

Legionella spp. are natural pathogens of protozoa and accidental pathogens of humans. Innate immunity in healthy individuals effectively controls Legionella infection due in part to rapid and robust production of proinflammatory cytokines resulting from detection of Dot/Icm-translocated substrates, including effectors. Here, we demonstrate that the effector LegC4 enhances proinflammatory host restriction of Legionella by macrophages. These data suggest that LegC4 may augment proinflammatory signaling or antimicrobial activity of macrophages, a function that has not previously been observed for another bacterial effector. Further insight into LegC4 function will likely reveal novel mechanisms to enhance immunity against pathogens.
KEYWORDS: LegC4, Legionella, effector functions, host-pathogen interactions, innate immunity, interferon gamma, tumor necrosis factor
ABSTRACT
Legionella pneumophila is ubiquitous in freshwater environments, where it replicates within unicellular protozoa. However, L. pneumophila is also an accidental human pathogen that can cause Legionnaires’ disease in immunocompromised individuals by uncontrolled replication within alveolar macrophages. To replicate within eukaryotic phagocytes, L. pneumophila utilizes a Dot/Icm type IV secretion system to translocate a large arsenal of over 300 effector proteins directly into host cells. In mammals, translocated effectors contribute to innate immune restriction of L. pneumophila. We found previously that the effector LegC4 is important for L. pneumophila replication within a natural host protist but is deleterious to replication in a mouse model of Legionnaires’ disease. In the present study, we used cultured mouse primary macrophages to investigate how LegC4 attenuates L. pneumophila replication. We found that LegC4 enhanced restriction of L. pneumophila replication within macrophages activated with tumor necrosis factor (TNF) or interferon gamma (IFN-γ). In addition, expression of legC4 was sufficient to restrict Legionella longbeachae replication within TNF- or IFN-γ-activated macrophages. Thus, this study demonstrates that LegC4 contributes to L. pneumophila clearance from healthy hosts by potentiating cytokine-mediated host defense mechanisms.
IMPORTANCE Legionella spp. are natural pathogens of protozoa and accidental pathogens of humans. Innate immunity in healthy individuals effectively controls Legionella infection due in part to rapid and robust production of proinflammatory cytokines resulting from detection of Dot/Icm-translocated substrates, including effectors. Here, we demonstrate that the effector LegC4 enhances proinflammatory host restriction of Legionella by macrophages. These data suggest that LegC4 may augment proinflammatory signaling or antimicrobial activity of macrophages, a function that has not previously been observed for another bacterial effector. Further insight into LegC4 function will likely reveal novel mechanisms to enhance immunity against pathogens.
INTRODUCTION
Legionella spp. are natural pathogens of unicellular protozoa and accidental pathogens of humans that can cause a severe inflammatory pneumonia called Legionnaires’ disease, which results from uncontrolled bacterial replication within alveolar macrophages. To replicate within eukaryotic phagocytes, Legionella spp. subvert normal endocytic signaling by establishing a specialized compartment called the Legionella-containing vacuole (LCV). To form the LCV and replicate intracellularly, Legionella spp. employ a Dot/Icm type IV secretion system (T4SS) to translocate virulence factors, called effector proteins, into host cells (1). Although >15 Legionella species are capable of causing human disease, the overwhelming majority of disease is caused by L. pneumophila (2, 3). In healthy individuals, L. pneumophila infection is efficiently controlled, and human-to-human transmission is incredibly rare (4). This is due to efficient detection and subsequent clearance of L. pneumophila by the mammalian innate immune system. Consequently, L. pneumophila is a well-established model pathogen used to characterize mechanisms of host defense against bacterial pathogens.
Innate immune detection of bacterial pathogens is facilitated by host pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs). Surface Toll-like receptors (TLRs) are PRRs that are critical for host defense against L. pneumophila. The majority of TLRs signal through the adaptor MyD88 to activate proinflammatory gene expression. Mice lacking MyD88 are highly susceptible to L. pneumophila infection, which is mostly due to lack of interleukin-1 (IL-1)- and TLR2-mediated signaling (5–7). Intracellular PRRs such as Nod1, Nod2, and inflammasomes also contribute to innate immune restriction of L. pneumophila in macrophages (reviewed in references 8 and 9). Leakage of PAMPs, such as peptidoglycan, lipopolysaccharide, and flagellin (FlaA monomers), into the macrophage cytosol amplifies cell autonomous restriction of L. pneumophila within immune phagocytes. Specifically, recognition of L. pneumophila FlaA by the NAIP5/NLRC4 inflammasome is sufficient to restrict L. pneumophila replication within macrophages (10, 11). Engagement of both extracellular and intracellular PRRs results in a robust proinflammatory response mediated by secretion of cytokines by infected and bystander immune phagocytes (5, 12–16). In particular, tumor necrosis factor (TNF) and interferon gamma (IFN-γ) are critical for restriction of pulmonary L. pneumophila infection (17–21). TNF and IFN-γ both promote cell autonomous defense against L. pneumophila within macrophages and mediate bacterial killing by increasing phagolysosmal fusion (22, 23).
In addition to canonical PAMPs, translocated effectors can augment proinflammatory responses in L. pneumophila-infected macrophages. For example, effector-mediated inhibition of host protein translation results in increased expression of proinflammatory genes in macrophages (24–27). In addition, macrophage proinflammatory gene expression was decreased during infection with legionellae that possess a functional Dot/Icm T4SS but are unable to translocate a subset of effectors due to a mutation in the icmS effector chaperone gene (28). These studies elaborate the concept of effector-triggered immunity in animal cells (29) and provide further evidence for the contribution of effectors to innate immune restriction of L. pneumophila.
We recently demonstrated that the effector LegC4 attenuates L. pneumophila fitness in a mouse model of Legionnaires’ disease (30). Loss-of-function mutation of the legC4 gene conferred a fitness advantage on L. pneumophila in the mouse lung as evidenced by increased pulmonary bacterial burden and the ability to outcompete the wild-type strain (30). However, the legC4 mutation had no effect on L. pneumophila replication in primary bone-marrow derived macrophages (BMDMs) and impaired replication within a natural amoeba host, Acanthamoeba castellanii (30). Moreover, expression of legC4 from a plasmid further attenuated L. pneumophila fitness in the mouse lung compared to that of the wild-type strain. Thus, we hypothesized that LegC4 is deleterious to L. pneumophila in the presence of cell-mediated innate immunity.
The present study was designed to determine how LegC4 augments restriction of L. pneumophila in mammalian hosts. Expression of legC4 from a plasmid was sufficient to attenuate L. pneumophila replication in BMDMs, which relied on TNF secretion and subsequent signaling. Moreover, a ΔlegC4 mutant exhibited increased replication in cytokine-activated BMDMs. Interestingly, expression of legC4 was sufficient to attenuate Legionella longbeachae replication within TNF- and IFN-γ-activated BMDMs. These results suggest that LegC4 enhances macrophage cell autonomous defense against Legionella by potentiating cytokine-mediated restriction.
RESULTS
LegC4 confers a fitness disadvantage on nonflagellated L. pneumophila in wild-type mice.
