

Abstract
Motivation
Skeletal diseases are prevalent in society, but improved molecular understanding is required to formulate new therapeutic strategies. Large and increasing quantities of available skeletal transcriptomics experiments give the potential for mechanistic insight of both fundamental skeletal biology and skeletal disease. However, no current repository provides access to processed, readily interpretable analysis of this data. To address this, we have developed SkeletalVis, an exploration portal for skeletal gene expression experiments.
Results
The SkeletalVis data portal provides an exploration and comparison platform for analysed skeletal transcriptomics data. It currently hosts 287 analysed experiments with 739 perturbation responses with comprehensive downstream analysis. We demonstrate its utility in identifying both known and novel relationships between skeletal expression signatures. SkeletalVis provides users with a platform to explore the wealth of available expression data, develop consensus signatures and the ability to compare gene signatures from new experiments to the analysed data to facilitate meta-analysis.
Availability and implementation
The SkeletalVis data portal is freely accessible at http://phenome.manchester.ac.uk.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Skeletal disease affects millions of the adult population, causing a huge burden on healthcare systems (Cross et al., 2014). This includes common polygenic forms of joint disease such as osteoarthritis (OA) and rheumatoid arthritis (RA) and the rare monogenic skeletal conditions. Despite years of research there are no disease modifying drugs for osteoarthritis and other skeletal diseases (Karsdal et al., 2016). There is a critical need to understand the underlying molecular mechanisms to find potential therapeutic targets. Transcriptomics analysis of diseased cells or tissues gives us insight into altered expression of genes which are potentially causing an individual disease. There is a large and growing amount of publicly available expression data from microarray and more recently RNA-Seq for skeletal disease (Steinberg and Zeggini, 2016). These datasets are analysed to produce lists of differentially expressed genes and derive broader functional information such as enriched pathways. For instance, we have previously used transcriptomics data to understand the altered processes in osteoarthritis cartilage damage and other researchers have characterized mouse models of rare skeletal diseases using transcriptomics (Cameron et al., 2011; Dunn et al., 2016; Soul et al., 2018). Despite the use of global transcriptomics, papers describing these experiments only focus on a fraction of the information within these datasets. Extended use of this existing data would allow exploration and mining for new overlooked features. With the increased coverage of annotations for pathways/transcription factors and improved methods of analysis, re-analysis of these datasets may identify new features (Wadi et al., 2016). Furthermore, consistent analysis and integration of the data would allow identification of similarities between new and existing datasets, allowing for sharing of knowledge between diseases and experimental models, identification of shared pathogenic mechanisms and giving the potential for re-purposing of therapeutics and identification of new experimental models.
The differentially expressed genes and downstream analysis (e.g. enriched pathways) generated from an experiment generally only exist as Supplementary Tables in the original publication creating a barrier to the reuse and integration of these datasets. Furthermore, the inconsistent methods used in analysis of the published transcriptomics data render robust direct comparison of datasets challenging. An increasing number of researchers are depositing their raw transcriptomic data in public transcriptomics repositories such as ArrayExpress and GEO that provide the transcriptomics data and meta-data needed to re-analyse the data (Edgar, 2002; Kolesnikov et al., 2015).The EBI Expression Atlas has begun to analyse existing transcriptomics data and offers exploration of selected public datasets with differential expression analysis and basic pathway analysis (Papatheodorou et al., 2018). However, as this database is unfocused on a particular area of biology it has poor coverage of the available skeletal disease data and offers no way to compare expression responses between experiments or generate consensus signatures for further analysis. The CREEDS portal allows comparison of query gene expression signatures against a large database of GEO-derived signatures, but does not allow assessment of the quality and exploration of the underlying datasets (Wang et al., 2016). Furthermore, this search engine relies on automatic identification of the perturbation and the control samples, which although very scalable, is less accurate than human curation. As with Expression Atlas there is poor coverage of skeletal disease-related datasets.
With the growing repository of skeletal disease transcriptomic data available there is now the opportunity to systematically analyse and integrate this data. Specialized repositories exist for diseases such as cancer, but none currently exist for skeletal disease to make use of this data (Bowman et al., 2017). We have therefore developed a web-application to allow exploration and comparisons of publicly available skeletal disease transcriptomics data in order to analyse the pathology and predict the active mechanisms driving skeletal disease. The SkeletalVis data-portal avoids the requirement for bench scientists to download raw data and re-analyse every dataset needed in a comparison. We highlight its utility in exploring this data, identifying the similarities between skeletal disease models and skeletal genetic perturbations and elucidation of potential therapeutic targets for groups of similar expression responses. The SkeletalVis data portal is freely available at phenome.manchester.ac.uk.
