

Abstract
Spermidine, a naturally occurring polyamine, has been recognized as a caloric restriction mimetic that confers health benefits, presumably by inducing autophagy. Recent studies have reported that oral administration of spermidine protects against liver fibrosis and hepatocarcinogenesis through activation of MAP1S-mediated autophagy. NRF2 is a transcription factor that mediates cellular protection by maintaining the cell’s redox, metabolic, and proteostatic balance. In this study, we demonstrate that spermidine is a non-canonical NRF2 inducer, and that MAP1S is a novel component of this non-canonical pathway of NRF2 activation. Mechanistically, MAP1S induces NRF2 signaling through two parallel mechanisms, both resulting in NRF2 stabilization: (i) MAP1S competes with KEAP1 for NRF2 binding via an ETGE motif, and (ii) MAP1S accelerates p62-dependent degradation of KEAP1 by the autophagy pathway. We further demonstrate that spermidine confers liver protection by enhancing NRF2 signaling. The importance of both NRF2 and p62-dependent autophagy in spermidine-mediated liver protection was confirmed using a carbon tetrachloride-induced liver fibrosis model in WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− mice, as the protective effect of spermidine was significantly reduced in NRF2 or p62 single knockout mice, and completely abolished in the double knockout mice. Our results demonstrate the pivotal role of NRF2 in mediating the health benefit of spermidine, particularly in the context of liver pathologies.
Keywords: NRF2, KEAP1, MAP1S, p62, autophagy, spermidine
Introduction
Liver fibrosis leads to liver scarring via excessive accumulation of extracellular matrix, which prevents the liver from performing its normal physiological functions (1). However, there are currently no effective treatments available for liver fibrosis except removal of the underlying etiology or ultimately liver transplantation. Therefore, the development of novel therapeutic strategies for prevention or reversal of liver fibrosis is urgently needed. Spermidine (SPD) is a polyamine compound (C7H19N3) originally isolated from semen. It was reported to promote longevity by upregulating autophagy in multiple models (2, 3). Furthermore, the beneficial effects of SPD in ameliorating liver diseases have been demonstrated in rodent models (4–6). Recently, Liu et al. reported that SPD conferred microtubule associated protein 1S (MAP1S)-dependent protection against carbon tetrachloride (CCl4)-induced liver fibrosis and N-nitrosodiethylamine (DEN)-induced hepatocellular carcinoma in mouse models through upregulation of MAP1S and autophagy (7).
MAP1S is the shortest member of the microtubule-associated protein (MAP) 1 family, and unlike the rest of the members of the family it is ubiquitously expressed (8). Additionally, MAP1S has been shown to interact with F-actin, microtubule-binding and stabilizing protein RASSF1A, Y-chromosome protein VCY2, mitochondrial LRPPRC, and MAP1LC3 (9–11). Therefore, MAP1S has emerged as a potential signal integrator that links the mitochondria, the autophagy pathway, and the cytoskeleton (9). Autophagy is a bulk degradation process that engulfs proteins, protein aggregates, lipids, or intracellular parasites into a double-membrane bound vesicle known as the autophagosome, which later fuses with the lysosome to form the autolysosome. Basal autophagy is required to recycle intracellular components and to eliminate unfolded proteins or defective organelles, such as mitochondria (12). Importantly, MAP1S has been demonstrated to activate autophagy to suppress tumorigenesis in renal, liver, and prostate cancers, and high levels of MAP1S are correlated with longer survival of patients (13–15).
The NRF2-KEAP1 signaling pathway is a cytoprotective and pro-survival pathway in mammalian cells (16, 17). NRF2 is a transcription factor that activates the expression of hundreds of genes, including phase II detoxification enzymes, antioxidant proteins, transporters, as well as carbohydrate and lipid metabolism enzymes (18). Under normal, basal conditions, NRF2 localizes to the cytosol where it binds to its repressor KEAP1, a substrate adaptor protein for a Cullin-3-based E3 ubiquitin ligase complex (19, 20). This association between NRF2 and KEAP1 results in ubiquitylation of NRF2 and subsequent degradation by the 26S proteasome (21). In its N-terminal NEH2 domain, NRF2 contains two motifs, DLG (weak binding) and ETGE (strong binding), that each interact with the Kelch domain of one KEAP1 molecule in what is known as the “hinge-and-latch” model of interaction (22, 23). There are two main mechanisms by which the NRF2 signaling pathway is activated. The canonical mechanism, which entails activation by cellular exposure to oxidants and electrophiles that modify key cysteine residues in KEAP1, results in a conformational change that releases binding to the weaker DLG motif and subsequent stabilization of NRF2 (24). The non-canonical mechanism involves autophagy deregulation, and it does not involve modification of KEAP1 cysteines (25–27). An important mediator of the non-canonical mechanism is p62 (also known as sequestosome 1, (SQSTM1)), which is a selective autophagy substrate that regulates the formation of protein aggregates and inclusion bodies by binding to ubiquitylated proteins and to the autophagy protein LC3 (28, 29). p62 contains an STGE motif in its C-terminus that binds the KEAP1 Kelch domain (25–27), and when autophagy flux is blocked, as in the case of arsenic-mediated disruption of autophagosome fusion with the lysosome, KEAP1 is sequestered into p62-positive inclusion bodies, resulting in stabilization of NRF2 (30). Conversely, in a scenario where autophagy is activated, KEAP1 is degraded together with p62 through the autophagy pathway, which also upregulates the protein level of NRF2 (31, 32).
In the present study, we demonstrate that SPD is a non-canonical NRF2 inducer and that MAP1S is a novel component of this non-canonical pathway of NRF2 activation. SPD induces the NRF2 pathway through two parallel MAP1S-dependent mechanisms, both resulting in NRF2 stabilization: (i) MAP1S competes with KEAP1 for NRF2 binding via an ETGE motif, and (ii) MAP1S accelerates p62-dependent KEAP1 degradation by the autophagy pathway. Furthermore, a pivotal role of NRF2 in SPD-mediated protection against liver fibrosis was demonstrated using a chemical-induced liver fibrosis model in WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− mice, as the protective effect of SPD was significantly reduced in Nrf2 or p62 single knockout mice, and completely lost in double knockout mice.
Experimental Procedures
Chemicals, reagents and antibodies
All chemicals were purchased from Sigma-Aldrich: Spermidine (S0266), Bafilomycin A (B1793), and Carbon tetrachloride (289116). The Amplite™ Colorimetric Aspartate Aminotransferase (AST) Assay Kit (13801) and Amplite™ Colorimetric Alanine Aminotransferase (ALT) Assay Kit (13803) were purchased from AAT Bioquest. Antibodies against KEAP1 (sc-15246), NRF2 (sc-13032), p62 (sc-28359), NQO1 (sc-32793), α-SMA (sc-53142), COL1A1 (sc-293182), c-Myc (sc-40), GAPDH (sc-32233), as well as the mouse, goat and rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) were purchased from Santa Cruz Biotechnology. The antibody against MAP1S (ab175503) was purchased from Abcam. The antibody against LC3 (L8918) was purchased from Sigma. The antibody against HA (901502) was purchased from BioLegend. The antibody against CBD (E8034S) was from New England Biolabs.
Cell culture
HEK293 cells (ATCC, CRL-1573) and NCI-H1299 cells (ATCC, CRL-5803) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro, 10–013-CV) supplemented with 10% fetal bovine serum (HyClone, SH30910.03), and 1% penicillin/streptomycin (Thermo Fisher Scientific, 15140122). Hepatic stellate cells (HSCs) were isolated from mouse liver and cultured as reported previously (33) The cell purity was determined using GFAP immunofluorescence staining.
