

Abstract
Background
Sarcopenia is the loss of muscle mass/function that occurs during the aging process. The links between mechanistic target of rapamycin (mTOR) activity and muscle development are largely documented, but the role of its downstream targets in the development of sarcopenia is poorly understood. Eukaryotic initiation factor 4E‐binding proteins (4E‐BPs) are targets of mTOR that repress mRNA translation initiation and are involved in the control of several physiological processes. However, their role in skeletal muscle is still poorly understood. The goal of this study was to assess how loss of 4E‐BP1 and 4E‐BP2 expression impacts skeletal muscle function and homeostasis in aged mice and to characterize the associated metabolic changes by metabolomic and lipidomic profiling.
Methods
Twenty‐four‐month‐old wild‐type and whole body 4E‐BP1/4E‐BP2 double knockout (DKO) mice were used to measure muscle mass and function. Protein homeostasis was measured ex vivo in extensor digitorum longus by incorporation of l‐[U‐14C]phenylalanine, and metabolomic and lipidomic profiling of skeletal muscle was performed by Metabolon, Inc.
Results
The 4E‐BP1/2 DKO mice exhibited an increase in muscle mass that was associated with increased grip strength (P < 0.05). Protein synthesis was higher under both basal (+102%, P < 0.05) and stimulated conditions (+65%, P < 0.05) in DKO skeletal muscle. Metabolomic and complex lipid analysis of skeletal muscle revealed robust differences pertaining to amino acid homeostasis, carbohydrate abundance, and certain aspects of lipid metabolism. In particular, levels of most free amino acids were lower within the 4E‐BP1/2 DKO muscle. Interestingly, although glucose levels were unchanged, differences were observed in the isobaric compound maltitol/lactitol (33‐fold increase, P < 0.01) and in several additional carbohydrate compounds. 4E‐BP1/2 depletion also resulted in accumulation of medium‐chain acylcarnitines and a 20% lower C2/C0 acylcarnitine ratio (P < 0.01) indicative of reduced β‐oxidation.
Conclusions
Taken together, these findings demonstrate that deletion of 4E‐BPs is associated with perturbed energy metabolism in skeletal muscle and could have beneficial effects on skeletal muscle mass and function in aging mice. They also identify 4E‐BPs as potential targets for the treatment of sarcopenia.
Keywords: Anabolism, mTOR, Protein synthesis, Proteolysis, Skeletal muscle
Introduction
The mechanistic target of rapamycin (mTOR) is a central regulator of cell growth and metabolism that responds to a wide range of different stimuli including nutrient availability, growth factors, and cellular energy status. In the last two decades, extensive research has established mTOR as a master regulator of many fundamental processes including protein synthesis and autophagy. Moreover, when deregulated, it has been found to be associated with the development of metabolic disorders such as obesity, type 2 diabetes, cancer progression, and the aging process (for review, see Saxton and Sabatini1).
The eukaryotic translation initiation factor 4E‐binding proteins (4E‐BPs) are a family of three proteins (4E‐BP1, 2, and 3)2, 3 that inhibit translation initiation by sequestering eIF4E.4 Their activity is controlled by the mTOR pathway. mTOR can form two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Under nutrient or growth factor deprivation, 4E‐BPs are hypophosphorylated and interact with eIF4E. In response to nutrients or growth factors, activation of the mTORC1 leads to phosphorylation of the 4E‐BPs and their dissociation from eIF4E, ultimately resulting in increased cap‐dependent translation initiation.4, 5 The physiological role of 4E‐BPs has been addressed in several studies. 4E‐BP1 knockout (KO) mice display reduced adipose tissue and increased energy expenditure.6 In contrast, deletion of both 4E‐BP1 and 4E‐BP2 is associated with increased body weight and increased sensitivity to diet‐induced obesity and insulin resistance.7, 8 Finally, overexpression of 4E‐BP1 is able to protect mice from age‐induced and diet‐induced obesity and insulin resistance.9, 10 So far, only a few studies have addressed the role of the 4E‐BPs in skeletal muscle function and metabolism. In a recent study, Tsai et al. further analysed the role of 4E‐BP1 in skeletal muscle. They demonstrated that muscle‐specific transgenic overexpression of a constitutively active form of 4E‐BP1 protects mice from diet‐induced obesity. They also observed that activation of 4E‐BP1 in skeletal muscle results in muscle fibre transformation and enhanced mitochondrial respiration.10 In response to diet‐induced obesity, we previously reported an increased ectopic accumulation of lipotoxic species (diacylglycerols, ceramides, and sphingomyelins) in skeletal muscle of mice lacking both 4E‐BP1 and 4E‐BP2 associated with increased systemic insulin resistance.8 Surprisingly, despite the development of a more severe insulin resistance, 4E‐BP1/2 double KO (DKO) mice displayed an increased muscle mass in response to diet‐induced obesity, which suggests that 4E‐BPs regulate muscle homeostasis regardless of insulin sensitivity.8 Altogether, these data demonstrate that 4E‐BPs are key regulators of muscle homeostasis and metabolism and play a critical role in the development of insulin resistance and the accumulation of ectopic lipids.
Aging is characterized by a progressive and involuntary loss of skeletal muscle, termed sarcopenia, associated with reduced muscle strength and exercise endurance.11, 12 Sarcopenia is associated with a high risk of adverse outcomes such as metabolic disorders, frailty, poor quality of life, and death.13 Potential mechanisms contributing to sarcopenia include inadequate or insufficient protein intake,14, 15 a decline in the mitochondrial function,16, 17 unbalanced protein turnover such as increased proteolysis18 and reduced protein synthesis,19, 20 and resistance to anabolic signals (i.e. amino acids and insulin).21, 22, 23 Anabolic resistance is associated with the ectopic accumulation of lipids in skeletal muscle, a process occurring frequently with age but also favoured by obesity.24, 25
Because 4E‐BPs control translation initiation, a critical step in the protein synthesis process, and regulate insulin sensitivity and the ectopic accumulation of lipotoxic compounds in skeletal muscle, we hypothesized that deletion of 4E‐BP1 and 4E‐BP2 might protect mice from aging‐associated loss of muscle mass and function.
Materials and methods
Animals and experimental design
All breeding and experimental protocols were reviewed and approved by the local ethics committee for animal experimentation (CREFA Auvergne, agreement #00782.01) and met the National Research Council's guideline for the care and use of laboratory animals. Congenic BALB/c Eif4ebp1−/− and Eif4ebp2−/− (4E‐BP1/2 DKO mice) as previously described7 and BALB/c wild‐type (WT) mice were provided by Dr Nahum Sonenberg (McGill University, Montréal, Canada). Sixty‐week‐old male mice were individually housed in plastic cages and maintained at 21–23°C with a 12 h dark–12 h light schedule, given free access to water and food, and followed up to age 24 months. Mice were fed a normal chow diet (25% protein, 61% carbohydrate, and 14% fat; Safe‐Diets, Augy, France). Twenty‐four‐month‐old mice were used for all the experiments in this study. Euthanasia was performed at 10 a.m. after overnight fasting during which mice had free access to water. Some mice received an intraperitoneal injection of insulin (1.2 mU/g body weight) 20 min before sacrifice. The genotype of each mouse was verified by PCR at birth and at the end of the experiment.
Indirect calorimetric studies and whole body composition analysis
Energy expenditure, volunteer home cage activity, and food and water intake were measured by using a PhenoMaster/LabMaster four‐cage TSE system (Bad Homburg, Germany). Spontaneous activity was measured using a three‐dimensional meshing of light beams. For whole body composition analysis, mice were placed in an EchoMRI‐100 analyser (Echo Medical Systems LLC, Houston, TX) to determine fat and lean body mass (g).
Grip strength and wire screen holding tests
Grip strength and wire screen holding were measured as previously described.26 Briefly, grip strength was assessed using a commercially available force gauge (Bioseb, Vitrolles, France) by the same investigator. The apparatus consisted of a metal grid connected to a force transducer. Each mouse was held by the base of its tail and lowered towards the grid of a grip strength meter and allowed to grasp it with its forepaws or both forepaws and hindpaws. The mouse was then pulled steadily away from the rod by its tail until its grip broke. Hindlimb grip strength in each mouse was measured five times. For wire screen holding (hang test), each mouse was placed on an aluminium mesh grid, which was then inverted and held approximately 2 ft over a mouse cage containing 2–3 in. of litter to avoid injury. The wire screen holding time is defined as the time elapsed before the mouse falls from the inverted grid. The procedure was repeated three times for each mouse with approximately 5 min between each test. The physical impulse (Fdt) is the holding time (ht) multiplied by the gravitational force of the mouse [Fdt (N·s) = body mass (g) × 0.00980665 N/g × ht (s)] and represents the minimal total sustained force exerted to oppose the gravitational force.