2 Materials and methods
2.1 Identification and annotation of skeletal transcriptomic datasets
The ArrayExpress and GEO databases and the linked European nucleotide archive (ENA) and sequence read archive (SRA) data repositories were searched for keywords relating to skeletal cell types and skeletal disease (Supplementary Table S1) (Leinonen et al., 2011a,b). These results were filtered to keep only those experiments using whole genome mRNA transcriptomics with raw data available for the commonly studied species of Cow, Human, Mouse, Pig and Rat. Experiments were annotated by the experimental platform, the tissue under study, the type of experimental perturbation and with a concise description of the experiment. Comparisons (contrasts) to perform within each experiment were identified manually through the provided meta-data and corresponding publication (if available) for each experiment.
2.2 Transcriptomics analysis pipeline
A Galaxy pipeline was used to analyse the identified experiments in a high-throughput manner. Where available existing tools were used from Galaxy tool-shed, otherwise RGalaxy (v1.22.0) was used to create bespoke tools (Afgan et al., 2016). These modules were linked together to create a flexible pipeline for the analysis of microarray or RNA-seq data. For RNA-seq the raw data and meta-data were downloaded from ENA/SRA. Pseudo-alignment and qualification of reads was performed with Kallisto (v0.43.0) using the Ensembl transcriptome reference (release 79) for the appropriate species (Bray et al., 2016). MultiQC (v1.4) was used to generate summary reports of the FastQC (v0.11.5) read statistics and Kallisto mapping logs for quality control (Ewels et al., 2016). Tximport (v1.6.0) was used to summarize the mapped transcript level counts to gene level (Soneson et al., 2016). For microarray experiments, the unnormalized data and meta-data were downloaded from GEO or ArrayExpress. Normalization was performed using the robust mean average for Affymetrix arrays and quantile normalization for Illumina and Agilent arrays. Probesets were collapsed to the median value to provide a representative level of expression. Poor quality samples that are either mentioned in the corresponding publication or based on quality control data were removed to ensure robust gene expression signatures.
Batch effect has previous been established as a confounding factor in differential expression analysis. For both RNA-seq and microarray data, unless experimental batches were explicitly stated in the experimental meta-data they were inferred using sva (v3.26.0) with automatic identification of the number of surrogate variables (Leek et al., 2012). For visualization of the experimental samples by principal component analysis (PCA) the surrogate variables were regressed from the expression matrix before PCA. Limma (v3.34.9) and DESeq2 (v1.18.1) were used to calculate fold-changes and P-values between the comparisons in each experiment for microarray and RNA-Seq experiments, respectively (Love et al., 2014; Ritchie et al., 2015). Where insufficient replicates (<3) were available in an experiment only the fold-changes were calculated. The surrogate variables were incorporated as co-variants in the limma or DESeq2 statistical models to correct for the identified batch effects. Independent filtering based on mean gene-wise microarray intensity or RNA-seq read count was used to minimize false-positive differentially expressed genes, as implemented in the genefilter package (the default procedure in DESeq2), with automatic selection of the expression filtering threshold based on the number of differentially expressed genes (Bourgon et al., 2010). Benjamini-Hochberg correction was performed on the resulting P-values to account for multiple testing. As an alternative method for identifying differentially expressed genes, the characteristic direction method as implemented in GeoDE (v1.0) was used with the batch effect corrected count/intensity matrix using a threshold of the top 500 influential genes as previously used (Clark et al., 2014). As a quality control check, where published, the transcriptomics results were checked to ensure they were broadly similar to the re-analysed data with differentially expressed genes mentioned in the corresponding publication dysregulated in our analysis.
Differentially expressed genes from RNA-seq and microarray experiments were further analysed in a common downstream pipeline. For enrichment-based methods, differentially expressed genes were defined with combinations of absolute fold-change (none, 1.5 or 2) and an adjusted P-value (none, 0.05) thresholds. Pathway (PathwayCommons) and gene ontology biological process (GOBP) enrichment was performed using goseq (v1.30.0) (Ashburner et al., 2000; Cerami et al., 2011; Young et al., 2010. Significant pathways and GOBP terms were defined with an adjusted P-value ≤ 0.05 threshold. Redundancy of the pathways was reduced with the set cover algorithm (Stoney et al., 2018). Redundancy reduction of the GOBP terms was performed using the Revigo algorithm with the Resnick semantic similarity threshold set to 0.4 (Supek et al., 2011). Significantly enriched transcription factors based on motif occurrence in the differentially expressed genes were identified by RcisTarget (v0.99.0) for mouse and human experiments (Aibar et al., 2017).
For network analysis the human STRINGDB (v10.5) and the BioGrid (v3.4.162) protein-protein interaction networks were used with Ensembl ortholog mapping for other species. The STRINGDB network was filtered using an edge confidence threshold of > 400 to remove low quality interactions and text-mining derived edges were removed. The largest connected components were retained for both networks (von Mering et al., 2003). Active sub-networks (de novo pathways) were identified using the ranked list of differential expression data and the GIGA algorithm with a maximum sub-network size of 10 (Breitling et al., 2004). GO enrichment of the genes in the sub-network was used to identify the function of the sub-network.
