signals were recorded using an Axopatch 200B (Molecular Devices). Stimulus delivery and data acquisition were performed using pClamp 10 software (Molecular Devices). Before electrophysiological recordings, the temperature threshold in response to cold stimuli was determined using calcium imaging.
Temperature stimulation.
Coverslip pieces with plated cells were placed in a microchamber and were continuously perfused (∼1 ml/min) with solutions warmed at ∼34°C. The temperature was adjusted with a homemade water-cooled computer-controlled Peltier device, with the outlet close to the imaging field and controlled by a feedback device. Cold sensitivity was investigated with ∼40 s duration ramp-like temperature drops to 19°C from basal temperature of 34°C, applied in control solution and in the presence of different compounds.
Experimental protocols.
The first step in the protocol was determining the temperature response threshold to cold using the elevation in [Ca2+]i. Thereafter, 100 μm 4-AP or 500 nm α-DTx was applied extracellularly, and the neurons were stimulated again with a cold ramp in the presence of the drug. Stimuli of 100 μm menthol and 100 μm menthol plus cold (agonists of TRPM8), 100 μm allyl isothiocyanate (AITC; agonist of TRPA1), and 200 nm capsaicin (agonist of TRPV1) were also used, to determine the expression of these thermo-TRP channels in CSNs. The ability to respond to these stimuli was correlated with the thermal threshold and the active and passive membrane properties (see below), to determine whether these neurons correspond functionally to canonical low- or high-threshold CSNs or to polymodal nociceptors.
For electrophysiological experiments, neurons were approached with a patch pipette and after attaining the seal and the whole-cell configuration they were recorded under current or voltage clamp. In current-clamp mode, neurons were held at −60 mV, and a series of negative and positive current steps of 500 ms duration (Δi = 10–100 pA depending on input resistance) were delivered at a rate of 0.1 Hz to the cell to determine resting membrane potential, input resistance, rheobase, spike duration, inward rectification index, and firing pattern. Under voltage clamp, neurons were initially held at −50 mV. A 500 ms hyperpolarizing pulse to −120 mV was used to remove the inactivation of IKD (Viana et al., 2002; Madrid et al., 2009) and to estimate Ih (Orio et al., 2009, 2012). The slow outward current, measured 1 s after the return to −40 mV, was taken as IKD (Madrid et al., 2009). Thereafter, cells were held at −60 mV and cooled from 34 ± 1°C to ∼20°C. The difference between holding current at both temperatures was considered as the cold-sensitive current (Icold). Maximal TRPM8-dependent current was taken as the inward current enhanced by 100 μm menthol at 20°C (Icold+menthol), as in Madrid et al. (2009).
To characterize IKD in DRG neurons in more detail, we used standard activation and inactivation pulse protocols combined with pharmacological manipulation as previously described by Madrid et al. (2009). In brief, the current–voltage (I–V) relationship for the total K+ current was determined from a series of depolarizing voltage steps (1.5 s duration) between −80 and 60 mV, in 20 mV increments, delivered at a rate of 0.1 Hz, in the presence of 0.5 μm tetrodotoxin citrate (TTx). Pulses were delivered from a holding potential of −50 mV following a prepulse of 500 ms to −120 mV. This protocol was repeated in the presence of 100 μm 4-AP. The inactivation protocol consisted of a fixed pulse to 0 mV (1.4 s) preceded by a variable prepulse (0.5 s duration) between −120 and −10 mV, in 10 mV increments, delivered at a rate of 0.1 Hz.
Electrophysiological data analysis.
The 4-AP-sensitive current (IKD) was obtained by digital subtraction of the records at the same membrane potentials before and after application of the blocker at 100 μm. Conductance-voltage (G–V) curves were constructed from the I–V curves of individual neurons by dividing the evoked current by the driving force, according to Equation 1 as follows:
where Vm is the testing membrane potential and Vrev is the theoretical reversal potential of the current determined from the Nernst equation for K+ ions.
Steady-state activation and inactivation data were fitted with a Boltzmann function as follows:
where G is the whole-cell conductance, Gmax is the fitted maximal conductance, V1/2 is the potential for half-maximal activation, and s is the slope factor. Data were then normalized to Gmax.
Data are reported as mean ± SEM. Statistical significance (p < 0.05) was assessed by Student's t test or two-way ANOVA test for normally distributed data; Fisher's (F) test was used to compare populations.
Mathematical model.
We have used the model of a CSN, including TRPM8 recently described (Olivares and Orio, 2015; Olivares et al., 2015). After the addition of IKD, the equation for the membrane potential is as follows:
 |
where Cm is the membrane capacitance, Isd and Isr are the slow depolarizing and repolarizing currents, respectively, that create an intrinsic oscillation of membrane potential. Id and Ir are Hodgkin and Huxley-type depolarizing and repolarizing currents for action potential firing, IM8 is the cold-activated current depending on TRPM8, Il is an ohmic leakage current, and Iwn is a noise term (Olivares et al., 2015). The brake potassium current IKD is described by the following equation:
being ρ(T) a function of temperature to adjust the conductance (Q10 = 1.3), and gKD the maximal conductance density of 4-AP-sensitive channels. mKD and hKD are variables for the fast activation and slow inactivation, respectively, that depend on voltage according to Equation 2, with values for s and V1/2 close to those reported in Figure 5B. Vm is the membrane potential, and EK is the reversal potential for potassium currents set to −90 mV. Activation time course of the conductance follows the differential equation as follows:
 |
where Φ(T) is a function of temperature to adjust the channel kinetics (Q10 = 3) and m∞(V) and τm(V) are functions of voltage for the steady-state activation level and the relaxation time constant, respectively. This approach was also used to model the slow inactivation kinetics of the 4-AP-sensitive potassium current of primary somatosensory neurons. m∞(V) and h∞(V) were described by the function (m, h)∞ = 1/(1 + exp((V1/2 − V)/s)), with V1/2 = −12 mV, s = 12 mV for activation (m) and V1/2 = −68 mV, s = −10 for inactivation (h). Values for τ at different potentials were obtained using symmetric bell-shaped functions of voltage, namely, τ(m, h) = a + (b * (ek(v+c)/(1 + e2k(v+c)))), where k = 0.12 and v is the membrane voltage, with a = 7 ms, b = 105, c = 12 mV for τm, and a = 5250 ms, b = 78750, c = 68 mV for τh.
Figure 5.

Biophysical properties of IKD and differential expression of IKD and ITRPM8 in CSNs from sham and CCI mice. A, 4-AP-sensitive currents (top), obtained from the digital subtraction of whole-cell currents in control and 100 μm 4-AP with an activation protocol in a CSN from a sham animal. The voltage protocol is shown at the bottom. Note the slow inactivation at negative membrane potentials. Dotted line indicates the 0 current level. B, Average activation (orange) and steady-state inactivation (blue) curves obtained from five control CSNs. Note the window current around the resting membrane potential. C, Whole-cell current in a representative CSNs from sham (left) and CCI (right) mice during a bipolar voltage protocol. The holding potential was −50 mV, and a hyperpolarizing pulse to −120 mV (see Materials and Methods) was applied before to reach the subthreshold membrane potential of −40 mV to reveal IKD. D, Mean IKD current density in sham (n = 34) and CCI (n = 36) CSNs: p = 0.003 (t test). E, Simultaneous recording of membrane current (top) and bath temperature (bottom) during a long cooling step (20°C) combined with application of menthol (100 μm) in a representative CSN from a sham animal. Dotted line indicates the 0 current level. F, Bar plot summarizing the mean Icold and Icold+menthol (ITRPM8) current density (measured at the pick of both currents) in 26 CSNs from sham and 20 CSNs from CCI animals (Vhold = −50 mV): p = 0.477 and p = 0.267 (t test), respectively. G, Dot plot of IKD and Icold+menthol current density in sham and CCI CSNs separated into HT- and LT-CSNs. The abscissa represents the thermal threshold, measured using [Ca2+]i imaging. *p < 0.05 (t test). **p < 0.01 (t test). Same as in C, IKD was measured at −40 mV 1 s after the return from a holding potential of −120 mV, in the presence of 0.5 μm TTx.
Values for the free parameters are detailed in Table 3. The rest of the equations and parameters of the model are as in (Olivares et al., 2015), with the sole exception that we used τr = 2 ms. The model was implemented in Neuron simulation environment (RRID: SCR_005393) controlled with Python scripts (RRID: SCR_008394) (Hines and Carnevale, 1997; Hines et al., 2009). Analysis of the simulations was performed in Python with the libraries Numpy (RRID: SCR_008633), Scipy (RRID: SCR_008058), and Matplotlib (RRID: SCR_008624).
Table 3.
Values for the free parameters of the model
| Parameter set | gKD (mS/cm2) | gTRPM8 (mS/cm2) | gsd (mS/cm2) | gsr (mS/cm2) | gd (mS/cm2) | gr (mS/cm2) | gl (mS/cm2) | τCa (ms) | tdV (ms) | pCa × 103 | δVmin (mV) | δVmax (mV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.220 | 3.40 | 0.345 | 0.195 | 4.30 | 4.67 | 0.310 | 950 | 27,500 | 5.00 | −144 | 245 |
| 2 | 0.240 | 2.56 | 0.343 | 0.207 | 4.05 | 4.72 | 0.292 | 1150 | 29,100 | 6.30 | −212 | 210 |
| 3 | 0.220 | 4.00 | 0.348 | 0.205 | 4.31 | 4.60 | 0.290 | 1000 | 39,000 | 9.95 | −190 | 230 |
| 4 | 0.220 | 3.20 | 0.306 | 0.193 | 3.15 | 4.51 | 0.282 | 850 | 26,000 | 6.50 | −190 | 220 |
| 5 | 0.225 | 2.50 | 0.351 | 0.209 | 4.98 | 4.77 | 0.300 | 970 | 27,900 | 5.22 | −145 | 256 |
Sets of parameters that satisfactorily reproduce the response to cold of cold-thermoreceptors. The rest of parameters of the model and their meanings are given in Olivares et al. (2015).
Reagents and drugs.
l-Menthol (266523), 4-AP (A78403), AITC (377430), and capsaicin (M2028) were purchased from Sigma-Aldrich. α-Dendrotoxin (α-DTx) and TTx were purchased from Alomone Labs. PBMC was provided by Pfizer through its Compound Transfer Program (to R.M.).