Ex vivo protein synthesis and proteolysis measurement
Protein synthesis and degradation were measured as previously described.27 Briefly, mouse extensor digitorum longus (EDL) muscles were preincubated for 30 min in Krebs–Henseleit buffer (KHB) (120 mM NaCl, 4.8 mM KCl, 25 mM NaHCO3, 2.5 mM CaCl2, 1.2 mM KH2PO4, and 1.2 mM MgSO4; pH 7.4), supplemented with 5 mM HEPES, 5 mM glucose, and 0.1% bovine serum albumin (BSA) and saturated with a 95% O2–5% CO2 gas mixture. Muscles were then transferred into fresh media of the same composition containing 200 μM leucine, 5 mM insulin, and 500 μM l‐[U‐14C]phenylalanine (0.5 μCi/mL) for 60 min. At the end of the incubation, muscles were quickly rinsed in KHB, blotted, and homogenized in 10% trichloroacetic acid (TCA). TCA insoluble material was washed three times with 10% TCA and solubilized in 1 N NaOH at 37°C for determination of protein‐bound radioactivity and protein content. For protein degradation, muscles were incubated as described earlier. Because tyrosine is neither synthesized nor degraded by muscle, the release of this amino acid from EDL muscle in the incubation medium reflects net proteolysis. Tyrosine was measured by fluorometry.28 Protein breakdown was calculated as the sum of net protein breakdown and protein synthesis, after converting the rate of phenylalanine incorporation in proteins into tyrosine equivalents.29
Chemicals
Protease inhibitor cocktail, primers, and the antibody against P38 were purchased from Sigma‐Aldrich (Saint‐Quentin‐Fallavier, France); antibodies against 4E‐BP1, Akt, Ser473‐phosphorylated Akt, S6K, and Thr389‐phosphorylated S6K were from Cell Signaling Technology; horseradish peroxidase‐conjugated secondary antibodies were from DAKO (Trappes, France); SuperScript® III reverse transcriptase, random hexamer, and oligo‐dT primers were from Invitrogen (Life Technologies, Saint‐Aubin, France); Tri reagent was from Euromedex (Mundolsheim, France); and Rotor‐Gene SYBR Green PCR Master Mix from Qiagen (Courtaboeuf, France).
Western blot analysis
Quadriceps were harvested in lysis buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EDTA, 10 mM NaPPi, 25 mM β‐glycerophosphate, 100 mM NaF, 2 mM Na orthovanadate, 10% glycerol, 1% Triton X‐100 containing 1% of protease inhibitor cocktail (Sigma‐Aldrich, Saint‐Quentin‐Fallavier, France). Homogenates were centrifuged at 13 000 g for 10 min at 4°C. Denatured proteins were separated by SDS–PAGE and transferred to a PVDF membrane (Millipore, Molsheim, France). Immunoblots were blocked with TBS‐Tween‐20 0.1% containing 5% dry milk and then probed overnight at 4°C with primary antibodies. After several washes with TBS‐Tween‐20 0.1%, immunoblots were incubated with a horseradish peroxidase‐conjugated secondary antibody (DAKO, Trappes, France) for 1 h at room temperature. The immune reactive strips or whole lanes were visualized by chemiluminescence (ECL Western Blotting Substrate, Thermo Fisher Scientific, Courtaboeuf, France). Luminescent secondary antibodies were visualized using an MF ChemiBis 2.0 camera (Fusion Solo, Vilber Lourmat, France). Band densities were quantified using MultiGauge 3.2 software (Fujifilm Corporation, distributor FSVT, Courbevoie, France). An internal control was used on each gel to normalize signal intensities between gels.
RNA extraction and quantitative real‐time PCR
Total RNA was extracted from frozen quadriceps using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA was quantified by measuring optical density at 260 nm. The concentrations of the mRNAs corresponding to the genes of interest were measured by reverse transcription followed by real‐time PCR using a Rotor‐Gene Q (Qiagen) system. One microgram of total RNA was reverse transcribed using SuperScript® III reverse transcriptase and a combination of random hexamer and oligo‐dT primers (Invitrogen). PCR amplification was performed in a 20 μL total reaction volume. The RT–PCR mixture contained 5 μL of diluted cDNA template, 10 μL of 2× Rotor‐Gene SYBR Green PCR Master Mix, and 0.5 μM of forward and reverse primers. The amplification profile was initiated by 5 min incubation at 95°C to activate HotStarTaq Plus DNA Polymerase, followed by 40 cycles of two steps: 95°C for 5 s (denaturation step) and 60°C for 10 s (annealing/extension step). Relative mRNA concentrations were analysed using Rotor‐Gene software. The relative abundance of mRNAs was calculated using the 2−ΔΔCT method with 36Β4 as housekeeping gene. The primers used in the PCR are described in Table 1.
Table 1.
Primer sequences
| Gene | Primers 5′–3′ | |
|---|---|---|
| Forward | Reverse | |
| 36B4 | ACTGGTCTAGGACCCGAGAAG | TCCCACCTTGTCTCCAGTCT |
| Atg5 | TCAACCGGAAACTCATGGAA | CGGAACAGCTTCTGGATGAA |
| Bcat2 | GCAGCCACACTAGGACAGGT | ACACATCTTTTGGACCCACAT |
| Bckdha | CTTGGAGCGCACAGACCT | GGTAGTCCCGGTACATGAGC |
| Bckdhb | CCCTTGTCTCAGGCTGAAGT | TCCCGGATGACATGAACC |
| Cd36 | TTGTACCTATACTGTGGCTAAATGAGA | CTTGTGTTTTGAACATTTCTGCTT |
| Cpt1b | TGGGACTGGTCGATTGCATC | TCAGGGTTTGTCGGAAGAAGAA |
| Ctsl | CAAATAAGAATAAATATTGGCTTGTCA | TGTAGCCTTCCATACCCCATT |
| Dbt | ACCGGAAGTAGCCATTGGT | AAACGTCTCCTTTCTGGTCAAA |
| Dld | GGGAGCACATATTCTAGGACCA | TCACAGGAAGCACCATATTCC |
| Fatp1 | TTCTCGGAGTCTGGAATGCT | CACAGAGGCTGTTCCTGCTC |
| Lpl | TCTAAATCCCACGAGCCCTA | TTGAACCAAGCAGGTCACAG |
| MAFbx | AAGCTTGTGCGATGTTACCCA | CACGGATGGTCAGTGCCCTT |
| MCAD | TTTCGGAGGCTATGGATTCAACAC | TCAATGTGCTCACGAGCTATGATC |
| MuRF1 | AGGTGTCAGGGCAAACAGT | CCTCCTTTGTCCTCTTGCTG |
| Lat1 | ATGGAGTGTGGCATTGGCTTC | AGCACCGTCACAGAGAAGATA |
| Lat2 | GACATCGGCCTCGTTGCT | TGTAAGGATCCACAAGTTCCTCAGT |
| Snat1 | CGAACCAGGACGGAGATAAAGG | TCGTAGATGACCAGAGGGATGC |
| Snat2 | CTCACTTACTGTGTCCCACA | AACACGGTAGGCAGGCGGAT |
Metabolomic analysis
Six samples per experimental group from overnight‐fasted WT and 4E‐BP1/2 DKO mice were included in the metabolomic analysis of quadriceps. Metabolomic and complex lipid profiling was performed by Metabolon (Morrisville, NC); detailed methods are provided in the Supporting Information.
Statistical analysis
Data are expressed as mean ± SEM. Differences between groups were analysed with two‐way analysis of variance, and Bonferroni post‐tests were used to compare replicate means. Differences between groups on a per‐metabolite basis were analysed with Welch's two‐sample t‐test. A p value of <0.05 was considered statistically significant.