To identify potential drugs that could reverse or mimic the observed differential expression the LINCS L1000 perturbation database, accessed with the L1000CDS2 API, was used to find overlap between the gene expression signatures (Duan et al., 2016). To annotate molecular targets of the enriched drugs the PubChem BioAssay Database was queried to find proteins which each drug has activity against (Wang et al., 2012).
Code for the pipeline and post-processing of the data can be found at www.github.com/soulj/SkeletalVis-Pipeline
2.3 Expression similarity
To allow comparison of the gene expression across species, all genes symbols were mapped to human gene symbols using Ensembl orthologs. Genes not measured in an experiment were regarded as NA. Four measures of gene expression similarity were calculated to allow comparison which considers the direction of the fold-change.
The signed Jaccard index for two signatures and is defined as:
where and refer to the up- and down-regulated genes respectively.
This measure was calculated with the gene expression signatures defined with (i) a 1.5-fold change threshold, (ii) a 1.5-fold change threshold and an adjusted P-value ≤ 0.05 threshold and (iii) the characteristic direction genes.
The cosine similarity measure (the cosine of the angle between two expression vectors) was also calculated using the fold-changes of each perturbation response as an alternative to the set overlap-based measures.
2.4 t-Distributed stochastic neighbour embedding visualization of signature similarity
t-Distributed stochastic neighbour embedding (t-SNE) implemented in Rtsne (v0.13) was run 1000 times with a perplexity of 10 and the clustering solution with the lowest KL divergence was selected (Maaten and Hinton, 2008). dbscan (v1.1.1) clustering with the size of the epsilon neighbourhood set to 2.5 was used to colour groups of density in the plot (Ester et al., 1996). The groups were labelled based on representative descriptions of the perturbations within the groups.
2.5 Identification of perturbation group consensus signatures and enriched drugs
Consensus signatures were generated for each of the t-SNE perturbation groups by applying the RankProd (v3.4) rank product method to perturbation gene log ratios in each group (Del Carratore et al., 2017). Genes with a percentage of false-positive predictions ≤ 0.05 were used to identify enriched mimic and reverse drugs signatures using the LINCS L1000 drug signatures as described earlier.
2.6 Web interface
SkeletalVis is an interactive web-based tool which can run on any common browser such as Chrome, Firefox and Safari. The web application is implemented using the R Shiny framework designed to display the output of the Galaxy pipeline as well as the processed expression similarity data. The application makes use of datatables (v0.2) for responsive tables to allowing fluid data exploration and enrichR (v1.0) to identify enriched pathways from user generated consensus signatures. Interactive visualization of graphs and networks was implemented with plotly (v4.7.1) and visNetworks (2.0.1). Single, global loading of the data ensures a responsive application. Code for the web interface can be found at www.github.com/soulj/SkeletalVis-Shiny
3 Results
3.1 Re-analysis of skeletal transcriptomics data
Our overall approach was to use a high-throughput, transcriptomics pipeline to analyse existing skeletal disease transcriptomics data. Searching for relevant datasets in ArrayExpress and GEO identified 287 experiments (Supplementary Table S2). Analysis of the raw data through the transcriptomics pipeline generated 739 expression response profiles with quality control and PCA plots, differential expression and comprehensive downstream analysis comprising of pathway, active sub-network, GO Term, drug, transcription factor enrichment (Supplementary Fig. S1). Annotation of these datasets revealed a variety of platforms, species and experimental design types (Fig. 1). The majority of datasets was from human and mouse reflecting the focus on use of both human tissue and mouse models for the study of skeletal disease and showing need for cross-species analysis. Affymetrix was the most common array type and there is a growing number of RNA-Seq (Illumina)-based datasets.
Fig. 1.

Summary of analysed expression perturbations
3.2 Recovery of prior knowledge and assessment of bias
Expression similarity is a useful tool to find related biological responses to perturbations thereby identifying diseases that potentially share similar mechanisms. A recurrent barrier to this type of analysis is the heterogeneous experimental platforms used to measure the gene expression (Leek et al., 2012). To investigate wherever there is strong bias in expression similarity of the analysed comparisons due to the experimental platform, we compared the similarity rank of experiments within the same platform compared with different platforms (Supplementary Fig. S2A). No strong global bias was observed in the similarity of perturbations within platforms. Likewise, no strong global similarity between perturbations within the tissue under study was observed, but a stronger bias was seen between species, possibly due to the nature of the experiments performed in the different species (Supplementary Fig. S2B and C).