We found previously that the L. pneumophila effector LegC4 is detrimental to bacterial replication in the mouse lung (30). Those experiments were performed using mice and macrophages deficient for production of the NLRC4 inflammasome (Nlrc4−/−) to prevent flagellin-mediated restriction of L. pneumophila replication (10, 30, 31). To confirm that LegC4-mediated phenotypes were not due to loss of NLRC4, we examined the fitness of a legC4-deficient (ΔlegC4) L. pneumophila strain in the lungs of wild-type C57BL/6 mice using competitive index (CI) experiments. To prevent NLRC4-mediated restriction of bacterial replication, we generated flagellin (flaA) loss-of-function mutations in our wild-type (ΔflaA) and legC4 mutant (ΔflaA ΔlegC4) strains (see Materials and Methods). We also used a previously generated flaA::Tn mutant to facilitate selective plating, since the transposon confers resistance to chloramphenicol (30). Mice were infected intranasally with a 1:1 mixture of L. pneumophila ΔflaA ΔlegC4 and flaA::Tn for 48 h. Lung tissue was subsequently homogenized and plated on selective media for CFU enumeration and calculation of CI values (see Materials and Methods). The ΔflaA ΔlegC4 strain significantly outcompeted the flaA::Tn mutant in the lungs of wild-type mice, as evidenced by average CI values significantly greater than 1.0 (P < 0.01) (Fig. 1A). Our previous study also revealed that expression of legC4 from a multicopy plasmid conferred a fitness disadvantage on L. pneumophila compared to the wild-type strain (30). To confirm these results in wild-type mice, we generated a strain of L. pneumophila ΔflaA ΔlegC4 harboring a plasmid carrying legC4 under the control of its endogenous promoter (pJB::plegC4). Plasmid expression of legC4(pJB::plegC4) resulted in significantly impaired fitness compared with the ΔflaA parental strain, which was not observed for a ΔflaA ΔlegC4 strain harboring vector alone (pJB) (P < 0.05) (Fig. 1B). These data demonstrate that NLRC4 does not affect LegC4-mediated attenuation of L. pneumophila replication in the mouse lung. To fully evaluate LegC4-mediated phenotypes, the remainder of our study was performed using flaA-deficient L. pneumophila strains and bone marrow-derived macrophages (BMDMs) derived from wild-type mice.
FIG 1.

LegC4 attenuates L. pneumophila fitness in wild-type mice. (A) Competitive index (CI) of L. pneumophila ΔflaA ΔlegC4 versus flaA::Tn (chloramphenicol resistant [Cmr]) from the lungs of wild-type mice. (B) CI of ΔflaA ΔlegC4(pJB::plegC4) or ΔflaA ΔlegC4(pJB) versus ΔflaA L. pneumophila from the lungs of wild-type mice. Each symbol represents an individual animal, and the line represents the mean CI values. Asterisks denote statistical significance by the Mann-Whitney U test (**, P < 0.01). Data are representative of at least two independent experiments.
Plasmid expression of legC4 attenuates L. pneumophila replication in BMDMs.
Since plasmid expression of legC4 attenuated L. pneumophila fitness in the mouse lung, we examined whether this also occurred in macrophages ex vivo using BMDMs derived from wild-type mice. We quantified bacterial replication within BMDMs over 72 h. Consistent with our previous study, loss of endogenous legC4 (ΔflaA ΔlegC4) did not affect replication of L. pneumophila within primary mouse BMDMs compared to the parental strain (ΔflaA) (30) (Fig. 2A). However, L. pneumophila ΔflaA ΔlegC4(pJB::plegC4) was significantly attenuated for replication in BMDMs at 48 and 72 h postinfection (p.i.) compared to the empty vector control strain (Fig. 2B). Furthermore, IPTG (isopropyl-β-d-1-thiogalactopyranoside)-induced expression of 3×FLAG epitope-tagged legC4 (3×FLAG-legC4) from a plasmid (plegC4) also resulted in impaired L. pneumophila replication compared to that of the control strain (carrying pEV) (P < 0.01) (Fig. 2C). Expression of 3×FLAG-legC4 was confirmed by Western blot analysis using an anti-FLAG antibody (Fig. 2D). Fitness defects associated with plasmid expression of legC4 were specific to intracellular replication, since replication in rich medium in vitro was unaffected (see Fig. S1 in the supplemental material). To determine if the intracellular replication defect observed for IPTG-induced expression of legC4 was specific, we cloned the effector gene lpg2505 into the same plasmid used to overexpress legC4, which resulted in an IPTG-inducible 3×FLAG gene fusion (3×FLAG-lpg2505) (see Materials and Methods). The growth attenuation observed for plasmid-encoded legC4 was not just due to overexpression of a Dot/Icm effector gene, since IPTG-induced expression of 3×FLAG-lpg2505 did not impair intracellular replication compared to that of the L. pneumophila ΔflaA strain (see Fig. S2A in the supplemental material). Expression of 3×FLAG-legC4 and -lpg2505 was visualized by Western blotting using an anti-FLAG antibody (Fig. S2B). Together, these data demonstrate that overexpression of legC4 is detrimental to L. pneumophila intracellular replication in macrophages.
FIG 2.

Plasmid expression of legC4 impairs L. pneumophila replication within BMDMs. (A to C) Fold replication of L. pneumophila ΔflaA and ΔflaA ΔlegC4 (A), ΔflaA ΔlegC4(pJB) and ΔflaA ΔlegC4(pJB::plegC4) (B), or ΔflaA ΔlegC4(pEV) and ΔflaA ΔlegC4(plegC4) (C) within wild-type BMDMs over 72 h. (D) L. pneumophila ΔflaA ΔlegC4(pEV) and ΔflaA ΔlegC4(plegC4) were lysed, and 3×FLAG-LegC4 was visualized by Western blotting. Expression of legC4 from plegC4 was induced with 1 mM IPTG as described in Materials and Methods. Data are shown as mean ± SD for samples in triplicate. Asterisks denote statistical significance by Student’s t test (**, P < 0.01; *, P < 0.05), and data are representative of at least two independent experiments.
LegC4-mediated restriction of L. pneumophila replication is dependent on cytokine production.
We subsequently investigated the mechanism by which plasmid expression of legC4 attenuates L. pneumophila replication. In BMDMs, L. pneumophila infection results in production of proinflammatory cytokines through engagement of TLRs by bacterial ligands. Indeed, we previously reported that BMDMs infected with L. pneumophila expressing legC4 secreted increased levels of interleukin-12 (IL-12) (30). However, increased levels of IL-12 would likely not be sufficient to attenuate L. pneumophila intracellular replication within BMDMs. Like IL-12, tumor necrosis factor (TNF) is a proinflammatory cytokine expressed downstream of Toll-like receptors (TLRs) in macrophages. TNF is important for host defense against L. pneumophila in mice and humans, and TNF-mediated signaling is sufficient to restrict L. pneumophila intracellular replication within macrophages (17–19, 23, 32). Thus, increased TNF signaling could account for LegC4-mediated attenuation of L. pneumophila intracellular replication.
We hypothesized that plasmid expression of legC4 would be sufficient to increase TNF secretion from L. pneumophila-infected BMDMs. Wild-type BMDMs were infected with L. pneumophila ΔflaA ΔlegC4(pEV) or ΔflaA ΔlegC4(plegC4) for 2 to 8 h, and secreted TNF was quantified by enzyme-linked immunosorbent assay (ELISA) (see Materials and Methods). Significantly greater concentrations of TNF were present in the supernatants of cells infected with L. pneumophila expressing legC4 from a plasmid (P < 0.05) (Fig. 3A). We also observed a significant increase in IL-6 production from macrophages infected with L. pneumophila(plegC4) compared to those infected with the isogenic empty vector control strain at 8 h p.i. (P < 0.05) (Fig. 3B). Increased cytokine production was not correlated with increased bacterial replication, since bacterial counts did not change during the course of infection (Fig. 3C). We also observed increased TNF secretion at 6 h p.i. in BMDMs infected with L. pneumophila strains constructed in the Lp02 background, which is metabolically active but does not replicate in the absence of exogenous thymidine (see Fig. S3 in the supplemental material). Consistent with our previous observations (30), these data demonstrate that LegC4 enhances cytokine secretion from L. pneumophila-infected macrophages.
FIG 3.