Generation of KEAP1−/− and p62−/− cells
KEAP1 knockout (KEAP1−/−) and p62 knockout (p62−/−) cells were generated using CRISPR-Cas9-mediated gene editing (34). A pair of single guide RNA (sgRNA) sequences was used to target coding sequences near the promoter region of each gene of interest. The sgRNA sequences used were as follows:
KEAP1: sgRNA-A 5’-AGCGTGCCCCGTAACCGCAT-3’
sgRNA-B 5’- GATCTACACCGCGGGCGGCT-3’
p62: sgRNA-A 5’-AATGGCCATGTCCTACGTGA −3’
sgRNA-B 5’-CGACTTGTGTAGCGTCTGCG −3’
Transfection
Cells were transfected with cDNA using Lipofectamine 3000 (Thermo Fisher Scientific, L3000150) according to manufacturer’s instructions. The plasmid containing full-length (FL) hMAP1S cDNA in pcDNA3 was a kind gift of Dr. Leyuan Liu (Texas A&M Health Science Center); the plasmid containing mRFP-GFP-LC3 was obtained from Dr. Eileen White (Cancer Institute of New Jersey, Rutgers). The human MAP1S-FL cDNAs were cloned into pCMV-HA vectors. KEAP1-CBD and c-Myc-p62 cDNAs were also cloned into pcDNA3 vectors. Generation of the hMAP1S-FL was done by site-directed mutagenesis as has been previously described (35).
Immunoprecipitation and immunoblot analyses
For immunoprecipitation, cells were harvested 24 hours after transfection in radio immunoprecipitation assay (RIPA) buffer. Cell lysates were incubated overnight at 4° C with HA beads (Sigma, A2095). Samples were eluted and boiled for 5 min. For immunoblots, cells were harvested in Sample buffer, and were immediately boiled for 5 min and then sonicated as our previous study (36). All samples were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane for immunoblot analysis.
Live cell immunofluorescence microscopy
NCI-H1299 cells or HSCs were grown in 35 mm glass-bottom dishes. Cells were transfected with different vectors using Lipofectamine 3000 for 24 hours, then either left untreated, or treated with 100 μM SPD for 4 or 16 hours. Finally, the cells were imaged in phenol red-free DMEM using a Zeiss Observer.Z1 microscope with the Slidebook 4.2.0.11 software (Intelligent Imaging Innovations).
Animal studies
All mice were handled according to the Guide for the Care and Use of Laboratory Animals, and the protocols were approved by the University of Arizona Institutional Animal Care and Use Committee. In addition, all animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985). Generation of the Map1s−/−, Nrf2−/− and p62−/− mice were described previously (9, 37, 38). Four genotypes of mice were used for experiments, Nrf2+/+;p62+/+ (wild type, WT), Nrf2−/−;p62+/+ (Nrf2−/−), Nrf2+/+;p62−/− (p62−/−), and Nrf2−/−;p62−/− mice, which were generated by breeding Nrf2+/− with p62+/− mice, both in the C57BL/6J background. Eight-week-old mice (25–27g) were randomly allocated to the Ctrl group, SPD group, CCl4 group and CCl4+SPD group. To induce liver fibrosis, the mice were injected with 5 μl [0% (Ctrl group and SPD group) or 10% (CCl4 group and CCl4+SPD group) CCl4 in corn oil]/g body weight twice a week for 5 weeks intraperitoneally. During this process, mice were fed with either normal water (Ctrl group and CCl4 group) or water containing 3 mM spermidine (SPD group and CCl4+SPD group). 5 weeks later, the mice in each group were weighed and sacrificed to collect liver tissue for RNA extraction, protein assays and tissue section as our previous study (36).
Statistical analysis
Results are presented as means ± SD for at least three independent experiments. Statistical analysis was performed using SPSS 17.0. Unpaired Student’s t-tests were applied to compare the means of two groups. One-way ANOVA with Bonferroni’s correction was used to compare the means of three or more groups. p<0.05 was considered statistically significant.
Results
SPD induces NRF2 in a KEAP1-dependent, but only partially p62-dependent manner
To determine the time and dose dependent effect of SPD on autophagy and NRF2 induction, immunoblot analysis was performed in H1299 cells treated with increasing doses of SPD for 4 hours (the optimal time for NRF2, MAP1S, LC3-I, and LC3-II; Figure 1A) and 16 hours (the optimal time for NRF2-target gene expression, such as NQO1 and FTL; Figure 1B). SPD resulted in a dose-dependent increase in the protein levels of MAP1S, p62, and NRF2, and a decrease in KEAP1 protein levels in WT cells. The expression of NQO1 and FTL was only upregulated at 16 hours, consistent with the timing of NRF2 target gene induction. In addition, the level of LC3-II, an autophagy marker, was also increased (Figure 1A–1B, Figure S1A–D), which is consistent with the previous reports that SPD upregulates autophagy. In p62−/− cells, SPD enhanced the MAP1S level to a similar degree as H1299-WT, however SPD-mediated upregulation of NRF2, NQO1 and FTL was diminished, and SPD had no effect on KEAP1 and LC3-II (Figure 1A–1B, Figure S1A–D). In KEAP1−/− cells, SPD increased MAP1S, p62 and LC3-II, but had no effect on NRF2, NQO1 and FTL (Figure 1A–1B, Figure S1A–D). These results suggest that SPD upregulates MAP1S upstream of KEAP1-mediated regulation of NRF2, and that the upregulation of NRF2 in response to SPD is KEAP1-dependent, but only partially p62-dependent.
Figure 1.
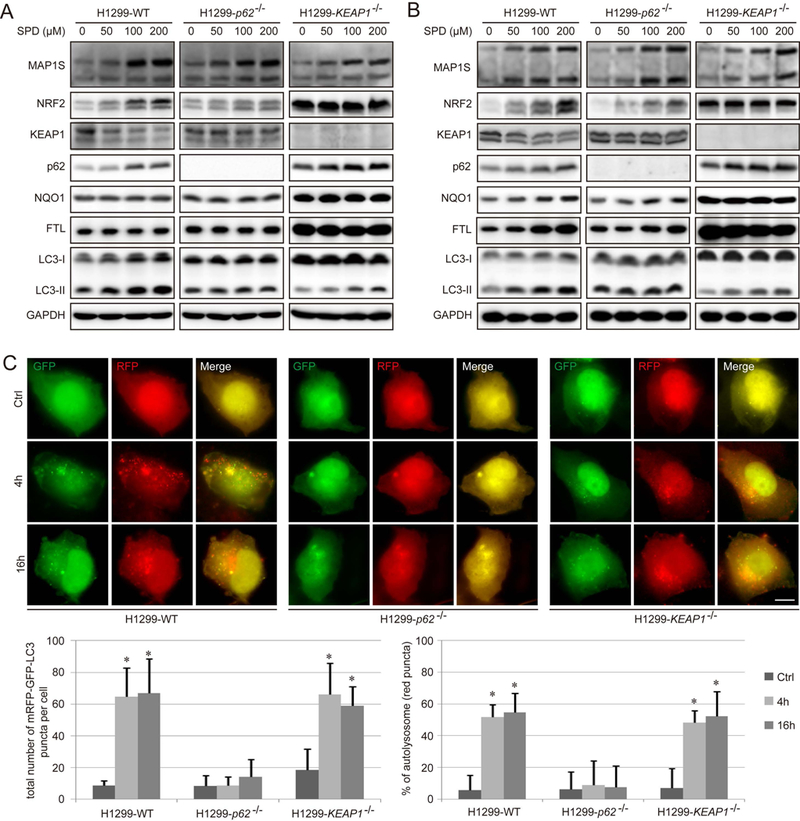
Spermidine increases the level of NRF2 via promoting autophagic degradation of KEAP1. A-B. Immunoblot analysis of NRF2 and autophagy in WT, p62−/−, and KEAP1−/− H1299 cells treated with the indicated concentrations of Spermidine (SPD) for 4 h (A) and 16 h (B). GAPDH was used as an internal loading control. Results are expressed as mean ± SD. A Student’s unpaired t-test was used to compare groups, and p<0.05 was considered statistically significant. *: p<0.05 compared with the control (0 μM) group. C. WT, p62−/−, and KEAP1−/− H1299 cells were transfected with 1 μg of mRFP-GFP-LC3 for 24 h, and then treated with SPD (100 μM) for 4 h and 16 h respectively, and imaged. Scale bar=10 μm. Bar graphs represent the total number of mRFP-GFP-LC3 positive puncta and % of autolysosomes (red puncta) per cell.