To validate the expression similarity analysis, sets of experiments examining well-defined, related experimental perturbations that would be expected to have a shared expression response were selected i.e. same gene perturbation, exogenous treatment or disease (Supplementary Table S3). For each set of experiments the rank based on the four genes expression similarity measures (see methods) of the annotated experiments was calculated (Supplementary Fig. S3). Despite differences in the experimental set-up of the related experiments, the annotated datasets were among the most similar in the database, suggesting we can recover prior biological knowledge. The characteristic direction measure showed the best performance with these datasets consistent with a previous assessment against limma-based differential expression analysis (Wang et al., 2016). These findings suggest that the strength of the biological signal from the perturbations is sufficient to identify related experiments with shared biological mechanisms.
3.3 Identifying signature associations in skeletal biology
Associations between single gene perturbations and other transcriptomic responses can imply upstream regulation or shared signalling cascades. Transcriptomic signatures from gene perturbation experiments were examined to find the top pairwise similar expression responses to highlight examples of association identification enabled by this re-analysis of the transcriptomic datasets (Supplementary Table S4). Several examples which demonstrate cross-species in vitro and in vivo response similarity are shown (Table 1). The perturbation signature from mouse Eed knockout in rib cartilage shows similarity to mouse Ezh2 knockout growth plate zones, both components are part of the Polycomb repressive Complex 2 which methylates target genes (Mirzamohammadi et al., 2016; Lui et al., 2016). The Eed knockout signature also shows similarity to the in vitro human chondrocyte de-differentiation expression responses. The androgen receptor knockout mice signature is similar to the oestrogen receptor knockout (Kondoh et al., 2014; Russell et al., 2012). Interestingly, two skeletal disease mouse models also show similarity to the androgen receptor knockout; the Phex-deficient Hyp mouse model of X-linked hypophosphatemia and the AHSG knockout model of model of slipped capital femoral epiphysis (SCFE) (Brylka et al., 2017). Likewise, inhibition of the DOTL1 methyltransferase shows similarity to several inflammatory datasets (Monteagudo et al., 2017). These results suggest that by examining the similarities between cross-species skeletal transcriptomic responses from gene perturbations we can both recover known relationships and identify novel associations for future experimental validation.
Table 1.
The top pairwise similar characteristic direction expression responses for selected gene perturbations. Unless stated perturbations are relative to wild type/control conditions
| Perturbation | Accession | Species | Signed Jaccard |
|---|---|---|---|
| Eed knockout | GSE66862 | Mouse | |
| Ezh knockout | GSE84198 | Mouse | 0.0574 |
| Superficial versus deep zone cartilage | E-GEOD-54216 | Rat | 0.0563 |
| Dedifferentiating chondrocytes | GSE42235 | Human | 0.0447 |
| mOL-AR knockout | E-MTAB-1123 | Mouse | |
| AhsgHET versus Ahsg WT | GSE105139 | Mouse | 0.0579 |
| Hyp females versus Wildtype females | GSE5657 | Mouse | 0.0574 |
| Estrogen Receptor alpha knock-out | GSE41997 | Mouse | 0.0495 |
| DOT1L inhibition | GSE77916 | Human | |
| Galectin 1 treatment | E-GEOD-68760 | Human | 0.0400 |
| Galectin3 treatment | GSE85254 | Human | 0.0315 |
| IL-1 and glucosamine treatment | E-GEOD-6119 | Rat | 0.0311 |
To further explore the gene expression signatures we applied the t-SNE algorithm to visualize the characteristic direction derived signatures distance matrix, enabling a global overview of the skeletal disease transcriptional landscape and exploration of groups of related experimental perturbations. The resulting plot shows the heterogeneous perturbations broadly separate into groups of related perturbations with similar expression responses (Fig. 2). Several of the groups demonstrate the ability of this analysis to highlight-related experiments (Supplementary Table S5). For instance, several rheumatoid arthritis datasets are clustered together (Group 8). Similarly many short-term cytokine stimulated tissue perturbations form a group (Group 6).This analysis also highlights the ability to identify cross-species groups of profiles. For example, the model OA group (Group 35) includes a diverse collection of cross-species osteoarthritis animal model perturbations such as the mouse surgical destabilization of the medial meniscus post-traumatic model of OA and the rat metabolic model of OA with monoiodoacetate treatment (Burleigh et al., 2012; Korostynski et al., 2018; Loeser et al., 2013).These results suggest similarity in the expression response and shared mechanisms of action in these different models of induced osteoarthritis leading to the degradation of cartilage.
Fig. 2.