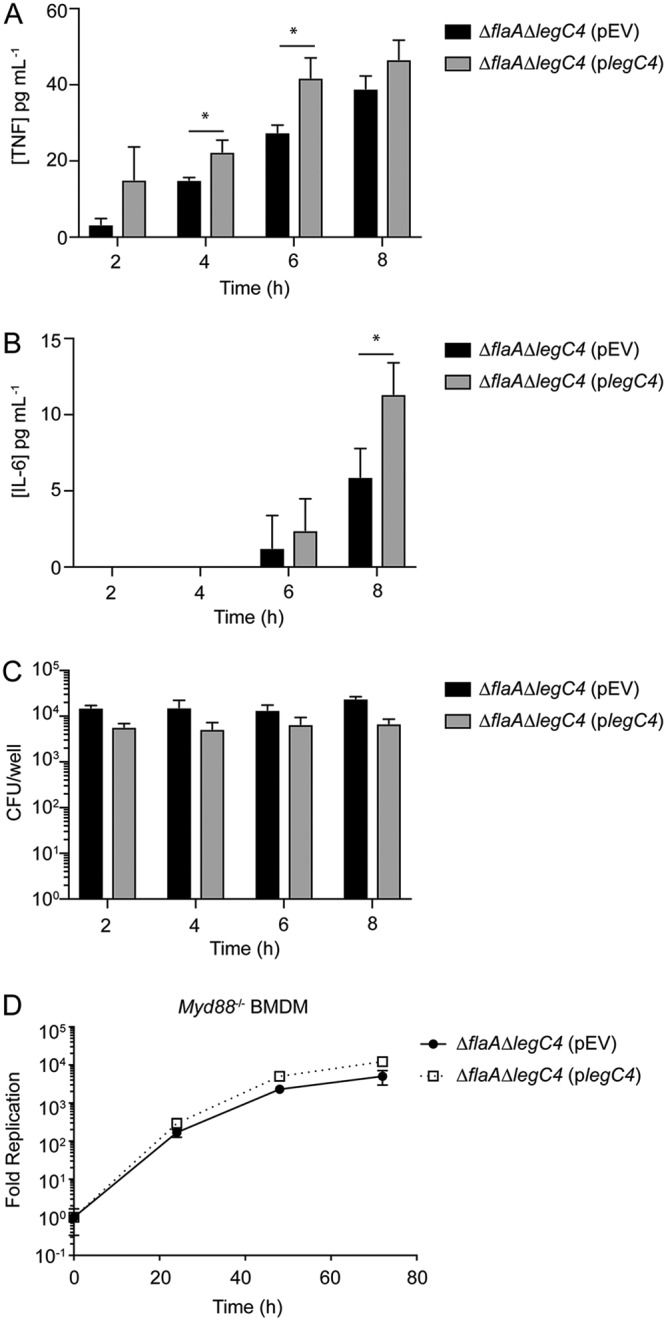
Role of TNF secretion in LegC4-mediated attenuation of L. pneumophila replication. (A and B) ELISA quantification of TNF (A) or IL-6 (B) secretion from wild-type BMDMs infected with L. pneumophila ΔflaA ΔlegC4(pEV) and ΔflaA ΔlegC4(plegC4) at the indicated time points. (C) Enumeration of L. pneumophila strains from BMDMs assayed for panels A and B. (D) Fold replication over 72 h of the indicated L. pneumophila strains within wild-type or Myd88−/− BMDMs. Expression of legC4 was induced with 1 mM IPTG. Data shown are mean ± SD for samples in triplicate. Asterisks denote statistical significance (*, P < 0.05; **, P < 0.01) by Student’s t test. Data are representative of at least two independent experiments.
To determine if TNF secretion contributed to LegC4-mediated attenuation of intracellular replication, intracellular replication of L. pneumophila within BMDMs deficient for production of MyD88 was quantified. We infected Myd88−/− BMDMs with L. pneumophila ΔflaA ΔlegC4(pEV) or ΔflaA ΔlegC4(plegC4) and quantified fold replication over 72 h. We found that loss of MyD88-mediated signaling abrogated LegC4-mediated attenuation of L. pneumophila intracellular replication in BMDMs (Fig. 2 and 3D). As expected, TNF was not secreted from Myd88−/− BMDMs under any of our experimental conditions (reference 33 and data not shown). Together, these data suggest that proinflammatory cytokine production is required for LegC4-mediated attenuation of L. pneumophila intracellular replication in BMDMs.
To further characterize LegC4-mediated restriction of L. pneumophila replication within BMDMs, L. pneumophila replication was evaluated in the absence of TNF signaling. To determine if TNF signaling contributed to legC4-mediated attenuation of L. pneumophila replication, we neutralized TNF in the supernatants of infected wild-type BMDMs using an anti-TNF antibody. Wild-type BMDMs were infected with L. pneumophila ΔflaA ΔlegC4 harboring plegC4 or pEV in the presence of either anti-TNF, a rat IgG isotype control antibody, or neither, and fold replication at 48 h p.i. was quantified. Plasmid expression of legC4 resulted in significantly attenuated L. pneumophila replication within untreated and rat IgG-treated BMDMs (P < 0.05); however, anti-TNF antibody neutralization of TNF restored replication of the legC4-overexpressing strain to wild-type levels (Fig. 4A). We subsequently examined replication of these strains in BMDMs deficient for signaling from TNF receptor 1 (TNFR1). Tnfr1−/− BMDMs were infected with L. pneumophila ΔflaA ΔlegC4 harboring either plegC4 or pEV, and fold replication was quantified over 72 h of infection. Overexpression of legC4 did not impair L. pneumophila intracellular replication within Tnfr1−/− BMDMs compared to that of the empty vector control strain (Fig. 4B). Interestingly, overexpression of legC4 resulted in significantly increased L. pneumophila replication within Tnfr1−/− BMDMs (P < 0.01) (Fig. 4B). These data demonstrate that TNF signaling contributes to LegC4-mediated attenuation of L. pneumophila replication within BMDMs.
FIG 4.

LegC4 augments TNF-mediated restriction of L. pneumophila replication. (A) Fold replication (48 h) of the indicated L. pneumophila strains within wild-type BMDMs treated with 50 ng ml−1 anti-TNF or an isotype control (rat IgG) or left untreated (UT) (see Materials and Methods). (B) Fold replication of the indicated L. pneumophila strains within Tnfr1−/− BMDMs over 72 h. Expression of legC4 was induced with 1 mM IPTG. (C) Fold replication (48 h) of L. pneumophila ΔflaA and ΔflaA ΔlegC4 within wild-type BMDMs the presence or absence of 50 ng ml−1 recombinant mouse TNF (rTNF). Data shown are mean ± SD for samples in triplicate. Asterisks denote statistical significance (*, P < 0.05; **, P < 0.01; n.s., not significant) by Student’s t test. Data are representative of at least two independent experiments. (D) CI of ΔflaA ΔlegC4(pJB::plegC4) or ΔflaA ΔlegC4(pJB) versus ΔflaA L. pneumophila from the lungs of wild-type mice. Each symbol represents an individual animal, and the lines represent the mean CI values. Asterisks denote statistical significance by the Mann-Whitney U test (*, P < 0.05). Data are representative of three independent experiments.
Endogenous LegC4 exacerbates TNF-mediated restriction of L. pneumophila from BMDMs.
To further characterize LegC4-mediated restriction of L. pneumophila from BMDMs, we examined replication of L. pneumophila in BMDMs activated with recombinant mouse TNF (rTNF). Wild-type BMDMs were infected with L. pneumophila ΔflaA or ΔflaA ΔlegC4 in the presence or absence of rTNF, and fold replication was quantified at 48 h p.i. L. pneumophila ΔflaA ΔlegC4 replicated to significantly higher levels than the parental ΔflaA strain in rTNF-treated BMDMs (P < 0.01) (Fig. 4C). As reported above, loss of endogenous legC4 does not affect L. pneumophila replication within untreated BMDMs (Fig. 4C). These data show that endogenous levels of LegC4 can augment TNF-mediated restriction of L. pneumophila replication.
Loss of TNFR1-mediated signaling is not sufficient for LegC4-mediated restriction of L. pneumophila replication in vivo.