Results
Muscle mass
In a previous study, we observed that hindlimb muscle mass was slightly higher in 5‐month‐old 4E‐BP1/2 DKO mice fed a regular diet than in WT mice.8 Because aging is associated with loss of muscle mass (sarcopenia), we measured body composition by EchoMRI and the mass of several hindlimb muscles of 24‐month‐old WT and 4E‐BP1/2 DKO mice. As previously described,7, 8 we observed that 4E‐BP1/2 DKO mice weighed significantly more than did the WT mice (Table 2). Food intake (5.12 ± 0.09 vs. 4.98 ± 0.08 g/day, NS) and energy expenditure (39.2 ± 1.3 vs. 42.3 ± 1.0 kJ/day, NS) were similar in WT and 4E‐BP1/2 DKO mice, respectively. Overnight‐fasted glucose (2.03 ± 0.17 and 2.13 ± 0.05 g/L in WT and DKO, respectively) and insulin (0.31 ± 0.03 and 0.37 ± 0.05 ng/mL in WT and DKO, respectively) levels were unchanged in the WT and 4E‐BP1/2 DKO mice. EchoMRI revealed that the difference in body weight was mainly attributable to an increase in lean mass in the 4E‐BP1/2 DKO mice as the amount of fat mass was similar between the WT and 4E‐BP1/2 DKO mice (Table 2). Several tissues were collected and weighed after sacrifice to further characterize this phenotype. No statistical difference was observed in weight for the adipose tissues (gonadal and subcutaneous), heart, or liver between the WT and 4E‐BP1/2 DKO mice. However, analysis of the hindlimb muscle revealed an increase in the weight of all the muscle collected from 4E‐BP1/2 DKO mice compared with WT mice (Table 2, P < 0.01).
Table 2.
Hindlimb skeletal muscle and tissue weight in WT and 4E‐BP1/2 DKO mice
| WT (n = 7) | DKO (n = 9) | |
|---|---|---|
| Weight (g) | 26.8 ± 0.6 | 29.7 ± 0.7* |
| Lean mass (g) | 22.8 ± 0.4 | 26.1 ± 0.7* |
| Fat mass (g) | 2.42 ± 0.28 | 2.63 ± 0.26 |
| Gastrocnemius (mg) | 99.3 ± 4.8 | 119.0 ± 3.4* |
| Soleus (mg) | 7.3 ± 0.3 | 10.2 ± 0.4* |
| Tibialis (mg) | 42.5 ± 2.4 | 52.6 ± 1.4** |
| Plantaris (mg) | 14.1 ± 0.5 | 17.1 ± 0.5* |
| Quadriceps (mg) | 163.7 ± 6.0 | 195.7 ± 3.8** |
| EDL (mg) | 13.5 ± 0.9 | 14.1 ± 0.5* |
| All muscles (mg) | 340.4 ± 9.4 | 409.1 ± 6.7** |
| GWAT (mg) | 217.3 ± 38.6 | 211.6 ± 30.9 |
| SWAT (mg) | 233.9 ± 46.2 | 234.2 ± 36.6 |
| Liver (mg) | 1160 ± 21 | 1328 ± 28 |
| Heart (mg) | 181.4 ± 8.4 | 205.6 ± 9.1 |
Mice were 24 months old. Results are expressed as mean ± SEM. p values were assessed using unpaired t‐test.
GWAT, gonadal white adipose tissue; SWAT, subcutaneous white adipose tissue.
P < 0.05.
P < 0.01 vs. WT.
Muscle function
We measured voluntary locomotor activity in calorimetric cages during the light and dark phases, and we performed several mechanical tests to evaluate the muscle function of 24‐month‐old WT and 4E‐BP1/2 DKO mice. We did not observe any difference in travelled distance between WT and 4E‐BP1/2 DKO mice regardless of the time of the day (Figure 1 A). We further analysed voluntary activity by measuring the time mice were standing. Both genotypes spent more time standing in the dark phase than in the light phase (Figure 1 A). However, despite the fact that the WT and 4E‐BP1/2 DKO mice ran the same distance, the 4E‐BP1/2 DKO mice spent about twice as much time standing than did WT mice in both the light and dark phases (Figure 1 B). To have a direct measurement of muscle function, we performed a grip strength test (to measure the strength of the forelimbs alone or forelimbs + hindlimbs) and a hang test (physical impulse, which measures the time during which a mouse opposes the gravitational force associated with body mass). We did not observe any difference in physical impulse between the WT and 4E‐BP1/2 DKO mice (Figure 1 E). Both mean (≈+50%, P < 0.01) and maximum grip strength (≈+35%, P < 0.01) were higher in 4E‐BP1/2 DKO mice than in WT mice (Figure 1 C and D).
Figure 1.
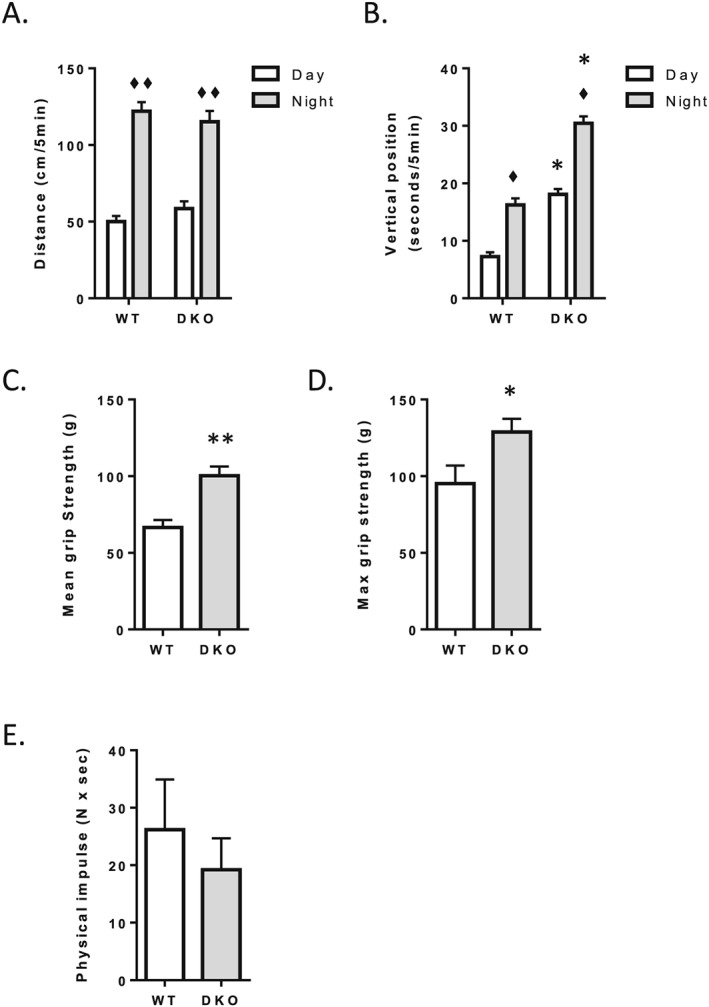
Spontaneous locomotor activity and muscle function in WT and 4E‐BP1/2 DKO mice. Spontaneous locomotor activity in WT and 4E‐BP1/2 DKO was measured using infrared sensor pairs arranged in strips for horizontal (A) and vertical (B) activities. Front forearm mean (C) and maximal (D) grip strength in WT and 4E‐BP1/2 DKO mice. (E) Mean holding impulse in the four limb wire grid holding test in WT and 4E‐BP1/2 DKO mice. n = 5–6 in each genotype, p values were assessed by two‐way analysis of variance, and Bonferroni post‐tests were used to compare replicate means by row (in A and B) or by unpaired t‐test (in C–E). ♦P < 0.05 vs. day, ♦♦P < 0.01 vs. day, * P < 0.05 vs. WT, ** P < 0.01 vs. WT.
Extensor digitorum longus protein turnover
Because 4E‐BPs are involved in the control of translation initiation, a critical step in the process of protein synthesis, protein turnover was measured in EDL muscle of WT and 4E‐BP1/2 DKO mice. Ex vivo stimulation of EDL by leucine and insulin led to genotype differences. In WT EDL, stimulation of protein synthesis by incubation with leucine and insulin induced a ≈50% increase in L‐[U‐14C]phenylalanine incorporation (Figure 2 A, P < 0.05). In EDL from 4E‐BP1/2 DKO mice, this stimulation led to a ≈25% increase in L‐[U‐14C]phenylalanine incorporation (P < 0.05), but its incorporation was ≈2‐fold higher under both basal and leucine/insulin‐stimulated conditions than in WT mice (Figure 2 A, P < 0.05). In response to insulin injection, phosphorylation of Akt on Ser473 and of S6K1 on Thr389 was increased in both WT and 4E‐BP1/2 DKO muscles, with there being no statistical significance based on genotype (Figure 2 B).
