

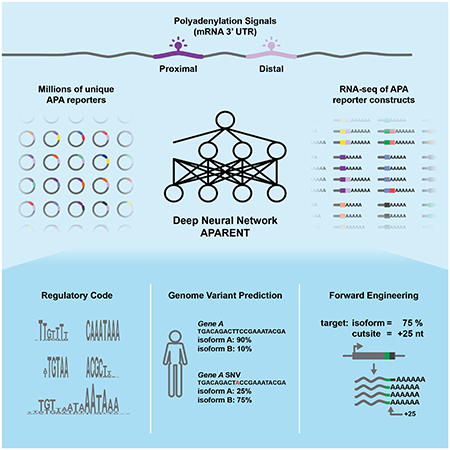
Abstract
Alternative polyadenylation (APA) is a major driver of transcriptome diversity in human cells. Here, we use deep learning to predict APA from DNA sequence alone. We trained our model (APARENT, APA REgression NeT) on isoform expression data from over three million APA reporters. APARENT’s predictions are highly accurate when tasked with inferring APA in synthetic and human 3’UTRs. Visualizing features learned across all network layers reveals that APARENT recognizes sequence motifs known to recruit APA regulators, discovers previously unknown sequence determinants of 3’-end processing, and integrates these features into a comprehensive, interpretable cis-regulatory code. We apply APARENT to forward engineer functional polyadenylation signals with precisely defined cleavage position and isoform usage and validate predictions experimentally. Finally, we use APARENT to quantify the impact of genetic variants on APA. Our approach detects pathogenic variants in a wide range of disease contexts, expanding our understanding of the genetic origins of disease.
Graphical Abstract
In brief
A deep neural network enables precise engineering of polyadenylation signals, identifies human genetic variants that act through mis-regulating APA, and learns a comprehensive model of the cis regulatory APA code.
Alternative polyadenylation (APA) is a ubiquitous regulatory process by which multiple RNA isoforms with distinct 3’-ends can be derived from a single gene (Figure 1A) (Di Giammartino et al., 2011; Elkon et al., 2013; Tian and Manley, 2017). APA is tightly regulated through a combination of cis-regulatory sequences -- most importantly a set of competing polyadenylation (polyA) signals (PAS) -- and trans-acting RNA-binding proteins (RBPs) that recognize these sequences. Each PAS is defined by the 6-base central sequence element (CSE), most commonly AATAAA, and its upstream and downstream sequence elements (USE, dSe) that recruit mRNA-processing machinery to cleave and polyadenylate a transcript. Although we have extensive information about individual regulators and some of their interactions, we still lack an interpretable and quantitative model that integrates this information to predict isoform abundance and cleavage position.
Figure 1. Massive Parallel Reporter Assay for Alternative Polyadenylation.
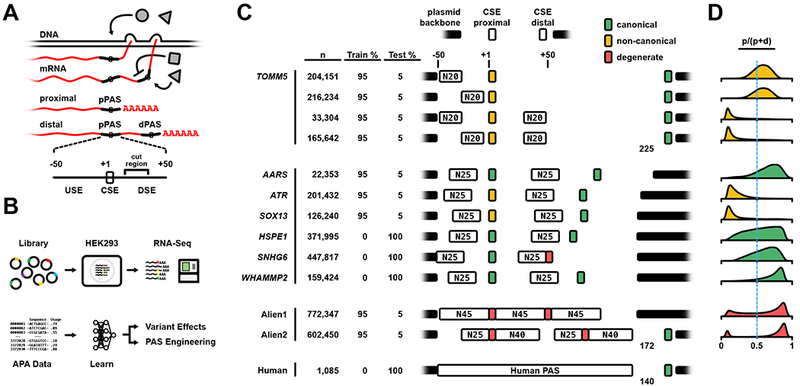
A) APA and the polyA signal. Newly-transcribed mRNA is targeted by multiple factors (grey) that enhance or suppress selection of APA sites. A PAS (below) is defined by the 6-base CSE and regions of approximately 50 bp both upstream and downstream.
(B) Massively parallel reporter assay for APA. Millions of unique reporters are cloned from degenerate oligos and transiently transfected in human cell culture where they are expressed and alternatively polyadenylated. RNA is extracted, sequenced and quantified for every reporter, and the data used to train a deep neural network.
(C) The library is comprised of multiple sublibraries that vary in structure and 3’UTR context (human italicized; CSE sequence denoted in legend; thick black bar = plasmid, thin = native 3’UTR sequence). Degenerate sequence was introduced up and/or downstream (N20-45) of the proximal PAS, and in some cases, within the CSE (degenerate, red). The CSE sequence is either the canonical AATAAA (green), the canonical with a single base substitution (yellow), or varied (red; SNHG6 = NNTAAA, Alien1 = AWTAAA, Alien2 = (95% A, 2% G/C, 3% T)).
(D) The distribution of relative proximal site usage per unique sequence for each library. Each histogram is color-coded to match the proximal CSE.
See also Figure S1.
Genetic variants that interfere with APA in cis have been implicated in disease (Danckwardt et al., 2008; Bennett et al., 2001; Wiestner et al., 2007) and a quantitative understanding of the APA code would dramatically improve our ability to identify such pathogenic variants. Experimentally characterizing every possible variant is not feasible even with high-throughput measurement techniques such as massively parallel reporter assays (MPRA) (Patwardhan et al. 2009; Melnikov et al. 2012; Findlay et al. 2014; Matreyek et al. 2018; Sharon et al. 2012; Smith et al. 2013; White et al. 2013) which are still limited to tens of thousands of variants and mostly targeted to specific genes. Statistical methods, such as genome-wide association studies (GWAS), have had great success in linking genetic variation to disease but require large sampling to characterize rare variants and provide no information for de novo variants. Furthermore, GWAS cannot predict why a variant is pathogenic. Similarly, models based on conservation cannot predict functional outcomes (Cooper et al., 2005; Fu et al., 2014; Pollard et al., 2010). Overcoming these limitations of scale and generality requires accurate and functionally-aware models that can be used to screen variants at genome scale and identify candidates likely to be most disruptive.
Perhaps a more intriguing application of a predictive sequence-function model for APA is the ability to engineer functional sequence elements. “Writing” functional DNA sequences that result in a specified level of gene expression and can be combined into gene circuits is the defining goal of synthetic biology (Purnick and Weiss 2009). Quantitative models that enable de novo design of cis-regulatory sequence elements could thus dramatically accelerate synthetic biology research and boost progress in metabolic engineering, synthetic immunology, mRNA therapeutics, gene therapy and related fields (Ruder et al. 2011; Sahin et al. 2014; Roybal and Lim 2017).
Deep learning has enabled important strides toward building predictive models that relate cis-regulatory sequence to molecular phenotype (Alipanahi et al., 2015; Jaganathan et al., 2019; Kelley et al., 2016; Lanchantin et al., 2016; Leung et al., 2017; Xiong et al., 2015; Zhou and Troyanskaya, 2015). Such models have shown promise for identifying disease-related variants and, when used together with genetic algorithms, are beginning to be deployed for de novo design of gene sequences (Biswas et al., 2018; Cuperus et al., 2017; Sample et al., 2018). Visualization techniques can provide insight into the regulatory motifs identified as important, thus making deep neural networks (DNNs) interpretable (Alipanahi et al., 2015; Kelley et al., 2016). Still, the datasets available for training neural networks in the context of gene expression are often limited in scale, which limits model quality. Moreover, visualization techniques have been limited to motifs learned in the lowest network layer rather than presenting a view of motif interactions across an entire regulatory region. Finally, engineering applications are only beginning to emerge and techniques that take full advantage of neural network approaches, such as those used in computer vision (e.g. DeepDream; Szegedy et al., 2014), still need to be developed.
Here we train a deep neural network, APARENT, on over 3 million synthetic APA reporter genes, overcoming size limitations inherent to traditional biological datasets. We demonstrate APARENT’s utility through three separate applications. First, we use it to identify and quantify regulatory interactions, furthering our understanding of the biology of APA. Second, we develop an optimization algorithm for forward engineering of synthetic PASs that result in precisely defined isoform expression levels and cleavage positions. Third, we apply APARENT to predict the impact and putative mechanism of genetic variants on APA and identify deleterious mutations that disrupt gene function by mis-regulating APA.
A Massively Parallel Reporter Assay for APA with over 3 Million Reporter Constructs
To generate a dataset large and diverse enough to represent the complexity of the APA code we constructed and transiently expressed minigene libraries of >3 million unique UTRs and obtained the isoform and cleavage data from the expressed RNA (Figure 1B). Each library expresses multiple PASs in a unique context -- 10 libraries were derived from seven different human 3’UTRs but with USE and DSE regions replaced by randomized sequence; another 2 libraries are fully degenerate with either a canonical (AWTAAA) or doped (95% A, 2% G/C, 3% T) CSE (Figure 1C). Finally, upstream of a bGH PAS, we cloned a reporter library with 1,085 PASs from the human reference genome. The 1,085 PASs come from a total of 817 genes and constitute the complete set of all PASs in the ClinVar database with at least one annotated variant. To quantify isoform expression, we transiently transfected all libraries into HEK293 cells, extracted the expressed library RNA, and sequenced it (Figure 1B). In total, we collected isoform expression data for 3,372,030 unique reporters with an average of 39 reads per reporter for random libraries and over 4000 reads per native human APA reporter. The majority of our reads, 53%, mapped to proximal isoforms while 28% mapped to distal isoforms. The remaining reads (19%) mapped to de novo PASs within the degenerate regions.