We subsequently investigated whether loss of TNFR1-mediated signaling would be sufficient to restore the fitness of legC4-expressing L. pneumophila strains in vivo. To test this hypothesis, we performed a CI experiment to quantify the fitness of legC4-deficient L. pneumophila strains in the lungs of mice deficient for production of Tnfr1. Tnfr1−/− mice were infected with a 1:1 mixture of L. pneumophila ΔflaA and ΔflaA ΔlegC4(pJB) or L. pneumophila ΔflaA and ΔflaA ΔlegC4(pJB::plegC4) for 48 h. Lung tissue was homogenized and plated on selective media for enumeration of CFU and calculation of CI values (see Materials and Methods). L. pneumophila ΔflaA ΔlegC4(pJB) had a significant fitness advantage over the parental ΔflaA strain compared to L. pneumophila ΔflaA ΔlegC4(pJB::plegC4) (P < 0.05) (Fig. 4D). Thus, LegC4-mediated augmentation of TNFR1 signaling is not sufficient for attenuation of L. pneumophila in vivo.
LegC4 impairs L. pneumophila replication in IFN-γ-activated BMDMs.
Since TNF signaling was not sufficient for LegC4-mediated restriction of L. pneumophila in the lungs of mice, we evaluated the contribution of LegC4 to interferon gamma (IFN-γ)-mediated restriction in BMDMs. IFN-γ plays a major role in host defense against L. pneumophila in the lung (20, 34). The fold replication of L. pneumophila ΔflaA ΔlegC4(pEV) or ΔflaA ΔlegC4(plegC4) within wild-type BMDMs activated with recombinant mouse IFN-γ (rIFN-γ) was quantified over 72 h. We found that overexpression of legC4 significantly attenuated L. pneumophila replication within IFN-γ-activated BMDMs at all time points examined (Fig. 5A). We also determined that the L. pneumophila ΔflaA ΔlegC4 strain replicated to significantly higher levels than the ΔflaA parental strain in IFN-γ-activated, but not untreated, BMDMs (P < 0.05) (Fig. 5B). Together, these data demonstrate that LegC4 also augments IFN-γ-mediated restriction of L. pneumophila in macrophages.
FIG 5.
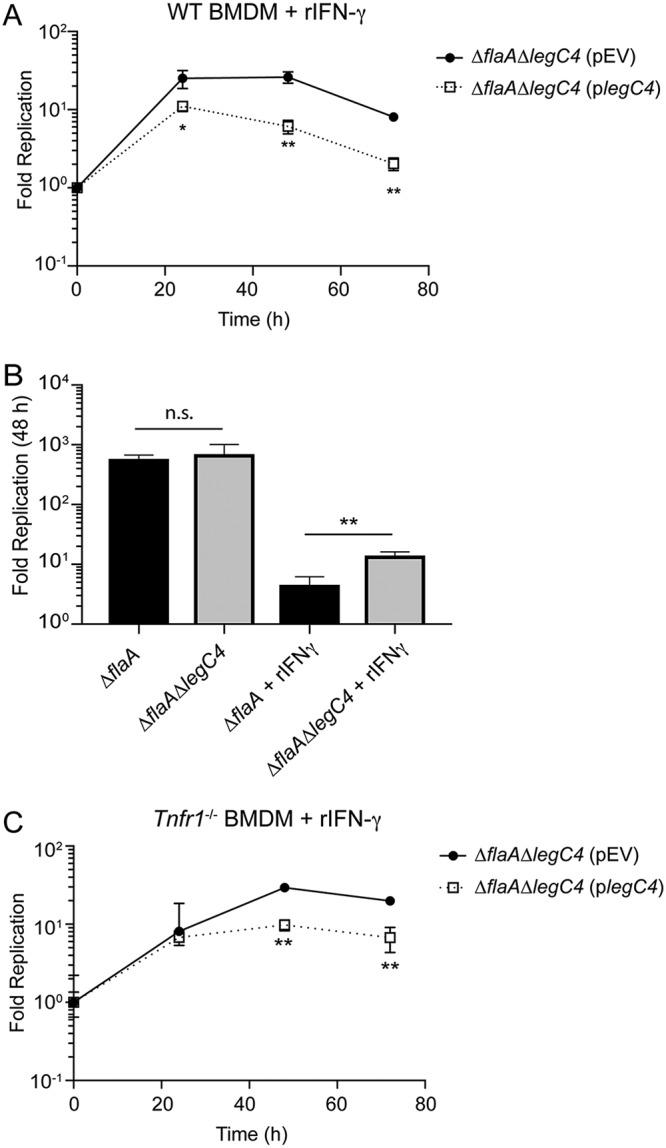
LegC4 enhances IFN-γ-mediated restriction of L. pneumophila replication. (A) Fold replication of the indicated L. pneumophila strains within wild-type (WT) BMDMs in the presence of 50 ng ml−1 rIFN-γ. (B) Fold replication (48 h) of the indicated strains within wild-type BMDMs in the presence or absence of 5 ng ml−1 rIFN-γ. (C) Fold replication of the indicated L. pneumophila strains within Tnfr1−/− BMDMs in the presence of 50 ng ml−1 rIFN-γ. Expression of legC4 was induced with 1 mM IPTG. Data shown are mean ± SD for samples in triplicate. Asterisks denote statistical significance (**, P < 0.01; n.s., not significant) by Student’s t test. Data are representative of at least two independent experiments.
IFN-γ-mediated signaling results in increased macrophage TNF production (35, 36). Thus, to determine if LegC4-mediated restriction of L. pneumophila within IFN-γ-activated macrophages was due to TNF signaling, we quantified L. pneumophila replication within Tnfr1−/− BMDMs treated with rIFN-γ. Overproduction of LegC4 resulted in significantly decreased L. pneumophila replication in IFN-γ-activated Tnfr1−/− BMDMs at 48 and 72 h p.i. compared to that of the control strain (Fig. 5C). Thus, LegC4 potentiates IFN-γ-mediated restriction of L. pneumophila intracellular replication in the absence of TNFR1-mediated signaling. Together, these data demonstrate that both IFN-γ- and TNF-mediated restriction of L. pneumophila are augmented by LegC4.
LegC4 impairs L. longbeachae replication within cytokine-activated BMDMs.
We next aimed to determine if LegC4-mediated restriction in macrophages was specific to L. pneumophila. Legionella longbeachae is the second-leading cause of Legionnaires’ disease and is dependent on Dot/Icm-mediated effector translocation for intracellular replication (37). The Dot/Icm secretion system is highly conserved between L. longbeachae and L. pneumophila; however, the effector repertoires are quite distinct, and L. longbeachae does not carry a homolog of legC4 (38, 39). Importantly, L. longbeachae is more virulent than L. pneumophila and is lethal in a mouse model of infection (40). To determine if LegC4 attenuates bacterial replication in a non-L. pneumophila Legionella species, we generated L. longbeachae strains either expressing IPTG-inducible 3×FLAG-tagged legC4 (plegC4) or harboring the empty vector (pEV). Expression of 3×FLAG-legC4 from L. longbeachae was confirmed by Western blot analysis (Fig. 6A). Wild-type BMDMs were infected with the indicated L. longbeachae strains in the presence or absence of rTNF or rIFN-γ as indicated, and fold replication at 48 h was quantified. Intracellular replication of the legC4-expressing strain of L. longbeachae was significantly attenuated within TNF- and IFN-γ-treated, but not untreated, BMDMs compared to that of the empty vector (pEV)-carrying control strain (P < 0.001) (Fig. 6B). These data demonstrate that LegC4 augments cytokine-mediated restriction of a non-L. pneumophila Legionella species within BMDMs.
FIG 6.

Replication of L. longbeachae producing LegC4 in cytokine-treated BMDMs. (A) Detection of 3×FLAG-LegC4 production by L. longbeachae (Llo) by Western blot analysis. (B) Fold replication (48 h) of L. longbeachae harboring the indicated plasmids within wild-type BMDMs the presence or absence of 50 ng ml−1 rTNF or rIFN-γ, as indicated. Expression of legC4 was induced with 1 mM IPTG. Data shown are mean ± SD for samples in triplicate. Asterisks denote statistical significance (**, P < 0.01; n.s., not significant) by Student’s t test. Data are representative of two independent experiments.