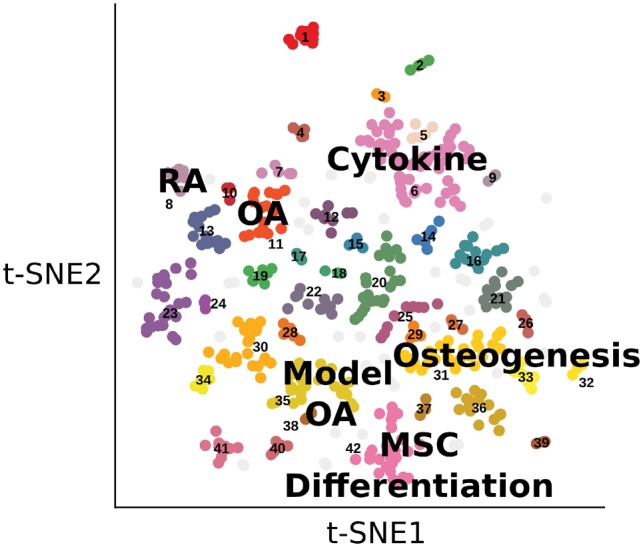
t-SNE visualization of skeletal expression signatures using the characteristic direction signature distance matrix. Groups of perturbations are labelled and coloured by regions of density identified using dbscan
To identify compounds potentially capable of mimicking or reversing the observed differential expression in the identified perturbation groups, we generated rank-product consensus signatures and performed drug enrichment analysis (Supplementary Tables S6–S8). A group of in vitro histone deacetylase (HDAC) inhibitor perturbations corresponded with several HDAC inhibitor drug signatures (Table 2). An activator of PKC, part of the cytokine signalling cascade, was predicted to mimic the cytokine stimulation responses. Several drugs were found to have an opposite transcriptomic response in skeletal disease-related groups. For instance, in the model osteoarthritis group MEK1/2 and PI3K inhibitors were identified among the top reverse drug signatures. These results suggest we can generate robust consensus signatures from groups of transcriptomic signatures to both recover known signalling pathways and predict potential therapeutic targets.
Table 2.
Top mimic or reverse drugs for selected perturbation groups. Enriched drugs with nominal targets and overlap scores were found using the LINCSL1000 CDS database for the t-SNE perturbation group consensus signatures. The enriched drugs act as inhibitors of the indicated targets unless otherwise stated
| In vitro group | Broad annotation | Top mimic drugs | Target | Score |
|---|---|---|---|---|
| 6 | Cytokine | Ingenol 3, 20-dibenzoate | PKC activator | 0.0534 |
| 33 | HDAC inhibition | Vorinostat | HDACs | 0.0933 |
|
| ||||
| Disease Group | Top reverse drugs | Nominal target | Score | |
|
| ||||
| 8 | RA | Curcubitacin I | JAK/STAT3 | 0.0389 |
| 15d-PGJ2 | PPARG activator | 0.0354 | ||
| Bortezomib | Proteasome | 0.0349 | ||
| 11 | Osteoarthritis | Narciclasine | Apoptosis | 0.0659 |
| Manumycin A | Ras | 0.0579 | ||
| Salermide | SIRT1/2 | 0.0568 | ||
| 35 | Animal models Osteoarthritis | Selumetinib | MEK1/2 | 0.0574 |
| TG101348 | PI3K | 0.0549 | ||
| BMS-536924 | IGF1R | 0.0544 | ||
3.4 SkeletalVis web portal
3.4.1 Exploration module
To enable future exploration and comparison of these data we constructed an interactive data-portal, SkeletalVis. SkeletalVis is composed of exploration and comparison modules as well as a detailed help section (Supplementary Fig. S4a). The exploration section of the data portal allows visualization of the detailed analysis (Supplementary Fig. S4b). To illustrate the utility of the exploration module we selected a well-characterized experiment investigating the expression profile of a Col10a1 knock-in mutation mouse, which is a model of the Metaphyseal chondrodysplasia type Schmid (MCDS) form of dwarfism (GSE30628) (Cameron et al., 2011). An experimental table shows the available experiments with the ability to sort, search and filter the table to find an experiment of interest (Supplementary Fig. S4c). From a chosen experiment the user can view and then select a comparison associated with that experiment to load the data in the other tabs (Supplementary Fig. S4d).
Once an experiment and comparison are loaded, quality control summaries including heatmaps allow the user to quickly assess the quality of the data (Fig. 3a). The data portal allows searching of the differential expression table with fold-changes and adjusted P-values (Fig. 3b). This table can be searched to find particular genes and can be filtered with a user defined thresholds to identify differentially regulated genes. All tables in the data portal can be copied or exported as text files for use with external tools. SkeletalVis provides detailed downstream analysis with enriched pathways, drugs, transcription factors, which can be viewed in interactive tables to identify the key dysregulated biological processes. As GO enrichment is often difficult to interpret, we use interactive multi-dimensional scaling plots based on the semantic similarity of the ontology terms to group-related terms together allowing a quick overview of the perturbed processes (Fig. 3c). Active sub-networks can be viewed as interactive networks coloured by fold change which often give more sensitive analysis compared with standard pathway enrichment analysis (Fig. 3d). For the MCDS experiment pathways, transcription factors and active sub-networks relating to endoplasmic reticulum stress and the Atf4 transcription factor are consistent with the findings reported in the corresponding publication.