Figure 2.
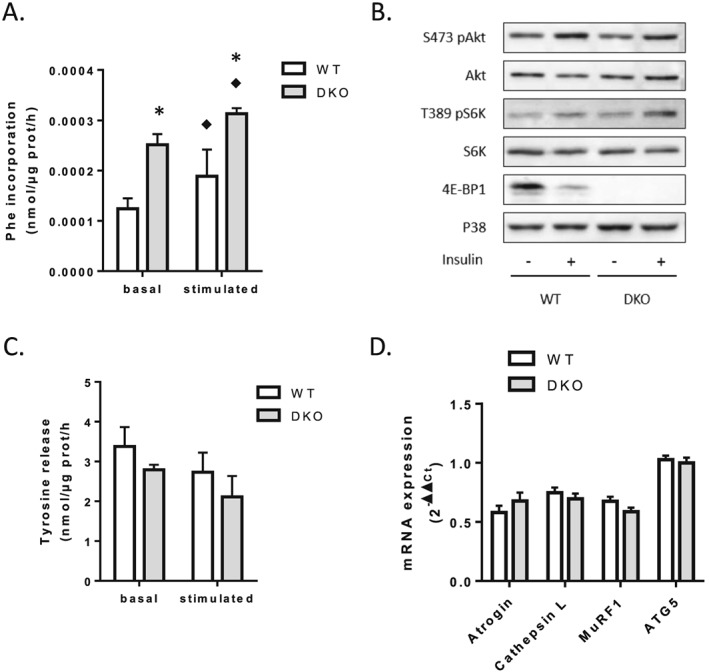
Loss of 4E‐BP1 and 4E‐BP2 alters protein homeostasis in skeletal muscle. (A) Protein synthesis in WT and 4E‐BP1/2 DKO skeletal muscles. Protein synthesis was measured ex vivo by radioactive phenylalanine incorporation in extensor digitorum longus with (stimulated) or without (basal) stimulation with a mixture of leucine and insulin. (B) Representative western blot levels of Akt, S6K, and 4E‐BP1 phosphorylation in WT and 4E‐BP1/2 DKO quadriceps. Mice were fasted overnight before receiving a 1.2 mU/g body weight intraperitoneal insulin injection. Mice were sacrificed 20 min later, and tissues were collected for western blot analysis. (C) Proteolysis in WT and 4E‐BP1/2 DKO skeletal muscles. Proteolysis was analysed ex vivo in extensor digitorum longus by measuring tyrosine release as described in the materials and methods Materials and methods. (D) Real‐time PCR quantification of Atrogin/MAFbx, Cathepsin L, MuRF1, and ATG5 mRNA expression in quadriceps from WT and 4E‐BP1/2 DKO mice. n = 5–6 in each genotype, p values were assessed by two‐way analysis of variance, and Bonferroni post‐tests were used to compare replicate means by row (in A–C) or by unpaired t‐test (in D). ♦P < 0.05 vs. basal, * P < 0.05 vs. WT.
We then measured protein breakdown from the release of tyrosine in the incubation medium as described in the incubation medium as described in materials and methods. Under basal conditions, tyrosine release was reduced by ≈20% in EDL from 4E‐BP1/2 DKO mice compared with WT mice (2.73 ± 0.12 vs. 3.38 ± 0.48 nmol/μg prot/h, Figure 2 C, NS). Incubation with leucine/insulin led to a similar ≈20% decrease in tyrosine release in EDL from both the 4E‐BP1/2 DKO and WT mice (2.11 ± 0.52 and 2.79 ± 0.49 nmol/μg prot/h, respectively, Figure 2 C, NS). The expression level of some proteolytic system mRNA (MuRF1, Atrogin/MAFbx, Cathepsin L, ATG5) was not affected by the deletion of the 4E‐BPs (Figure 2 D). These changes in protein synthesis and degradation could explain the increased muscle mass in 4E‐BP1/2 DKO male mice.
Global metabolomics and complex lipid profile of skeletal muscle
To further analyse the metabolic alterations underpinning the increased muscle mass and improved function in the 4E‐BP1/2 DKO mice, we performed metabolomic and lipid profile analysis on quadriceps derived from overnight‐fasted WT and 4E‐BP1/2 DKO mice (n = 6 per group). We detected 548 and 939 biochemicals in the metabolomic and complex lipid analyses, respectively, of which 496 and 435 matched known chemical structures in the Metabolon chemical reference library. A summary of the numbers of biochemicals reaching statistical significance (P ≤ 0.05), and of those approaching significance (0.05 < P < 0.10), is shown in the Supporting Information (Figure S1).
Amino acid metabolism in skeletal muscle
When comparisons were made between the DKO and WT groups, we observed that global free amino acid abundance tended to be lower in 4E‐BP1/2 DKO muscle (Figure 3 A), with significant reductions being present in non‐essential (i.e. glutamine, cysteine, arginine, and proline, P < 0.05) as well as essential amino acids (i.e. threonine, phenylalanine, tryptophan, and valine, P < 0.05). We also observed a 37% decrease in 3‐methylhistidine (Figure 3 A, P = 0.072), an indicator of skeletal muscle protein breakdown30 in the 4E‐BP1/2 DKO muscle. In addition, the intracellular content of several dipeptides, which are incomplete breakdown products of protein catabolism, was also significantly reduced in 4E‐BP1/2 DKO muscle (Figure S2). To address whether amino acid transport was modified by the deletion of the 4E‐BP1 and 4E‐BP2, we measured mRNA levels of L‐type amino acid transporters (LAT1 and 2) and sodium‐coupled neutral amino acid transporters (SNAT1 and 2). Expression levels of LAT1, LAT2, SNAT1, and SNAT2 were unaffected within the 4E‐BP1/2 DKO muscle (Figure 3 B). To understand whether amino acid catabolism was altered in 4E‐BP1/2 DKO muscle, mRNA levels of genes involved in branched‐chain amino acid catabolism (BCAT2, BCKDHA, BCKDHB, DBT, and DLD) were measured. mRNA levels of these genes, likewise, were also unaltered in the 4E‐BP1/2 DKO muscle (Figure 3 C).
Figure 3.
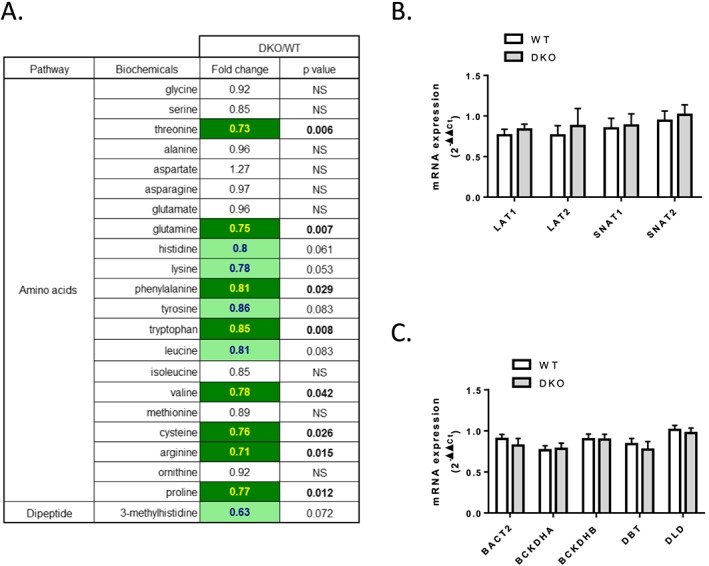
Free amino acid content is reduced in 4E‐BP1/2 DKO skeletal muscle. (A) Statistical heat map displaying the fold‐change values observed when amino acid abundances were compared between the WT and 4E‐BP1/2 DKO quadriceps of 24‐month‐old mice. The dark green colour is used to indicate statistically significant decreases P < 0.05, while the light green indicates differences that trended towards significance with a 0.05 < P < 0.1. (B) Real‐time PCR quantification of amino acid transporter mRNA expression in quadriceps from WT and 4E‐BP1/2 DKO 24‐month‐old mice. (C) Real‐time PCR quantification of mRNA expression of genes involved in branched‐chain amino acid catabolism in quadriceps from WT and 4E‐BP1/2 DKO 24‐month‐old mice. n = 6 in each genotype.