Next, the effects of SPD on autophagy induction were further tested using the tandem mRFP-GFP-LC3 reporter construct. The mRFP-GFP-LC3 reporter allows analysis of autophagy completion, as yellow puncta indicate autophagosomes; whereas red puncta indicate autolysosomes due to the quenching of GFP in this acidic autolysosomal environment. WT, p62−/−, and KEAP1−/− H1299 cells were transfected with the mRFP-GFP-LC3 construct and treated with SPD for 4 hours or 16 hours. Following treatment, red puncta together with some yellow puncta were observed in both WT and KEAP1−/− H1299 cells, but not p62−/− cells, confirming the important function of p62 in promoting SPD-mediated autophagosome formation (Figure 1C).
MAP1S binds to KEAP1 and p62
To further characterize the nature of the MAP1S, p62, and KEAP1 interaction, HEK293 cells were co-transfected with HA-MAP1S, CBD-KEAP1, and cMyc-p62, and protein-protein interactions were examined using immunoprecipitation/immunoblot analyses. HA-MAP1S co-immunoprecipitated with both CBD-KEAP1 and cMyc-p62 (Figure 2A). Furthermore, the binding between HA-MAP1S and CBD-KEAP1 was enhanced by overexpression of p62 (Figure 2A). However, MAP1S binding to p62 was not affected by overexpression of KEAP1 (Figure 2A). MAP1S contains an ETGE motif comprising amino acids 124 to 127 that is identical to the ETGE motif in NRF2 through which it binds to the Kelch domain of KEAP1. Thus, to investigate if the interaction between MAP1S and KEAP1 depends on this domain, an ETGE to EAGE MAP1S mutant (HA-MAP1S-Mu) was generated, as this single amino acid substitution in NRF2 is sufficient to disrupt NRF2 binding with the Kelch domain of KEAP1. Next, WT and p62−/− H1299 cells were cotransfected with CBD-KEAP1 and either HA-MAP1S-WT or HA-MAP1S-Mu for immunoprecipitation/immunoblot analysis. Both MAP1S-WT and MAP1S-Mu interact with KEAP1 in H1299-WT, although there was a reduction in co-immunoprecipitated KEAP1 with the mutant MAP1S (Figure 2B). However, in p62−/− H1299 cells, EAGE mutation completely abolished the binding between MAP1S and KEAP1 (Figure 2B). Furthermore, the interaction between MAP1S and p62 was detected not only in WT but also in KEAP1−/− cells, indicating that there is a KEAP1-independent interaction between MAP1S and p62. The EAGE mutation in MAP1S also weakened the binding between MAP1S and p62 in WT cells, but had no effect in KEAP1−/− cells (Figure 2C). Taken together, these results indicate that MAP1S interacts with KEAP1 through both a direct ETGE (in MAP1S)-Kelch (in KEAP1) interaction, and a p62-dependent indirect interaction (MAP1S-p62-KEAP1) (see Figure 8 for the interaction model).
Figure 2.
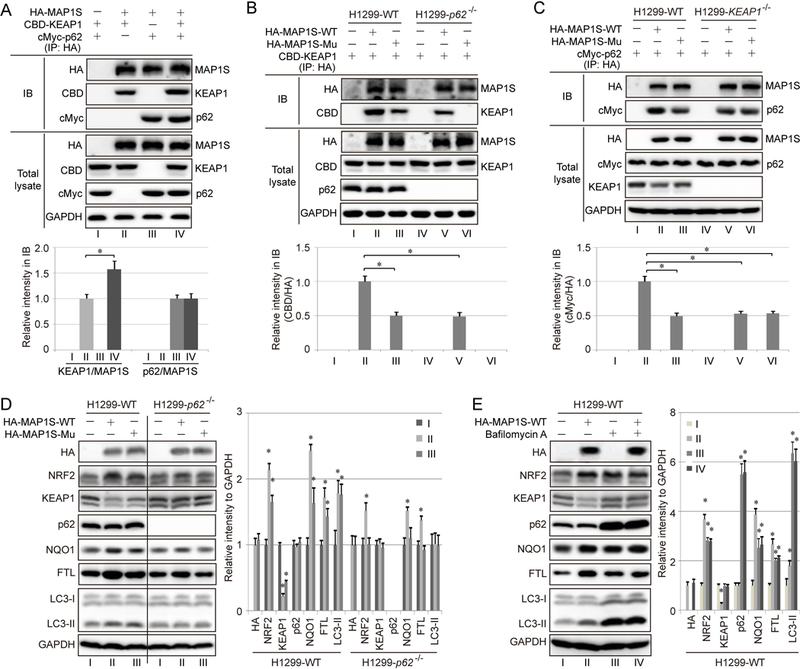
MAP1S binds to KEAP1 and p62. A. HEK293 cells were transfected with 1 μg of HA-MAP1S, 1 μg of CBD-KEAP1, and 1 μg of c-Myc-p62, and cell lysates were immunoprecipitated using anti-HA beads, resolved by SDS-PAGE, and subjected to immunoblot analysis. B-C. Immunoprecipitation analysis of cell lysates from WT and p62−/− (B) or WT and KEAP1−/− (C) H1299 cells transfected with CBD-KEAP1 alone or in combination with HA-MAP1S containing either wild type ETGE (HA-MAP1S-WT) or ETGE mutated to EAGE (HA-MAP1S-Mu). Complexes were co-immunoprecipitated using anti-HA beads, resolved by SDS-PAGE, and subjected to immunoblot analysis. D. WT and p62−/− H1299 cells were transfected with either HA-MAP1S-WT or HA-MAP1S-Mu and subjected to immunoblot analysis of key NRF2 and autophagy pathway proteins. E. WT H1299 cells were transfected with HA-MAP1S-WT and treated with 40 nM bafilomycin A1 (BAF) for 24 h. GAPDH was used as an internal loading control. Results are expressed as mean ± SD. *: p<0.05 compared with the control group.
Figure 8.