DISCUSSION
The data presented in this study support the hypothesis that LegC4 potentiates cytokine-mediated host defense against Legionella. Our previous work (30) identifying LegC4 as contributing to L. pneumophila clearance from the lung was performed using flagellated L. pneumophila in an NLRC4-deficient (Nlrc4−/−) mouse model. To fully evaluate the mechanisms of LegC4-mediated clearance, we utilized flagellin-deficient (ΔflaA) L. pneumophila and wild-type mice and BMDMs. Consistent with our previous study (30), we found that a loss-of-function mutation in the legC4 gene (ΔlegC4) conferred a fitness advantage on L. pneumophila ΔflaA within the wild-type mouse lung. Moreover, complementation of the ΔlegC4 mutation by a plasmid encoding legC4 in trans conferred a fitness disadvantage on L. pneumophila compared to the parental strain. Also consistent with our previous report, L. pneumophila ΔflaA ΔlegC4 replication within BMDMs did not differ from replication of the ΔflaA strain. However, in the present study, we found that plasmid expression of legC4 was sufficient to attenuate L. pneumophila replication within BMDMs. Although legC4 was expressed downstream of its endogenous promoter, an exaggerated phenotype likely occurred due to expression from a multicopy plasmid, suggesting a potential dose response. Importantly, this strain provided us with a tool to increase the magnitude of LegC4-mediated fitness attenuation within cultured cells. These phenotypes were corroborated by the observation that endogenous LegC4 was deleterious in cytokine-activated BMDMs. We further found that the fitness disadvantage associated with plasmid expression of legC4 was abolished in macrophages deficient for TNF-mediated signaling, suggesting that LegC4 is able to exacerbate cytokine-mediated antimicrobial responses. Finally, we determined that LegC4 could impair replication of L. longbeachae in cytokine-activated macrophages. Together, these data suggest that LegC4 potentiates cytokine-mediated restriction of L. pneumophila within macrophages.
Inflammation is mediated primarily through cytokine secretion, which is critical for restriction of L. pneumophila replication in vivo. This has been evidenced by the inability of Myd88−/− mice to control L. pneumophila replication. Specifically, Myd88−/− BMDMs will not secrete TNF during infection. The inability of plasmid-expressed legC4 to attenuate L. pneumophila replication in Myd88−/− BMDMs is likely due to lack of TNF signaling. In addition, LegC4-mediated increases in TNF secretion may amplify Tnf expression, which would further restrict L. pneumophila replication.
Proinflammatory cytokines contribute to host defense against L. pneumophila in vivo and in cultured macrophages (8, 41). Mice deficient for TNF-mediated signaling have increased pulmonary bacterial burdens and can succumb to infection (23, 32). TNF can signal through both TNFR1 and TNFR2; however, TNFR1-mediated signaling is primarily responsible for L. pneumophila restriction within alveolar macrophages in vivo (23) and is potentiated by LegC4. In the lung, multiple cell types contribute to TNF production, a consequence of which would be higher local TNF concentrations (16, 18, 23, 42). In addition, production of IFN-γ during L. pneumophila infection in vivo is mediated primarily by circulating natural killer (NK) cells (34, 43).
Our observation that the L. pneumophila ΔlegC4 mutant had a fitness advantage compared to the wild type in the mouse lung but not in cultured macrophages suggested that LegC4 was detrimental to replication under specific environmental conditions. This was supported by the observation that attenuated L. pneumophila replication was correlated with increased TNF secretion from BMDMs. Since the L. pneumophila-infected lung is an inflammatory environment, we examined whether cytokine-mediated restriction was exacerbated by LegC4. Abrogation of signaling from TNFR1 was sufficient to alleviate LegC4-mediated restriction of intracellular replication. Increased replication of the ΔlegC4 mutant within rTNF-treated BMDMs strongly suggests that proinflammatory responses are exacerbated by LegC4. This conclusion was corroborated by the observation that the ΔlegC4 mutant consistently replicated to higher levels within IFN-γ-activated macrophages. The observation that a legC4-deficient L. pneumophila strain still outcompeted the parental strain in Tnfr1−/− mice further suggests that LegC4-mediated growth attenuation occurs through multiple pathways. We are currently investigating whether additional factors are important for LegC4-mediated attenuation of L. pneumophila intracellular replication.
Similar to the case for L. pneumophila, L. longbeachae replicates within an LCV by employing a Dot/Icm secretion system and a repertoire of translocated effector proteins (37). Despite high levels of homology between the Dot/Icm secretion systems of these two organisms, the effector repertoires are quite diverse, and L. longbeachae does not carry a homolog of legC4 (38, 39). In contrast to L. pneumophila, L. longbeachae is highly virulent in a mouse model of Legionnaires’ disease (40, 44). Lethality in mice is likely due to L. longbeachae being poorly immunostimulatory and failing to induce substantial levels of proinflammatory cytokines during infection. However, proinflammatory cytokines contribute to the host defense against L. longbeachae in BMDMs and in vivo (40). Since interspecies translocation of Dot/Icm effectors by Legionella has been previously observed (37, 45), we introduced legC4 into L. longbeachae. Production of LegC4 by L. longbeachae resulted in significantly attenuated replication within cytokine-treated, but not untreated, BMDMs. These data reinforce our previous observations and demonstrate that LegC4-mediated restriction is not specific to L. pneumophila. Since L. longbeachae infection does not induce appreciable TNF secretion from BMDMs (40), it is likely that the concentration of TNF secreted by these cells is too low to permit LegC4-mediated restriction. Together with relatively low levels of effector translocation by L. longbeachae compared to L. pneumophila (37), the amount of translocated LegC4 may be insufficient to restrict bacterial replication within untreated BMDMs. However, LegC4 is sufficient to attenuate L. longbeachae replication within BMDMs activated with either rTNF or rIFN-γ. Whether LegC4 can protect mice from L. longbeachae-mediated lethality will be the subject of a future study.
Multiple effectors contribute to the innate immune response to L. pneumophila infection (reviewed in reference 46). Together with our data, these studies point to a complex interplay between effectors during Legionella infection of mammalian hosts. The effectors LnaB and LegK1 enhance NF-κB activation, which augments immune signaling (47, 48). Since mammals are a dead-end host for Legionella, the evolutionary basis for effector modulation of NF-κB is intriguing. Interestingly, the effector EnhC enhances L. pneumophila replication in TNF-activated macrophages (49), the opposite of what we have observed for LegC4. Thus, it is tempting to speculate that there may be interplay between EnhC and LegC4 within L. pneumophila-infected cells. Future investigations will reveal whether LegC4-mediated phenotypes are dependent on other Dot/Icm-translocated effectors.
In summary, we found that the Dot/Icm effector LegC4 can augment cytokine-mediated restriction of Legionella replication within macrophages. These data add to the growing body of literature on effector-triggered immunity in animal cells. As an accidental pathogen that did not coevolve under the selective pressure of an innate immune system, L. pneumophila continues to provide insight into novel mechanisms of innate immunity toward intracellular bacterial pathogens. Consequently, further understanding of LegC4 function will reveal strategies to augment proinflammatory signaling. Thus, this study has provided the foundation for future investigations into the molecular mechanism by which LegC4 enhances host defense against intracellular bacterial pathogens.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and growth conditions.