Carbohydrate metabolism in skeletal muscle
Several carbohydrates were found to differ across the WT and 4E‐BP1/2 DKO muscles (Figure S3). Although glucose levels were unchanged between WT and 4E‐BP1/2 DKO muscles, there were appreciable differences in select metabolites reporting on glycolysis and sugar metabolism. We observed a 52% reduction in glucose 6‐phosphate levels (P < 0.05) and a slight decrease in lactate (P = 0.08). In addition, compared with WT muscle, the 4E‐BP1/2 DKO muscle also exhibited decreases in the levels of galactose 1‐phosphate (−47%, P < 0.05), galacticol (−14%, P < 0.05), and mannose 6‐phosphate (P < 0.05). Only minor changes in the TCA cycle were observed, with citrate (+14%, P = 0.07) and aconitate (+21%, P = 0.07) being slightly elevated. Lastly, we observed a 33‐fold increase in the accumulation of sugar alcohols (maltitol/lactitol/cellobiotol/palatinol, P < 0.001) in the 4E‐BP1/2 DKO muscle. This compound is isobaric in nature in that we cannot distinguish between maltitol/lactitol/cellobiotol/palatinol on the basis of their mass spectrometry signatures. Therefore, this measurement can be contributed by one or all the above sugars. Altogether, these changes may suggest potential changes in carbohydrate intake and utilization in muscle.
Fatty acid metabolism
The sum of free fatty acid, diacylglycerol, and triacylglycerol contents was unchanged between the WT and 4E‐BP1/2 DKO muscles. However, compared with WT muscle, the 4E‐BP1/2 DKO muscle showed increased levels of several diacylglycerol and triacylglycerol species (Figure S4) and a specific increase in polyunsaturated free fatty acid (PUFA) content (Figure 4 A) as exemplified by the increases observed in arachidonic acid (Free Fatty Acid (FFA) 20:4, +87%, P < 0.05), docosapentaenoic acid (FFA 22:5, +44%, P < 0.05), and docosahexaenoic acid (FFA 22:6, +34%, P < 0.05). mRNA expression levels of CD36, FATP1, and LPL, involved in fatty acid transport, were unchanged in 4E‐BP1/2 DKO muscle compared with WT muscle (Figure 4 B). Despite unchanged carnitine levels, 4E‐BP1/2 DKO muscle showed increased levels of several short‐chain and medium‐chain acylcarnitines (Figure 5 A). We observed an accumulation of C5, C6, C8, and C10 species and a reduced C2 (acetylcarnitine) to carnitine ratio (Figure 5 B), indicative of a lower rate of β‐oxidation.31 The 4E‐BP1/2 DKO mice demonstrated a significantly higher respiratory quotient (calculated as the ratio of VO2 to carbon dioxide production) than did the WT mice (0.863 ± 0.018 vs. 0.808 ± 0.013, respectively, P < 0.05, Figure 5 D), which also indicate a reduced utilization of fat as fuel source. No differences in any of the long‐chain acylcarnitine species were noted between the WT and 4E‐BP1/2 DKO muscles (Figure 5 A). mRNA expression level of CPT1B was unchanged, whereas MCAD mRNA level was reduced by ≈30% (P < 0.05) in 4E‐BP1/2 DKO muscle compared with WT muscle (Figure 5 C).
Figure 4.
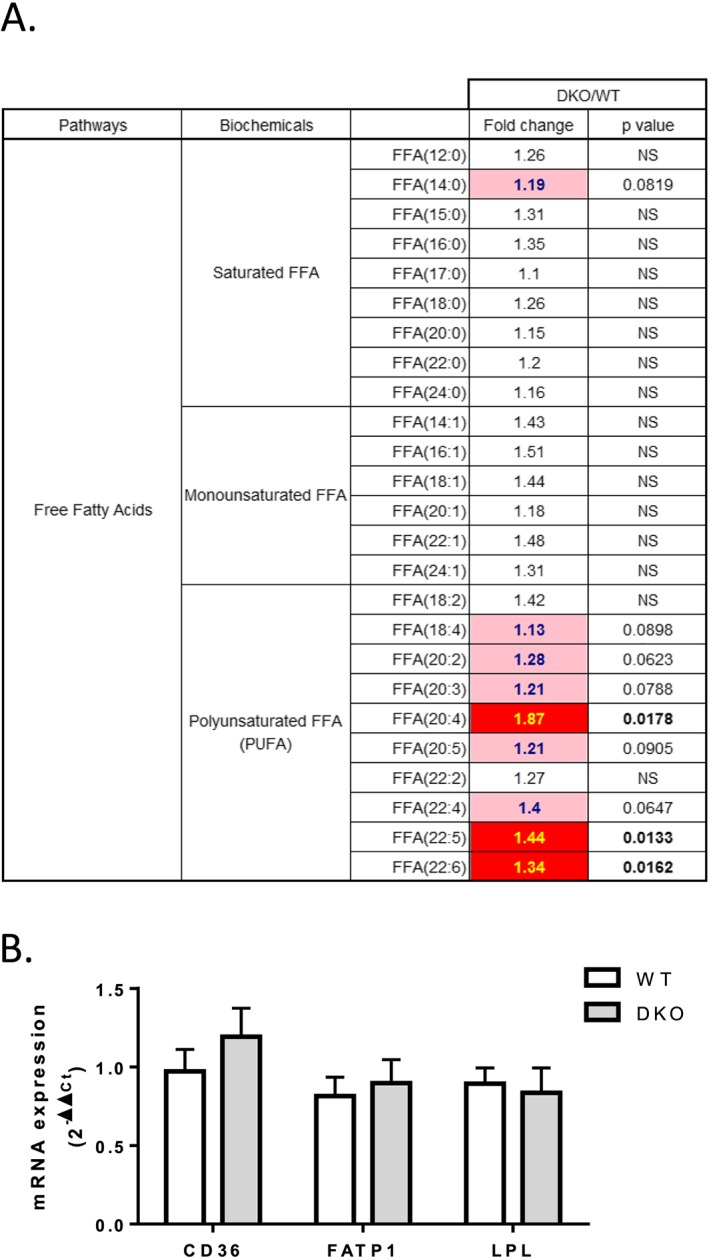
Loss of 4E‐BP1 and 4E‐BP2 induces accumulation of PUFA in skeletal muscle. (A) Statistical heat map displaying the fold‐change values observed when saturated, monounsaturated, and polyunsaturated free fatty acid abundance was compared between the quadriceps of 24‐month‐old WT and 4E‐BP1/2 DKO mice. The dark red colour is used to indicate statistically significant increases P < 0.05, while the light pink indicates differences that trended towards significance with a 0.05 < P < 0.1. (B) Real‐time PCR quantification of fatty acid transporters mRNA expression in quadriceps from WT and 4E‐BP1/2 DKO 24‐month‐old mice. n = 6 in each genotype.
Figure 5.