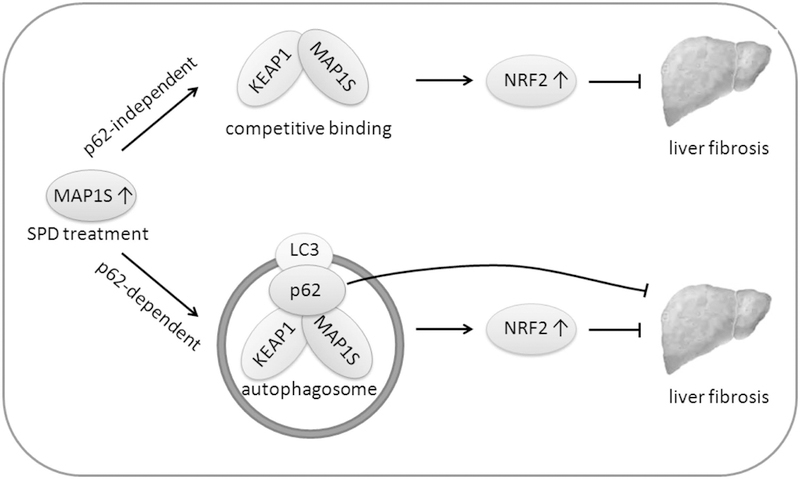
Proposed model for the therapeutic action of SPD against liver fibrosis. MAP1S levels can be increased by SPD treatment. The increased MAP1S can bind to KEAP1 via an ETGE motif. This competitive binding influences the NRF2-KEAP1 interaction resulting in a direct increase in NRF2 levels that is independent of p62. Upregulation of MAP1S also promotes autophagy resulting in the p62-dependent autophagic degradation of KEAP1, leading to the upregulation of NRF2. These pathways may act in parallel to promote liver protection.
To further evaluate the effect of MAP1S on NRF2, both H1299-WT and H1299-p62−/− cells were transfected with HA-MAP1S-WT or HA-MAP1S-Mu. Both MAP1S-WT and MAP1S-Mu increased the level of NRF2, NQO1 and FTL, possibly by promoting autophagic degradation of KEAP1, with the effect of MAP1S-Mu on NRF2 and NQO1 protein levels being weaker than MAP1S-WT. In addition, mutation of ETGE to EAGE in MAP1S weakened its effects on KEAP1, NRF2, NQO1 or FTL levels in WT cells, but completely abolished the effects in p62−/− cells (Figure 2D), supporting that the direct binding between MAP1S and KEAP1 requires an intact ETGE motif in MAP1S (Figure 2D, and the model in Figure 8). To validate that MAP1S promotes KEAP1 degradation via autophagy, Bafilomycin A1 (BAF, increases lysosomal pH) was used to block autophagy. As expected, there was a robust increase in the protein levels of p62 and LC3-II following BAF treatment. Importantly, BAF upregulated NRF2 protein levels compared to the untreated control, with MAP1S overexpression not causing a further increase in BAF-treated cells (Figure 2E).
SPD-mediated NRF2 induction depends on MAP1S
To confirm that SPD is able to induce NRF2 through upregulation of MAP1S, liver tissue was isolated from Map1s+/+ (WT) and Map1s −/− mice that received regular drinking water or drinking water with SPD (3 mM) for 5 weeks, and activation of NRF2 and the autophagy pathway was determined. As shown in Figure 3, SPD significantly upregulated MAP1S levels, however, SPD-mediated NRF2 induction and upregulation of NRF2 target genes (NQO1 and FTL) were only observed in WT, but not Map1s−/− mice. Similarly, SPD enhanced autophagy, as measured by increased LC3-I and LC3-II levels, in a MAP1S-dependent manner. These in vivo results are consistent with the data observed using cultured cells (Figure 1A–1B). However, unlike the short-term SPD treatment in the cell-based assays (Figure 1A–1B), the level of p62 in the liver tissues from mice treated with SPD for five weeks had no detectable changes (Figure 3A–3B).
Figure 3.
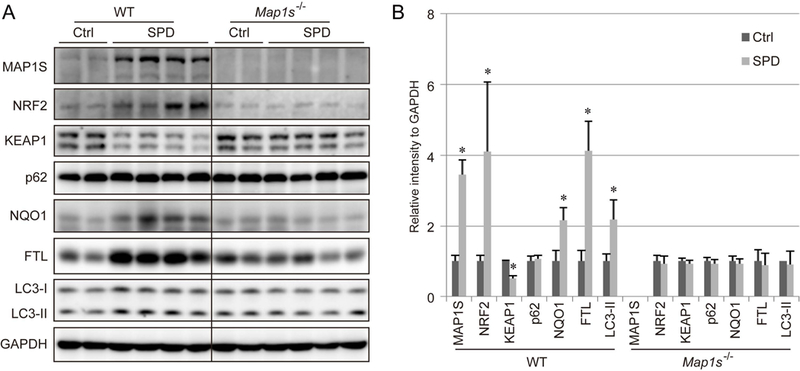
SPD-mediated NRF2 induction depends on MAP1S. A-B. Immunoblot analysis of MAP1S, as well as key NRF2 and autophagy pathway proteins in liver tissue isolated from Map1s+/+ (WT) and Map1s−/− mice that received regular drinking water or drinking water with SPD (3 mM) for 5 weeks. GAPDH was used as an internal loading control. The data (divided by black line) were from the same gel. Results are expressed as mean ± SD.
SPD ameliorates hepatic injury and liver fibrosis in vivo
Since increased levels of MAP1S resulted in the formation of a MAP1S-p62-KEAP1 complex, and we recently demonstrated that SPD ameliorates liver fibrosis via MAP1S-dependent autophagy (7), the role of p62 and NRF2 in mediating the protective effects of SPD in liver fibrosis was further assessed. Mice with four different genotypes (WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/−) received SPD (3 mM) through the drinking water and 10% CCl4 via intraperitoneal injection, twice a week for 5 weeks (Figure 4A). Following treatment, liver tissue was harvested and morphological changes were observed in sections stained by H&E. While SPD-treated groups had no morphological changes compared to the untreated Ctrl group, CCl4 exposure induced the formation of thick fibrotic septa (Figure 4B). Similar to the previous study mentioned above, SPD treatment showed an obvious protective effect against fibrosis in WT mice. In the Nrf2−/− and p62−/− mice, SPD still slightly alleviated the fibrotic alterations, but the protective effects of SPD were completely lost in Nrf2−/−;p62−/− mice (Figure 4B). CCl4 increased the liver/body weight ratio in all four genotypes, and SPD treatment only showed a protective effect in the WT and p62−/− groups (Figure 4C). SPD also reduced both alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, which were elevated by CCl4 exposure, in WT and p62−/− mice, while only ALT levels were slightly decreased in Nrf2−/− mice (Figure 4D–4E). Similar to the morphological changes, the protective effect of SPD was completely abolished in Nrf2−/−;p62−/− mice, as measured by liver/body weight ratio, AST and ALT (Figure 4C–4E).
Figure 4.