Isoform ratios exhibited a wide range of variation within and between APA libraries (Figure 1D, S1A). Variation could be attributed to the CSE but also the surrounding regulatory sequences. In UTRs with two strong signals - both AATAAA - the proximal site was slightly preferred, consistent with the “first come, first served” hypothesis of kinetic coupling (Bentley, 2014; DeZazzo and Imperiale, 1989) (Figure 1D; AARS, HSPE1, WHAMMP2). A weaker, proximal CSE can be favored, especially if the stronger, distal PAS is relatively far downstream, as with the TOMM5 library. However, expression of the proximal isoform is nearly completely suppressed when the TOMM5 DSE is randomized, underscoring the importance of regulatory sequence outside of the CSE.
Predicting Alternative Polyadenylation with APARENT
We then trained a DNN, APARENT (Figure 2A), to predict PAS usage from DNA sequence. Assuming that competing PASs are independently and identically regulated when they do not physically overlap, the DNN should learn a general model of APA from our minigene libraries even though only the proximal PAS was randomized.
Figure 2. Model Architecture, Performance and Layer-by-Layer Feature Analysis.
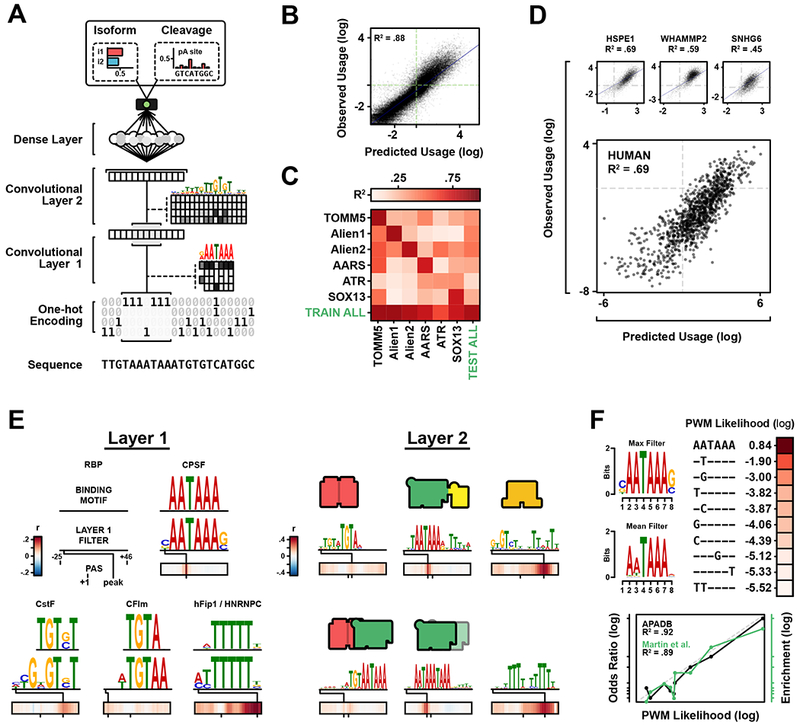
(A) APARENT takes a 1-Hot-coded PAS sequence as input to predict % proximal isoform and % cleavage at each position.
(B) Predicted vs. observed proximal isoform log odds of the test set.
(C) Cross-library confusion matrix when predicting proximal isoform use. Diagonal entries are tests on the training library. Off-diagonal entries are models trained on one library but tested on another.
(D) Predicted vs. observed proximal use on held-out random libraries (HSPE1, SNHG6, and WHAMMP2; mean R2 = .58), and on the held-out native human PAS library (HUMAN; R2 = .69).
(E) RBP motif logos and per-position effects (Pearson’s r between filter activation and proximal use) learned in the first and second convolutional layers. Layer 2 filters are shown with their proposed effector interactions. Additional RBP motifs of Layer 1 and 2 are shown in Figure S2E–F.
(F) Left: Sequence Logos for the CSE detector filter. Right: Ranked CSE variants extracted from the filter. Below: Comparison of variant scores against previously-published data.
APARENT was trained to predict the proximal APA isoform ratio of each variant UTR given its sequence as input. We used 95% of the data from 9 out of 13 libraries for training (~2.4M variants), 2% for validation (~50,000 variants) and 3% for testing (~80,000). Four libraries (including the 1,085 human reference PASs) were held out entirely, allowing for tests on generalization (Figure 1C). The best-performing model architecture consisted of two convolutional layers interlaced with subsampling layers, a fully connected layer, and a logistic regression output node (Figure 2A).
To evaluate APARENT, we tested its ability to infer the proportion (log odds) of proximal isoform expression (isoform use). The DNN performed remarkably well at predicting the isoform use of the combined test set (R2 = .88; Figure 2B). To show that joint training on all libraries improves generalization, separate DNNs were trained on each individual library and cross-tested by predicting isoform use of every other library. The DNN trained on the combined data performed at least as well as, and in some cases better than, any individual network on its corresponding library (min library R2 = .50, mean library R2 = .68, total R2 = .88; Figure 2C). The degree of correlation changes depending on UTR library because of differences in variance; libraries with low or high variance in isoform use are coupled with lower or higher R2 respectively, as the mean prediction error remains almost constant. As a baseline test, we also compared APARENT to a 6-mer linear logistic regression model and found that APARENT outperforms this simpler model, suggesting that positional and non-linear effects are important for accurately predicting APA (Figure S2A).
Next, we asked whether APARENT could generalize to entirely new UTR contexts. First, we predicted proximal isoform use of the three held-out libraries (HSPE1, SNHG6 and WHAmMp2). The predictions had a mean correlation and error comparable to the trained-on libraries (mean library R2 = .58, Figure 2D, S2B). Second, we evaluated APARENT’s performance on the fully held-out library of >1,000 reference human PASs. APARENT performed remarkably well (R2 = .69, Figure 2D) confirming that the model generalizes from completely random to evolved human 3’ UTRs.
Sequence Determinants of Isoform Selection
To visualize sequence determinants of PAS preference learned in the first convolutional layer we extracted filter position weight matrices (PWMs) following (Alipanahi et al., 2015) (Figure 2e, S2D–E, Table S1). The position-specific effect on PAS usage of each filter was quantified by measuring the correlation between activations and isoform use at each position across the PAS (Cuperus et al., 2017). We cross-referenced the PWMs with published binding data, as well as the Compendium of RNA Binding Protein motifs (Ray et al., 2013) using the Tomtom comparison tool (Gupta et al., 2007), and surveyed the top-scoring results for APA mediators.
We identified motifs matching known binding sites for all components of the core polyA machinery (Figure 2E, Layer 1). One filter clearly resembles the canonical CSE, AATAAA, which can directly bind WDR33/CPSF30, subunits of CPSF (Schönemann et al., 2014). We also identified filters corresponding to the CstF-binding motif. CstF functions in cooperation with CPSF to direct cleavage and polyA in the DSE (Pérez Cañadillas and Varani, 2003; Sun et al., 2018). TGTA is a regulatory motif commonly found in studies of APA and identified as a target of CFIm25 (Masamha et al., 2014; Zhu et al., 2018). Additionally, the motif recognized by hFip1 – another subunit of CPSF and an enhancer of CPSF-CstF interactivity (Kaufmann et al., 2004; Martin et al., 2012) -- was also represented. Importantly, the position-specific enhancing or suppressing effect assigned to each filter by APARENT is consistent with the known preferred binding position of its matched RBP: CFIm25 in the USE, CPSF at the CSE, hFip1 adjacent to the CSE, and CstF in the DSE (Figure 2E) (Hu et al. 2005). We identified additional RBP binding motifs in the filters of this layer, many previously implicated in APA regulation (Figure S2E and Table S1).
Variants of the consensus hexamer AATAAA are common (Gruber et al., 2016). These variants exhibit reduced affinity for recruiting the CPSF complex through WDR33/CPSF30, with the degree of affinity determined by the nucleotide substitution. We generated scaled filter PWMs and compared the filter response with published data (Gruber et al., 2016; Müller et al., 2014), confirming the impact of different CSE variants on APA (Figure 2F). We further validated motifs identified by the DNN directly in the data using a 6-mer log odds ratio analysis (Figure S2C) (Rosenberg et al., 2015). As additional validation, we identified the same set of convolutional filter motifs when training a new network instance with an independent random weight initialization (Figure S2G).