Results
Nocifensive behavior of injured mice in response to innocuous cold stimulation is associated with an increase in cold sensitivity in primary sensory neurons
Nocifensive responses to innocuous cold stimulation by acetone evaporation in CCI animals appear 1 d after constrictive nerve ligation (Fig. 1A); 92% (33 of 36) of the CCI mice showed nociceptive responses to innocuous cold indicative of cold allodynia. These responses reached a near maximal value by the third day and remained stable at least 14 d after CCI (Fig. 1A). As it was shown in a previous study, the pharmacological suppression of IKD in nerve terminals of intact animal enhances cold-evoked nocifensive behaviors (Madrid et al., 2009). Seven days after surgery, local injection of the IKD blocker 4-AP into the hindpaw in sham mice produced a significantly large acute nocifensive response to acetone evaporation, compared with preinjection of vehicle (Fig. 1B). In contrast, in CCI animals, acetone evaporation evokes large nocifensive responses, which are only slightly potentiated by IKD blockage (Fig. 1B). The sensitization induced by 4-AP injected into the hindpaw suggests that a reduction of IKD in intact animals can induce cold-evoked nocifensive behaviors, similar to those observed in response to cold in mice with chronically injured nerves. Because CCI mice also appear to be less sensitive to IKD blockage by 4-AP, we hypothesize that the functional expression of the molecular target of this blocker could be reduced in injured animals.
Figure 1.

Nocifensive behavior of sham and injured mice in response to innocuous cold stimulation. A, Time course of the cold-evoked nocifensive behavior assessed by acetone response net score in mice, evaluated at 0 (basal), 1, 3, 7, and 14 d after injury (see Materials and Methods). Red line and red dots represent CCI animals. Black line and dots represent sham operated mice; n = 6 for each group. B, Bar graph of the nocifensive behavior after application of acetone to the plantar surface of the hindpaw in sham and CCI animals, before and after pharmacological suppression of IKD by intraplantar injection of 10 mm 4-AP (n = 6). A, B, Intergroup analyses of nociceptive behavior net scores were performed by means of two-way ANOVA followed by the Bonferroni post hoc multiple-comparisons test: **p < 0.01; ***p < 0.001. B, Intragroup analyses were assessed by means of paired t test: ##p < 0.01; ###p < 0.001.
CCI induces both an increase in the population size of CSNs and a shift of their cold threshold to higher temperatures
To compare cold sensitivity of primary sensory neurons from CCI and sham mice, we used intracellular Ca2+ imaging in dissociated L3-L5 DRG neurons during rapid temperature reductions. Similar to our findings in trigeminal ganglion neurons (Madrid et al., 2009), cold sensitivity varies among individual DRG neurons. Figure 2A shows a representative response to cold stimulation of a CSN from a control mouse. In Figure 2B, we represent the cumulative population of CSNs activated by lowering temperature. At a temperature recruiting 50% of the CSNs in sham-operated animals, nearly 75% are recruited following injury. We found that the mean cold threshold of CSNs from the sham group was 27.9 ± 0.3°C (n = 135) (ranging from 20.0°C to 34.7°C), whereas the mean temperature threshold of cold responses of CSNs from injured animals was 29.9 ± 0.3°C (n = 140) (ranging from 22.0°C to 35.1°C) (p < 0.001, unpaired t test), an average shift of 2.0°C to higher temperatures in the injured animals compared with the sham group (Fig. 2C). The amplitude of cold-evoked responses was also augmented in 15%, with a mean value of 227 ± 12 nm (n = 135) in control neurons and 261 ± 12 nm (n = 140) in CSNs from CCI animals (p = 0.040, t test) (Fig. 2D). Because [Ca2+]i rises in response to cold are mainly due to the activation of voltage-gated Ca2+ channels during repetitive action potential firing in CSNs (Viana et al., 2002; Madrid et al., 2006; González et al., 2015), this increment is consistent with an increase in the maximal firing frequency in response to cold in CSNs from CCI mice compared with sham animals. In Figure 2E, we show the analysis of the sensitivity of the entire population of primary sensory neurons studied by calcium imaging. We found an increase in the percentage of CSNs in the CCI group compared with sham animals (8.6%, n = 135 of 1571 control DRG neurons, vs 11.9%, n = 140 of 1174 CCI neurons; p = 0.005, F test) (Fig. 2E, top, pie charts).
Figure 2.

Altered cold sensitivity of primary sensory neurons in response to chronic nerve damage. A, Ratiometric [Ca2+]i response to cooling (middle) in one representative low-threshold CSNs from a sham animal, recorded simultaneously with the temperature of the bath (bottom). Top, Transmitted (a) and pseudocolor ratiometric [Ca2+]i images of this neuron, that correspond with b (control) and c (cold) points in the middle panel; the fluorescence images in b and c correspond with the time points marked in magenta. B, Percentage of active population recruited during a cooling ramp in CSNs from sham versus CCI mice. C, Dot plot of temperature thresholds exhibited by CSNs from sham (n = 135) and CCI (n = 140) mice. Temperature thresholds were compared using a two-tailed unpaired Student's t test: ***p < 0.001. D, Summary bar plot of the magnitude of the cold-induced responses in CSNs from sham (black bar) and CCI (red bar) animals. Intracellular calcium increases in CSNs were compared using unpaired Student's t test: *p = 0.040. All values in this figure that include error bars indicate mean ± SEM. E, Pie plots showing the percentage of the different populations of CSNs in sham and CCI condition. CINs are cold-insensitive neurons. **p = 0.005 (F test). ***p < 0.001 (F test).
CSNs present a wide range of temperature sensitivities both in vivo and in vitro and can be operationally classified as low-threshold and high-threshold CSNs (Thut et al., 2003; Madrid et al., 2009). We have considered low-threshold CSNs (LT-CSNs) those neurons with a threshold temperature >26.5°C (Madrid et al., 2009). In sham animals, LT-CSNs represented 68.9% of the whole population of cold-sensitive primary sensory neurons (mean threshold = 29.6 ± 0.2°C). The rest of them (31.1%) correspond to the high-threshold CSNs (HT-CSNs), with a mean temperature threshold of 24.1 ± 0.3°C. In CCI animals, 85.7% of CSNs correspond to low-threshold ones (mean threshold = 30.7 ± 1.2°C), whereas the HT-CSNs were less frequent (14.3%; mean threshold = 24.7 ± 0.3°C) (p < 0.001, F test) (Fig. 2E, bottom pie charts).
Together, these results suggest that the peripheral mechanisms of painful hypersensitivity to cold in CCI animals include not only a higher temperature sensitivity of individual primary sensory neurons, but also an increase in the population size of CSNs in response to this form of axonal damage.
Reduced IKD is associated with lower temperature thresholds observed in CSNs from CCI animals
Temperature threshold of cold-sensitive neurons is determined to a large extent by the outward potassium current IKD, acting as an excitability brake (Viana et al., 2002; Madrid et al., 2009). We wondered whether changes in this current contribute to the altered cold sensitivity observed in injured neurons. Using intracellular calcium imaging, we studied the effect of the pharmacological suppression of IKD on cold-evoked responses of DRG neurons from control and CCI groups. To achieve this, we evaluated the effect of 100 μm 4-AP and 500 nm α-DTx, two known and well-characterized IKD blockers in CSNs (Viana et al., 2002; Madrid et al., 2009) on the temperature threshold of their cold-induced response. In Figure 3A, D, we show the effect of the suppression of IKD by 4-AP on the cold-induced calcium rise of a representative neuron from a sham animal and in a representative neuron from CCI group, respectively. Temperature threshold of CSNs from control animals was shifted to higher temperatures in 83.3% of the cells (20 of 24) in the presence of 4-AP (Fig. 3B,C). We have previously shown that this shift is fully reversible (Madrid et al., 2009), suggesting that this effect would not be related to alterations of the thermal threshold due to repetitive cold application (see below). In 1 of 24 CSNs depicted in Figure 3B, 4-AP shifted the threshold of cold response to a lower temperature. In control CSNs, mean cold threshold was 27.6 ± 0.6°C (n = 24), and in the presence of the inhibitor of the brake current the mean temperature threshold was shifted to a higher value (30.0 ± 0.5°C; p < 0.001, paired t test) (Fig. 3C). Interestingly, CSNs from CCI animals were less sensitive to the suppression of IKD by 4-AP (Fig. 3D,E). Thus, in 25 CSNs from CCI group studied in the same conditions, the mean control temperature threshold remained practically unaltered after 4-AP (30.5 ± 0.5 vs 29.9 ± 0.6°C; p = 0.354, paired t test) (Fig. 3C,E). In 5 of 25 CSNs, 4-AP shifted the threshold of cold responses to higher temperatures (ΔT>1°C), and in six neurons the temperature thresholds were shifted to lower values (ΔT>−1°C). Very similar results were obtained using α-DTx in a smaller population of CSNs from both groups of animals (Fig. 3F).
Figure 3.

Effect of the pharmacological suppression of IKD on thermal threshold of CSNs from sham and CCI mice. A, Ratiometric [Ca2+]i response in a representative CSN from sham group during two consecutive cooling stimuli in control solution and the presence of 100 μm 4-AP. In this neuron, the cold threshold was shifted to higher temperatures, from 26.6°C to 29.2°C (black arrowheads). B, Summary of effect of 100 μm 4-AP on temperature threshold of cold-evoked responses in 24 CSNs from sham group. C, Mean values of the temperature threshold of CSNs in sham (black bar), control + 4-AP (dashed black), CCI (red bar), and CCI in the presence of the inhibitor of IKD. The mean threshold of CCI CSNs was unaffected by the pharmacological suppression of IKD. D, Ratiometric [Ca2+]i response in a CSN from CCI group during two consecutive cooling ramps in control solution and in the presence of 100 μm 4-AP. In this neuron, the cold threshold was unaffected by the drug (31.9°C vs 31.1°C, red arrowheads). E, Summary of the effect of 100 μm 4-AP on temperature threshold of cold-evoked responses in 25 CSNs from CCI mice. F, Mean values of the temperature threshold of CSNs in control (black bar), control + α-DTx (dashed black bar), CCI (red bar), and CCI in the presence of the inhibitor of IKD (dashed red bar). The mean threshold of CCI CSNs was unaffected by the suppression of IKD by this toxin. Temperature threshold shifts after treatment with 4-AP (C) or α-DTx (F) were assessed using paired Student's t test: ***p < 0.001 and **p = 0.008, respectively. C, F, Temperature thresholds exhibited by CSNs from sham and CCI mice were compared using a Student's t test: **p = 0.001; *p = 0.044. All values in this figure that include error bars indicate mean ± SEM.