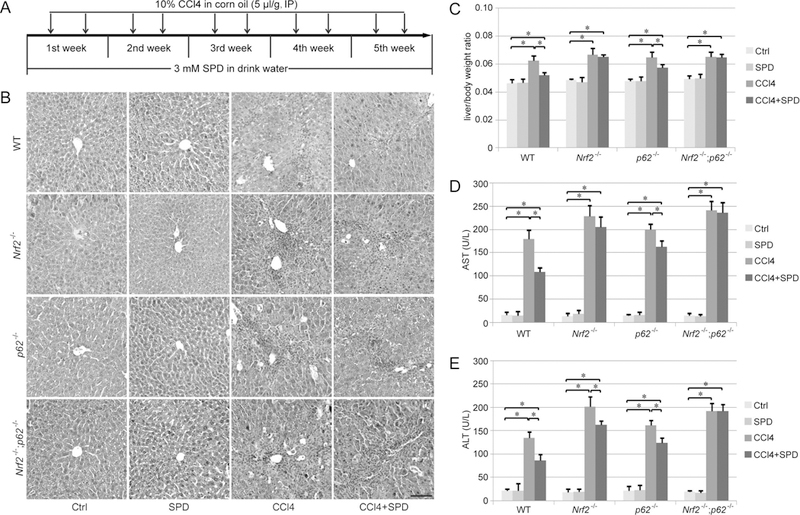
SPD protects mice against CCl4-induced liver injury. A. Schematic indicating CCl4 and SPD treatment schedule. B. Hematoxylin and eosin staining of liver sections from WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− mice that received the indicated treatments. Scale bar=50 μm. C-E. The ratio of body weight to liver weight (C), as well as serum aspartate aminotransferase (AST, D) and alanine aminotransferase (ALT, E) levels of each genotype following treatment were measured. AST and ALT levels are expressed as units per liter (U/L). Results are expressed as mean ± SD (n=5). *: p<0.05 compared between different groups.
To exclude the possibility that the observed difference in hepatic injury is due to variation in CCl4-detoxification across the four different genotype groups, the level of CYP2E1, a critical P450 enzyme that metabolizes CCl4, was measured. Importantly, knockout of Nrf2, p62 or both did not affect CYP2E1 expression (Figure S2), indicating that CCl4 metabolism should be similar between groups.
The protective effect of SPD in mitigating liver fibrosis was further evaluated using sirius red staining (for collagen I and III fibers) and α-smooth muscle actin (α-SMA) IHC staining (for myofibroblast-like cells, a marker for hepatic fibrosis). Morphometric analysis of sirius red staining and α-SMA IHC staining demonstrated that CCl4 caused a remarkable accumulation of collagen and α-SMA in the liver that can be significantly reduced by SPD in WT mice, but only slightly reduced by SPD in Nrf2−/− and p62−/− mice, with no apparent reduction in the Nrf2−/−;p62−/− mouse group (Figure 5A–5B). To further evaluate liver fibrosis quantitatively, both COL1A1 and α-SMA protein levels were measured using immunoblot analysis. CCl4 increased the level of COL1A1 and α-SMA dramatically, and SPD treatment attenuated hepatic fibrosis significantly in the WT group, slightly in the Nrf2−/− and p62−/− groups, but had no effect in the Nrf2−/−;p62−/− group (Figure 5C). Collectively, these results indicate that the protective effect of SPD on hepatic fibrosis is dependent on NRF2 and p62.
Figure 5.
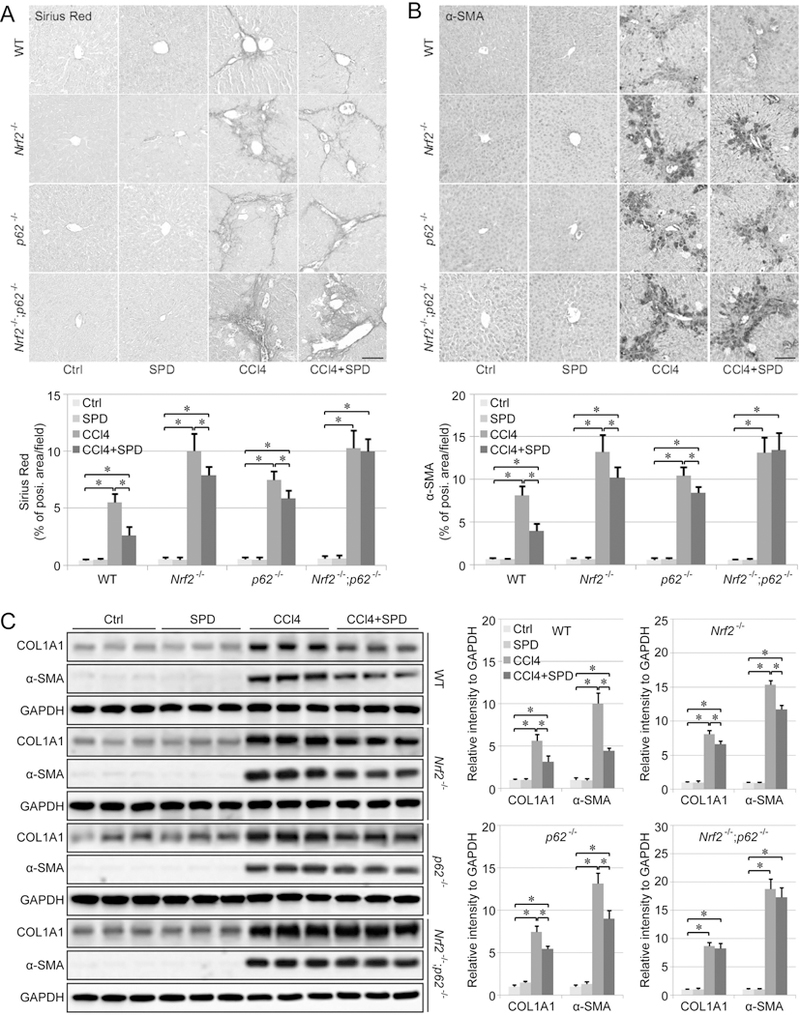
SPD promotes the recovery of CCl4-induced liver fibrosis. A-B. Liver fibrosis assessed by Sirius Red staining (A) and α-smooth muscle actin (α-SMA) IHC staining (B). Scale bar=50 μm. Bar graphs represent the %positive area of staining per field. C. Immunoblot analysis of type I collagen (COL1A1) and α-SMA protein levels in liver tissue isolated from WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− mice that received the indicated treatments. GAPDH was used as an internal loading control. Results are expressed as mean ± SD. *: p<0.05 compared between different groups.
SPD upregulates both the NRF2 and autophagy pathways in a liver fibrosis model
SPD increased MAP1S protein levels in all four genotypes of mice (Figure 6A–6D, Figure S3A), supporting our model (Figure 8) that MAP1S is upstream of KEAP1-mediated regulation of NRF2. In fibrotic liver tissues, NRF2 expression was reduced in both WT and p62−/− mice, which is consistent with our previous study (36). SPD increased NRF2, NQO1, and FTL expression significantly in WT mice, but only slightly in p62−/− mice (Figure 6A and 6C, Figure S3B–S3D), which correlates with a decrease in oxidative DNA damage in fibrotic liver tissues from WT mice, as detected by 8-deoxyguanosine levels (8-oxo-dG) (Figure S3E). However, SPD showed no detectable protective effects against DNA damage in Nrf2−/−, p62−/−, and Nrf2−/−;p62−/− mice (Figure S3E).
Figure 6.