Fig. 3.
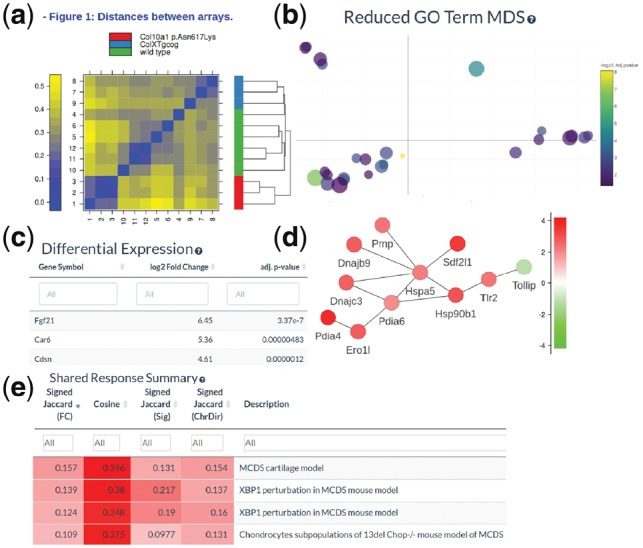
Analysis of MCDS mouse model with SkeletalVis. SkeletalVis provides quality control (a), differential expression analysis (b) with detailed downstream analysis including GO term enrichment (c) and network analysis (d). The shared gene expression responses can be readily examined (e)
The shared response tool shows pre-calculated expression similarities to every other comparison in the data portal using the four above described measures. This module is of use for both quality assurance in ensuring the expression response is similar to a related experiment, and for discovery of novel associations between disease and genetic/exogenous perturbations. The genes overlapping with each expression measure are shown and histograms show the distribution of the similarity scores. In the MCDS comparison data, among the top hits is an independent profile from mice with the same causative mutation knock-in and a profile from an alternative transgenic mouse model in the same causative Col10a1 gene, allowing consensus signatures to be developed (Fig. 3e).
3.4.2 Comparison module
The comparisons module allows for comparison of newly generated data to the experiments analysed in SkeletalVis. Gene signatures (up- and down-regulated genes) identified from an experiment outside the data portal can be compared with the differential expression and characteristic direction signatures within the portal. Overlap of the genes between the signatures is shown to allow focus on shared genes, allowing identification of novel associations between datasets. To illustrate the utility of this module the gene expression signature reported in the recent paper examining bone lesions in osteoarthritis was queried against the signatures in the data portal (Supplementary Fig. S5) (Kuttapitiya et al., 2017). Among the most similar experiments is a study on subchondral bone in an osteoarthritis surgical mouse model suggesting that cross-species similarities in the signatures can be observed. The module highlights the overlapping genes including THBS4 known to be involved in pain sensitization which is highlighted in the corresponding publication. The comparison module can also be used identify experiments where a particular gene is dysregulated, to identify exogenous perturbations that modulate the expression of that gene or finding other diseases where that gene is dysregulated. For example, searching for THBS4 reveals human osteoarthritis datasets where THBS4 is also up-regulated illustrating how SkeletalVis allows rapid cross-species comparison of newly available gene expression responses against this existing repository of knowledge.
4 Discussion
With the expanding expression data available for the study of skeletal disease, SkeletalVis collates 287 cross-species skeletal transcriptomic experiments and is an intuitive data-portal to allow exploration and meta-analysis. This consolidation of complex data to an accessible format is crucial to gaining meaningful information from the large numbers of datasets. Through our analysis of gene perturbation response associations we have highlighted several examples of links between cross-species in vitro and in vivo experiments. For instance, our findings suggest that modulation of the polycomb repressive Complex 2 role could be targeted to modulate the chondrocyte de-differentiation gene expression signature that occurs with culturing chondrocytes for future cell therapy to treat cartilage degeneration (Ma et al., 2013). The comparisons of expression profiles from the SCFE model, Hyp and hormone signalling perturbations have not been previously reported but AHSG is a known transcriptional target of ERα and these findings are consistent with suggestions it is a hormonal balance driven skeletal disorder (Qiu et al., 2014; Witbreuk et al., 2013).