The pharmacological suppression of IKD using 4-AP (or α-DTx) not only causes a shift in the thermal threshold to higher temperatures in CSNs but also induces cold sensitivity in neurons from other somatosensory modalities expressing high levels of IKD (Viana et al., 2002; Madrid et al., 2009). In HT-CSNs, this reversible shift (mean cold threshold = 24.0 ± 0.6°C in control condition vs 27.3 ± 0.3°C in 4-AP, p = 0.006, paired t test) is accompanied by an increase in the firing frequency of the neuron during the cold-induced response (Fig. 4). Figure 4A, B shows the effect of 100 μm 4-AP on the thermal threshold and on the action potential firing frequency of HT-CSNs from sham animals. In a group of LT-CSNs studied in the same conditions, the thermal threshold was virtually unaffected by the pharmacological suppression of IKD (mean threshold = 31.4 ± 0.6°C in control condition vs 30.7 ± 0.8°C in 4-AP; data not shown), consistent with the low expression of the brake current in these neurons. Figure 4C shows the appearance of cold-induced discharge in a cold-insensitive neuron in response to IKD blockage by 4-AP, and the quantification of this firing in nine transformed neurons (Fig. 4D).
Figure 4.

Reversible shift of thermal threshold induced by pharmacological suppression of IKD in primary sensory neurons. A, Simultaneous recording of membrane potential (top) and bath temperature (bottom) during three consecutive cooling ramps in a cold-sensitive DRG neuron recorded in current-clamp mode (Ihold = 0 pA). Application of 100 μm 4-AP reversibly shifted the temperature threshold and enhanced the firing of action potentials during the cooling ramp. B, Left, Response-threshold temperatures of five CSNs during cold applications in control solution, 100 μm 4-AP and after washing. Colors of the symbols represent each neuron studied using this protocol. Right, Bar plot of mean firing frequency during the cold-induced responses measured for the neurons in B. C, Simultaneous recording of membrane potential (top) and bath temperature (bottom) during three consecutive cooling ramps in a cold-insensitive DRG neuron recorded in current-clamp mode (Ihold = 0 pA). Note the reversible firing response induced by the pharmacological suppression of IKD by 4-AP during the cooling ramp. D, Left, Response-threshold temperatures of nine cold-insensitive neurons during cold applications in control solution supplemented with 100 μm 4-AP. Colors of the symbols represent each CIN studied using this protocol. Right, Bar plot of mean firing frequency during cold ramps measured for the neurons in D. B, D, Temperature threshold and firing frequencies were compared using paired Student's t test: **p = 0.002 and *p = 0.022, respectively. All values in this figure that include error bars indicate mean ± SEM.
Together, these results are consistent with the hypothesis that the increase in thermal sensitivity of CCI animals depends on a reduction of the functional expression of a 4-AP- and α-DTx-sensitive target, such as IKD, in response to axonal damage.
The mean IKD current density is reduced in CSNs from CCI animals
CSNs from injured animals present a mean temperature threshold of cold responses shifted ∼2°C to higher temperatures. This difference is very similar to the temperature shift induced by pharmacological suppression of IKD at control conditions (see above). Moreover, a significantly large subgroup of CSNs from CCI animals appears to be insensitive to 4-AP (and α-DTx), suggesting that the brake IKD current could be diminished in these cells in response to injury. To explore this possibility, we studied the biophysical properties of IKD in CSNs in control conditions, and then we determined the IKD current density in CSNs from sham and CCI animals at physiologically relevant membrane potentials. Using the whole-cell configuration of the patch-clamp technique, and conventional activation and inactivation protocols in combination with pharmacological isolation of IKD using 4-AP (see Materials and Methods), we determined the biophysical properties of IKD in control CSNs. Figure 5A shows an example of the IKD current obtained by digital subtraction of whole-cell currents in the presence of 4-AP and in extracellular control solution. In these neurons, we found that the voltage for half-maximal activation (V1/2) of IKD averaged −12 ± 2 mV, with a slope factor of 12 ± 1 mV (n = 5), while the V1/2 for steady-state inactivation curve averaged −68 ± 4 mV with a slope factor of −10 ± 2 mV (n = 5) (Fig. 5B). IKD current presented a significant overlap in its activation and inactivation curves, giving rise to a window current around the resting membrane potential (Fig. 5B). These properties are very similar to those described for IKD in trigeminal CSNs (Viana et al., 2002; Madrid et al., 2009).
Next, we determined the mean current density of IKD at −40 mV in both groups, a membrane potential subthreshold to the action potential firing in these neurons, and where the brake current exerts its functional role. Figure 5C presents recordings of two representative CSNs from the sham (Fig. 5C, left) and CCI group (Fig. 5C, right). We found that mean IKD current density is reduced in CSNs from CCI animals compared with control neurons (3.9 ± 0.5 pA/pF, n = 34, vs 2.5 ± 0.3 pA/pF, n = 36, in CSNs from CCI animals; p = 0.003, t test) (Fig. 5D). These results suggest that a reduction of the IKD current density in injured animals contributes to the increased cold sensitivity observed in CSNs after nerve damage, explaining both the shift in thermal threshold and its lower sensitivity to 4-AP and α-DTx.
Changes in the functional expression of TRPM8 channels are not necessary for cold allodynia induced by chronic peripheral nerve injury
TRPM8 channels are the main entity responsible for the excitatory current in response to cold in primary somatosensory neurons (for review, see Babes et al., 2011; McCoy et al., 2011; Almaraz et al., 2014; Madrid and Pertusa, 2014; González et al., 2015). It has been suggested, with some controversy, that an increased expression of TRPM8 could be linked to developing and maintaining cold allodynia and hyperalgesia after peripheral nerve damage (Katsura et al., 2006; Colburn et al., 2007; Frederick et al., 2007; Xing et al., 2007; Namer et al., 2008; Caspani et al., 2009; Staaf et al., 2009; Knowlton et al., 2010; Su et al., 2011). We asked whether changes in the functional expression of TRPM8 influences the differences in temperature sensitivity observed among CSNs from control and CCI mice. A direct determination of functional TRPM8 channels was obtained using whole-cell voltage-clamp recording at −60 mV, during stimulation with cold (Icold) and a combination of cold plus 100 μm menthol (Icold+menthol), to reach the maximal activation of TRPM8 channels (Malkia et al., 2007; Madrid et al., 2009), in CSNs from both groups of mice (Fig. 5E). Notably, we found that ITRPM8 current density in CSNs from CCI animals was, on average, indistinguishable to the one obtained in neurons from sham animals, even showing a downward trend. Icold and Icold+menthol current densities were −2.8 ± 0.5 and −18.8 ± 3.3 pA/pF, respectively, in control condition (n = 26), versus −2.2 ± 0.6 and −13.6 ± 3.3 pA/pF, respectively (n = 20), in CSNs from CCI animals (p = 0.477 and 0.267, t test) (Fig. 5F). A comparison of IKD and ITRPM8 (Icold+menthol) in HT- and LT-CSNs is shown in Figure 5G. The differences in the IKD current density are only significant in HT-CSNs.
Together, these results show that the IKD current density is strongly reduced in CSNs from the CCI group compared with sham mice, with no significant differences in the mean excitatory current dependent on TRPM8 channels in CSNs from both groups of animals.
The proportion of cold-insensitive neurons transformed into CSNs by suppression of IKD is reduced in CCI animals
It has been previously shown that pharmacological suppression of IKD induces cold sensitivity in a subpopulation of cold-insensitive primary sensory neurons (Viana et al., 2002) and peripheral axons (Roza et al., 2006). We reasoned that a reduction of the expression of the brake K+ current in response to axonal damage could imply a reduction in the incidence of this functional phenotype. Thus, the increase in the percentage of CSNs in CCI group could be related to a recruitment of cold-insensitive neurons (CINs) that became cold-sensitive due to a reduction of the IKD expression. In control conditions, we have found that 100 μm 4-AP induced the recruitment of a subpopulation of CINs that were transformed into cold-sensitive ones after pharmacological suppression of IKD (Fig. 6). As in CSNs, the intracellular calcium rise in response to cold in transformed neurons is the result of the action potential firing of the neuron, in this case under the suppression of IKD by 4-AP (Figs. 4A,C, 6D,E). In Figure 6A, we show a representative trace of [Ca2+]i in a CIN that responded to cold in the presence of 100 μm 4-AP. The mean amplitude of cold-induced responses in transformed (recruited) neurons was 148 ± 11 nm versus −7 ± 1 nm, measured as the peak of the response during cold stimulation compared with the response before the drug (n = 38) (p < 0.001, paired t test) (Fig. 6A, right, inset), with a mean thermal threshold of 24.0 ± 0.5°C. In Figure 6B, we show the mean IKD current recorded in 23 recruited neurons, compared with the IKD measured in 21 CSNs recorded under the same conditions. 8.1% (38 of 471) of CINs from sham animals were transformed into CSNs in the presence of the IKD blocker (Fig. 6C). Notably, in the CCI group, the application of 100 μm 4-AP only transformed 2.5% (3 of 120) of CINs into cold-sensitive ones (p = 0.042, F test) (Fig. 6C). In CCI animals, the mean amplitude of cold-induced responses of the recruited neurons in the presence of 4-AP was 194 ± 23 nm, versus −18 ± 23 nm previous to the application of the blocker (n = 3), with a mean cold threshold of 24.6 ± 1.2°C (data not shown). Similar results were obtained using 500 nm α-DTx as the IKD current blocker (data not shown). Thus, the population of CINs transformed into CSNs appears to be strongly reduced in injured animals, suggesting that the functional downregulation of IKD also affects the thermal sensitivity of CINs that have the potential to respond to cold in the absence of this brake current. These neurons would become cold-sensitive in injured mice, contributing to the nociceptive phenotype in response to axonal damage.