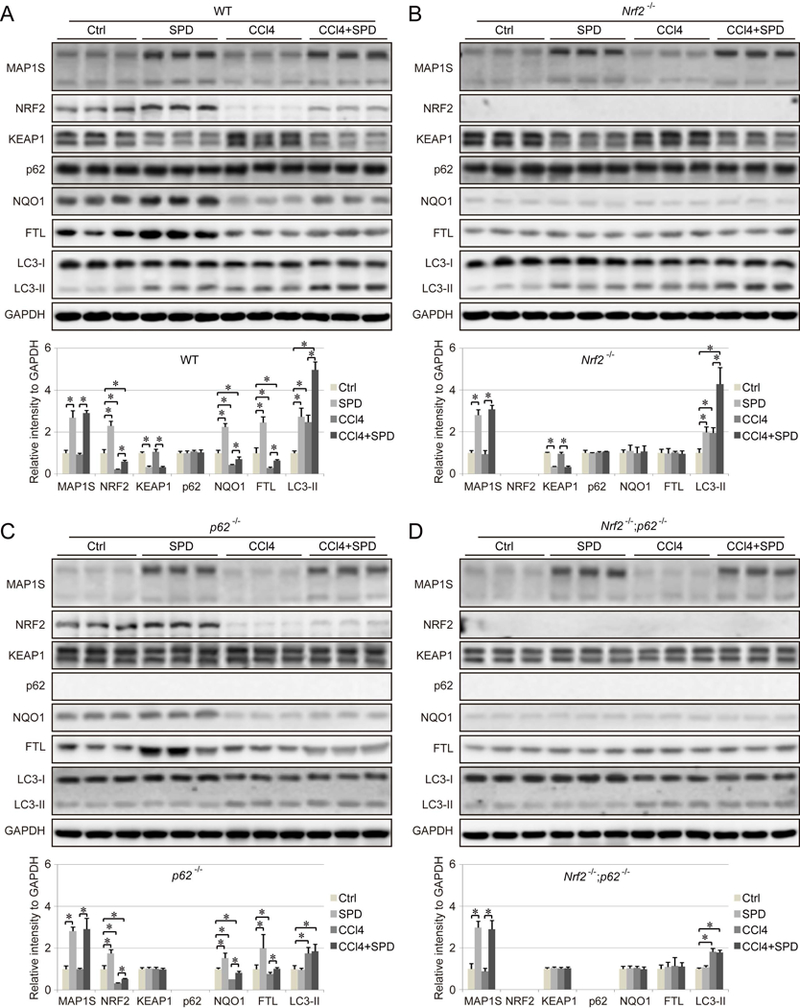
Effect of SPD on NRF2 expression and autophagy in a liver fibrosis model. A-D. Immunoblot analysis of MAP1S, as well as key NRF2 and autophagy pathway components in liver tissue from WT (A), Nrf2−/− (B), p62−/− (C) and Nrf2−/−;p62−/− (D) mice that received the indicated treatments. GAPDH was used as an internal loading control. Results are expressed as mean ± SD. *: p<0.05 compared between different groups.
Consistent with the results obtained with WT and Map1s−/− mice (Figure 3), SPD-mediated induction of p62 was not detected in vivo, which may due to high basal levels of p62 in the liver (Figures 6A and 6B, Figure S3F). CCl4-induced fibrosis increased LC3-II and decreased LC3-I in liver tissues, as reported previously (39, 40). SPD treatment promoted autophagy, and further increased LC3-II levels in both WT and Nrf2−/− mice, but not p62−/− and Nrf2−/−;p62−/− mice. SPD decreased KEAP1 levels in both WT and Nrf2−/− mice, but not p62−/− and Nrf2−/−;p62−/− mice, which is most likely due to the fact that SPD-induced autophagic degradation of KEAP1 is dependent on p62. Therefore, SPD upregulates NRF2 by accelerating autophagic degradation of KEAP1 bound to p62. In addition, SPD-induced upregulation of MAP1S increased NRF2 levels via ETGE-mediated competitive binding with KEAP1, which explains the weak increase of NRF2 in p62−/− mice (Figure 6C).
SPD reduces TGF-β-mediated activation of HSCs in an NRF2-dependent manner in ex vivo models
Activated HSCs are the predominant type of myofibroblast that drive the fibrogenic process, and the degree of HSC activation is mostly paralleled by the degree of hepatic fibrosis in human clinical samples (41). Thus, primary HSCs were isolated from the liver of all four genotypes of mice, with the quality of isolation being confirmed using GFAP immunostaining (Figure S4A–B). TGF-β was then used to induce HSC activation, which was measured by α-SMA. SPD increased MAP1S levels in HSCs from all groups (Figure 7A), consistent with the in vitro and in vivo results obtained in this study. In addition, SPD increased NRF2, p62, and LC3-II, but decreased KEAP1 in WT HSCs (Figure 7A). SPD also enhanced autophagy in WT and Nrf2−/− HSCs (Figure 7A–7B). Both mechanisms: (i) MAP1S competing with KEAP1 for NRF2 binding, and (ii) MAP1S accelerating KEAP1 degradation, were observed as determined by changes in NRF2 or autophagy signaling in HSCs from single Nrf2−/− or p62−/− mice, as well as the complete loss of SPD effects in modulating these pathways in HSCs from Nrf2−/−;p62−/− mice. More importantly, SPD alleviated α-SMA expression induced by TGF-β significantly in WT HSCs, only slightly in p62 HSCs, but not at all in Nrf2−/− HSCs and Nrf2−/−;p62−/− HSCs (Figure 7A), highlighting the essential role of NRF2 in mediating the observed protective effects of SPD.
Figure 7.
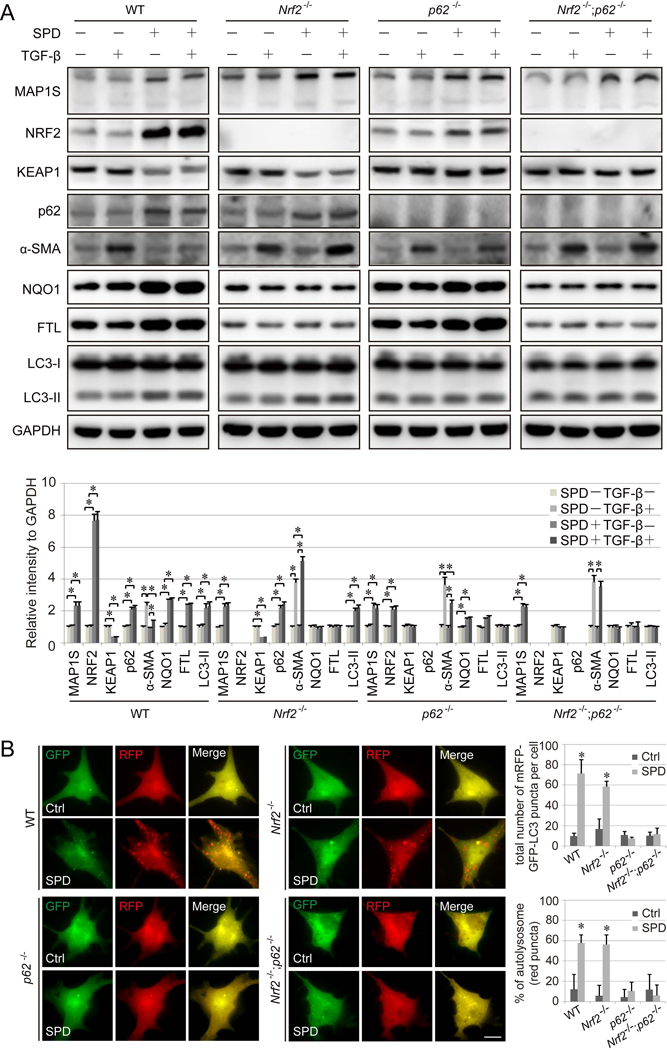
SPD inhibition of hepatic stellate cell (HSC) activation is dependent on NRF2. A. WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− HSCs were pretreated with SPD (100 μM) for 16 h, followed by TGF-β treatment (10 ng/ml for 8 h), then harvested for immunoblot analysis. GAPDH was used as an internal loading control. Results are expressed as mean ± SD. *: p<0.05 compared between different groups. B. WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− HSCs cells were transfected with mRFP-GFP-LC3 for 24 h, and then treated with SPD (100 μM) for 16 h, and imaged. Scale bar=10 μm. Bar graphs represent the total number of mRFP-GFP-LC3 positive puncta and % of autolysosomes (red puncta) per cell.