We identified groups of similar signatures and identified enriched drug responses in consensus signatures to demonstrate the ability of this analysis to find regulators of core differentially expressed genes in groups of transcriptomic responses. Although the drug response signatures are derived from treated cancer cell lines rather than skeletal cell types, several of the highlighted top drug predictions are support by previous studies. In the RA group, the predicted reverse drug targets JAK/STAT3 and the proteosome are known inflammatory mediators (Elliott et al., 2003; Schwartz et al., 2017). PPARγ has previously been suggested as a potential therapeutic target in RA (Ormseth et al., 2013). In the model OA group, pharmacological MEK and PI3K inhibition protected against cartilage damage in rabbit and mouse OA models respectively (Lin et al., 2018; Pelletier et al., 2003). In the OA cartilage group, the enriched drug narciclasine reduced joint destruction in a rat model of arthritis (Lubahn et al., 2012). Interestingly, Salermide a Sirt1/2 inhibitor is an enriched reverse drug in the OA cartilage group consensus signature and in several individual studies examining human intact OA versus non-OA cartilage within that group. Evidence from in vivo mouse studies suggests that that Sirt1/2 activity is protective in OA (Matsuzaki et al., 2014). This expression overlap may therefore represent activation of the protective Sirt1/2 pathway in the intact OA cartilage. These data therefore allow development of many hypotheses to be followed up with new data and functional studies.
The aim of SkeletalVis is not to replace existing tools such as ExpressionAtlas, but to provide a more specialized repository for skeletal disease researchers with extended downstream data analysis. For example, searching for the common skeletal disease osteoarthritis in ExpressionAtlas returns only 3 experiments compared with 30 in SkeletalVis. Several existing cancer expression specific databases such as GlioVis focus on integration of survival and somatic mutation data with expression data which is not generally applicable for skeletal diseases (Bowman et al., 2017). Instead, SkeletalVis offers added value in coverage of skeletal datasets and considerably more in-depth down-stream analysis with network analysis and integration with databases such as LINCS L1000 and also includes the ability to compare expression profiles. This data portal is of wide use to skeletal biology/disease researchers as it can be used to rapidly screen for evidence of target gene dysregulation in skeletal development and disease, and also to identify perturbations that can be modulated to altered expression activity of these targets in a skeletal cellular context. The portal will be useful for prioritization of identified differentially expressed genes for experimental validation and for initial functional characterization of novel disease-associated genes identified through genome-wide association studies, allowing understanding of potential functions of the genes in context of skeletal tissues. Not all included studies have sufficient replicates to calculate the statistical significance of the altered expression. Although these studies can be readily filtered from the tables, these studies often describe unique perturbations in the database and may be useful for researchers interested in finding shared perturbations for further experimental validation. Curation efforts to make a context-specific database are likely to produce more immediately relevant results for users than generic databases. Although gene expression responses can be shared with non-skeletal tissues, the specialized nature of the skeletal tissues makes investigating perturbations within the same biological system more useful. The approach herein is likely to be of interest to many investigators building context-specific omics databases. The developed pipeline and app can be deployed in other areas of biological interest and this collection of data with known perturbations will be useful for development and validation of methods for analysing skeletal transcriptomics data.
As new expression data becomes available in public repositories this data can readily be analysed and integrated into the web platform. Similarly, as new methods of analysis are developed these can be performed on these collection of datasets. Future updates could include integration of further omics data such as non-coding transcription, proteomics and epigenetic data so to investigate the interplay of multiple regulatory layers.
Supplementary Material
Acknowledgements
We thank Certus Technology for assistance with implementation of the bioinformatics pipeline.
Funding
This work has been supported by the European Community’s Seventh Framework Programme grant [602300, SYBIL]. The Wellcome Centre for Cell-Matrix Research, University of Manchester, is supported by core funding from the Wellcome Trust [grant number 203128/Z/16/Z].
Conflict of Interest: none declared.