Visualizing Motif Interactions
Next, we generalized the method of (Alipanahi et al., 2015; Cuperus et al., 2017) to visualize features learned in the second layer. By estimating PWMs and position-dependent effects from sequences that give rise to Layer 2 activations, we capture longer motifs or combinations of short motifs (Figure 2E, S2F, Table S2). Combinatorial effects of cis-elements have been suggested in earlier bioinformatics studies of PASs (Cheng et al., 2006). Filters that capture known motif interactions align well with the current understanding of APA regulation (Figure 2E, Layer 2, Top row). For example, APARENT identifies the core hexamer AATAAA in combination with a downstream polyT motif to upregulate selection compared to the average effect of AATAAA, reflecting known interactions with hFip1 (Kaufmann et al., 2004). Another filter is sensitive to dual TGTA motifs, supporting the notion that CFIm25 binds as a dimer (Yang et al., 2010, 2011). A third example identifies strong combinatorial effects of DSE determinants, such as GT-rich motifs (CstF-binding) preceding a polyT motif further downstream.
We also find novel motif interactions with strong net effect sizes (Figure 2E, Layer 2, Bottom row). One of these filters is sensitive to a “multi-CSE” consisting of two overlapping canonical hexamers forming the 10-mer AAWAAAWAAA, estimated to substantially increase proximal selection compared to a single AATAAA. Another filter is sensitive to TGTAWTAAA, an overlapping binding site for both CFIm25 and CPSF. As detailed below, we find evidence that competitive binding of CFIm25 and CPSF can result in both strong up and downregulation depending on sequence context. Finally, one of the filters recognizes multiple polyT islands in the DSE to be preferential for APA selection, possibly due to the vast number of enhancing RBPs that bind to this motif.
Predicting Cleavage Distribution with APARENT
Since RNA-seq provides information about the precise cut position, we asked whether APARENT could be trained to predict the probability of cleavage occurring at any given position. To directly learn the cleavage distribution for each PAS we used a DNN almost identical to the isoform-based model, but with a final network layer that outputs a multinomial probability distribution of cleavage at any position across the sequence (Figure 3A).
Figure 3. Cutsite Predictions and Regulatory Determinants Learned by APARENT.

(A) APARENT’s output layer predicts the probability of cleavage at each of the 186 input nucleotides.
(B) Average predicted cut position on the Alien1 test set. X-axis denotes average position. The Y-axis sorts each sequence on observed cut position.
(C) Predicted isoform abundance using the DNN of Figure 2A vs. integration of cleavage distribution using the DNN of Figure 3A.
(D) Example Alien1 sequences with their predicted (blue) and observed (red) cut distribution. The panel also shows the average Alien1 cut distribution.
(E) Selection of layer 1 filters validating known and newly discovered cleavage determinants. Each plot measures correlation between a filter activating at a certain position and cleavage occurring at some other position. See Figure S3B for additional identified motifs.
(F) % sequences from the Alien1 library with significantly folded USEs (black) or CSE-Cut regions (red), as a function of cut position relative to the CSE. “USE” refers the 50 nt region upstream of the CSE, and “CSE-Cut” refers to the region from the end of the CSE to the cut position.
(G) Predicted vs. observed cut magnitude at positions near (left) or far from (right) the CSE. The color encodes DSE MFE.
See also Figure S3.
We evaluated this generalized version of APARENT in two ways. First, we compared the predicted average cleavage position with the observed average position of every test set sequence (Figure 3B). The two quantities correlated well (R2 = .82 for Alien1, R2 = .55 for the held-out WHAMMP2 library). Second, we compared the area under the proximal region of the predicted cleavage curve, which corresponds to total isoform abundance, against the previously predicted proximal isoform ratios (Figure 3C). The two predictions show strong agreement (R2 = .95).
Encouraged by the substantial cleavage variation observed in the libraries, we searched for UTR variants with large deviation in cleavage sites (Figure 3D). These UTRs reveal a rich landscape of possible cleavage distributions ranging from unimodal cuts, to bimodal distributions with cut sites separated by as much as 10 nucleotides, to a cluster of positions where cleavage is initiated at many different sites. APARENT is able to predict these sites with high precision, suggesting the choice of cleavage site itself is governed by a deterministic regulatory code.
The Determinants of Cleavage Site Selection
To identify cleavage site determinants, we applied APARENT to a random sample of 120,000 sequences from the Alien1 library, predicting their entire cleavage distribution. Each Layer 1 filter activation at every position was recorded and correlated with the magnitude of cleavage at every position, resulting in set of two-dimensional plots that together with their consensus sequence logos describe the regulatory impact of each filter motif on every cleavage site as a function of position (Figure 3E, S3A–B).
Consistent with earlier studies, the DNN filter heatmap characterizes the CA dinucleotide as the most favorable substrate for cleavage (Derti et al., 2012). However, TA and GA are also identified as functional, albeit weaker cleavage sites (Chen et al., 1995; Li and Du, 2013). We validated these findings by calculating the odds ratio of cleaving at every possible dinucleotide in the fully random Alien1 UTR library (Figure S3C) and found that cleaving at CA has a preferential bias (odds ratio = 4.7) compared to GA and TA (odds ratio = 2.7 and 3.3, respectively). This observation conflicts with recent studies suggesting that there is no bias towards cA (Li and Du, 2013; Wang et al., 2018) but these studies are based on enrichment analyses of conserved native PASs, whereas our observations build on probing a uniformly random DSE for a functional response. The GT-rich motif, typically identified as the CstF binding site (Pérez Cañadillas and Varani, 2003), is found to enhance cleavage when located immediately downstream of the cleavage site (Figure S3B). Interestingly, the motif only appears to be functional for cleavage sites located ~15-30 bp downstream of the first nucleotide of the CSE.
Beyond cut site dinucleotides and CstF, APARENT revealed an extensive regulatory code governing the choice of cut site. For example, polyG and polyT motifs are highly effective cleavage site regulators; polyG preceding a cut site suppresses its usage, while a polyT motif enhances usage. PolyG and polyT strongly enhance or suppress cleavage adjacent to or far away from the CSE, locations at which the dinucleotides CA, TA, and GA have no measurable impact.
Recently, 3’ mRNA secondary structure was implicated in guiding cleavage site position (Wu and Bartel, 2017). We computationally folded the sequence upstream (−45 to −1) and downstream (+7 to cut site) of over 100k Alien1 PASs and evaluated the significance of structure relative to each cut position (Freyhult et al., 2005). We found that cleavage position indeed correlates with increased secondary structure (Figure 3F). We then confirmed that APARENT predicts cleavage accurately even at distances >40 bases from the CSE, where cut magnitude is highly correlated with MfE (Figure 3G).
Precise Forward-Engineering of PASs with Stochastic Sequence Backpropagation
We then asked if APARENT could computationally engineer PASs according to target specifications and constraints. Previous approaches to forward engineering typically employ genetic algorithms, but because these algorithms rely on random chance and selection to generate new sequences instead of predicting improvements, they can easily get stuck in local minima. We took a different approach, by extending a computer vision method (Szegedy et al., 2014), previously used in genomics to visualize TF binding sites (Lanchantin et al., 2016), to the task of forward-engineering PASs (Figure 4A, STAR methods). We randomly initialized a PWM and send it as input to APARENT and specified an objective function in terms of APARENT’s outputs (isoform or cut). Since a neural net is differentiable, we computed the gradient of the objective w.r.t the PwM (with backpropagation). We then iteratively optimized the PWM with gradient ascent to perform better at the objective, generating sequences that conform to target APA ratios or cleave at target positions. The resulting algorithm, SeqProp, can accurately construct PASs that conform to a wide range of target objectives. To evaluate APARENT’s engineering capabilities, we synthesized, assembled, and expressed the engineered PASs in our APA assay (Figure 4B, STAR methods). For comparison, we used data from our human wild type reporter library as a reference for the native range of alternative isoform use.
Figure 4. Forward-Engineering of PASs by Backpropagation.
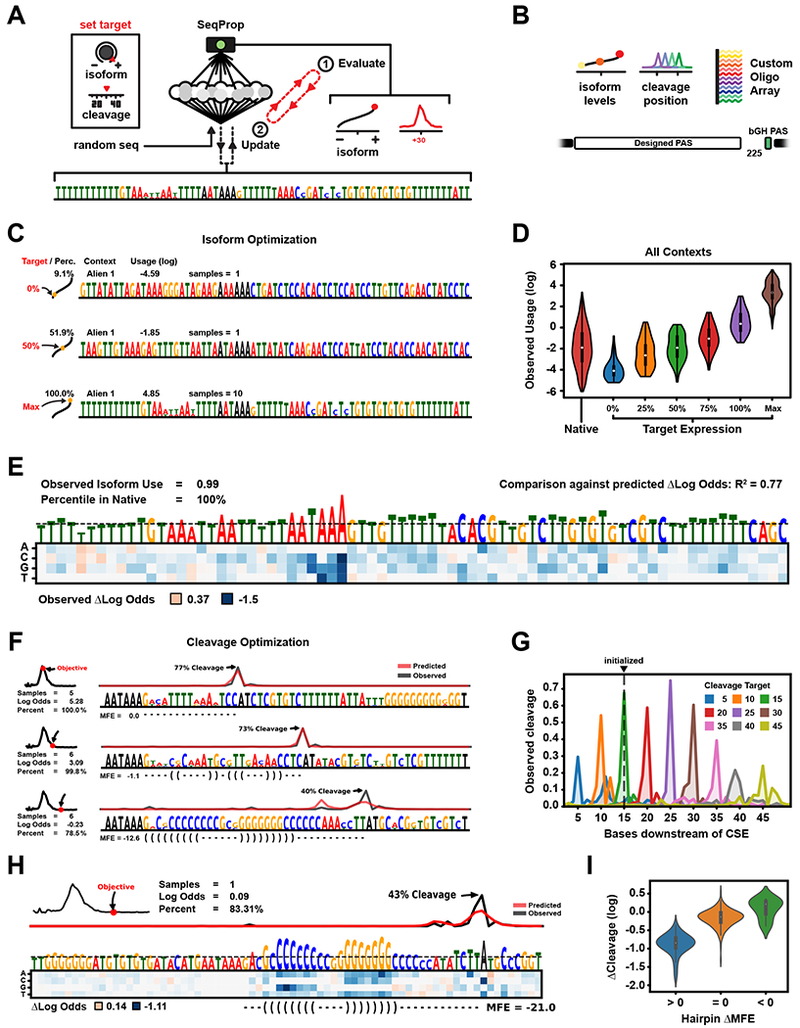
(A) A sequence PWM is iteratively optimized against a polyA objective by gradient ascent through APARENT.