Figure 6.

Thermal modulation of cold-insensitive neurons by suppression of IKD. A, Time course of the [Ca2+]i in a CIN from a sham animal transformed in CSN in response to the pharmacological suppression of IKD by 4-AP. Inset, Histogram representing the mean value of the Ca2+ response of the population of recruited neurons from sham mice: ***p < 0.001 (paired Student's t test). B, IKD current density in recruited (n = 23) and CSNs (n = 21), recorded in the same conditions: **p = 0.003 (unpaired Student's t test). C, Stacked bar graph representing the percentage of CSNs, CINs, and 4-AP-recruited neurons in sham and CCI conditions. Note the reduction of the population of 4-AP-recruited neurons in response to injury: *p = 0.042 (F test). D, E, Simultaneous recording of membrane potential (top) and bath temperature (bottom) during two consecutive cooling ramps in two control cold-insensitive DRG neurons recorded under current-clamp (Ihold = 0 pA; Vrest= −50 and −49 mV, respectively), transformed into cold-sensitive ones by pharmacological suppression of IKD by 4-AP. D, E, Insets, First action potential in both neurons in response to a depolarizing current pulse at 34°C. Note the hump in the repolarizing phase in the action potential from the nociceptor-like neuron in D and its lower firing frequency compared with the cold thermoreceptor-like neuron in E.
We have also studied electrophysiological properties of recruited neurons in control animals, to shed light on its neural phenotype, using whole-cell configuration of the patch-clamp technique under current-clamp condition. Canonical cold-thermoreceptor neurons fire short duration action potentials (∼1 ms at the half-amplitude of the depolarizing phase) (Reid et al., 2002; Viana et al., 2002; Madrid et al., 2009). Nociceptors, on the other hand, fire wide action potentials often with an inflection (or hump) in the falling phase (∼2 ms at the half-amplitude) (Fang et al., 2005). We asked whether transformed neurons correspond to canonical CSNs with very low sensitivity to cold, or to nociceptors transformed into CSNs due to a functional reduction of IKD expression. To clarify this, we first identified transformed neurons in control animals using calcium imaging, and then we determined the passive and active membrane properties of these cells under current clamp. Two subpopulations were distinguishable in terms of their electrophysiological properties (Fig. 6D,E; Table 1); 80% (24 of 30) of the transformed neurons (mean cold threshold 22.8 ± 0.4°C, n = 24) fired action potentials with a hump in the repolarizing phase (Fig. 6D, inset). These neurons would correspond to nociceptive neurons damped by the functional expression of IKD. The firing frequency of current-induced action potentials at twice the rheobase in nociceptor-like cells is nearly half of that observed in cold thermoreceptor-like neurons (Fig. 6D,E; Table 1). The rest of the neurons (6 of 30) (Fig. 6E) (mean cold threshold in calcium imaging 25.4 ± 1.4°C, n = 6) fired action potentials with the shape of canonical cold thermoreceptor neurons (Fig. 6E, inset), suggesting that these neurons correspond to damped cold thermoreceptors expressing high levels of IKD and low levels of cold-sensitive excitatory current.
Table 1.
Active and passive membrane properties of recruited neurons from control animalsa
| Cold-insensitive (recruited) neurons | Temperature threshold (°C) | Temperature threshold 4-AP (°C) | Resting potential (mV) | Input resistance (mΩ) | Rheobase current (pA) | AP duration (ms) | Inward rectification index (%) | Firing frequency at 2 × rheobase (Hz) |
|---|---|---|---|---|---|---|---|---|
| Cold thermoreceptor-like (n = 6) | — | 25.4 ± 1.4 | −50.0 ± 5.0 | 308 ± 99 | 130 ± 29 | 0.54 ± 0.10 | 45.2 ± 3.8 | 46 ± 7 |
| Nociceptor-like (n = 24) | — | 22.8 ± 0.4* | −45.4 ± 1.4 | 490 ± 54 | 99 ± 24 | 1.39 ± 0.07*** | 43.0 ± 2.5 | 27 ± 4** |
aTemperature thresholds were determined in intact neurons by calcium imaging. AP, Action potential. Input resistance was measured from the slope of peak voltage response to a series of negative current steps. AP duration was measured at half-amplitude. Inward rectification index was measured as 100 × (Vpeak − Vsteady-state)/Vpeak, during hyperpolarizing voltage responses that reached a voltage peak value ∼−120 mV. The number of spikes at 2 × rheobase current were counted in a 500 ms period.
*p = 0.014;
**p = 0.022;
***p < 0.001; unpaired t test.
Thus, these results are consistent with the idea that IKD is downregulated in primary sensory neurons from CCI animals, and suggest that a subpopulation of nociceptive-like CINs transformed into CSNs by reduction of IKD could be participating in the development of cold allodynia in injured animals.
Nociceptive-like phenotype is more often found in CSNs from CCI group compared with control neurons
If the change in cold sensitivity observed in allodynic animals involves not only the participation of cold thermoreceptors with an exacerbated cold sensitivity (i.e., HT-CSNs transformed in LT-CSNs signaling cold discomfort), but also recruiting of polymodal nociceptors transformed into HT-CSNs due to a reduction of IKD, this should be reflected in the electrophysiological properties of CSNs from CCI mice. To explore this possibility, we studied the passive and active membrane properties of CSNs from both groups, under current clamp conditions. We found that 94.1% (32 of 34) CSNs form control animals fire action potentials of short duration (∼1 ms), with the canonical shape and width of action potentials found in cold-thermoreceptor neurons (Fig. 7A; Table 2). On the other hand, a subpopulation of CSNs from CCI animals (29.7%, n = 11 of 37) fire wide action potentials, with a hump in the repolarizing phase, and a mean duration of ∼1.8 ms, similar to those observed in conventional C-type polymodal nociceptors (p = 0.013, F test) (Fig. 7B; Table 2). Figure 7C shows a plot of the action potential firing frequency in response to external current in both populations of CSNs from sham and CCI mice. These results support the idea that a subpopulation of nociceptive neurons has been recruited as CSNs in injured mice, most probably due to the functional downregulation of IKD in response to injury.
Figure 7.

Electrophysiological properties of CSNs from sham and injured mice. A, B, Voltage responses to 500 ms hyperpolarizing and depolarizing current (Iext) pulses in two CSNs from injured mice, showing the phenotype of a canonical cold thermoreceptor (A) or broad action potentials similar to nociceptive neurons (B). Note the hump in the repolarizing phase and the lower firing frequency in the nociceptor-like CSN; both characters are often found in CSNs from injured animals. Also note the rebound firing of the neuron in A (orange trace). Insets, First action potential of neurons in A and B (black and blue arrowheads). C, Plot of the firing frequency versus depolarizing current in neurons of both phenotypes in sham and CCI mice. Left, Cold thermoreceptor-like (CT-like) neurons. Black circles represent control CT-like neurons (n = 20). Red circles represent CCI CT-like neurons (n = 7). Right, Nociceptor-like (N-like) neurons. Black circles represent control N-like neurons (n = 2). Red circles represent CCI N-like neurons (n = 8).
Table 2.
Active and passive membrane properties of CSNs from control and injured animalsa
| Cold-sensitive neurons | Temperature threshold (°C) | Resting potential (mV) | Input resistance (mΩ) | Rheobase current (pA) | AP duration (ms) | Inward rectification index (%) | Firing frequency at 2 rheobase (Hz) | Phenotype incidence (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Control (n = 36) | Cold thermoreceptor-like (n = 34) | 27.8 ± 0.6 | −46.0 ± 1.3 | 536 ± 42 | 67 ± 7 | 1.01 ± 0.04 | 51.4 ± 1.7 | 58.9 ± 4.5 | 94.4 |
| Nociceptor-like (n = 2) | 23.6 ± 1.6 | −45.0 ± 0.0 | 392 ± 90 | 68 ± 3 | 1.55 ± 0.15 | 57.9 ± 0.7 | 27.0 ± 5.0 | 5.6 | |
| Injured (n = 37) | Cold thermoreceptor-like (n = 26) | 27.5 ± 0.7 | −45.8 ± 2.0 | 394 ± 34 | 58 ± 5 | 0.91 ± 0.04 | 55.0 ± 2.0 | 67.8 ± 7.0 | 70.3 |
| Nociceptor-like (n = 11) | 26.5 ± 0.9 | −44.3 ± 2.1 | 494 ± 62 | 75 ± 7 | 1.75 ± 0.20*** | 53.1 ± 3.4 | 28.9 ± 5.9** | 29.7* |
aTemperature thresholds were determined in intact neurons by calcium imaging. AP, Action potential. Input resistance was measured from the slope of peak voltage response to a series of negative current steps. AP duration was measured at half-amplitude. Inward rectification index was measured as 100 × (Vpeak − Vsteady-state)/Vpeak, during hyperpolarizing voltage responses that reached a voltage peak value ∼−120 mV. The number of spikes at 2 × rheobase current were counted in a 500 ms period.
*p = 0.013 (F test).
**p = 0.002;
***p < 0.001; unpaired t test.
To further characterize CSNs in both conditions, we also studied the chemical sensitivity of these neurons using calcium imaging. To this end, we explored the responsiveness of CSNs from both groups of animals to activators of TRPM8 and two other thermo-TRP channels considered as molecular markers of nociceptive neurons: TRPA1 and TRPV1. The responses to the chemical activator of TRPM8, menthol, included an increase in the [Ca2+]i at 34°C or a shift in the thermal threshold of the response to cold toward higher temperatures. In the case of TRPA1 and TRPV1, positive responses were considered as an increase in the [Ca2+]i of the neuron at 34°C (Fig. 8A). We found that 87.2% of CSNs from sham group (34 of 39) responded to the application of menthol, and a positive response was observed in 88.1% (37 of 42) CSNs from CCI group (p = 0.442, F test) (Fig. 8B). Although an upward trend was observed, the percentage of CSNs responding to AITC in control (4 of 34) and CCI CSNs (11 of 36) were similar (p = 0.081, F test) (Fig. 8B). On the other hand, 44.1% of CSNs (15 of 34) responded to TRPV1 activator capsaicin in control group, and in CSNs from CCI animals 70.9% of the tested neurons responded to the vanilloid (22 of 31) (p = 0.045, F test) (Fig. 8B), indicating a significant increase in the sensitivity to this TRPV1 activator among CSNs from CCI mice compared with sham animals. The analysis of the Δ[Ca2+]i in response to each chemical agonist revealed no differences in the mean maximal response to these thermo-TRP activators in both conditions (Fig. 8C). The percentage of capsaicin-induced responses in control conditions is consistent with previous reports of several laboratories, showing that this vanilloid activates near to 50% of CSNs (Reid et al., 2002; Viana et al., 2002; Dhaka et al., 2008), or even more (Parra et al., 2010; Yudin et al., 2016). Low functional coexpression of TRPA1 channels in CSNs, revealed by AITC, is also in agreement with previous reports (Story et al., 2003; Parra et al., 2010; Pogorzala et al., 2013).