Legionella pneumophila Philadelphia-1 SRS43 (30), SRS43 flaA::Tn (30), Lp02 ΔflaA (10), and Lp03 (50) and Escherichia coli strains were gifts from Craig Roy (Yale University). L. longbeachae NSW 150 was a gift from Hayley Newton (University of Melbourne). Escherichia coli strains used for cloning (Top10; Invitrogen) and L. pneumophila mutagenesis (DH5α-λpir [51]) were maintained in Luria-Bertani (LB) medium supplemented with 25 μg ml−1 chloramphenicol (pJB1806 and pSN85) or 50 μg ml−1 kanamycin (pSR47s). Legionella strains were cultured on supplemented charcoal–N-(2-acetamido)-2-aminoethanesulfonic acid (ACES)-buffered yeast extract (CYE) and grown at 37°C as described previously (52). L. pneumophila Lp02 strains were maintained on CYE supplemented with 100 μg ml−1 thymidine. Liquid cultures were grown at 37°C with aeration in supplemented ACES-buffered yeast extract (AYE) as described previously (52, 53). When necessary, media were supplemented with 10 μg ml−1 chloramphenicol (for plasmid maintenance), 10 μg ml−1 kanamycin (for allelic exchange), or 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG).
Unless otherwise indicated, recombinant mouse interferon-γ (rIFN-γ) (Thermo Fisher Scientific), recombinant mouse tumor necrosis factor (rTNF) (Gibco), rat anti-mouse TNF antibody (R&D Systems), or normal rat IgG control (R&D Systems) were used at a concentration of 50 ng ml−1. In all cases, antibodies and recombinant cytokines were added at the time of infection.
A complete list of oligonucleotide primers used in this study is provided in Table 1.
TABLE 1.
Oligonucleotide primers used in this study
| Name | Sequencea |
|---|---|
| legC4KO-up | TTGTGGACAATAGCTCTTGG |
| legC4KO-down | ATACGCTGGCTATAGCACC |
| flaAKO-up | CCAGTTCAGTACTGTAAAGC |
| flaAKO-down | TATTTCTGCCGTGACTATCG |
| Lpg2505BglII-F | TGGAGATCTTTATGATAAAAGGAAAACTTATGC |
| Lpg2505PstI-R | GGCTGCAGTTATAAAATAATTGGTCGAG |
| LegC4BglII-F | TGGAGATCTAATGAGTTAAGAAAGGATCCG |
| LegC4BamHI-F | TGGGGATCCTTTTGATTCATTATGTATCCTTG |
| LegC4XbaI-R | ATTTCTAGATTATAGCTTAATATCAAAAG |
Restriction endonuclease cleavage sites are in bold.
Molecular cloning, plasmid construction, and generation of Legionella strains.
In-frame deletions of legC4 were generated by allelic exchange. Plasmids pSR47s::ΔlegC4 and pSR47s::ΔflaA, gifts from Craig Roy, were conjugated into SRS43 or Lp02, followed by selection for L. pneumophila deletions as described previously (54, 55). Sucrose-resistant, kanamycin-sensitive colonies were screened by PCR using the legC4KO-up/legC4KO-down and flaAKO-up/flaAKO-down primer pairs for ΔlegC4 and ΔflaA deletions, respectively.
To express legC4 on a plasmid under control of its endogenous promoter, legC4 plus the 300-bp upstream region was amplified from L. pneumophila genomic DNA (gDNA) with the LegC4BglII-F/LegC4XbaI-R primer pair and cloned as a BglII/XbaI fragment into BamHI/XbaI-digested pJB1806 (pJB) (56).
For IPTG-mediated expression of legC4, the legC4 gene was amplified using the primer pair LegC4BamHI-F/LegC4XbaI-R and cloned as a BamHI/XbaI fragment into BamHI/XbaI-digested pSN85, a gift from Craig Roy (pEV; N-terminal 3×FLAG epitope tag fusion [57]). For IPTG-mediated expression of lpg2505, the lpg2505 gene was amplified using the primer pair Lpg2505BglII-F/Lpg2505PstI-R and cloned as a BglII/PstI fragment into BamHI/PstI-digested pSN85. Sequence-confirmed pJB1806::plegC4 (pJB::plegC4), pSN85::legC4 (plegC4), and pSN85::lpg2505 (plpg2505) plasmids and empty vectors were transformed into Legionella strains as previous described (37).
Mice and BMDMs.
C57BL/6 wild-type, Myd88−/−, and Tnfr1−/− breeding pairs were purchased from the Jackson Laboratories (Bar Harbor, ME), and in-house colonies were maintained under specific-pathogen-free conditions at Kansas State University. All experiments involving animals were approved by the Kansas State University Institutional Animal Care and Use Committee (protocol number 4022) and performed in compliance with the Animal Welfare Act and NIH guidelines.
Bone marrow was harvested from mice as previous described (58). Bone marrow-derived macrophages were generated by differentiation in RPMI supplemented with 20% heat-inactivated fetal bovine serum (HI-FBS) (Gibco) and 15% L929 cell supernatant for 6 days prior to seeding for infection.
CI experiments in mice.
Six- to 10-week-old age- and sex-matched C57BL/6 wild-type or Tnfr1−/− mice were infected for competitive index (CI) experiments as previously described (30). Mixed bacterial inoculums (1:1) were diluted and plated on selective medium (5 μg ml−1 chloramphenicol for flaA::Tn and 10 μg ml−1 chloramphenicol for plasmid selection). At 48 h p.i., mice were euthanized and whole lung tissue was harvested. Lung tissue was homogenized in 300 μl of sterile water using a Bullet Blender (Next Advance) as described previously (59). Dilutions were plated on selective medium as described above. CFU were enumerated and used to calculate CI values [(CFUcmR48h/CFUwt48h)/(CFUcmRIN/CFUwtIN)].
Quantification of Legionella replication within macrophages.
Differentiated BMDMs were maintained in RPMI supplemented with 10% HI-FBS (Gibco) and 7.5% L929 cell supernatant. BMDMs were seeded at 2.5 × 105/well in 24-well plates 1 day prior to infection. BMDMs were infected with the indicated strains of L. pneumophila or L. longbeachae at a multiplicity of infection (MOI) of 1 in the presence or absence of 1 mM IPTG and/or recombinant cytokine as indicated. At 1 h p.i., cell monolayers were washed three times with phosphate-buffered saline (PBS), and fresh supplemented medium was added. Infections were allowed to proceed for up to 72 h or for 48 h, as indicated. To enumerate CFU, BMDMs were lysed in sterile water for 8 min, followed by repeat pipetting. Lysates were diluted as appropriate and plated on CYE agar plates, which were then incubated at 37°C for 4 days. For growth curve experiments, bacteria were enumerated after 1 h of infection and every 24 h thereafter for up to 72 h. To quantify fold replication, BMDMs were infected for 1 h and 48 h, and fold replication was enumerated by normalization of CFU counts at the indicated time points to the 1-h CFU counts.
Quantification of L. pneumophila replication in rich media in vitro.
The Legionella pneumophila ΔflaA, ΔflaA ΔlegC4, ΔflaA ΔlegC4(pEV), and ΔflaA ΔlegC4(plegC4) strains were streaked on CYE agar plates for single colonies, followed by generation of a 48-h heavy patch, which was harvested from plates and resuspended in supplemented AYE liquid medium (52, 53). Media for strains harboring plasmids were supplemented with 10 μg ml−1 chloramphenicol, and 1 mM IPTG was used to induce expression of plasmid-carried legC4. Liquid cultures for each strain in triplicate were started at an optical density at 600 nm (OD600) of 0.3 and grown for 24 h. The OD600 was quantified at the indicated time points.
ELISA.
BMDMs were seeded in a 24-well plate at 2.5 × 105/well 1 day prior to infection. The indicated Lp02 or SRS43 strains were used to infect the BMDMs (n = 3) at an MOI of 30 or 10, respectively, for the indicated times. For SRS43 strains, CFU were enumerated at each time point examined. Lp02 infections were performed in the absence of exogenous thymidine to prevent bacterial replication. At 1 h p.i., media were aspirated and cells were washed 3 times with PBS. Media were replaced, and supernatants were collected at the indicated time points. Expression of legC4 was induced by addition of 1 mM IPTG to the culture medium. Supernatants were either used fresh or stored at −20°C for up to 1 week, followed by quantification of TNF or IL-6 using a mouse TNF or IL-6 enzyme-linked immunosorbent assay (ELISA) kit (BioLegend) following the manufacturer’s instructions.