(B) The sequences were synthesized in an oligo array, expressed in HEK293 and measured by sequencing.
(C) PWMs engineered for target isoform abundance. Shown are the target objectives, the measured percentile among human sequences, the number of PWM samples, and measured proximal use.
(D) Measured isoform use per isoform objective. The ‘Native’ category displays human sequences.
(E) Saturation mutagenesis of a PWM maximized for proximal use. The heatmap shows measured isoform fold change (in log-scale) as a result of each SNV. APARENT’s predicted log fold changes strongly agree with these measurements (R2 = 0.77). (F) PWMs engineered for cleavage at target positions. Shown are the target objectives, the number of PWM samples, measured/predicted cleavage profiles and predicted MFE structures of the DSE.
(G) Average cut profile of all synthesized sequences separated by objective cut position.
(H) Saturation mutagenesis of a PWM optimized for cleavage at CSE+35. The heatmap shows measured change in cleavage proportion as a result of each SNV.
(I) The measured SNV effects of Figure 4H grouped by the effect on DSE hairpin folding. The Y-axis displays the measured fold change (in log-scale) of cleavage proportion at the target cut site (CSE+35) due to each SNV.
Engineering Isoform Expression
We first optimized PASs for a range of target isoform use: 0%, 25%, 50%, 75% and 100%. We generated 10 sequences per target expression for 6 different UTR contexts. We noted that for the 100% target objective, sequences usually converged to ~95-98% predicted usage. To get as close to 100% usage as possible, we directly maximized the proximal class score (the log odds) just before the sigmoid output. We optimized 20-50 PWMs per library for maximal proximal preference and sampled up to 10 sequences from each PWM for a total of 1,200 “Max” PASs (Figure 4C, S4D–E). The predicted isoform use of all generated sequences were highly accurate when compared to experimental measurements (Figure 4D, S4A–B, Movie S1, R2 = .90). The “Max” sequences had a mean usage higher than any native PAS measured in the array, and the strongest sequence had a usage of 99.7% (against a strong distal PAS). We also optimized sequences for maximal preference with previously unseen UTR library backgrounds (Figure S4C). Again, the sequences were regulated as predicted.
The USE and DSE regions exhibit similar characteristics across UTR contexts, suggesting there is a “consensus” template for a strong PAS (Figure 4C, S4D–E). Strong USEs typically include polyT and possibly tAtA, polyC, or TGTA motifs. Strong DSEs contain a stretch of polyT, an A-rich stretch, followed by GT- or CT-rich content and finally another stretch of polyT. While the generated patterns share many similarities, there are also differences due to the UTR context. An example is the polyG track found in the Alien2 DSE, a repressor motif that SeqProp uses to suppress a downstream competing PAS.
We performed saturation mutagenesis of one of the sequences maximized for proximal isoform (Figure 4E). The vast majority of variants were measured to have a down-regulatory effect on isoform abundance, and the few upregulatory SNVs had low measured net effects. These results indicate that SeqProp indeed converges sequences to (local) optima.
Visualizing Motif Interactions Across an Entire PAS
When visualizing higher layer features we cannot resort to simply sampling the data, as we did for visualizing motifs in the first and second layer, because the very long length of the dense layer neurons (186 nt) make it exceedingly unlikely that we find maximally activating examples by chance. However, SeqProp can be used for visualization of higher-layer features in a DeepDream-style procedure whereby dense neurons are maximized by gradient ascent (Olah et al., 2017). The resulting sequence logos combine functional motifs identified in lower layers and describe global sequence determinants of PAS strength by (Figure S2H–I). The neurons specialize on different sequence subspaces, where G- and GC-rich sites are much less favored compared to sites containing various conserved T- and GT-rich stretches.
Engineering cleavage position
Next, we optimized randomly initialized PASs for cleavage across a wide range of target positions downstream of the CSE hexamer, while also maximizing site preference (Figure 4F, S4I, Movie S2). SeqProp could successfully generate cut distributions with high specificity, even at extreme distances (Figure 4G, S4F–G, total R2 = .94 between predicted and measured average cut positions). For cleavage sites within 30 nt, the optimized DSEs converged to sequences with no predictable secondary structure. Rather, APARENT positioned core processing elements (CstF, various RBP binding sites, etc.) in optimal locations relative to the cleavage site (Figure 4F, S4I). For cleavage beyond 30 nt, SeqProp built sequences upstream of the cleavage site that were predicted to form stable hairpins with stem lengths that increase with site distance (Figure S4H).
To validate the hypothesis that DSE hairpin folding is a crucial determinant for the engineered sequences with distant cuts, we performed saturation mutagenesis of an engineered sequence targeted for cleavage +35 nt downstream of the CSE (Figure 4H). SNVs outside the hairpin had very low effect on cleavage magnitude at the target cut site, while nearly any interference with the hairpin structure had a down-regulatory effect (Figure 4I). Interestingly, a few SNVs further stabilized the hairpin structure, and a majority were measured to have slightly upregulatory effects at the target cut site.
APARENT Accurately Predicts Native APA
Next, we turned to APADB, a dataset of APA events from multiple human tissues. We extracted every pair of adjacent 3’ UTR PAS sequences and used the corresponding RNA-seq read counts to estimate relative isoform proportions (see STAR Methods). For this part of the analysis, we pooled the isoform read counts across all tissues. Building on our assumption that individual PAS strengths are independent when signals are well-separated, we used APARENT to score each of the two PASs for every neighboring pair. We then used logistic regression with these scores and the logarithm of the distance between the sites as the only features to predict isoform use in APADB (Figure 5A).
Figure 5. Performance of APARENT on Endogenous APA.
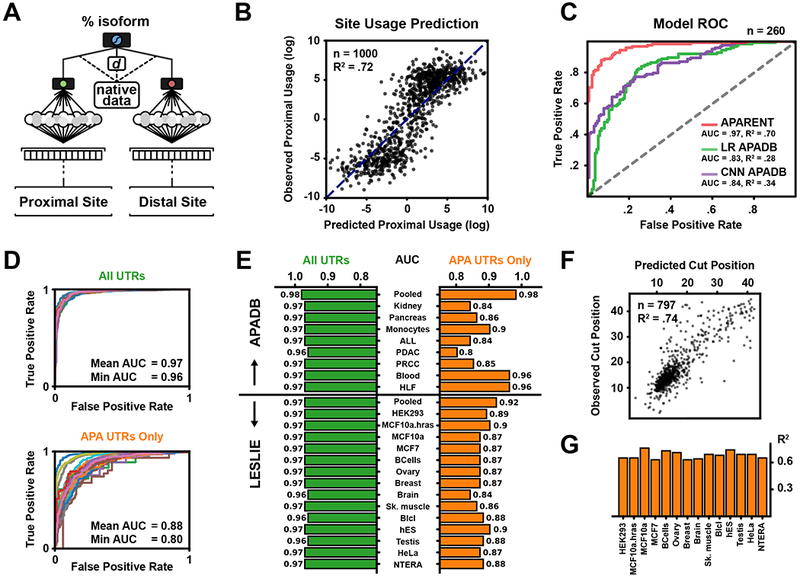
(A) The extended model for predicting isoform ratio between adjacent APA sites of the APADB and Leslie datasets.
(B) Leave-one-out Cross-validation of predicted vs. observed proximal use on the 1,000 Pooled-tissue APADB PASs with highest measured total read count.
(C) ROC curves obtained from classifying preferential site usage on held-out APADB data. APARENT is compared against a DNN and a linear 6-mer model trained only on APADB.
(D) Preferential site prediction ROC curves in tissues and cell types from APADB and Leslie datasets. (Top) All pairs of adjacent PASs. (Bottom) only pairs where both sites have supporting reads in the tissue.
(E) Bar chart of preferential site classification scores (AUC) per tissue.