Figure 8.

Evaluation of the responses to chemical agonist of thermo-TRP channels of cold-sensitive neurons from sham and injured mice. A, Protocol in calcium imaging used to evaluate the responses to cold, menthol (100 μm), cold plus menthol, AITC (100 μm; TRPA1 agonist), and capsaicin (200 nm; TRPV1 agonist) in cold-sensitive neurons from sham and CCI animals. A, The CSN corresponds to a LT control neuron, which is insensitive to AITC but sensitive to capsaicin. B, Pie plots summarizing the percentage of menthol-, AITC-, and capsaicin-sensitive CSNs from sham and CCI animals. Menthol responses included an increase in the [Ca2+]i at 34°C or a shift (>1°C) in the cold threshold toward higher temperatures. C, Bar histograms represent the maximal response to menthol at 34°C and/or cold plus menthol (left panel), AITC at 34°C (central panel), and capsaicin at 34°C (right panel) in CSNs responding to these agonists, isolated from sham (black bars) and CCI animals (red bars). *p = 0.045 (F test).
Together, these results reveal an increase in the presence of CSNs with an electrophysiological phenotype similar to nociceptors signaling pain in injured animals, with no differences in the response to the activators of thermo-TRP channels TRPM8 and TRPA1 compared with the sham group. This would be accompanied by an increase in the incidence of the response to the TRPV1-activator capsaicin in CSNs from injured animals compared with control mice. These results support the proposal that a decrease in IKD current density in response to axonal damage induces cold sensitivity in CINs of nociceptive routes, contributing to the allodynic phenotype in CCI mice.
TRPM8 has a key role in cold-induced responses in both control and injured mice
We also wondered whether TRPM8 is the main responsible of cold-induced responses in CSNs from CCI animals. To study this, we have tested the TRPM8 antagonist PBMC on cold-induced responses of primary sensory neurons from control and allodynic animals. This compound has been described as a strong antagonist of TRPM8 channels both in vitro and in vivo (Knowlton et al., 2011; Gardiner et al., 2014). To determine an effective concentration of this compound to fully block the TRPM8-dependent response in these neurons, we first tested PBMC on cold-induced responses of HEK293 stably transfected with mTRPM8 channel using calcium imaging (Fig. 9A,B). At 1 μm, the blocking effect was complete (Fig. 9A), with an IC50 value of 92 nm (Fig. 9B). In our hands, the suppression by PBMC of cold-induced responses in these cells also appears to be dependent on the resting temperature preceding the cold stimuli because PBMC shows a higher inhibitory effect at lower basal temperatures (data not shown). This could be related to the effect of resting temperature on the thermal threshold and amplitude of TRPM8-dependent currents observed in these systems (Fujita et al., 2013). In any case, our results fully support the idea that PBMC is a strong and reversible inhibitor of TRPM8-dependent cold responses in heterologously expressed channels. The blockade of mTRPM8 by PBMC was also verified on cold-evoked whole-cell currents in these cells (data not shown).
Figure 9.

PBMC blocks TRPM8-mediated responses in TRPM8(+)-HEK293 cells and suppress generator potentials in CSNs from sham and injured mice. A, Ratiometric [Ca2+]i response in a TRPM8(+)-HEK 293 cell during three consecutive cooling ramps to 18°C, in control solution, in the presence of PBMC at 1 μm, and after 5 min washing this TRPM8 blocker. B, Percentage of inhibition of cold-evoked calcium signals in TRPM8(+)-HEK293 cells by different concentrations of PBMC. Gray curve indicates the dose–response fit to the Hill equation, with an IC50 of 92.7 nm and a Hill coefficient of 1.3 ± 0.2 (n > 20 cells for each concentration). C, D, Representative simultaneous recordings of [Ca2+]i (top) and bath temperature (bottom), during two consecutive cooling drops from 34°C to 20°C, in a CSN isolated from DRG of a sham (C, black open circles) and an injured mouse (D, red open circles). PBMC at 1 μm produced a large reduction in the cold-evoked response, with only minor effects on the response induced by a 30 mm elevation in extracellular K+. E, F, Normalized [Ca2+]i responses to cold and to elevated K+ in control extracellular solution (Cold and KCl), in extracellular solution supplemented with 1 μm PBMC (Cold + PBMC), and with 30 mm KCl (KCl + PBMC) for both sham (C, black filled and stripped bars) and injured mice (D, red filled and stripped bars) (n = 6 CSNs neurons for each condition. **p = 0.003 (paired t test). ***p < 0.001 (paired t test).
Unlike the [Ca2+]i elevations observed in mTRPM8-HEK293 cells in response to temperature drops, cold-evoked [Ca2+]i rises in CSNs largely depend on the action potential firing in response to the depolarizing receptor potential induced by TRPM8 activation (Viana et al., 2002; Madrid et al., 2006). To study the effect of PBMC on cold-evoked responses, and to rule out the possibility of an inhibitory effect of PBMC resulting from a nonspecific effect on voltage-gated channels responsible for the spike generation, we used a combination of cold-induced and K+-induced depolarization in control condition and in the presence of the blocker. Using this strategy, we tested the inhibitory effect and the specific nature of PBMC blockade on cold-induced activity in CSNs from both control and injured mice neurons using calcium imaging. In Figure 9C, we show the effect of saturating concentration of PBMC (1 μm) on the [Ca2+]i elevation evoked by two consecutive cold stimuli, followed by two applications of elevated extracellular K+ (30 mm). Whereas cold-evoked responses were almost fully suppressed by PBMC, the depolarization-evoked [Ca2+]i increases in response to high K+ were well preserved. Figure 9E (bar graph) summarizes the normalized results obtained in cold-sensitive neurons from sham animals. In parallel experiments using this protocol, we found similar results in cold-sensitive neurons from allodynic animals (Fig. 9D,F). These results suggest that the inhibitory effect of saturating concentrations of PBMC are selective to cold-induced depolarizing currents in both populations on CSNs, with probably minimal effects on the voltage-dependent currents underlying action potential firing. Our results suggest that TRPM8 channels are critical components of the cold-activated excitatory machinery in both normal and allodynic mice, and that the response of transformed neurons in CCI animals most probably relies on this thermo-TRP channel.
Mathematical modeling of CSNs shows that IKD is a critical element to the tuning of thermal threshold in primary somatosensory neurons
To further support the hypothesis that changes in the functional expression of IKD detected in the soma of DRG neurons can underlie the changes in their cold detection thresholds, we used computer simulations of a conductance-based model of cold thermoreceptors. In this model, sensitivity to temperature drops is due to a depolarizing cold- and voltage-dependent current (TRPM8-like), which generates large changes in the firing rate of the cold thermoreceptor (Olivares and Orio, 2015; Olivares et al., 2015). A calcium-dependent desensitization causes this response to be transient, and this property has been included in the model. Steady temperatures are coded as a change in the discharge patterns (Olivares and Orio, 2015; Olivares et al., 2015). We have introduced into this model a current resembling the IKD recorded in CSNs, and obtained several sets of parameters that reproduced a satisfactory dynamic and static response to temperature drops (Table 3). Then, we systematically varied the IKD and ITRPM8 conductance densities and determined the basal firing rates, temperature response thresholds, and maximum firing rates. Figure 10 shows sample responses to a cooling stimulus with different parameter combinations. Voltage-clamp simulation of the IKD current inserted into the model is shown in Figure 10A. A response resembling a canonical low-threshold CSN is obtained with a high gTRPM8 and a low gKD density (Fig. 10B, a). An intermediate density of TRPM8 conductance yields a high-threshold CS neuron (Fig. 10C, b1), which upon reduction of gKD drastically reduces its temperature threshold (Fig. 10C, b2). Finally, with a very low gTRPM8 density and high gKD density (Fig. 10D, c1), the model resembles a cold-insensitive (CI) neuron. However, upon reduction of gKD (Fig. 10D, c2), the model neuron becomes responsive to temperature reductions into the mild-cold range.
Figure 10.
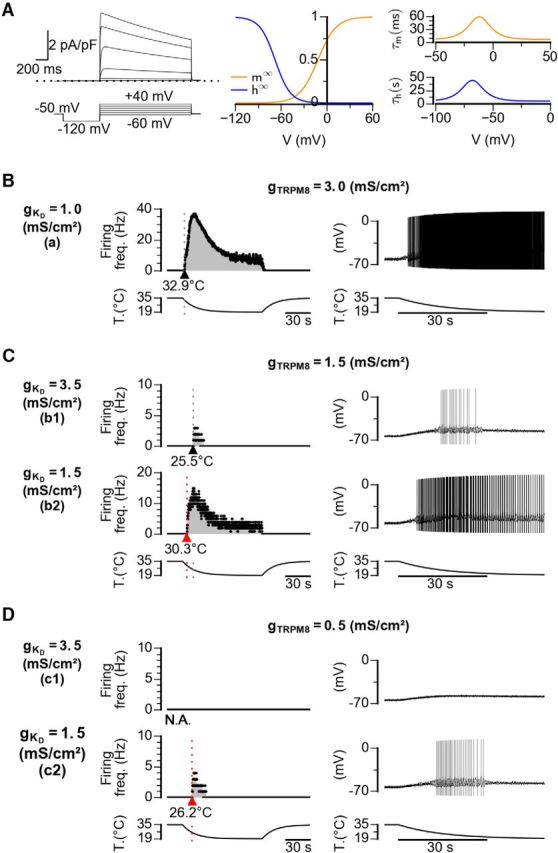
Mathematical model of CSNs with different densities of IKD and ITRPM8. A, Left, Voltage-clamp simulation of the IKD current inserted into the model. Voltage protocol is shown at the bottom. The maximum density (gKD) is 0.1 mS/cm2. Middle, Activation (m∞) and inactivation (h∞) curves. Right, Activation time constants at different voltages were obtained with the equation described in Materials and Methods. B–D, Firing rate (left) and voltage trace (right) of model CSNs with different densities of IKD current and ITRPM8 in response to the cooling stimulus depicted at the bottom. The Kv1.1–1.2 and TRPM8 maximum conductance configurations are indicated at the left (Kv1.1–1.2) and top (TRPM8) in mS/cm2. Note the shift of the thermal threshold to higher temperatures induced by a reduction of IKD density (C) and the response to cold induced in a cold-insensitive neuron when IKD density is reduced, even with a very low density of ITRPM8 (D).