References
- Afgan E. (2016) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res., 44, W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S., et al. (2017) SCENIC: single-cell regulatory network inference and clustering. Nat. Methods, 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgon R., et al. (2010) Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. USA, 107, 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman R.L., et al. (2017) GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol., 19, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L., et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Breitling R., et al. (2004) Graph-based iterative group analysis enhances microarray interpretation. BMC Bioinformatics, 5, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brylka L.J., et al. (2017) Post-weaning epiphysiolysis causes distal femur dysplasia and foreshortened hindlimbs in fetuin-A-deficient mice. PLoS One, 12, e0187030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh A., et al. (2012) Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum., 64, 2278–2288. [DOI] [PubMed] [Google Scholar]
- Cameron T.L., et al. (2011) Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS One, 6, e24600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Carratore F., et al. (2017) RankProd 2.0: a refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics, 33, 2774–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E.G., et al. (2011) Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res., 39, D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark N.R., et al. (2014) The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics, 15, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross M., et al. (2014) The global burden of hip and knee osteoarthritis: estimates from the Global Burden of Disease 2010 study. Ann. Rheum. Dis., 73, 1323–1330. [DOI] [PubMed] [Google Scholar]
- Duan Q., et al. (2016) L1000CDS2: lINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl., 2, 16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S.L., et al. (2016) Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthr. Cartil., 24, 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., et al. (2002) Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P.J., et al. (2003) Proteasome inhibition: a new anti-inflammatory strategy. J. Mol. Med., 81, 235–245. [DOI] [PubMed] [Google Scholar]
- Ester M., et al. (1996) A density-based algorithm for discovering clusters a density-based algorithm for discovering clusters in large spatial databases with noise. Proc. Second Int. Conf. Knowl. Discov. Data Min., 226–231. [Google Scholar]
- Ewels P., et al. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsdal M.A., et al. (2016) Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: lessons learned from failures and opportunities for the future. Osteoarthr. Cartil., 24, 2013–2021. [DOI] [PubMed] [Google Scholar]
- Kolesnikov N., et al. (2015) ArrayExpress update-simplifying data submissions. Nucleic Acids Res., 43, D1113–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh S., et al. (2014) Estrogen receptor α in osteocytes regulates trabecular bone formation in female mice. Bone, 60, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostynski M., et al. (2018) Cell-type-specific gene expression patterns in the knee cartilage in an osteoarthritis rat model. Funct. Integr. Genomics, 18, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttapitiya A., et al. (2017) Microarray analysis of bone marrow lesions in osteoarthritis demonstrates upregulation of genes implicated in osteochondral turnover, neurogenesis and inflammation. Ann. Rheum. Dis., 76, 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T., et al. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R., et al. (2011a) The European Nucleotide Archive. Nucleic Acids Res., 39, D28–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R., et al. (2011b) The sequence read archive. Nucleic Acids Res., 39, D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., et al. (2018) Blocking PI3K/AKT signaling inhibits bone sclerosis in subchondral bone and attenuates post-traumatic osteoarthritis. J. Cell Physiol., 233, 6135–6147. [DOI] [PubMed] [Google Scholar]
- Loeser R.F., et al. (2013) Disease progression and phasic changes in gene expression in a mouse model of osteoarthritis. PLoS One, 8, e54633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubahn C., et al. (2012) Preclinical efficacy of sodium narcistatin to reduce inflammation and joint destruction in rats with adjuvant-induced arthritis. Rheumatol. Int., 32, 3751–3760. [DOI] [PubMed] [Google Scholar]
- Lui J.C., et al. (2016) EZH1 and EZH2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy. Nat. Commun., 7, 13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B., et al. (2013) Gene expression profiling of dedifferentiated human articular chondrocytes in monolayer culture. Osteoarthr. Cartil., 21, 599–603. [DOI] [PubMed] [Google Scholar]
- Matsuzaki T., et al. (2014) Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis., 73, 1397–1404. [DOI] [PubMed] [Google Scholar]
- Mirzamohammadi F., et al. (2016) Polycomb repressive complex 2 regulates skeletal growth by suppressing Wnt and TGF-β signalling. Nat. Commun., 7, 12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteagudo S., et al. (2017) DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat. Commun., 8, 15889. Doi: 10.1038/ncomms15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormseth M.J., et al. (2013) Peroxisome proliferator-activated receptor γ agonist effect on rheumatoid arthritis: a randomized controlled trial. Arthritis Res. Ther., 15, R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodorou I., et al. (2018) Expression Atlas: gene and protein expression across multiple studies and organisms. Nucleic Acids Res., 46, D246–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J.-P., et al. (2003) In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum., 48, 1582–1593. [DOI] [PubMed] [Google Scholar]
- Qiu C., et al. (2014) Estrogen increases the transcription of human α2-Heremans-Schmid-glycoprotein by an interplay of estrogen receptor α and activator protein-1. Osteoporos. Int., 25, 1357–1367. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., et al. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P.K., et al. (2012) Identification of gene pathways altered by deletion of the androgen receptor specifically in mineralizing osteoblasts and osteocytes in mice. J. Mol. Endocrinol., 49, 1–10. [DOI] [PubMed] [Google Scholar]
- Schwartz D.M., et al. (2017) JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., et al. (2016) Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. Version 2. F1000Res., 4, 1521. Doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul J., et al. (2018) Stratification of knee osteoarthritis: two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann. Rheum. Dis., 77, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg J., Zeggini E. (2016) Functional genomics in osteoarthritis: past, present, and future. J. Orthop. Res, 34, 1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoney R.A., et al. (2018) Using set theory to reduce redundancy in pathway sets. BMC Bioinformatics, 19, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., et al. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L., Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- von Mering C., et al. (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res., 31, 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadi L., et al. (2016) Impact of outdated gene annotations on pathway enrichment analysis. Nat. Methods, 13, 705–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., et al. (2012) PubChem’s BioAssay Database. Nucleic Acids Res., 40, D400–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. (2016) Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun., 7, 12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witbreuk M., et al. (2013) Slipped capital femoral epiphysis and its association with endocrine, metabolic and chronic diseases: a systematic review of the literature. J. Child Orthop., 7, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., et al. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol., 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.