(F) Predicted vs. observed mean cut position relative to the CSE of individual human PAS sequences. The observed mean cut position per PAS was estimated from the Lianoglou et al. RNA-Seq data, pooled across cell types. Only high-quality estimated PASs with a minimum pooled read count of 500 were included (n=797, R2 = .74).
(G) Bar chart showing the correlation (R2) between predicted and observed mean cut position of high-quality estimated PASs with a minimum read count of 200, separated by cell type of the Lianoglou et al. data.
See also Figure S5.
Predicted isoform use agreed well with APADB measurements (R2 = .70, Figure 5B). Performance also increased , monotonically with APADB read coverage, as deeper sequencing results in higher quality estimates of the true isoform ratios (Figure S5A). APARENT also outperformed a DNN trained exclusively on APADB at predicting preferential site usage (binary APA classification, AUC=.97, Figure 5C, S5B). Consistent with earlier studies (Lackford et al. 2014; Li et al. 2015), the model learned a positive relationship between increased distance and use of the proximal site (Figure S5C).
APARENT Predicts APA across different tissue and cell types
Next, we tested how well APARENT’s predictions compared with measurements from individual tissues and cell types. We used RNA-seq data from APADB (Müller et al., 2014) and (Lianoglou et al., 2013) to estimate true isoform proportions per cell type for all 3’ UTR APA events (see STAR Methods). When considering all pairs of adjacent PASs, APARENT could predict the preferred PAS almost perfectly (Figure 5D–E, mean AUC = .97 across tissues). When narrowing the data to only pairs where both isoforms have at least one supporting read in a given tissue, performance only slightly decreased (mean AUC = .88). Due to the low average read count (10-50 reads per isoform and tissue type), obtaining reliable estimates of more balanced PASs is fundamentally more challenging than detecting extreme events (Figure S5D). To understand why APAReNt achieves high classification rates across tissues and cell types (without having trained on cell type-specific data), we compared the measured APA isoform proportions between pairs of tissues and observed very small differences for the majority of pairs (Figure S5E; mean R2 = .97, mean difference in isoform proportion = 0.031). We do find subsets of APA sites which are significantly differential, but these sets are small (on average 3.3% of all events have an isoform difference > 0.25).
Finally, we used APARENT to predict the cut distribution of every individual human PAS sequence. We compared the mean predicted cut position of each PAS against the mean cut position estimated from the RNA-Seq data of (Lianoglou et al., 2013) and found strong correlation across all cell types (Figure 5F–G).These results suggest that a non-tissue-specific cis-regulatory model such as APARENT is efficient for 3’UTR APA inference in general, regardless of tissue.
APARENT Predicts SNVs Linked to APA Misregulation
Nucleotide variants near PASs in human 3’ UTRs have been implicated in genetic disease, including IPEX syndrome, Thrombophilia and Alpha and Beta Thalassaemia. Most disease-implicated APA variants identified so far act by disrupting the CSE hexamer (Bennett et al., 2001; Gehring et al., 2001; Higgs et al., 1983). While a number of cryptic pathogenic variants have been identified in the USE and DSE, it is largely unknown how many high-impact non-CSE mutations exist in disease-implicated PASs. Using APARENT, we set out to characterize the frequency and complexity of cryptic APA variants across the human genome. We curated and synthesized >1,000 ClinVar variants -- SNVs and InDels linked to medical phenotypes (Landrum et al., 2018) -- occurring within 50 nt upstream and 100 nt downstream of an annotated PAS, as well as >10,000 variants from saturation mutagenesis of PASs in disease-implicated UTRs (ACMG, ClinVar, HGMD; Figure 6A; STAR methods) (Kalia et al., 2017; Landrum et al., 2014, 2016; Stenson et al., 2017).
Figure 6. Predicting the human APA variants and the existence of complex variation.
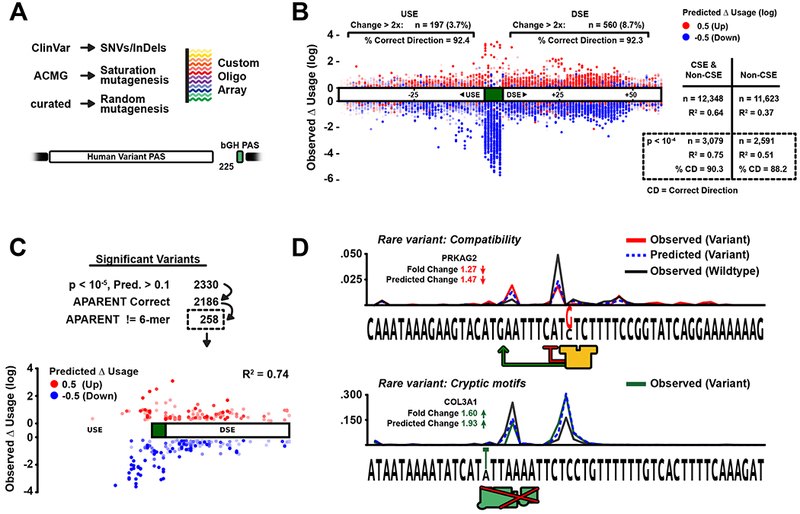
(A) MPRA used to measure the effect of human genomic APA variants on APA from ClinVar, hGmD, and ACMG genes.
(B) Measured isoform fold changes of all variants (Y-axis). X-axis denotes PAS position. The color indicates predicted fold change (blue/red = −/+ log odds ratio). All variants: R2 = 0.64. Significant variants (p < 0.0001): R2 = 0.75. Correctly predicted direction of change in isoform abundance (increase or decrease) = 90.3% of all variants. The figure is also annotated with the total number and % correctly predicted direction of fold change for USE and DSE variants with at least a 2-fold change in isoform abundance.
(C) Variants with significant measured fold change (p < 0.00001) where APARENT and a linear 6-mer model disagree on the direction of change.
(D) Two complex variant examples discovered in Figure 6C. (Top) A CSTF binding site variant was measured to decrease total RNA isoform abundance 1.27-fold. (Bottom) A cryptic CSE hexamer variant was measured to increase total RNA isoform abundance 1.6-fold.
See also Figure S6.
APARENT’s predictions agreed well with the measured fold changes of variant isoforms and could accurately classify the direction of change (Figure 6B, S6A, R2 = .75 / Correctly predicted direction = 90.3% for CSE and non-CSE variants, R2 = .51 / Correctly predicted direction = 88.2% for non-CSE variants only). Of the 12,348 measured variants, 757 non-CSE variants resulted in at least a 2-fold change in isoform abundance. The occurrence rate of such variants was about 3.7% in the USE and 8.7% in the DSE.
We further validated APARENT on variants found in the 1000 genomes data (1000 Genomes Project Consortium et al., 2012), comparing our predictions against transcriptomic measurements from the GEUVADIS project (Lappalainen et al., 2013). Since few insertions or deletions were found near PASs in the 1000 genomes variant annotation, we focused our analysis on SNVs. APARENT could infer isoform fold changes that agreed well with measured fold changes averaged over individuals carrying a variant (R2 = .68, Figure S6B, see STAR Methods).
APA SNVs Can Act Through Complex Regulatory Mechanisms
To identify variants that modulate APA through a non-linear regulatory interaction, we searched for SNVs whose experimentally measured direction of isoform expression change is incorrectly predicted by a linear 6-mer model, but correctly predicted by APARENT. While we found >90% agreement with APARENT on the predicted direction, the 6-mer model was considerably worse at predicting the magnitude of change. We identified 293 variants with a significant disagreement between the two model’s predictions, comprising ~2.3% of all the variants assayed (Figure 6C). Of those, 258 were predicted correctly by APARENT and only 35 by the 6-mer model (Figure S6C).
Figure 6D (top) displays a variant, correctly predicted by APARENT but not the 6-mer model, where the creation of a CstF site in the DSE results in a 1.27-fold decrease in isoform abundance rather than an increase, as might naively be expected. The measured distribution reveals that the main cut site is in fact only 1 nt upstream of the CstF site, consequently shifting cleavage to a less used cut site further upstream, all of which APARENT correctly predicts. The variant impact is particularly difficult to predict as there are 4 valid possible cut dinucleotides upstream of the main cut site. In our array we find additional cases of complex interaction between PAS components for variants causing both gain and loss of CstF binding sites (Figure S6D–E).
Many human PASs contain additional CSE hexamers in either the USE or DSE. Commonly, we would expect these hexamers to recruit CPSF and initiate independent polyA. Interestingly, Figure 6D (bottom) illustrates an example where the extra CSE hexamer instead takes on the role of a cut site rather than CPSF recruiting site. When the variant knocks out the extra CSE, cleavage downstream of both hexamers is upregulated rather than downregulated, resulting in a 1.6-fold increase in isoform abundance. Again, APARENT is able to predict the cut alteration accurately. In our collection of assayed variants, we find many more examples of cryptic CSE variation that affects APA non-trivially (Figure S6I). In addition, we find rare cases where a competitive CFIm25 binding site overlapping the CSE can have both up-regulatory and down-regulatory effects, largely dependent on the global strength of the wild type PAS (Figure S6F–H). We also find that variants altering folding structures can either enhance or repress enclosed cut sites (Figure S6J), and that variants interfering with canonical cleavage sites can both upregulate and downregulate total isoform levels (Figure S6K). In Figure S6K, we observe an example in the ARSA PAS where cleavage at a cryptic cut site is more efficient than the native CA dinucleotide, as knocking out the CA element results in a net-increase in isoform abundance. Clearly, it is insufficient to merely consider the creation or destruction of local motifs when assessing a variant. Rather, we must consider the composition of the entire PAS.