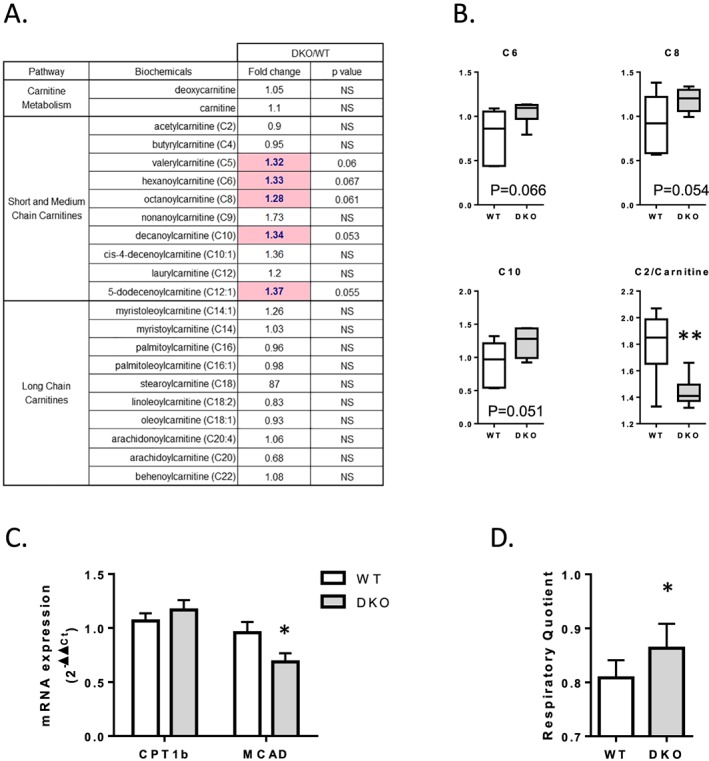
Loss of 4E‐BP1 and 4E‐BP2 alters β‐oxidation in skeletal muscle. (A) Statistical heat map displaying the fold‐change values observed when short‐chain, medium‐chain, and long‐chain acylcarnitine abundance was compared between the quadriceps of 24‐month‐old WT and 4E‐BP1/2 DKO mice. The light pink indicates increases that trended towards significance with a 0.05 < P < 0.1. (B) Boxplots are shown for medium‐chain acylcarnitines and for the C2/carnitine ratio. (C) Real‐time PCR quantification of CPT1b and MCAD mRNA expression in quadriceps from WT and 4E‐BP1/2 DKO 24‐month‐old mice. (D) Respiratory quotient calculated as the ratio of VO2 to carbon dioxide production in WT and 4E‐BP1/2 DKO 24‐month‐old mice. *P < 0.05 vs. WT, **P < 0.01 vs. WT. n = 6 in each genotype.
Discussion
Aging is characterized by a progressive loss of physiological functions with particular changes in the locomotor apparatus. Impairment of the musculoskeletal system is characterized by loss of muscle mass and function, commonly known as sarcopenia.13 The pathophysiology of sarcopenia is complex and, amongst other phenomena, involves increased muscle proteolysis,18 reduced muscle protein synthesis,19, 20, 32 and resistance to anabolic stimuli including insulin and amino acids.21, 23, 33 In the present study, we demonstrate that deletion of the 4E‐BP1 and 4E‐BP2 is associated with increased muscle mass and function as well as protein synthesis in 24‐month‐old mice. Global metabolomic and complex lipid analysis of WT and 4E‐BP1/2 DKO skeletal muscle revealed robust differences in amino acid homeostasis, carbohydrate abundance, and certain aspects of lipid metabolism.
In previous studies, it was observed that deletion of 4E‐BP1 and 4E‐BP2 resulted in reduced energy expenditure,7, 8 whereas energy expenditure is unaltered in the present study. An important difference between these previous studies and the current one is the age of the mice (24 vs. 6 months), and this difference might in part explain the absence of modification of energy expenditure we are observing. Indeed, in humans, it is known that total energy expenditure is declining from adulthood to old age.34, 35 This decline is closely linked with the decrease in whole body fat‐free mass, which is composed of metabolically active tissues, i.e. using a large part of body energy.36 Amongst them, skeletal muscle is the main energy consuming tissue, and aging is associated with a loss of muscle mass, called sarcopenia. In a recent study,8 we measured muscle mass in 6‐month‐old mice. Total hindlimb skeletal mass was 418 ± 5 and 432 ± 3 mg in WT and 4E‐BP1/2 DKO mice, respectively. Compared with the present results in 24‐month‐old mice, i.e. 340 ± 9 and 409 ± 6 mg in WT and 4E‐BP1/2 DKO mice, respectively, we can observe that the aging‐associated loss of muscle is more pronounced in WT (−20%) than in 4E‐BP1/2 DKO mice (−6%). Therefore, the maintained muscle mass observed in aged 4E‐BP1/2 DKO mice, as compared with WT mice, might have slowed down the energy expenditure decline.
The link between mTOR activity and muscle development is largely documented. Hyperactivation of mTORC1 by loss of negative regulators, such as TSC1 or NPRL2, is able to induce muscle hypertrophy.37, 38 Conversely, muscle‐specific inactivation of mTOR leads to severe myopathy,39 and inactivation of mTORC1 activity by deletion of Raptor or S6K1 is able to prevent muscle growth and enhance atrophy.38, 40 In a previous study, we observed that under high fat diet feeding, despite the development of a more severe insulin resistance, 4E‐BP1/2 DKO adult mice showed increased muscle mass.8 In the present study, using EDL muscles, we observed that protein synthesis was increased under basal conditions and after stimulation with a mix of leucine and insulin in 4E‐BP1/2 DKO mice. This result is in line with previous results where global protein synthesis was found to be increased in vitro in 4E‐BP1/2 DKO haematopoietic stem cells41 and primary fibroblasts.8 In another study using 4E‐BP1/2 DKO mice, Steiner et al. observed that basal skeletal muscle protein synthesis was unchanged in 4E‐BP1/2 DKO mice.42 The discrepancy between this last study and ours is unclear but could be due to the different models used. We measured protein synthesis ex vivo in 24‐month‐old mice, whereas Steiner et al. measured it in vivo in young mice. As impaired protein synthesis is known to be associated with the development of sarcopenia, it is conceivable that the increased protein synthesis we measured in 4E‐BP1/2 DKO muscle in aged mice reflects life‐long maintained protein synthesis in 4E‐BP1/2 DKO mice compared with WT mice in which aging induces a gradual decline in protein synthesis. A longitudinal animal study designed to characterize protein synthesis throughout the life course would be required to validate this hypothesis. In a recent study, Tsai et al.10 demonstrated that the overexpression of a constitutively active form of 4E‐BP1 was associated with reduced running capacity on treadmill and reduced muscle fibre size. This data are in agreement with the increased muscle strength and suggestive of an increased muscle fibre size in aged 4E‐BP1/2 DKO mice. However, the increased muscle mass and grip strength that we documented in aged 4E‐BP1/2 DKO mice are correlative in nature. No measures of muscle‐specific force production, muscle cross‐sectional area, or fibre‐type composition are presented and would be required to further understand the mechanisms responsible for this muscle phenotype.
To further analyse the muscle changes associated with aging, we conducted global metabolomic profiling of WT and 4E‐BP1/2 DKO quadriceps muscle derived from overnight‐fasted mice. This analysis revealed alterations in free amino acid content, as well as in metabolites reporting on energy metabolism. We observed an accumulation of polyunsaturated free fatty acids in 4E‐BP1/2 DKO muscle together with increased levels of short‐chain and medium‐chain acylcarnitines (Figure 5 A and B). In skeletal muscle, fatty acid synthesis is practically absent. Therefore, the increased polyunsaturated fatty acid content in 4E‐BP1/2 DKO skeletal muscle could be reflective of increased fatty acid uptake or reduced β‐oxidation. Short‐term fasting, such as an overnight fast, is known to increase lipolysis and β‐oxidation43; acylcarnitines are markers of mitochondrial lipid overload and high rates of mitochondrial β‐oxidation but are also indicative of incomplete β‐oxidation. In myotubes from donors with high fasting respiratory quotient, the production and release of medium‐chain acylcarnitines are higher, while the C2 (acetylcarnitine) to carnitine ratio, an index of β‐oxidation,31 is lowered.44 The accumulation of C5–C10 acylcarnitines, the lower C2/carnitine ratio in 4E‐BP1/2 DKO muscle, and the increased respiratory quotient of 4E‐BP1/2 DKO mice are consistent with such reduced β‐oxidation capacity. CPT1B is the rate‐controlling enzyme in the long‐chain fatty acid β‐oxidation pathway in muscle mitochondria, and MCAD is responsible for oxidizing C4 to C12 acyl‐CoA. CPT1B mRNA level was unchanged in 4E‐BP1/2 DKO muscle, suggesting that transport of fatty acids from the cytosol to the mitochondrial matrix, where β‐oxidation takes place, is unimpaired by the deletion of 4E‐BPs. The reason for the reduced expression mRNA level of MCAD in 4E‐BP1/2 DKO muscle is not clear, but it may account for the increased accumulation of medium‐chain acylcarnitines and the reduced rate of β‐oxidation. In vitro, inhibition of mTORC1 with rapamycin stimulates β‐oxidation in L6 muscle cell line.45 Conversely, constitutive activation of mTORC1 in mouse liver reduces the ability of hepatocytes to stimulate β‐oxidation in response to fasting, whereas loss of raptor (mimicking an mTORC1 inactivation) has the opposite effect.46 In liver, mTORC1 inhibits the activity of PPARα, a key transcription factor in the regulation of β‐oxidation, by promoting the nuclear accumulation of nuclear receptor corepressor 1 (NCoR1), which in turn interacts with nuclear receptors to inhibit their activity. When activated, mTORC1 favours the recruitment of NCoR1 to the nucleus leading to inhibition of PPARα activity.47 These data are in agreement with the reduction of β‐oxidation that we observed in 4E‐BP1/2 DKO skeletal muscle of aged mice. Whether this reduced β‐oxidation is mediated by altered PPARα activity and NCoR1 recruitment to the nucleus remains to be studied.