Discussion
The health benefits of SPD have been well-recognized, and dietary supplementation of SPD has been investigated as a preventive or intervening approach to promote healthy aging by ameliorating aging-associated diseases, including cancer, neurodegeneration, and diabetes (42). The molecular mechanisms underlying the protective effects associated with SPD have been extensively investigated, as SPD has been shown to improve mitochondrial function, stem cell function, and tissue regeneration (43). To this point, it has been widely believed that most of SPD’s effects were associated with its function as an autophagy inducer, and several studies indicate that the protective effects of SPD were abolished in animals deficient for key Atg genes (42, 44). In this study, we identify SPD as a novel non-canonical NRF2 inducer. SPD was able to induce NRF2 though two parallel mechanisms, both of which require upregulation of MAP1S: (i) MAP1S competitive binding to KEAP1, and (ii) autophagic degradation of a MAP1S-KEAP1-p62 complex (Figure 8). Through the utilization of WT, Nrf2−/−, p62−/− and Nrf2−/−;p62−/− mice, we demonstrated a pivotal role of NRF2 in SPD-mediated liver protection, as evidenced by the fact that the protective effect of SPD was significantly reduced in Nrf2 or p62 single knockout mice, and completely lost in the double knockout mice. It is worth mentioning that p62−/− mice are still proficient in p62-independent autophagy pathways, but NRF2 induction in these mice was significantly reduced compared to WT mice. It is also interesting to note that in some cases SPD-mediated protection was abolished even in Nrf2 single knockout mice as seen with the liver/body weight ratio, AST levels, α-SMA induction (in HSCs), but not by ALT levels, Sirius Red staining, α-SMA induction (tissues), or COL1A1 protein levels. However, SPD had no protective effects in any of the utilized assays in Nrf2−/−;p62−/− mice, even though the p62-independent autophagy pathways should still be intact in these mice. Collectively, our results could support that SPD ameliorates the fibrotic phenotype via parallel pathways, one of which is p62-dependent and one of which is p62-independent. Thus, it is possible that SPD can elicit p62-independent autophagy activation to provide some protection in the p62−/− and Nrf2−/− single knockout mice. However, the effects of SPD from p62-independent autophagy activation are too minimal to be detected in the double knock out mice. In addition, these results argue that activation of the NRF2 pathway may play a more important role than the activation of p62-independent autophagy pathways, at least in mediating the beneficial effects of SPD. Importantly, autophagy is intimately associated with the NRF2 pathway. Autophagy deficient mice, such as Atg5−/− and Atg7−/− mice, exhibit higher basal levels of NRF2 (27, 45). Furthermore, pharmacological activation or inhibition of autophagy has been demonstrated to induce NRF2 (30, 46). Therefore, it is reasonable to assume that the previously reported SPD protective effects being a result of autophagy upregulation may actually be due to activation of NRF2. Further investigation of SPD effects in an autophagy-deficient setting, such as Atg knockout mice, and Atg and NRF2 double knockout mouse models will deepen our understanding of the relative contribution of autophagy vs. NRF2 in mediating the protective effects of SPD.
In our newly proposed SPD-mediated model of NRF2 induction (Figure 8), MAP1S plays an essential role. As a protein with a role in autophagy activation that contains an ETGE motif capable of interacting with KEAP1, it is not surprising that MAP1S regulates NRF2 signaling. In cells that have low basal autophagy, NRF2 signaling is suppressed, as it is ubiquitylated and degraded by the proteasome in a KEAP1-dependent manner. However, SPD treatment increases MAP1S levels, which in turn upregulates NRF2 through two mechanisms: (i) MAP1S competes with NRF2 for KEAP1 binding; and (ii) MAP1S promotes autophagosome formation and sequestration of LC3, p62, KEAP1 and MAP1S into the autophagosome, which results in degradation of these proteins (Figure 8). Interestingly, SPD enhanced the protein levels of MAP1S, which induced autophagy in a p62-dependent manner in all the in vitro, in vivo and ex vivo models tested (using LC3-II protein levels and the tandem mRFP-GFP-LC3 reporter as indicators). It is currently unclear why SPD- or MAP1S-induced autophagy is p62-dependent. p62 is considered a selective autophagy substrate that regulates the formation of protein aggregates and inclusion bodies by binding to ubiquitylated proteins and to the autophagy protein LC3 (28, 29). A wide variety of interacting partners for MAP1S have been identified and in many cases the specific regions mediating this interaction have been determined. Some MAP1S protein-protein interactions might be mutually exclusive or restricted to certain cellular conditions (10). The function of MAP1S in promoting autophagy may also be associated with its interaction with LRPPRC, RASSF1A, and p62 (7, 47). Therefore, it is possible that p62 affects the dynamic interaction between MAP1S and other proteins important for the autophagy pathway, which are essential for MAP1S-mediated upregulation of autophagy.
The protective role of NRF2 in liver fibrosis via ameliorating liver injury and inhibiting HSC activation has been reported previously (48–50). It is possible that the beneficial effects of SPD and NRF2 activation on ameliorating CCl4-induced liver fibrosis could be independent of the effects on HSCs. Due to the fact that SPD is capable of activating both autophagy and NRF2 in multiple cell types, it is reasonable to infer that the protective effects are more universal than just reducing HSC activation. However, based on the prominent role that HSC activation plays in the pathology of liver fibrosis, as well as the clear anti-fibrotic effects that resulted in WT cells upon SPD treatment, we believe that SPD-dependent activation of NRF2 in HSCs is a key contributor to lessening the fibrotic phenotype.
Importantly, we have identified SPD as a new non-canonical NRF2 inducer, and MAP1S as a new component of the non-canonical pathway of NRF2 activation. SPD treatment increased the level of both MAP1S and NRF2, thus conferring protection against liver fibrosis (Figure 8). Our results demonstrate the pivotal role of NRF2 in mediating the health benefits associated with SPD. SPD is a naturally occurring polyamine produced in our body, but its levels decrease during aging, and its use as a dietary supplement has been gaining more and more attention due to extremely low cytotoxicity and its cytoprotective effects. Dietary supplementation of SPD has been reported to ameliorate many pathological processes associated with aging by enhancing immune- and anti-inflammatory- responses, improving glucose tolerance, and suppressing stem cell senescence (42). Therefore, future investigations on SPD’s beneficial effects in other disease models, as well as its NRF2 dependency, is warranted.
Supplementary Material
Acknowledgments
Financial Support
This work was supported by the following NIH grants: R01 DK109555, R01 ES026845, and P42 ES004940, P30 ES006694 to D.D.Z.