Many ClinVar SNVs Act Through Modulation of APA
More than half of ClinVar variants within PASs that are annotated as pathogenic occur in the CSE hexamer, however, a number of pathogenic variants in the DSE were both measured and predicted to have large effects on APA (Figure 7A, HBA2 104 G>T, F2 97 G>A, etc.). Importantly, APARENT also predicts low effects on APA for those pathogenic USE variants known to be deleterious through other mechanisms than APA (Gazda et al., 2008; Jenkinson et al., 2016; Poller et al., 1999). Across all assayed ClinVar and HGMD variants, we observe the fraction of variants with a strong effect on APA to increase with clinical significance (Figure 7B), supporting the link between misregulation of APA and disease. Similar to the analysis of (Huang et al., 2017) for assessing a model’s utility in prioritizing variants, we obtain an aUc of 0.916 at prioritizing pathogenic over benign variants based solely on predicted APA fold change. We also emphasize the vast number of variants of uncertain significance (VUS) that remain to be clinically classified, ~11% of which are associated with at least a 2-fold change on isoform abundance.
Figure 7. Characterization of pathogenic variants and PASs linked to disease.
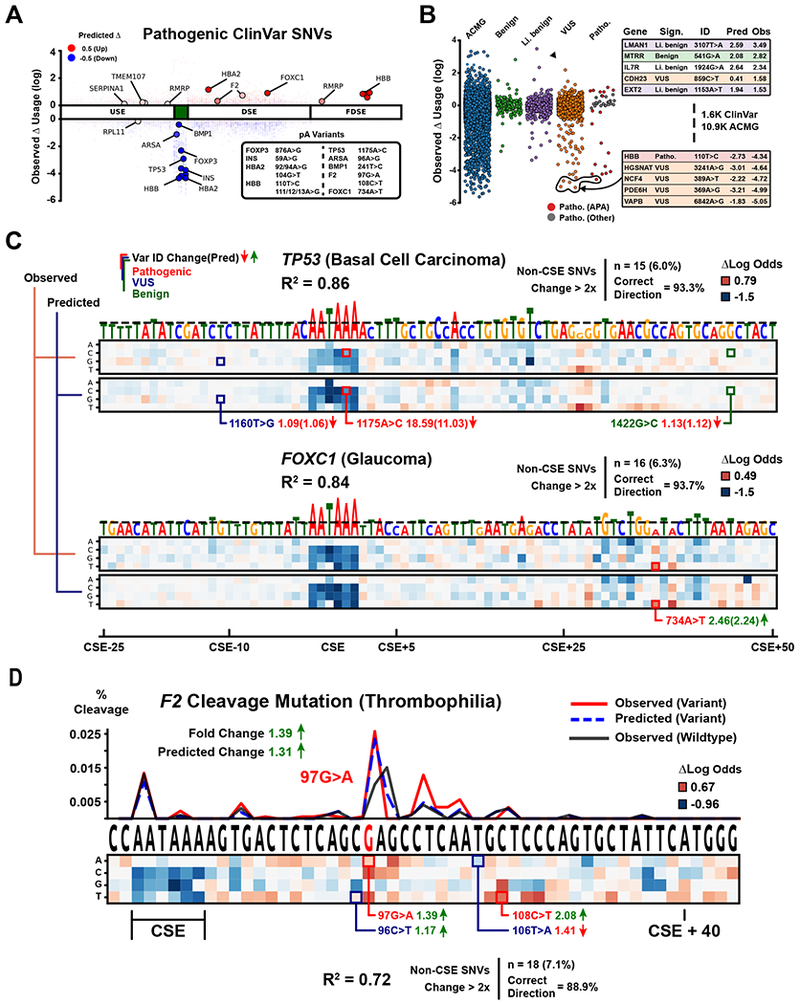
(A) Measured fold changes of pathogenic variants in ClinVar and HGMD (Y-axis). X-axis denotes PAS position. Color indicates APARENT predictions. The table lists all variant identifiers.
(B) Strip plot of all assayed variants categorized by clinical significance in ClinVar. The ‘ACMG’ category contains all of the unannotated variants obtained from saturation mutagenesis. The ‘Benign’ set contains 170 variants, ‘Likely benign’ contains 419 variants, ‘VUS’ contains 946 variants and ‘Pathogenic’ contains 19 variants. AUC = 0.916 for classifying the pathogenic set from the benign set based only on APARENT’s predicted log isoform fold change.
(C) Saturation mutagenesis of the TP53 (Basal Cell Carcinoma) and FOXC1 (Glaucoma) PASs. The heatmaps visualize measured and predicted APA isoform fold changes and are annotated with variants from ClinVar and HGMD. Shown above the heatmaps are correlations (R2) between predicted and measured isoform log fold changes, the total number of variants with at least 2-fold changes in isoform abundance and % correctly predicted direction of fold change.
(D) Saturation mutagenesis of the F2 (Thrombophilia) PAS. Shown are also the measured wildtype and variant cleavage distributions (black and red lines), and the predicted variant cleavage distribution (blue dashed line), for the pathogenic 97G>A variant (measured / predicted RNA isoform abundance increase = 1.39 / 1.31-fold).
See also Figure S7.
We also find variants annotated in ClinVar as of “conflicting interpretations of pathogenicity” which have significant predicted and measured effects on APA (Figure S7A). For example, variants found in the TYMP UTR (3C>T, 10G>A) and a variant found in HBA2 (98T>C). These results suggest APA misregulation is a functional consequence either causing or contributing to these phenotypes and that these variants are likely pathogenic. Besides variants found in ClinVar, we observe >1,000 currently unannotated mutations having at least 2-fold APA change in disease-implicated PASs.
Saturation Mutagenesis of Disease-Relevant PASs Reveals Putative Pathogenic Variants
We then studied the composition of known pathogenic PASs through experimental and computational saturation mutagenesis (Figure 7C). The TP53 1175A>C mutation transforms the canonical CSE AATAAA into the weaker AATACA and has been implicated in Basal Cell Carcinoma (Stacey et al., 2011). We identified two important regulatory regions in the DSE: the CstF binding site TGT[C/G]T and an HNRNPH binding site. We also find an SNV in the DSE that results in a 9-fold isoform change, as it interferes with CstF binding and cut site secondary structure (Figure S7B). The PAS contains two additional annotated ClinVar variants, a benign variant 1422G>C further downstream in the DSE and a VUS 1160T>G in the USE, both of which are predicted and measured to have negligible effects. Given that Basal Cell Carcinoma is caused by APA misregulation in the TP53 UTR, our results suggest there are many more mutations that could lead to disease (6.0% of USE and DSE variants are estimated to have at least a 2-fold change). APARENT’s predictions agree well with the measured fold changes in TP53 (R2 = .86 for all variants, correctly predicted direction of fold change for >2-fold variants = 93.3%).
Next, we performed saturation mutagenesis of the FOXC1 PAS, which contains a pathogenic DSE variant 734A>T implicated in Glaucoma (Medina-Trillo et al., 2016) (Figure 7C). This variant is known to alter a miRNA target site, potentially affecting both APA site preference (as a DSE regulator) and isoform stability. Indeed, both the predictions and experiments confirm ~2-fold upregulation. Roughly 6.3% of additional DSE nucleotide substitutions within this site are measured to have at least a 2-fold effect on APA, and APARENT predicts these effects accurately (R2 = .84 for all variants, correctly predicted direction of fold change for >2-fold variants = 93.7%).
Finally, we highlight the impact of a variant 97G>A (20210G>A) at the cleavage site in the prothrombin gene (F2) 3’UTR (Figure 7D). This gain-of-function variant is found in nearly 2% of the population and has been directly linked to Thrombophilia by increasing total RNA abundance 1.4-fold (Danckwardt et al., 2008; Gehring et al., 2001). APARENT accurately identifies the weak wildtype CG dinucleotide as a functional cleavage site, and furthermore, correctly predicts that the CA variant increases isoform abundance about 1.3-fold. We find that a considerably rarer variant 108C>T (also linked to thrombosis) (Stapenhorst et al., 2001) has a more pronounced upregulatory effect (>2-fold), possibly by enhanced binding of CstF (Danckwardt et al., 2004). The VUS 96C>T has a low isoform fold change compared to other pathogenic variants and is likely benign, while the VUS 106T>A results in a 1.41-fold change and may be deleterious.