In addition to their role in the control of translation initiation, it was demonstrated that 4E‐BPs are important mediators of mTORC1‐dependent control of mitochondrial biogenesis and function.48 Indeed, mTORC1 inhibition is associated with impaired mitochondrial function, glycolysis (i.e. intracellular accumulation of glucose and decreased levels of pyruvate and lactate), and a significant decrease in several TCA cycles intermediates (i.e. citrate, α‐ketoglutarate, and succinate) in MCF7 cells.48 Although mitochondrial respiration is unaffected in 4E‐BP‐depleted untreated MCF7 cells, loss of 4E‐BPs relieves the effects of mTORC1 inhibition on mitochondrial respiration, glycolysis, and Krebs cycle. In the present study, mTORC1 activity was not inhibited by rapamycin, and mitochondrial respiration was not measured in skeletal muscle. However, we observed a slight reduction in intracellular glucose and pyruvate and reduced levels of lactate, which are suggestive of an increased glycolysis rate in 4E‐BP1/2 DKO skeletal muscle. We also observed a slight increase in TCA cycle intermediates (i.e. citrate, isocitrate, and aconitate). Altogether, these data are in agreement with the 4E‐BP‐mediated role of mTORC1 in the control of glycolysis and Krebs cycle.
Skeletal muscle content of almost all free amino acids was reduced in the 4E‐BP1/2 DKO mice. GATOR1 is a multi‐protein complex consisting of NPRL2, NPRL3, and DEPDC5, which inhibits mTORC1 activity during amino acid limitation.49, 50 In a recent study where NPRL2 was specifically deleted in skeletal muscle (NPRL2 mKO), Dutchak et al. showed that loss of NPRL2 and GATOR1 function in skeletal muscle induces constitutive activation of mTORC1 and results in larger muscle fibres.37 Muscle deletion of NPRL2 induces aerobic glycolysis, suppresses glucose entry into the TCA cycle, and is associated with reduced level of several amino acids. In that study, proteomic analysis revealed increased abundance of enzymes required for glutamine and aspartate synthesis (precursors of purine and pyrimidine synthesis) and a striking enrichment in proteins involved in amino acid catabolism and fatty acid oxidation in NPRL2 mKO soleus. These data indicate that activation of mTORC1 promotes aerobic glycolysis and mitochondrial activity for biosynthetic purposes, such as synthesis of the non‐essential amino acids. This ultimately leads to a compensatory response in the fatty acid and amino acid catabolism pathways to replenish the TCA cycle intermediates for mitochondrial energy production.37 It is unlikely that reduction of free amino acids in 4E‐BP1/2 DKO muscle comes from a reduced amino acid uptake from the blood, because food intake and mRNA expression levels of several amino acid transporters in skeletal muscle were similar between the WT and 4E‐BP1/2 DKO mice (Figure 3 B). Furthermore, such a decrease in free amino acid content was also observed in 4E‐BP1/2 DKO MEFs in vitro (personal data). The increased protein synthesis we measured in 4E‐BP1/2 DKO muscle may explain the decreased amino acid content, but because protein synthesis is a process requiring large amounts of energy, it is also conceivable that a significant proportion of these amino acids is used as substrate to maintain energy production and compensate for the decreased β‐oxidation of fatty acids.
A limitation of our study was the use of a whole body KO model for 4E‐BP1 and 4E‐BP2 deletion. Therefore, we cannot exclude that some of the metabolic changes we documented in skeletal muscles are indirect as changes in other tissues may have an effect on muscle metabolism. Indeed, it is largely documented that organ cross‐talk is important to consider in the control of physiological and metabolic processes. For example, circulating lipids such as free fatty acids, from adipose tissue and short‐chain fatty acids from the gut, are able to act on liver and skeletal muscle to regulate insulin sensitivity.51 In addition to these circulating factors, emerging evidence also indicates that exosomes (small extracellular vesicles produced by tissues) play a central role in the inter‐organ dialogue by transporting bioactive proteins, lipids, mRNAs, and miRNAs that can be transferred to adjacent cells or to distant organs.52 Further experiments, using muscle‐specific deletion or muscle‐specific overexpression of 4E‐BPs, will be required to fully understand the role of these proteins in the maintenance of muscle mass/function in aging.
In conclusion, we demonstrated that a deficiency of both 4E‐BP1 and 4E‐BP2 contributes to the maintenance of muscle mass and function in aging through increased protein synthesis. Through metabolomic and global lipid analysis, we also demonstrated that 4E‐BPs play a role in the control of energy and amino acid metabolism in the skeletal muscle of 24‐month‐old mice. Thus, 4E‐BPs may be potential drug targets in the treatment of sarcopenia.
Conflict of interest
The authors declare they have no conflict of interest.
Supporting information
Data S1 Supporting Information
Acknowledgements
This study was supported by funding from the Région Auvergne and from the AlimH Department, INRA, to O.L.B. We thank Dr Nahum Sonenberg (McGill University, Montréal, Canada) for providing the 4E‐BP1/2 DKO mice. We thank Julien Hermet and Medhi Djelloul From the Unité d'Expérimentation en Nutrition (INRA Clermont‐Ferrand‐Theix, France) for excellent assistance with animal care and for maintaining the mouse colonies. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.53
Le Bacquer O., Combe K., Patrac V., Ingram B., Combaret L., Dardevet D., Montaurier C., Salles J., Giraudet C., Guillet C., Sonenberg N., Boirie Y., and Walrand S. (2019) 4E‐BP1 and 4E‐BP2 double knockout mice are protected from aging‐associated sarcopenia, Journal of Cachexia, Sarcopenia and Muscle, 10, 696–709, doi: 10.1002/jcsm.12412.