List of Abbreviations
- NRF2
Nuclear factor (erythroid-derived 2)-like 2
- KEAP1
Kelch-like ECH-associated protein 1
- NQO1
NAD(P)H dehydrogenase quinone1
- FTL
Ferritin light chain
- MAP1S
Microtubule associated protein 1S
- MAP1LC3
Microtubule-associated proteins 1 light chain 3
- CCl4
Carbon tetrachloride
- DEN
N-nitrosodiethylamine
- MAP
Microtubule-associated protein
- RASSF1A
Ras association domain family 1 isoform A
- VCY2
Y-chromosome protein variable charge Y-linked 2
- LRPPRC
Leucine-rich PPR-motif containing protein
- SQSTM1
sequestosome 1
- HSCs
Hepatic stellate cells
- sgRNA
Single guide RNA
- FACS
Fluorescence-activated cell sorting
- FL
Full-length
- RIPA
Radio immunoprecipitation assay
- WT
Wild type
- SPD
Spermidine
- MU
Mutant
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- α-SMA
α-smooth muscle actin
- 8-oxo-dG
8-deoxyguanosine levels
Footnotes
Financial Disclosure
Nothing to disclose
References
- 1.Guha IN, Myers RP, Patel K, Talwalkar JA. Biomarkers of Liver Fibrosis: What Lies Beneath the Receiver Operating Characteristic Curve? Hepatology 2011;54:1454–1462. [DOI] [PubMed] [Google Scholar]
- 2.Morselli E, Galluzzi L, Kepp O, Criollo A, Maiuri MC, Tavernarakis N, Madeo F, et al. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging (Albany NY) 2009;1:961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016;22:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao M, Zhao W, Li C, Xie X, Li M, Bi Y, Fang F, et al. Spermidine ameliorates non-alcoholic fatty liver disease through regulating lipid metabolism via AMPK. Biochem Biophys Res Commun 2018;505:93–98. [DOI] [PubMed] [Google Scholar]
- 5.Zahedi K, Lentsch AB, Okaya T, Barone S, Sakai N, Witte DP, Arend LJ, et al. Spermidine/spermine-N1-acetyltransferase ablation protects against liver and kidney ischemia-reperfusion injury in mice. Am J Physiol Gastrointest Liver Physiol 2009;296:G899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okumura S, Teratani T, Fujimoto Y, Zhao X, Tsuruyama T, Masano Y, Kasahara N, et al. Oral administration of polyamines ameliorates liver ischemia/reperfusion injury and promotes liver regeneration in rats. Liver Transpl 2016;22:1231–1244. [DOI] [PubMed] [Google Scholar]
- 7.Yue F, Li W, Zou J, Jiang X, Xu G, Huang H, Liu L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res 2017;77:2938–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orban-Nemeth Z, Simader H, Badurek S, Trancikova A, Propst F. Microtubule-associated protein 1S, a short and ubiquitously expressed member of the microtubule-associated protein 1 family. J Biol Chem 2005;280:2257–2265. [DOI] [PubMed] [Google Scholar]
- 9.Xie R, Nguyen S, McKeehan K, Wang F, McKeehan WL, Liu L. Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J Biol Chem 2011;286:10367–10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong EY, Tse JY, Yao KM, Lui VC, Tam PC, Yeung WS. Identification and characterization of human VCY2-interacting protein: VCY2IP-1, a microtubule-associated protein-like protein. Biol Reprod 2004;70:775–784. [DOI] [PubMed] [Google Scholar]
- 11.Zou J, Yue F, Jiang X, Li W, Yi J, Liu L. Mitochondrion-associated protein LRPPRC suppresses the initiation of basal levels of autophagy via enhancing Bcl-2 stability. Biochem J 2013;454:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- 13.Xie R, Wang F, McKeehan WL, Liu L. Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res 2011;71:7537–7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu G, Jiang Y, Xiao Y, Liu XD, Yue F, Li W, Li X, et al. Fast clearance of lipid droplets through MAP1S-activated autophagy suppresses clear cell renal cell carcinomas and promotes patient survival. Oncotarget 2016;7:6255–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang X, Zhong W, Huang H, He H, Jiang F, Chen Y, Yue F, et al. Autophagy defects suggested by low levels of autophagy activator MAP1S and high levels of autophagy inhibitor LRPPRC predict poor prognosis of prostate cancer patients. Mol Carcinog 2015;54:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaramillo MC, Zhang DD. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes & Development 2013;27:2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 2007;47:89–116. [DOI] [PubMed] [Google Scholar]
- 18.Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in Disease: Timing Is Everything. Annu Rev Pharmacol Toxicol 2019, 59:555–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 2004;24:10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal 2010;13:1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 2006;26:2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 2006;281:24756–24768. [DOI] [PubMed] [Google Scholar]
- 24.Baird L, Lleres D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci U S A 2013;110:15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, Foster BJ, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem 2010;285:16782–16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010;30:3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010;12:213–223. [DOI] [PubMed] [Google Scholar]
- 28.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res 2012;66:457–462. [DOI] [PubMed] [Google Scholar]
- 29.Nezis IP, Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid Redox Signal 2012;17:786–793. [DOI] [PubMed] [Google Scholar]
- 30.Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol 2013;33:2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhee SG, Bae SH. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic Biol Med 2015;88:205–211. [DOI] [PubMed] [Google Scholar]
- 32.Lee SB, Sellers BN, DeNicola GM. The Regulation of NRF2 by Nutrient-Responsive Signaling and Its Role in Anabolic Cancer Metabolism. Antioxid Redox Signal 2018; 29:1774–1791. [DOI] [PubMed] [Google Scholar]
- 33.Chen M, Liu J, Yang W, Ling W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017;13:1813–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol 2007;27:6334–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev 2014;28:708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 1996;93:13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okada K, Yanagawa T, Warabi E, Yamastu K, Uwayama J, Takeda K, Utsunomiya H, et al. The alpha-glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol Res 2009;39:490–500. [DOI] [PubMed] [Google Scholar]
- 39.Shi H, Han W, Shi H, Ren F, Chen D, Chen Y, Duan Z. Augmenter of liver regeneration protects against carbon tetrachloride-induced liver injury by promoting autophagy in mice. Oncotarget 2017;8:12637–12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.San-Miguel B, Crespo I, Sanchez DI, Gonzalez-Fernandez B, de Urbina Ortiz JJ, Tunon MJ, Gonzalez-Gallego J. Melatonin inhibits autophagy and endoplasmic reticulum stress in mice with carbon tetrachloride-induced fibrosis. J Pineal Res 2015;59:151–162. [DOI] [PubMed] [Google Scholar]
- 41.Washington K, Wright K, Shyr Y, Hunter EB, Olson S, Raiford DS. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol 2000;31:822–828. [DOI] [PubMed] [Google Scholar]
- 42.Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science 2018;359. [DOI] [PubMed] [Google Scholar]
- 43.Atiya Ali M, Poortvliet E, Stromberg R, Yngve A. Polyamines in foods: development of a food database. Food Nutr Res 2011;55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong D, Hill JA. Spermidine Promotes Cardioprotective Autophagy. Circ Res 2017;120:1229–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, et al. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci 2012;127:438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013;51:618–631. [DOI] [PubMed] [Google Scholar]
- 47.Liu L, Vo A, Liu G, McKeehan WL. Putative tumor suppressor RASSF1 interactive protein and cell death inducer C19ORF5 is a DNA binding protein. Biochem Biophys Res Commun 2005;332:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Husain H, Latief U, Ahmad R. Pomegranate action in curbing the incidence of liver injury triggered by Diethylnitrosamine by declining oxidative stress via Nrf2 and NFkappaB regulation. Sci Rep 2018;8:8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li JP, Gao Y, Chu SF, Zhang Z, Xia CY, Mou Z, Song XY, et al. Nrf2 pathway activation contributes to anti-fibrosis effects of ginsenoside Rg1 in a rat model of alcohol- and CCl4-induced hepatic fibrosis. Acta Pharmacol Sin 2014;35:1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma X, Luo Q, Zhu H, Liu X, Dong Z, Zhang K, Zou Y, et al. Aldehyde dehydrogenase 2 activation ameliorates CCl4 -induced chronic liver fibrosis in mice by up-regulating Nrf2/HO-1 antioxidant pathway. J Cell Mol Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.