We carried out saturation mutagenesis to functionally characterize many more PASs and genes implicated in disease (HBA2, HBB, INS, BRCA1, TPMT; Figure S7C–E). In many cases we were able to validate negligible effects on APA for annotated benign variants or VUSs, but we also identified many VUSs with considerable impact on APA, providing evidence for misregulated APA as the functional cause for disease. In general, APARENT is able to call variants with high sensitivity and specificity, and we find that most disease-implicated PASs contain approximately 5-10% variants outside of the CSE with at least 2-fold changes to APA isoform abundance, but the frequency of such high impact variants can reach 15% (e.g. HBA2, Fig. S7C). Importantly, high-impact variants in disease-implicated PASs may violate general trends of APA regulation, supporting the need for a non-linear functional model of APA to predict the impact of such variants. For example, a TA inserted between two neighboring canonical CA cut sites in the BRCA1 UTR directs all cleavage to the TA site, even though CA on average is the preferred element (Figure S7E).
DISCUSSION
We introduced APARENT, a DNN capable of accurately predicting APA in human 3’UTRs. Many of the sequence features identified as important by APARENT could be mapped to known cis-regulatory sequence elements, including known binding sites of core components of the polyA and cleavage machinery (CFIm25, CstF, CPSF and hFip1). APARENT also identified regulatory features that have not previously been described. For example, we found that the choice of cleavage site is regulated deterministically by sequence features besides the favored CA dinucleotide. APARENT identified GA and TA as almost equally functional, but also found that cleavage site selection is regulated by a more extensive cis-regulatory code combining RBP binding motifs (e.g. polyG/polyT/CstF) with secondary structure.
Recent work on visualizing the motifs learned by the first layer of a CNN has begun to address latent skepticism about the interpretability of dNns (Alipanahi et al., 2015; Kelley et al., 2016). We expanded on this work and showed that our network identifies important regulatory features of widely varying complexity, ranging from short motifs learned in the first layer, to longer and spatially connected combinations of motifs in the second layer, to full-length PAS compositions, including secondary structure, learned in the deeper layers.
We then built on our visualization algorithm to develop SeqProp, a method for engineering PASs. We applied SeqProp to design PASs targeting a range of isoform usage and cleavage positions. An experimental validation of over thousand designed sequences showed excellent agreement between targeted and measured characteristics. Previously, computational engineering of functional cis-regulatory DNA sequences has mostly been done with genetic algorithms. However, in genetic algorithms, the search naively generates new sequences by making random changes, instead of generating those that are predicted to improve the target objective. Moreover, a discrete nucleotide-swapping search may easily get stuck in local minima, especially if the search space is very large and the objective is met by only a narrow subset of sequences.
We expect that SeqProp will be applied to forward-engineer of functional 3’UTRs, enabling control over mRNA-stability, possibly even in a tissue-specific manner. In mammalian synthetic biology, such engineered PASs could provide an additional layer of control for improving molecular circuits that rely on miRNAs or RBPs binding to the 3’UTR (Xie et al. 2011; Ausländer et al. 2012; Strovas et al. 2014; Wroblewska et al. 2015). While initial work in fields such as gene therapy, mRNA therapeutics or cell therapy with chimeric antigen receptors primarily focused on optimizing the coding regions of therapeutic genes, it is likely that precise control over untranslated regions can enhance therapeutic efficacy and help minimize off-target effects (Ruder et al. 2011; Sahin et al. 2014; Roybal and Lim 2017). Moreover, although we here focused on APA, SeqProp can be generalized to other cis-regulatory regions such as the 5’UTR, introns or even promoters and enhancers, providing an approach for generating transcripts or genes with tailor-made properties.
We applied APARENT to predict the impact of 3’UTR variants in ClinVar and HGMD within 50 base pairs of the CSE and performed saturation mutagenesis on disease-implicated PASs. We found that many variants annotated as pathogenic also had a strong impact on APA, suggesting a molecular mechanism for disease. Model predictions were validated experimentally through massively parallel measurements of the impact on APA of over 12,000 human PAS variants. We identified a large number of VUSs that nonetheless resulted in strong shifts in the APA isoform distribution, making them intriguing candidates for future investigation. Approximately 4% of USE and 9% of DSE variants resulted in an at least 2-fold change in isoform abundance, confirming the importance of regulatory information outside the CSE. APARENT was able to identify and correctly predict the impact of complex variants that could not be explained by a simpler linear model, enabling us to detect novel regulatory interactions, such as variants interfering with RNA folding, cryptic cut sites or combinatorial effects.
Though we focused exclusively on APA in the 3’UTR, PASs are also found in terminal introns. Although predictive models of alternative splicing are available, more work is required to integrate them with the APA model introduced here. Second, it is likely that some of the cis-regulatory sequences identified in our assays differentially control transcript stability rather than regulating APA directly. Such sequence elements could be identified in an independent MPRA focused on mRNA stability (Zhao et al. 2014; Rabani et al., 2017). Still, the work presented here provides a comprehensive view of the regulatory code guiding APA and has direct implications for both basic biology and genomic medicine.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal bovine serum | Atlanta Biologicals | Cat # S11150 |
| DMEM, high glucose, pyruvate | ThermoFisher Scientific | SKU # 11995-065 |
| Critical Commercial Assays | ||
| Oligonucleotide Library Synthesis (244K) | Agilent | Part # G7223A |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat # E7490S |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat # 20024906 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Cat # 20024907 |
| NextSeq 500/550 Mid Output v2 kit (150 cycles) | Illumina | Cat # FC-404-2001 |
| MiSeq Reagent Kit v3 (150-cycle) | Illumina | Cat # MS-102-3001 |
| Deposited Data | ||
| Random 3’ UTR APA MPRA | This study | GSE113849 / https://github.com/johli/aparent |
| Designed 3’ UTR APA MPRA | This study | GSE113849 / https://github.com/johli/aparent |
| ClinVar Database | Landrum et al., 2014 | ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/variant_summary.txt.gz |
| APADB, Pooled-Tissue | Müller et al., 2014 | http://tools.genxpro.net/apadb/download/track/hg19.apadb_v2_final.bed/ |
| APADB, Tissue-specific | Müller et al., 2014 | http://tools.genxpro.net/apadb/browse/ |
| Leslie APA Atlas, Tissue-speciic | Lianoglou et al., 2013 | http://cbio.mskcc.org/public/Leslie/ApA/atlas-lianoglou/alignments/unique/clean-final/unified-atlas.bam |
| GEUVADIS | Lappalainen et al., 2013 | https://www.ebi.ac.uk/arrayexpress/files/E-GEUV-1/processed/ |
| Experimental Models: Cell Lines | ||
| HEK293FT | Invitrogen | Cat # R70007 |
| Software and Algorithms | ||
| APARENT Software | This study | https://github.com/johli/aparent |
| APARENT Model Training | This study | https://github.com/johli/aparent |
| SeqProp Software | This study | https://github.com/johli/aparent |
| 6-mer Logistic Regression Model | This study | https://github.com/johli/aparent |
| Random 3’ UTR Data Processing Software | This study | https://github.com/johli/aparent |
| Designed 3’ UTR Data Processing Software | This study | https://github.com/johli/aparent |
| APADB Data Processing Software | This study | https://github.com/johli/aparent |
| Leslie Data Processing Software | This study | https://github.com/johli/aparent |
| ClinVar Data Processing Software | This study | https://github.com/johli/aparent |
| GEUVADIS Data Processing Software | This study | https://github.com/johli/aparent |
Highlights.