To show a more complete picture, Figure 11A shows the detailed results of a systematic exploration of the parameter space for these two conductances, whereas Figure 11B shows a summary of their behavior with the different high/low density combination. Regardless of the parameter sets of the model (two samples of five explored are shown in Fig. 11A), we see that the gTRPM8/gKD ratio defines the temperature detection threshold and the maximum firing rate during cooling. However, gKD appears to have a greater effect on the threshold than gTRPM8; we thus predict that a reduction of Kv1.1–1.2 channels can produce more recruited CSNs than an increase of TRPM8 channels. Thus, thermal thresholds of the models are fully correlated to TRPM8-like and Kv1.1–1.2-like conductance densities, in agreement with functional studies in CSNs. LT-CSNs model, with the characteristic features of both the dynamic and static response of CSNs activated by innocuous cold, are correlated to high TRPM8 and low Kv1.1–1.2 conductance levels. Conversely, high detection thresholds and firing properties of HT-CSNs and recruitable neurons are associated with high Kv1.1–1.2 and low TRPM8 conductance densities.
Figure 11.

Systematic exploration of gKD and gTRPM8 parameters. A, Basal firing (Hz), thermal threshold (°C), and maximum firing rate (Hz) at different densities of gTRPM8 and gKD. Top and bottom rows represent parameter sets 1 and 2, respectively, from Figure 10 and Table 3. The points indicated as a, b1, b2, c1, and c2 are the parameter combinations used in Figure 10. Black represents complete absence of action potentials. B, Average thermal threshold and maximum firing rate of the model with the gKD and gTRPM8 combinations that were used in Figure 10. These data are the mean value of five combinations of the remaining free parameters of the model (Fig. 10; Table 3). p values were obtained using a t test for related samples. ***p < 0.001 in both cases.
A representation of our results regarding thermal sensitivity of primary sensory neurons before and after peripheral injury is shown in Figure 12.
Figure 12.

Hypothetical model of cold detection mechanisms and thermal sensitivity of primary sensory neurons in sham and CCI conditions. Left, Schematic representation of cold detection mechanisms in primary sensory neurons under physiological conditions. Boxes in nerve endings represent the ion channels that are involved in the detection of cold stimuli by the functional subtypes of peripheral sensory neurons. The temperature ranges in which these nerve endings would be excited by cold are shown by the colored thermometers at left of each box. These neurons may have excitatory (+) or inhibitory (−) (indirect) actions on higher-order neurons of central sensory pathways, leading ultimately to distinct cold and cold-induced pain sensations. The size of labels reflects the relative density of these channels in the different subclasses of neurons. Right, Schematic representation of these mechanisms in injured mice, emphasizing the functional variations in response to peripheral nerve damage. The relative change of the size of the labels reflects the expected changes in functional expression levels for the different ion channels after injury. After axonal damage, the reduction in the functional expression of IKD would recruit polymodal nociceptors normally activated by noxious cold temperatures inducing pain that will respond to mild cold temperatures in CCI animals (red arrows indicate this transition). HT-CSNs will respond to innocuous cold temperatures signaling cold discomfort, represented by the cyan neuron (cyan arrows indicate this transition). LT-CSNs expressing low levels of IKD would remain unaffected. Thus, after injury, the reduction in the functional expression of IKD increases the cold sensitivity of HT-CSNs signaling cold discomfort and recruits nociceptors normally activated by extremely cold temperatures that cause pain.
Discussion
Several ion channels have been proposed as important elements in the development of painful hypersensitivity to cold in different models of axonal damage, including cold-activated thermo-TRP channels TRPM8 and TRPA1, voltage-gated and background K+ channels, voltage-gated Na+ channels, and HCN channels, among others (for review, see Abrahamsen et al., 2008; Patapoutian et al., 2009; Takeda et al., 2011; Emery et al., 2012; Du and Gamper, 2013; Tsantoulas and McMahon, 2014; Yin et al., 2015). Regarding K+ channels, its functional downregulation could not only increase the membrane excitability but also maintain this exacerbated state for long periods of time. In the case of the Kv1 channels underlying IKD, it has been also proposed that Kv1.1 also acts as a critical brake in both mechanical and pain sensitivity (Hao et al., 2013), and that Kv1.1 knock-out mice display cold-induced discharges in myelinated motor fibers (Zhou et al., 1998). Peripheral nerve damage induces the downregulation of Kv1.2 through the nerve injury-induced promotion of Kv1.2 antisense RNA expression (Zhao et al., 2013; Fan et al., 2014; Li et al., 2015), and the overexpression of Kv1.2 can revert the nociceptive phenotype (Fan et al., 2014). It has also been shown that the expression of Kv1.1 and Kv1.2 is markedly decreased in neuromas of myelinated axons following injury (Calvo et al., 2016) and that 4-AP induces an enhancement of thermal sensitivity in mechanosensitive neuromas (Roza et al., 2006). Because heteromultimeric Kv1.1–1.2 channels are the most probable molecular counterparts of the brake current IKD in CSNs (Madrid et al., 2009), our rationale included the idea that injury-induced downregulation of these Kv1 channels could produce a similar effect on painful cold sensitivity than selective inhibition of IKD. Accordingly, we found that CCI of the sciatic nerve induces an important reduction of the IKD current density in both HT-CSNs and in nociceptive neurons that became sensitive to innocuous and mild cold after nerve injury, respectively.
In our model, both HT-CSNs signaling cold discomfort and a subgroup of nociceptors are transformed into LT-CSNs and HT-CSNs, respectively, after nerve damage, providing a molecular and neural mechanism to explain the damage-gated painful hypersensitivity to cold. Because PBMC blocks cold-induced responses in DRG neurons from both damage and sham mice, and that systemic application of this blocker reduces cold allodynia (Knowlton et al., 2011), it is reasonable to think that in this model of nerve injury, TRPM8(+) cold thermoreceptors and TRPM8(+) nociceptors shift their thermal sensitivity to higher temperatures due to the reduction of this brake current, contributing to the painful phenotype. HT-CSNs correspond to neurons signaling unpleasant cold discomfort (Mauderli et al., 2003). Nociceptive fibers that respond to mild cold in injured animals would correspond to polymodal nociceptors eliciting burning cold pain (Simone and Kajander, 1997). A relatively high expression of TTx-resistant Na+ channels, most likely Nav1.8, allows HT-CSNs and polymodal nociceptors to sustain cold-induced responses at lower temperatures than canonical cold thermoreceptors (Zimmermann et al., 2007). Because the expression of Nav1.8 channels decreases in several forms of neuropathic pain (Waxman et al., 1999), a compensatory reduction of a brake current, such as IKD, could also contribute to expedite cold-induced firing in these neurons.
Developing cold hypersensitivity induced by CCI is strongly reduced in TRPM8−/− animals (Colburn et al., 2007), and an increase in the expression of TRPM8 has been linked to the development and maintenance of cold hypersensitivity in response to axonal damage (Frederick et al., 2007; Xing et al., 2007; Knowlton et al., 2011, 2013; Su et al., 2011). Using CCI in rats, Xing et al. (2007) found that peripheral damage resulted in a significant increase in the percentage of menthol- and cold-sensitive-neurons, and an enhancement in the responsiveness of these neurons to both menthol and cold. On the other hand, Caspani et al. (2009) found that CCI of the sciatic nerve in mice result in cold allodynia with a reduction of the TRPM8 and TRPA1 transcripts. We did not find changes in the functional expression level of TRPM8 in response to this form of chronic damage. Nevertheless, we found that cold responses in CSNs from both control and injured animals are strongly affected by the blockage of TRPM8 using PBMC. These results are consistent with the observation that cold allodynia is largely reduced by systemic blockage of TRPM8 using PBMC, and by conditional ablation of TRPM8(+) neurons in mice (Knowlton et al., 2011, 2013). Moreover, it has been suggested that the development of cold pain induced by inflammation, nerve injury, and chemotherapy is mainly mediated by a subpopulation of TRPM8(+) neurons expressing the neurotrophic factor receptor GFRα3 (Lippoldt et al., 2013, 2016). These results suggest that this sensory alteration requires the activation of TRPM8-expressing sensory neurons connected to nociceptive pathways, observations that are in line with our results.
After spinal nerve ligation, the expression of TRPM8 and TRPA1 is reduced in injured ganglia (Katsura et al., 2006), and pharmacological inhibition of TRPA1 reverses cold-induced hyperalgesia caused by inflammation and peripheral nerve damage (Obata et al., 2005). On the other hand, Frederick et al. (2007) found slight increases in TRPA1, TRPV2, and TRPM8 mRNAs following unilateral sciatic nerve chronic constriction. Interestingly, in humans, hypersensitivity to cold after cold-induced peripheral damage does not seem to be related to an altered presence of TRPM8 or TRPA1 channels (Namer et al., 2008). In the same line, we did not see significant changes in the responses to the TRPM8-activator menthol to the TRPA1-activator AITC in CSNs from injured mice compared with sham animals, suggesting that changes in the functional expression of these channels are not necessarily involved in cold allodynia induced by CCI of the sciatic nerve.