Western blot.
To confirm production of 3×FLAG fusion proteins from Legionella, suspensions of strains harboring either pSN85 alone (pEV), pSN85::legC4 (plegC4), or pSN85::lpg2505 (plpg2505) induced with IPTG were lysed by boiling in 3× Laemmli buffer. Proteins were separated by SDS-PAGE, followed by transfer to a polyvinylidene difluoride (PVDF) membrane (Thermo Fisher) using a wet transfer cell (Bio-Rad). Membranes were incubated in blocking buffer (5% nonfat milk dissolved in Tris-buffered saline–0.1% Tween 20 [TBST]). Anti-FLAG (clone M2; Sigma) was diluted at 1:1,000 in blocking buffer and incubated with membranes either overnight at 4°C or for 3 h at ambient temperature with rocking. Wash steps were performed 3 times for 10 min each in TBST. Horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Sigma) was diluted in blocking buffer at 1:5,000 and incubated with membranes for 1 to 2 h at room temperature with rocking. Membranes were washed, incubated with ECL substrate (Amersham), and imaged by chemiluminescence using a c300 Azure Biosystems Darkroom Replacer.
Statistical analysis.
Statistical analysis was performed with GraphPad Prism software using either the Mann-Whitney U test or Student’s t test, as indicated, with a 95% confidence interval. For all experiments, error bars denote ±standard deviations (SD) of samples in triplicate.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. A. Montelone and A. L. Passarelli for critical reading of the manuscript. The idea for a portion of this work was conceived in Craig Roy’s laboratory at the Yale School of Medicine.
This work was supported by start-up funds and a Mentoring Award from Kansas State University (to S.R.S.), an NIH K-INBRE Developmental Research Project Award (P20GM103418; to S.R.S.), an NIH NIGMS CoBRE Research Project Award (P20GM113117; to S.R.S.), and an NIH K-INBRE Semester Scholar Award (to A.J.H.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00755-18.
REFERENCES
- 1.Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol 7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton HJ, Ang DKY, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev 23:274–298. doi: 10.1128/CMR.00052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu VL, Plouffe JF, Pastoris MC, Stout JE, Schousboe M, Widmer A, Summersgill J, File T, Heath CM, Paterson DL, Chereshsky A. 2002. Distribution of Legionella species and serogroups isolated by culture in patients with sporadic community-acquired legionellosis: an international collaborative survey. J Infect Dis 186:127–128. doi: 10.1086/341087. [DOI] [PubMed] [Google Scholar]
- 4.Borges V, Nunes A, Sampaio DA, Vieira L, Machado J, Simões MJ, Gonçalves P, Gomes JP. 2016. Legionella pneumophila strain associated with the first evidence of person-to-person transmission of Legionnaires’ disease: a unique mosaic genetic backbone. Sci Rep 6:26261. doi: 10.1038/srep26261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mascarenhas DPA, Pereira MSF, Manin GZ, Hori JI, Zamboni DS. 2015. Interleukin 1 receptor-driven neutrophil recruitment accounts to MyD88-dependent pulmonary clearance of Legionella pneumophila infection in vivo. J Infect Dis 211:322–330. doi: 10.1093/infdis/jiu430. [DOI] [PubMed] [Google Scholar]
- 6.Archer KA, Roy CR. 2006. MyD88-dependent responses involving Toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires’ disease. Infect Immun 74:3325–3333. doi: 10.1128/IAI.02049-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neild AL, Shin S, Roy CR. 2005. Activated macrophages infected with Legionella inhibit T cells by means of MyD88-dependent production of prostaglandins. J Immunol 175:8181–8190. doi: 10.4049/jimmunol.175.12.8181. [DOI] [PubMed] [Google Scholar]
- 8.Shin S. 2012. Innate immunity to intracellular pathogens: lessons learned from Legionella pneumophila. Adv Appl Microbiol 79:43–71. doi: 10.1016/B978-0-12-394318-7.00003-6. [DOI] [PubMed] [Google Scholar]
- 9.Mascarenhas DPA, Zamboni DS. 2017. Inflammasome biology taught by Legionella pneumophila. J Leukoc Biol 101:841–849. doi: 10.1189/jlb.3MR0916-380R. [DOI] [PubMed] [Google Scholar]
- 10.Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog 2:e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molofsky AB, Shetron-Rama LM, Swanson MS. 2005. Components of the Legionella pneumophila flagellar regulon contribute to multiple virulence traits, including lysosome avoidance and macrophage death. Infect Immun 73:5720–5734. doi: 10.1128/IAI.73.9.5720-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Case CL, Shin S, Roy CR. 2009. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun 77:1981–1991. doi: 10.1128/IAI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A 110:1851–1856. doi: 10.1073/pnas.1211521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frutuoso MS, Hori JI, Pereira MSF, Junior DSL, Sônego F, Kobayashi KS, Flavell RA, Cunha FQ, Zamboni DS. 2010. The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect 12:819–827. doi: 10.1016/j.micinf.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Opitz B, Vinzing M, van Laak V, Schmeck B, Heine G, Günther S, Preissner R, Slevogt H, N'Guessan PD, Eitel J, Goldmann T, Flieger A, Suttorp N, Hippenstiel S. 2006. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J Biol Chem 281:36173–36179. doi: 10.1074/jbc.M604638200. [DOI] [PubMed] [Google Scholar]
- 16.Copenhaver AM, Casson CN, Nguyen HT, Duda MM, Shin S. 2015. IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis. Proc Natl Acad Sci U S A 112:7557–7562. doi: 10.1073/pnas.1501289112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skerrett SJ, Bagby GJ, Schmidt RA, Nelson S. 1997. Antibody-mediated depletion of tumor necrosis factor-alpha impairs pulmonary host defenses to Legionella pneumophila. J Infect Dis 176:1019–1028. doi: 10.1086/516530. [DOI] [PubMed] [Google Scholar]
- 18.Brieland JK, Remick DG, Freeman PT, Hurley MC, Fantone JC, Engleberg NC. 1995. In vivo regulation of replicative Legionella pneumophila lung infection by endogenous tumor necrosis factor alpha and nitric oxide. Infect Immun 63:3253–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanternier F, Tubach F, Ravaud P, Salmon D, Dellamonica P, Bretagne S, Couret M, Bouvard B, Debandt M, Gueit I, Gendre J-P, Leone J, Nicolas N, Che D, Mariette X, Lortholary O, Research Axed on Tolerance of Biotherapies Group. 2013. Incidence and risk factors of Legionella pneumophila pneumonia during anti-tumor necrosis factor therapy: a prospective French study. Chest 144:990–998. doi: 10.1378/chest.12-2820. [DOI] [PubMed] [Google Scholar]
- 20.Brieland J, Freeman P, Kunkel R, Chrisp C, Hurley M, Fantone J, Engleberg C. 1994. Replicative Legionella pneumophila lung infection in intratracheally inoculated A/J mice. A murine model of human Legionnaires’ disease. Am J Pathol 145:1537–1546. [PMC free article] [PubMed] [Google Scholar]
- 21.Shinozawa Y, Matsumoto T, Uchida K, Tsujimoto S, Iwakura Y, Yamaguchi K. 2002. Role of interferon-gamma in inflammatory responses in murine respiratory infection with Legionella pneumophila. J Med Microbiol 51:225–230. doi: 10.1099/0022-1317-51-3-225. [DOI] [PubMed] [Google Scholar]