References
- 1. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017;168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC Jr, et al. Insulin‐dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′‐cap function. Nature 1994;371:762–767. [DOI] [PubMed] [Google Scholar]
- 3. Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. 4E‐BP3, a new member of the eukaryotic initiation factor 4E‐binding protein family. J Biol Chem 1998;273:14002–14007. [DOI] [PubMed] [Google Scholar]
- 4. Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap‐dependent translation by 4E‐binding protein 1: competition with p220 for binding to eukaryotic initiation factor‐4E. EMBO J 1995;14:5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E‐BP1 phosphorylation: a novel two‐step mechanism. Genes Dev 1999;13:1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsukiyama‐Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E‐BP1. Nat Med 2001;7:1128–1132. [DOI] [PubMed] [Google Scholar]
- 7. Le Bacquer O, Petroulakis E, Paglialunga S, Poulin F, Richard D, Cianflone K, et al. Elevated sensitivity to diet‐induced obesity and insulin resistance in mice lacking 4E‐BP1 and 4E‐BP2. J Clin Invest 2007;117:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Bacquer O, Combe K, Montaurier C, Salles J, Giraudet C, Patrac V, et al. Muscle metabolic alterations induced by genetic ablation of 4E‐BP1 and 4E‐BP2 in response to diet‐induced obesity. Molecular nutrition & food research 2017;61: 10.1002/mnfr.201700128. [DOI] [PubMed] [Google Scholar]
- 9. Tsai SY, Rodriguez AA, Dastidar SG, Del Greco E, Carr KL, Sitzmann JM, et al. Increased 4E‐BP1 expression protects against diet‐induced obesity and insulin resistance in male mice. Cell Rep 2016;16:1903–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsai S, Sitzmann JM, Dastidar SG, Rodriguez AA, Vu SL, McDonald CE, et al. Muscle‐specific 4E‐BP1 signaling activation improves metabolic parameters during aging and obesity. J Clin Invest 2015;125:2952–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forbes GB, Reina JC. Adult lean body mass declines with age: some longitudinal observations. Metabolism: Clinical and Experimental 1970;19:653–663. [DOI] [PubMed] [Google Scholar]
- 12. Nair KS. Aging muscle. Am J Clin Nutr 2005;81:953–963. [DOI] [PubMed] [Google Scholar]
- 13. Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010;39:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boirie Y. Physiopathological mechanism of sarcopenia. J Nutr Health Aging 2009;13:717–723. [DOI] [PubMed] [Google Scholar]
- 15. Deutz NE, Bauer JM, Barazzoni R, Biolo G, Boirie Y, Bosy‐Westphal A, et al. Protein intake and exercise for optimal muscle function with aging: recommendations from the ESPEN Expert Group. Clin Nutr 2014;33:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Short KR, Bigelow ML, Kahl J, Singh R, Coenen‐Schimke J, Raghavakaimal S, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 2005;102:5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson ML, Robinson MM, Nair KS. Skeletal muscle aging and the mitochondrion. Trends in endocrinology and metabolism: TEM 2013;24:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Combaret L, Dardevet D, Bechet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Curr Opin Clin Nutr Metab Care 2009;12:37–41. [DOI] [PubMed] [Google Scholar]
- 19. Dardevet D, Sornet C, Balage M, Grizard J. Stimulation of in vitro rat muscle protein synthesis by leucine decreases with age. J Nutr 2000;130:2630–2635. [DOI] [PubMed] [Google Scholar]
- 20. Katsanos CS, Kobayashi H, Sheffield‐Moore M, Aarsland A, Wolfe RR. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. Am J Clin Nutr 2005;82:1065–1073. [DOI] [PubMed] [Google Scholar]
- 21. Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, et al. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:422–424. [DOI] [PubMed] [Google Scholar]
- 22. Guillet C, Boirie Y. Insulin resistance: a contributing factor to age‐related muscle mass loss? Diabetes & Metabolism 2005;31 Spec:5S20–5S26. [DOI] [PubMed] [Google Scholar]
- 23. Dardevet D, Remond D, Peyron MA, Papet I, Savary‐Auzeloux I, Mosoni L. Muscle wasting and resistance of muscle anabolism: the “anabolic threshold concept” for adapted nutritional strategies during sarcopenia. The Scientific World Journal 2012;2012:269531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tardif N, Salles J, Guillet C, Tordjman J, Reggio S, Landrier JF, et al. Muscle ectopic fat deposition contributes to anabolic resistance in obese sarcopenic old rats through eIF2alpha activation. Aging Cell 2014;13:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masgrau A, Mishellany‐Dutour A, Murakami H, Beaufrere AM, Walrand S, Giraudet C, et al. Time‐course changes of muscle protein synthesis associated with obesity‐induced lipotoxicity. J Physiol 2012;590:5199–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carlson CG, Rutter J, Bledsoe C, Singh R, Hoff H, Bruemmer K, et al. A simple protocol for assessing inter‐trial and inter‐examiner reliability for two noninvasive measures of limb muscle strength. J Neurosci Methods 2010;186:226–230. [DOI] [PubMed] [Google Scholar]
- 27. Dardevet D, Sornet C, Taillandier D, Savary I, Attaix D, Grizard J. Sensitivity and protein turnover response to glucocorticoids are different in skeletal muscle from adult and old rats. Lack of regulation of the ubiquitin‐proteasome proteolytic pathway in aging. J Clin Invest 1995;96:2113–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waalkes TP, Udenfriend S. A fluorometric method for the estimation of tyrosine in plasma and tissues. J Lab Clin Med 1957;50:733–736. [PubMed] [Google Scholar]
- 29. Tischler ME, Desautels M, Goldberg AL. Does leucine, leucyl‐tRNA, or some metabolite of leucine regulate protein synthesis and degradation in skeletal and cardiac muscle? J Biol Chem 1982;257:1613–1621. [PubMed] [Google Scholar]
- 30. Long CL, Dillard DR, Bodzin JH, Geiger JW, Blakemore WS. Validity of 3‐methylhistidine excretion as an indicator of skeletal muscle protein breakdown in humans. Metabolism 1988;37:844–849. [DOI] [PubMed] [Google Scholar]
- 31. Fingerhut R, Roschinger W, Muntau AC, Dame T, Kreischer J, Arnecke R, et al. Hepatic carnitine palmitoyltransferase I deficiency: acylcarnitine profiles in blood spots are highly specific. Clin Chem 2001;47:1763–1768. [PubMed] [Google Scholar]
- 32. Dardevet D, Sornet C, Bayle G, Prugnaud J, Pouyet C, Grizard J. Postprandial stimulation of muscle protein synthesis in old rats can be restored by a leucine‐supplemented meal. J Nutr 2002;132:95–100. [DOI] [PubMed] [Google Scholar]
- 33. Guillet C, Prod'homme M, Balage M, Gachon P, Giraudet C, Morin L, et al. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. FASEB J 2004;18:1586–1587. [DOI] [PubMed] [Google Scholar]
- 34. Manini TM. Energy expenditure and aging. Ageing Res Rev 2010;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poehlman ET. Regulation of energy expenditure in aging humans. J Am Geriatr Soc 1993;41:552–559. [DOI] [PubMed] [Google Scholar]
- 36. Poehlman ET, Goran MI, Gardner AW, Ades PA, Arciero PJ, Katzman‐Rooks SM, et al. Determinants of decline in resting metabolic rate in aging females. Am J Physiol 1993;264:E450–E455. [DOI] [PubMed] [Google Scholar]
- 37. Dutchak PA, Estill‐Terpack SJ, Plec AA, Zhao X, Yang C, Chen J, et al. Loss of a negative regulator of mTORC1 induces aerobic glycolysis and altered fiber composition in skeletal muscle. Cell Rep 2018;23:1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bentzinger CF, Lin S, Romanino K, Castets P, Guridi M, Summermatter S, et al. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skeletal muscle 2013;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 2009;187:859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, et al. Atrophy of S6K1(−/−) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol 2005;7:286–294. [DOI] [PubMed] [Google Scholar]
- 41. Signer RA, Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, et al. The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E‐BPs. Genes Dev 2016;30:1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steiner JL, Pruznak AM, Deiter G, Navaratnarajah M, Kutzler L, Kimball SR, et al. Disruption of genes encoding eIF4E binding proteins‐1 and ‐2 does not alter basal or sepsis‐induced changes in skeletal muscle protein synthesis in male or female mice. PloS one 2014;9:e99582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soeters MR, Sauerwein HP, Duran M, Wanders RJ, Ackermans MT, Fliers E, et al. Muscle acylcarnitines during short‐term fasting in lean healthy men. Clin Sci (Lond) 2009;116:585–592. [DOI] [PubMed] [Google Scholar]
- 44. Wolf M, Chen S, Zhao X, Scheler M, Irmler M, Staiger H, et al. Production and release of acylcarnitines by primary myotubes reflect the differences in fasting fat oxidation of the donors. J Clin Endocrinol Metab 2013;98:E1137–E1142. [DOI] [PubMed] [Google Scholar]
- 45. Sipula IJ, Brown NF, Perdomo G. Rapamycin‐mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism: clinical and experimental. 2006;55:1637–1644. [DOI] [PubMed] [Google Scholar]
- 46. Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting‐induced ketogenesis and its modulation by ageing. Nature 2010;468:1100–1104. [DOI] [PubMed] [Google Scholar]
- 47. Kim K, Pyo S, Um SH. S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator‐activated receptor alpha activity in the liver. Hepatology 2012;55:1727–1737. [DOI] [PubMed] [Google Scholar]
- 48. Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab 2013;18:698–711. [DOI] [PubMed] [Google Scholar]
- 49. Bar‐Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340:1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Panchaud N, Peli‐Gulli MP, De Virgilio C. Amino acid deprivation inhibits TORC1 through a GTPase‐activating protein complex for the Rag family GTPase Gtr1. Science signaling 2013;6:ra42. [DOI] [PubMed] [Google Scholar]
- 51. Gancheva S, Jelenik T, Alvarez‐Hernandez E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev 2018;98:1371–1415. [DOI] [PubMed] [Google Scholar]
- 52. Guay C, Regazzi R. Exosomes as new players in metabolic organ cross‐talk. Diabetes Obes Metab 2017;19:137–146. [DOI] [PubMed] [Google Scholar]
- 53. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Information