TREK-1, TREK-2, and TRAAK channels have also been linked to neuropathic cold-induced pain (Noël et al., 2009; Descoeur et al., 2011). Recently, Morenilla-Palao et al. (2014) have shown that the background K+ channel TASK-3 is highly expressed in TRPM8(+) neurons. Notably, the pharmacological blockage of this channel reduces thermal threshold of TRPM8(+) CSNs, and mice lacking TASK-3 display hypersensitivity to cold (Morenilla-Palao et al., 2014). Whether this channel contributes to cold allodynia following injury is still unknown. On the other hand, McNaughton and his group (Emery et al., 2011) have demonstrated that selective deletion of the HCN2 channel from nociceptive neurons abolishes diverse forms of neuropathic pain, reinforcing the idea that nociceptive afferents are also critically modulated by the activity of HCN channels. Among Nav channels, Nav 1.7 and Nav 1.8 are important for cold allodynia in CCI models of neuropathic pain (Minett et al., 2014), and Nav1.6 plays a role in ciguatoxin- and oxaliplatin-induced cold hypersensitivity (Sittl et al., 2012; Zimmermann et al., 2013). These studies, to name a few, show the real intricacy of the phenomenon behind sensing cold as pain in response to nerve damage (Lolignier et al., 2016).
We have delimited our approach to a well-known form of neural injury by chronic constriction of the sciatic nerve in mice, and we have studied the variations of cold sensitivity, IKD and Icold density 7 d after surgery. We do not discard any other important variations in molecular entities other than those we explored or under these conditions. Nevertheless, it is clear that 4-AP and α-DTx sensitivity is reduced in CSNs from injured animals, an observation in line with the idea that their molecular targets are functionally suppressed in a large proportion of CSNs from allodynic mice. Interestingly, intraplantar injection of 4-AP (and α-DTx) (Madrid et al., 2009) induces cold allodynia, and painful hypersensitivity to cold induced by CCI remains poorly altered after intraplantar pharmacological suppression of IKD. Consistent with the reduction of IKD density in response to axonal damage, the subpopulation of nociceptive-like recruitable neurons is reduced in injured mice, a population that we suggest will become part of the CSN population in CCI animals. Interestingly, capsaicin sensitivity is increased in CSNs from injured animals, an observation in line with the idea of nociceptive-like neurons being part of the cold-responding neurons population after injury. TRPV1-dependent responses in control conditions are consistent with results of several laboratories showing activation of near to 50% of CSNs by capsaicin (Reid et al., 2002; Viana et al., 2002; Dhaka et al., 2008), or even higher (Parra et al., 2010; Yudin et al., 2016).
Finally, here we have also provided the first comprehensive mathematical model of CSNs, including the brake current IKD. Making use of this, we also show that neuropathic painful hypersensitivity to cold can be largely explained as a consequence of a reduction in the functional expression of a brake potassium current with the biophysical properties of IKD. Thus, any decrease in the IKD/ITRPM8 ratio is enough to transform a cold nociceptor-like model neuron into a cold thermoreceptor-like one, and a high-threshold thermoreceptor into a low-threshold one. Moreover, this model revealed that very low densities of TRPM8 current are enough to generate cold-induced responses in neurons expressing low levels of IKD. Interestingly, thermal threshold is more influenced by IKD current density than by the density of TRPM8 current, in strong coincidence with our experimental findings.
Footnotes
This work was supported by FONDECYT Grants 1161733 and 1131064 to R.M., 1130862 to P.O., 11130144 to M.P., and 3150431 to A.G., CONICYT Anillo Grant ACT-1113 to R.M., P.O., M.P., and G.U., and the Advanced Center for Electrical and Electronic Engineering CONICYT FB0008 to P.O. R.M. thanks Pfizer Inc. (WI177114) and VRIDEI-USACH. G.H. holds a CONICYT PhD fellowship. The Centro Interdisciplinario de Neurociencia de Valparaíso is a Millennium Science Institute funded by the Ministry of Economy, Chile. The funders had no role in study conception, design, data collection, and interpretation or in the decision to submit the manuscript for publication. We thank Drs. F. Viana and C. Belmonte for comments on the manuscript; and J. Salas, M. Campos, and R. Pino for excellent technical assistance.
The authors declare no competing financial interests.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321:702–705. 10.1126/science.1156916 [DOI] [PubMed] [Google Scholar]
- Almaraz L, Manenschijn JA, de la Peña E, Viana F (2014) Trpm8. Handb Exp Pharmacol 222:547–579. 10.1007/978-3-642-54215-2_22 [DOI] [PubMed] [Google Scholar]
- Babes A, Ciobanu AC, Neacsu C, Babes RM (2011) TRPM8, a sensor for mild cooling in mammalian sensory nerve endings. Curr Pharm Biotechnol 12:78–88. 10.2174/138920111793937835 [DOI] [PubMed] [Google Scholar]
- Belmonte C, Brock JA, Viana F (2009) Converting cold into pain. Exp Brain Res 196:13–30. 10.1007/s00221-009-1797-2 [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–107. 10.1016/0304-3959(88)90209-6 [DOI] [PubMed] [Google Scholar]
- Calvo M, Richards N, Schmid AB, Barroso A, Zhu L, Ivulic D, Zhu N, Anwandter P, Bhat MA, Court FA, McMahon SB, Bennett DL (2016) Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. Elife 5:e12661. 10.7554/eLife.12661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL (2010) Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem 114:1460–1475. 10.1111/j.1471-4159.2010.06863.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspani O, Zurborg S, Labuz D, Heppenstall PA (2009) The contribution of TRPM8 and TRPA1 channels to cold allodynia and neuropathic pain. PLoS One 4:e7383. 10.1371/journal.pone.0007383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn RW, Lubin ML, Stone DJ Jr, Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54:379–386. 10.1016/j.neuron.2007.04.017 [DOI] [PubMed] [Google Scholar]
- Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, Couette B, Busserolles J, Courteix C, Noël J, Lazdunski M, Eschalier A, Authier N, Bourinet E (2011) Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med 3:266–278. 10.1002/emmm.201100134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaka A, Earley TJ, Watson J, Patapoutian A (2008) Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J Neurosci 28:566–575. 10.1523/JNEUROSCI.3976-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan KZ, Xu Q, Zhang XM, Zhao ZQ, Mei YA, Zhang YQ (2012) Targeting A-type K(+) channels in primary sensory neurons for bone cancer pain in a rat model. Pain 153:562–574. 10.1016/j.pain.2011.11.020 [DOI] [PubMed] [Google Scholar]
- Du X, Gamper N (2013) Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 11:621–640. 10.2174/1570159X113119990042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA (2011) HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333:1462–1466. 10.1126/science.1206243 [DOI] [PubMed] [Google Scholar]
- Emery EC, Young GT, McNaughton PA (2012) HCN2 ion channels: an emerging role as the pacemakers of pain. Trends Pharmacol Sci 33:456–463. 10.1016/j.tips.2012.04.004 [DOI] [PubMed] [Google Scholar]
- Fan L, Guan X, Wang W, Zhao JY, Zhang H, Tiwari V, Hoffman PN, Li M, Tao YX (2014) Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Mol Pain 10:8. 10.1186/1744-8069-10-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, McMullan S, Lawson SN, Djouhri L (2005) Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. J Physiol 565:927–943. 10.1113/jphysiol.2005.086199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick J, Buck ME, Matson DJ, Cortright DN (2007) Increased TRPA1, TRPM8, and TRPV2 expression in dorsal root ganglia by nerve injury. Biochem Biophys Res Commun 358:1058–1064. 10.1016/j.bbrc.2007.05.029 [DOI] [PubMed] [Google Scholar]
- Fujita F, Uchida K, Takaishi M, Sokabe T, Tominaga M (2013) Ambient temperature affects the temperature threshold for TRPM8 activation through interaction of phosphatidylinositol 4,5-bisphosphate. J Neurosci 33:6154–6159. 10.1523/JNEUROSCI.5672-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner JC, Kirkup AJ, Curry J, Humphreys S, O'Regan P, Postlethwaite M, Young KC, Kitching L, Ethell BT, Winpenny D, McMurray G (2014) The role of TRPM8 in the guinea-pig bladder-cooling reflex investigated using a novel TRPM8 antagonist. Eur J Pharmacol 740:398–409. 10.1016/j.ejphar.2014.07.022 [DOI] [PubMed] [Google Scholar]
- González A, Ugarte G, Piña R, Pertusa M, Madrid R (2015) TRP channels in cold transduction. In: TRP channels in sensory transduction (Madrid R, Bacigalupo J eds), pp 187–209. Cham, Switzerland: Springer. [Google Scholar]
- Hao J, Padilla F, Dandonneau M, Lavebratt C, Lesage F, Noël J, Delmas P (2013) Kv1.1 channels act as mechanical brake in the senses of touch and pain. Neuron 77:899–914. 10.1016/j.neuron.2012.12.035 [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT (1997) The NEURON simulation environment. Neural Comput 9:1179–1209. 10.1162/neco.1997.9.6.1179 [DOI] [PubMed] [Google Scholar]
- Hines ML, Davison AP, Muller E (2009) NEURON and Python. Front Neuroinform 3:1. 10.3389/neuro.11.001.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Sakagami M, Noguchi K (2006) Antisense knock down of TRPA1, but not TRPM8, alleviates cold hyperalgesia after spinal nerve ligation in rats. Exp Neurol 200:112–123. 10.1016/j.expneurol.2006.01.031 [DOI] [PubMed] [Google Scholar]
- Kim DS, Choi JO, Rim HD, Cho HJ (2002) Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res 105:146–152. 10.1016/S0169-328X(02)00388-1 [DOI] [PubMed] [Google Scholar]
- Knowlton WM, Bifolck-Fisher A, Bautista DM, McKemy DD (2010) TRPM8, but not TRPA1, is required for neural and behavioral responses to acute noxious cold temperatures and cold-mimetics in vivo. Pain 150:340–350. 10.1016/j.pain.2010.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD (2011) Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One 6:e25894. 10.1371/journal.pone.0025894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Palkar R, Lippoldt EK, McCoy DD, Baluch F, Chen J, McKemy DD (2013) A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J Neurosci 33:2837–2848. 10.1523/JNEUROSCI.1943-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R, Brauchi S, Madrid R, Orio P (2011) A cool channel in cold transduction. Physiology (Bethesda) 26:273–285. 10.1152/physiol.00004.2011 [DOI] [PubMed] [Google Scholar]