- 22.Santic M, Molmeret M, Abu Kwaik Y. 2005. Maturation of the Legionella pneumophila-containing phagosome into a phagolysosome within gamma interferon-activated macrophages. Infect Immun 73:3166–3171. doi: 10.1128/IAI.73.5.3166-3171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziltener P, Reinheckel T, Oxenius A. 2016. Neutrophil and alveolar macrophage-mediated innate immune control of Legionella pneumophila lung infection via TNF and ROS. PLoS Pathog 12:e1005591. doi: 10.1371/journal.ppat.1005591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontana MF, Shin S, Vance RE. 2012. Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect Immun 80:3570–3575. doi: 10.1128/IAI.00557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barry KC, Ingolia NT, Vance RE. 2017. Global analysis of gene expression reveals mRNA superinduction is required for the inducible immune response to a bacterial pathogen. Elife 6:e1004229. doi: 10.7554/eLife.22707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asrat S, Dugan AS, Isberg RR. 2014. The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines. PLoS Pathog 10:e1004229. doi: 10.1371/journal.ppat.1004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hempstead AD, Isberg RR. 2013. Host signal transduction and protein kinases implicated in Legionella infection, p 249–269. In Hilbi H. (ed), Molecular mechanisms in Legionella pathogenesis. Springer, Berlin, Germany. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin S, Case CL, Archer KA, Nogueira CV, Kobayashi KS, Flavell RA, Roy CR, Zamboni DS. 2008. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog 4:e1000220. doi: 10.1371/journal.ppat.1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart LM, Paquette N, Boyer L. 2013. Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat Rev Immunol 13:199–206. doi: 10.1038/nri3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shames SR, Liu L, Havey JC, Schofield WB, Goodman AL, Roy CR. 2017. Multiple Legionella pneumophila effector virulence phenotypes revealed through high-throughput analysis of targeted mutant libraries. Proc Natl Acad Sci U S A 114:E10446–E10454. doi: 10.1073/pnas.1708553114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, Swanson MS. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med 203:1093–1104. doi: 10.1084/jem.20051659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujita M, Ikegame S, Harada E, Ouchi H, Inoshima I, Watanabe K, Yoshida S-I, Nakanishi Y. 2008. TNF receptor 1 and 2 contribute in different ways to resistance to Legionella pneumophila-induced mortality in mice. Cytokine 44:298–303. doi: 10.1016/j.cyto.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 33.Coers J, Vance RE, Fontana MF, Dietrich WF. 2007. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol 9:2344–2357. doi: 10.1111/j.1462-5822.2007.00963.x. [DOI] [PubMed] [Google Scholar]
- 34.Archer KA, Alexopoulou L, Flavell RA, Roy CR. 2009. Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell Microbiol 11:21–36. doi: 10.1111/j.1462-5822.2008.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collart MA, Belin D, Vassalli JD, de Kossodo S, Vassalli P. 1986. Gamma interferon enhances macrophage transcription of the tumor necrosis factor/cachectin, interleukin 1, and urokinase genes, which are controlled by short-lived repressors. J Exp Med 164:2113–2118. doi: 10.1084/jem.164.6.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vila-del Sol V, Punzón C, Fresno M. 2008. IFN-gamma-induced TNF-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol 181:4461–4470. doi: 10.4049/jimmunol.181.7.4461. [DOI] [PubMed] [Google Scholar]
- 37.Wood RE, Newton P, Latomanski EA, Newton HJ. 2015. Dot/Icm effector translocation by Legionella longbeachae creates a replicative vacuole similar to that of Legionella pneumophila despite translocation of distinct effector repertoires. Infect Immun 83:4081–4092. doi: 10.1128/IAI.00461-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cazalet C, Gomez-Valero L, Rusniok C, Lomma M, Dervins-Ravault D, Newton HJ, Sansom FM, Jarraud S, Zidane N, Ma L, Bouchier C, Etienne J, Hartland EL, Buchrieser C. 2010. Analysis of the Legionella longbeachae genome and transcriptome uncovers unique strategies to cause Legionnaires’ disease. PLoS Genet 6:e1000851. doi: 10.1371/journal.pgen.1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burstein D, Amaro F, Zusman T, Lifshitz Z, Cohen O, Gilbert JA, Pupko T, Shuman HA, Segal G. 2016. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat Genet 48:167–175. doi: 10.1038/ng.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Massis LM, Assis-Marques MA, Castanheira FVS, Capobianco YJ, Balestra AC, Escoll P, Wood RE, Manin GZ, Correa VMA, Alves-Filho JC, Cunha FQ, Buchrieser C, Borges MC, Newton HJ, Zamboni DS. 2016. Legionella longbeachae is immunologically silent and highly virulent in vivo. J Infect Dis 215:440–451. doi: 10.1093/infdis/jiw560. [DOI] [PubMed] [Google Scholar]
- 41.Massis LM, Zamboni DS. 2011. Innate immunity to Legionella pneumophila. Front Microbiol 2:109. doi: 10.3389/fmicb.2011.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown AS, Yang C, Hartland EL, van Driel IR. 2017. The regulation of acute immune responses to the bacterial lung pathogen Legionella pneumophila. J Leukoc Biol 101:875–886. doi: 10.1189/jlb.4MR0816-340R. [DOI] [PubMed] [Google Scholar]
- 43.Spörri R, Joller N, Albers U, Hilbi H, Oxenius A. 2006. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J Immunol 176:6162–6171. doi: 10.4049/jimmunol.176.10.6162. [DOI] [PubMed] [Google Scholar]
- 44.Pereira MSF, Marques GG, Dellama JE, Zamboni DS. 2011. The Nlrc4 inflammasome contributes to restriction of pulmonary infection by flagellated Legionella spp. that trigger pyroptosis. Front Microbiol 2:33. doi: 10.3389/fmicb.2011.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carey KL, Newton HJ, Lührmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo Z-Q. 2012. Legionella secreted effectors and innate immune responses. Cell Microbiol 14:19–27. doi: 10.1111/j.1462-5822.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Losick VP, Haenssler E, Moy M-Y, Isberg RR. 2010. LnaB: a Legionella pneumophila activator of NF-kappaB. Cell Microbiol 12:1083–1097. doi: 10.1111/j.1462-5822.2010.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, Liu L, Shao F. 2009. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci U S A 106:13725–13730. doi: 10.1073/pnas.0907200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu M, Conover GM, Isberg RR. 2008. Legionella pneumophila EnhC is required for efficient replication in tumour necrosis factor alpha-stimulated macrophages. Cell Microbiol 10:1906–1923. doi: 10.1111/j.1462-5822.2008.01180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol 7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 51.Zuckman DM, Hung JB, Roy CR. 1999. Pore-forming activity is not sufficient for Legionella pneumophila phagosome trafficking and intracellular growth. Mol Microbiol 32:990–1001. doi: 10.1046/j.1365-2958.1999.01410.x. [DOI] [PubMed] [Google Scholar]
- 52.Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J Clin Microbiol 10:437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saito A, Rolfe RD, Edelstein PH, Finegold SM. 1981. Comparison of liquid growth media for Legionella pneumophila. J Clin Microbiol 14:623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo Z-Q, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A 101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagai H, Roy CR. 2001. The DotA protein from Legionella pneumophila is secreted by a novel process that requires the Dot/Icm transporter. EMBO J 20:5962–5970. doi: 10.1093/emboj/20.21.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bardill JP, Miller JL, Vogel JP. 2005. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol Microbiol 56:90–103. doi: 10.1111/j.1365-2958.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- 57.Folly-Klan M, Alix E, Stalder D, Ray P, Duarte LV, Delprato A, Zeghouf M, Antonny B, Campanacci V, Roy CR, Cherfils J. 2013. A novel membrane sensor controls the localization and ArfGEF activity of bacterial RalF. PLoS Pathog 9:e1003747. doi: 10.1371/journal.ppat.1003747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Case CL, Roy CR. 2013. Analyzing caspase-1 activation during Legionella pneumophila infection in macrophages. Methods Mol Biol 954:479–491. doi: 10.1007/978-1-62703-161-5_29. [DOI] [PubMed] [Google Scholar]
- 59.Ivanov SS, Roy CR. 2013. Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat Immunol 14:1219–1228. doi: 10.1038/ni.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.