Trained a neural network to predict APA using data from over 3 million reporters
Visualized learned features to reveal a rich cis regulatory code for APA
Developed and tested an algorithm to accurately engineer polyadenylation signals
Predicted and experimentally characterized over 12,000 human APA variants
Acknowledgements
We thank W.L. Ruzzo, B. Wang and E. Wilson for their valuable comments and discussions. This work was supported by NIH R01HG009136 to GS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, and McVean GA (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipanahi B, Delong A, Weirauch MT, and Frey BJ (2015). Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol 33, 831–838. [DOI] [PubMed] [Google Scholar]
- Ausländer S, Ausländer D, Müller M, Wieland M, and Fussenegger M (2012). Programmable single-cell mammalian biocomputers. Nature 487, 123–127. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Brunkow ME, Ramsdell F, O’Briant KC, Zhu Q, Fuleihan RL, Shigeoka AO, Ochs HD, and Chance PF (2001). A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics 53, 435–439. [DOI] [PubMed] [Google Scholar]
- Bentley DL (2014). Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet 15, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Kuznetsov G, Ogden PJ, Conway NJ, Adams RP, and Church GM (2018). Toward machine-guided design of proteins (bioRxiv). [Google Scholar]
- Chen F, MacDonald CC, and Wilusz J (1995). Cleavage site determinants in the mammalian polyadenylation signal. Nucleic Acids Res. 23, 2614–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Miura RM, and Tian B (2006). Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 22, 2320–2325. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, and Sidow A (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus JT, Groves B, Kuchina A, Rosenberg AB, Jojic N, Fields S, and Seelig G (2017). Deep learning of the regulatory grammar of yeast 5’ untranslated regions from 500,000 random sequences. Genome Res. 27, 2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danckwardt S, Gehring NH, Neu-Yilik G, Hundsdoerfer P, Pforsich M, Frede U, Hentze MW, and Kulozik AE (2004). The prothrombin 3′ end formation signal reveals a unique architecture that is sensitive to thrombophilic gain-of-function mutations. Blood 104, 428–435. [DOI] [PubMed] [Google Scholar]
- Danckwardt S, Hentze MW, and Kulozik AE (2008). 3’ end mRNA processing: molecular mechanisms and implications for health and disease. EMBO J. 27, 482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, and Babak T (2012). A quantitative atlas of polyadenylation in five mammals. Genome Res. 22, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeZazzo JD, and Imperiale MJ (1989). Sequences upstream of AAUAAA influence poly(A) site selection in a complex transcription unit. Mol. Cell. Biol 9, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, and Manley JL (2011). Mechanisms and consequences of alternative polyadenylation. Mol. Cell 43, 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Ugalde AP, and Agami R (2013). Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet 14, 496–506. [DOI] [PubMed] [Google Scholar]
- Findlay GM, Boyle EA, Hause RJ, Klein JC, and Shendure J (2014). Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyhult E, Gardner PP, and Moulton V (2005). A comparison of RNA folding measures. BMC Bioinformatics 6, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, and Gerstein M (2014). FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 15, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue M-F, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, et al. (2008). Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet 83, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Frede U, Neu-Yilik G, Hundsdoerfer P, Vetter B, Hentze MW, and Kulozik AE (2001). Increased efficiency of mRNA 3’ end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat. Genet 28, 389–392. [DOI] [PubMed] [Google Scholar]
- Gruber AJ, Schmidt R, Gruber AR, Martin G, Ghosh S, Belmadani M, Keller W, and Zavolan M (2016). A comprehensive analysis of 3’ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 26, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol. 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs DR, Goodbourn SEY, Lamb J, Clegg JB, Weatherall DJ, and Proudfoot NJ (1983). α-Thalassaemia caused by a polyadenylation signal mutation. Nature 306, 398–400. [DOI] [PubMed] [Google Scholar]
- Hu J, Lutz CS, Wilusz J, and Tian B (2005). Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11, 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-F, Gulko B, and Siepel A (2017). Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet 49, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548.e24. [DOI] [PubMed] [Google Scholar]
- Jenkinson EM, Rodero MP, Kasher PR, Uggenti C, Oojageer A, Goosey LC, Rose Y, Kershaw CJ, Urquhart JE, Williams SG, et al. (2016). Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat. Genet 48, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG sF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med 19, 249–255. [DOI] [PubMed] [Google Scholar]
- Kaufmann I, Martin G, Friedlein A, Langen H, and Keller W (2004). Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 23, 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Snoek J, and Rinn JL (2016). Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi E, Xie X, Wan J, Xing Y, Freudenberg JM, et al. (2014). Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J. 33, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanchantin J, Singh R, Lin Z, and Qi Y (2016). Deep Motif: Visualizing Genomic Sequence Classifications.
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, and Maglott DR (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MKK, Delong A, and Frey BJ (2017). Inference Of The Human Polyadenylation Code. [DOI] [PMC free article] [PubMed]
- Li X-Q, and Du D (2013). RNA polyadenylation sites on the genomes of microorganisms, animals, and plants. pLoS One 8, e79511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, et al. (2015). Systematic Profiling of Poly(A) Transcripts Modulated by Core 3’ End Processing and Splicing Factors Reveals Regulatory Rules of Alternative Cleavage and Polyadenylation. PlOS Genetics 11, e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, and Mayr C (2013). Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 27, 2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, and Zavolan M (2012). Genome-wide analysis of pre-mRNA 3’ end processing reveals a decisive role of human cleavage factor I in the regulation of 3’ UTR length. Cell Rep. 1, 753–763. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu A-B, Li W, and Wagner EJ (2014). CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ, et al. (2018). Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Trillo C, Aroca-Aguilar J-D, Méndez-Hernández C-D, Morales L, García-Antón M, García-Feijoo J, and Escribano J (2016). Rare FOXC1 variants in congenital glaucoma: identification of translation regulatory sequences. Eur. J. Hum. Genet 24, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG Jr, Kinney JB, et al. (2012). Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol 30, 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S, Rycak L, Afonso-Grunz F, Winter P, Zawada AM, Damrath E, Scheider J, Schmäh J, Koch I, Kahl G, et al. (2014). APADB: a database for alternative polyadenylation and microRNA regulation events. Database 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah C, Mordvintsev A, and Schubert L (2017). Feature visualization. Distill 2, e7. [Google Scholar]
- Patwardhan RP, Lee C, Litvin O, Young DL, Pe’er D, and Shendure J (2009). High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol 27, 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez Cañadillas JM, and Varani G (2003). Recognition of GU-rich polyadenylation regulatory elements by human CstF-64 protein. EMBO J. 22, 2821–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poller W, Merklein F, Schneider-Rasp S, Haack A, Fechner H, Wang H, Anagnostopoulos I, and Weidinger S (1999). Molecular characterisation of the defective α1-antitrypsin alleles PI Mwürzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68lle). Eur. J. Hum. Genet 7, 321. [DOI] [PubMed] [Google Scholar]
- Purnick PEM, and Weiss R (2009). The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 10, 410–422. [DOI] [PubMed] [Google Scholar]
- Rabani M, Pieper L, Chew G-L, and Schier AF (2017). A Massively Parallel Reporter Assay of 3’ UtR Sequences Identifies In Vivo Rules for mRNA Degradation. Mol. Cell 68, 1083–1094.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Patwardhan RP, Shendure J, and Seelig G (2015). Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163, 698–711. [DOI] [PubMed] [Google Scholar]
- Roybal KT, and Lim WA (2017). Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu. Rev. Immunol 35, 229–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruder WC, Lu T, and Collins JJ (2011). Synthetic biology moving into the clinic. Science 333, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Sahin U, Karikó K, and Tóreci Ö (2014). mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759. [DOI] [PubMed] [Google Scholar]
- Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen I, Morris DR, and Seelig G (2018). Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönemann L, Kühn U, Martin G, Schäfer P, Gruber AR, Keller W, Zavolan M, and Wahle E (2014). Reconstitution of CPSF active in polyadenylation: recognition of the polyadenylation signal by WDR33. Genes Dev. 28, 2381–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, and Segal E (2012). Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol 30, 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, and Ahituv N (2013). Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet 45, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Sulem P, Jonasdottir A, Masson G, Gudmundsson J, Gudbjartsson DF, Magnusson OT, Gudjonsson SA, Sigurgeirsson B, Thorisdottir K, et al. (2011). A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet 43, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapenhorst L, Wielckens K, Wylenzek M, Geisen C, and Klingler K (2001). A Novel Point Mutation in the 3’ Region of the Prothrombin Gene at Position 20221 in a Lebanese/Syrian Family. Thrombosis and Haemostasis 85, 943–944. [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, and Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Human Genetics 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strovas TJ, Rosenberg AB, Kuypers BE, Muscat RA, and Seelig G (2014). MicroRNA-based single-gene circuits buffer protein synthesis rates against perturbations. ACS Synth. Biol. 3, 324–331. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang Y, Hamilton K, Manley JL, Shi Y, Walz T, and Tong L (2018). Molecular basis for the recognition of the human AAUAAA polyadenylation signal. Proc. Natl. Acad. Sci. U. S. A. 115, E1419–E1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedy C, Liu W, Jia Y, Sermanet P, Reed S, Anguelov D, Erhan D, Vanhoucke V, and Rabinovich A (2014). Going Deeper with Convolutions. [Google Scholar]
- Tian B, and Manley JL (2017). Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 18, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng D, Yehia G, and Tian B (2018). A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res. 28, 1427–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, and Cohen BA (2013). Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc. Natl. Acad. Sci. U. S. A. 110, 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, Liu H, Rosenwald A, Muller-Hermelink HK, Ott G, et al. (2007). Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 109, 4599–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska L, Kitada T, Endo K, Siciliano V, Stillo B, Saito H, and Weiss R (2015). Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat. Biotechnol. 33, 839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Bartel DP (2017). Widespread Influence of 3’-End Structures on Mammalian mRNA Processing and Stability. Cell 169, 905–917.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Wroblewska L, Prochazka L, Weiss R, and Benenson Y (2011). Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 333, 1307–1311. [DOI] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Gilmartin GM, and Doublié S (2010). Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3’ processing. Proc. Natl. Acad. Sci. U. S. A. 107, 10062–10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Coseno M, Gilmartin GM, and Doublie S (2011). Crystal structure of a human cleavage factor CFI(m)25/CFI(m)68/RNA complex provides an insight into poly(A) site recognition and RNA looping. Structure 19, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Pollack JL, Blagev DP, Zaitlen N, McManus MT, and Erle DJ (2014). Massively parallel functional annotation of 3’ untranslated regions. Nature Biotechnology 32, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, and Troyanskaya OG (2015). Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang X, Forouzmand E, Jeong J, Qiao F, Sowd GA, Engelman AN, Xie X, Hertel KJ, and Shi Y (2018). Molecular Mechanisms for CFIm-Mediated Regulation of mRNA Alternative Polyadenylation. Mol. Cell 69, 62–74.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.