- Li Z, Gu X, Sun L, Wu S, Liang L, Cao J, Lutz BM, Bekker A, Zhang W, Tao YX (2015) Dorsal root ganglion myeloid zinc finger protein 1 contributes to neuropathic pain after peripheral nerve trauma. Pain 156:711–721. 10.1097/j.pain.0000000000000103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippoldt EK, Elmes RR, McCoy DD, Knowlton WM, McKemy DD (2013) Artemin, a glial cell line-derived neurotrophic factor family member, induces TRPM8-dependent cold pain. J Neurosci 33:12543–12552. 10.1523/JNEUROSCI.5765-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippoldt EK, Ongun S, Kusaka GK, McKemy DD (2016) Inflammatory and neuropathic cold allodynia are selectively mediated by the neurotrophic factor receptor GFRalpha3. Proc Natl Acad Sci U S A 113:4506–4511. 10.1073/pnas.1603294113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolignier S, Gkika D, Andersson D, Leipold E, Vetter I, Viana F, Noël J, Busserolles J (2016) New insight in cold pain: role of ion channels, modulation, and clinical perspectives. J Neurosci 36:11435–11439. 10.1523/JNEUROSCI.2327-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid R, Pertusa M (2014) Intimacies and physiological role of the polymodal cold-sensitive ion channel TRPM8. Curr Top Membr 74:293–324. 10.1016/B978-0-12-800181-3.00011-7 [DOI] [PubMed] [Google Scholar]
- Madrid R, Donovan-Rodríguez T, Meseguer V, Acosta MC, Belmonte C, Viana F (2006) Contribution of TRPM8 channels to cold transduction in primary sensory neurons and peripheral nerve terminals. J Neurosci 26:12512–12525. 10.1523/JNEUROSCI.3752-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid R, de la Peña E, Donovan-Rodriguez T, Belmonte C, Viana F (2009) Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J Neurosci 29:3120–3131. 10.1523/JNEUROSCI.4778-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkia A, Madrid R, Meseguer V, de la Peña E, Valero M, Belmonte C, Viana F (2007) Bidirectional shifts of TRPM8 channel gating by temperature and chemical agents modulate the cold sensitivity of mammalian thermoreceptors. J Physiol 581:155–174. 10.1113/jphysiol.2006.123059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauderli AP, Vierck CJ Jr, Cannon RL, Rodrigues A, Shen C (2003) Relationships between skin temperature and temporal summation of heat and cold pain. J Neurophysiol 90:100–109. 10.1152/jn.01066.2002 [DOI] [PubMed] [Google Scholar]
- McCoy DD, Knowlton WM, McKemy DD (2011) Scraping through the ice: uncovering the role of TRPM8 in cold transduction. Am J Physiol Regul Integr Comp Physiol 300:R1278–R1287. 10.1152/ajpregu.00631.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD. (2013) The molecular and cellular basis of cold sensation. ACS Chem Neurosci 4:238–247. 10.1021/cn300193h [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416:52–58. 10.1038/nature719 [DOI] [PubMed] [Google Scholar]
- Minett MS, Falk S, Santana-Varela S, Bogdanov YD, Nassar MA, Heegaard AM, Wood JN (2014) Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep 6:301–312. 10.1016/j.celrep.2013.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morenilla-Palao C, Luis E, Fernández-Peña C, Quintero E, Weaver JL, Bayliss DA, Viana F (2014) Ion channel profile of TRPM8 cold receptors reveals a role of TASK-3 potassium channels in thermosensation. Cell Rep 8:1571–1582. 10.1016/j.celrep.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namer B, Kleggetveit IP, Handwerker H, Schmelz M, Jorum E (2008) Role of TRPM8 and TRPA1 for cold allodynia in patients with cold injury. Pain 139:63–72. 10.1016/j.pain.2008.03.007 [DOI] [PubMed] [Google Scholar]
- Noël J, Zimmermann K, Busserolles J, Deval E, Alloui A, Diochot S, Guy N, Borsotto M, Reeh P, Eschalier A, Lazdunski M (2009) The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J 28:1308–1318. 10.1038/emboj.2009.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K (2005) TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest 115:2393–2401. 10.1172/JCI25437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares E, Orio P (2015) Mathematical modeling of TRPM8 and the cold thermoreceptors. In: TRP channels in sensory transduction (Madrid R, Bacigalupo J eds), pp 210–223. Cham, Switzerland: Springer. [Google Scholar]
- Olivares E, Salgado S, Maidana JP, Herrera G, Campos M, Madrid R, Orio P (2015) TRPM8-dependent dynamic response in a mathematical model of cold thermoreceptor. PLoS One 10:e0139314. 10.1371/journal.pone.0139314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Madrid R, de la Peña E, Parra A, Meseguer V, Bayliss DA, Belmonte C, Viana F (2009) Characteristics and physiological role of hyperpolarization activated currents in mouse cold thermoreceptors. J Physiol 587:1961–1976. 10.1113/jphysiol.2008.165738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Parra A, Madrid R, González O, Belmonte C, Viana F (2012) Role of Ih in the firing pattern of mammalian cold thermoreceptor endings. J Neurophysiol 108:3009–3023. 10.1152/jn.01033.2011 [DOI] [PubMed] [Google Scholar]
- Parra A, Madrid R, Echevarria D, del Olmo S, Morenilla-Palao C, Acosta MC, Gallar J, Dhaka A, Viana F, Belmonte C (2010) Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat Med 16:1396–1399. 10.1038/nm.2264 [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ (2009) Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov 8:55–68. 10.1038/nrd2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002) A TRP channel that senses cold stimuli and menthol. Cell 108:705–715. 10.1016/S0092-8674(02)00652-9 [DOI] [PubMed] [Google Scholar]
- Pogorzala LA, Mishra SK, Hoon MA (2013) The cellular code for mammalian thermosensation. J Neurosci 33:5533–5541. 10.1523/JNEUROSCI.5788-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS (2001) Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A 98:13373–13378. 10.1073/pnas.231376298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Babes A, Pluteanu F (2002) A cold- and menthol-activated current in rat dorsal root ganglion neurones: properties and role in cold transduction. J Physiol 545:595–614. 10.1113/jphysiol.2002.024331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roza C, Belmonte C, Viana F (2006) Cold sensitivity in axotomized fibers of experimental neuromas in mice. Pain 120:24–35. 10.1016/j.pain.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Simone DA, Kajander KC (1997) Responses of cutaneous A-fiber nociceptors to noxious cold. J Neurophysiol 77:2049–2060. [DOI] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P, Carr RW (2012) Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc Natl Acad Sci U S A 109:6704–6709. 10.1073/pnas.1118058109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf S, Oerther S, Lucas G, Mattsson JP, Ernfors P (2009) Differential regulation of TRP channels in a rat model of neuropathic pain. Pain 144:187–199. 10.1016/j.pain.2009.04.013 [DOI] [PubMed] [Google Scholar]
- Storm JF. (1988) Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature 336:379–381. 10.1038/336379a0 [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829. 10.1016/S0092-8674(03)00158-2 [DOI] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL (2011) Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci 12:120. 10.1186/1471-2202-12-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S (2011) Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain 7:5. 10.1186/1744-8069-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thut PD, Wrigley D, Gold MS (2003) Cold transduction in rat trigeminal ganglia neurons in vitro. Neuroscience 119:1071–1083. 10.1016/S0306-4522(03)00225-2 [DOI] [PubMed] [Google Scholar]
- Tsantoulas C, McMahon SB (2014) Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci 37:146–158. 10.1016/j.tins.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana F, de la Peña E, Belmonte C (2002) Specificity of cold thermotransduction is determined by differential ionic channel expression. Nat Neurosci 5:254–260. 10.1038/nn809 [DOI] [PubMed] [Google Scholar]
- Vriens J, Nilius B, Voets T (2014) Peripheral thermosensation in mammals. Nat Rev Neurosci 15:573–589. 10.1038/nrn3784 [DOI] [PubMed] [Google Scholar]
- Wang XC, Wang S, Zhang M, Gao F, Yin C, Li H, Zhang Y, Hu SJ, Duan JH (2016) α-Dendrotoxin sensitive Kv1 channels contribute to conduction failure of polymodal nociceptive C-fibers from rat coccygeal nerve. J Neurophysiol 115:947–957. 10.1152/jn.00786.2014 [DOI] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA (1999) Sodium channels and pain. Proc Natl Acad Sci U S A 96:7635–7639. 10.1073/pnas.96.14.7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Chen M, Ling J, Tan W, Gu JG (2007) TRPM8 mechanism of cold allodynia after chronic nerve injury. J Neurosci 27:13680–13690. 10.1523/JNEUROSCI.2203-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang EK, Takimoto K, Hayashi Y, de Groat WC, Yoshimura N (2004) Altered expression of potassium channel subunit mRNA and α-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience 123:867–874. 10.1016/j.neuroscience.2003.11.014 [DOI] [PubMed] [Google Scholar]
- Yin K, Zimmermann K, Vetter I, Lewis RJ (2015) Therapeutic opportunities for targeting cold pain pathways. Biochem Pharmacol 93:125–140. 10.1016/j.bcp.2014.09.024 [DOI] [PubMed] [Google Scholar]
- Yudin Y, Lutz B, Tao YX, Rohacs T (2016) Phospholipase C δ4 regulates cold sensitivity in mice. J Physiol 594:3609–3628. 10.1113/JP272321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, Gao YJ, Hoffman PN, Cui H, Li M, Dong X, Tao YX (2013) A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci 16:1024–1031. 10.1038/nn.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhang CL, Messing A, Chiu SY (1998) Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J Neurosci 18:7200–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW (2007) Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 447:855–858. 10.1038/nature05880 [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Deuis JR, Inserra MC, Collins LS, Namer B, Cabot PJ, Reeh PW, Lewis RJ, Vetter I (2013) Analgesic treatment of ciguatoxin-induced cold allodynia. Pain 154:1999–2006. 10.1016/j.pain.2013.06.015 [DOI] [PubMed] [Google Scholar]