

Abstract
The research presented here describes the development of two analytical methods for use in the certification of a ginsenoside calibration solution Standard Reference Material (SRM) 3389 consisting of seven ginsenosides: Rg1, Re, Rf, Rb1, Rc, Rb2, and Rd. The new methods utilized the liquid chromatographic (LC) separation of ginsenoside mixtures with absorbance detection (UV) and mass spectrometry (MS). Ginsenosides Rb3, Rg2, Rg3, Rh1, and Rh2 were evaluated for use as internal standards for LC/MS measurements. The 12 ginsenosides were baseline resolved by gradient elution LC/UV, with an initial mobile phase composition of 22 % acetonitrile and 78 % water, flow rate of 0.7 mL/min, and column temperature of 25 °C. The work presented here includes a detailed investigation into the optimization of the chromatographic conditions to minimize measurement biases that result from unresolved constituents. Temperature and mobile phase composition are known to play a significant role in column selectivity; however, flow rate is expected to influence primarily the separation efficiency and detection sensitivity. In the current study, column selectivity changed with changes in flow rate and the relative retention of ginsenoside Rg2 and Rh1 changed as the flow rate increased from 0.6 mL/min to 1.0 mL/min.
Keywords: Liquid chromatography – ultraviolet/visible absorbance, Liquid chromatography – mass spectrometry, Method development, Ginsenoside Calibration Solution
1. Introduction
Ginsenosides are a class of triterpene saponins primarily found in the plant genus Panax. Ginsenosides can be isolated from different parts of the Panax plant, but the root is the primary source in commerce. More than 150 naturally occurring ginsenosides have been isolated from roots, leaves, stems, fruits, and/or flower heads of various Panax species. Due to their purported physiological activity and their unique distribution among species and plant parts, separation and determination of each ginsenoside is required. The majority of these ginsenosides are in the dammarane family. The 12 ginsenosides used in this study are reported to exhibit a variety of beneficial properties that include neuroprotective (Rb1, Rd, Rg1, Rg2, and Rg3) [1–4], anti-cancer (Rb2 and Rg3) [5, 6], cardio protective (Rb3) [7], anti-diabetic (Rc) [8], antinociception (Rf) [9], anti-oxidant (Re, Rg3, Rh1, and Rh2) [10–12], vasodilating (Rg3) [13], and hepatoprotective effects (Rg3 and Rh2) [14]. These ginsenosides consist of a four-ring steroid aglycone main structure with multiple available hydroxyl (OH) groups, as illustrated in Fig. S1 – S2, which may be glycosylated with one or two carbohydrate units.
LC based methods are widely employed for the analysis of ginsenoside mixtures. A variety of LC methods have been developed that are compatibility with various detection techniques such as UV [15–20], evaporative light scattering (ELS) [21], fluorescence (FL) [22], pulsed amperometry (PA) [23], mass spectrometry (MS) [15, 19, 24, 25], and tandem mass spectrometry (MS/MS) [26–29]. UV detection is frequently used for simple sample matrixes because of its inexpensive cost and simplicity, but for ginsenosides, detection is limited to of the low UV region (200 nm to 205 nm). MS and MS/MS provide a complementary and selective detection of ginsenosides that may reduce biases from coeluting interferences and facilitate rapid separations. Wang et al. [29] has recently demonstrated this ability by combining ultra-high pressure liquid chromatography with quadrupole/time of flight MS/MS for the qualitative analysis of 131 ginsenosides in root and rhizomes samples of Panax ginseng.
The National Institute of Standards and Technology (NIST) regularly develops new calibration standards and natural matrix Standard Reference Materials (SRMs) for use in validating new and current analytical methods [30–32]. Certified calibration solutions are of importance to natural product laboratories for use in standardization and quality control. A ginsenoside calibration solution (SRM 3389) has been developed with the following constituents: ginsenosides Rg1, Re, Rf, Rb1, Rc, Rb2, and Rd. Prior to certification, new LC/UV and LC/MS methods were developed for the separation and detection of the seven ginsenosides present in SRM 3389 and five additional ginsenosides (Rb3, Rg2, Rg3, Rh1, and Rh2) typically present in ground root and root extract matrices. LC/UV and LC/MS techniques were utilized to provide independent analytical approaches required for the certification of reference materials, and to facilitate method reproduction in natural product laboratories. Several key parameters including flow rate, initial mobile phase composition, and column temperature were evaluated for the best separation. Ginsenosides Rb3, Rg2, Rg3, Rh1, and Rh2 were investigated as potential internal standards for the new LC/MS method.
2. Material and methods
2.1. Chemicals
Ginsenosides Rb1, Rb2, Rc, Rd, Re, and Rg1 reference standards were obtained from the National Research Council (NRC) of Canada (Ottawa, ON, Canada) with purity values of 96.7 %, 88.8 %, 92.5 %, 89.7 %, 97.8 %, and 96.5%, respectively. Ginsenosides Rb3, Rf, Rg2, Rg3, Rh1, and Rh2 reference standards were obtained from Cerilliant (Round Rock, TX, USA) with purity values of 91 %, 94 %, 94 %, 93 %, 93 %, and 85 %, respectively. Certified Reference Material MIGS-1 (Ginsenoside Mixture) was obtained from NRC for use as a measurement control. SRM 3389 was gravimetrically prepared from neat standards. HPLC grade acetonitrile (ACN), methanol (MeOH), water (H2O), and 0.1 % (v/v) formic acid (FA) in H2O were purchased from Fisher Scientific (Pittsburgh, PA, USA).
2.2. Sample Preparation
Small portions of each compound were dissolved in 1 mL aliquots of MeOH for evaluation of component identities. The mixture of the 12 ginsenosides used for method development was prepared by combining small quantities (< 100 µL) of each ginsenoside in MeOH. Single ampoules of MIGS-1 and SRM 3389 were analyzed without dilution.
For the LC/UV certification measurements, one ampoule of the MIGS-1, two ampoules of the single component SRM 3389, and five ampoules of the six-component SRM 3389 were transferred to LC autosampler vials for analysis. Only two ampoules of the single component SRM 3389 was used because of a limited number in comparison to the six-component material. The ampoules of SRM 3389 were randomly selected for analysis. For the LC/MS certification measurements, small quantities of the internal standard (Rh1) solution were weighed out and mixed with masses of the three calibrant solutions, MIGS-1 solution, and the SRM 3389 solutions.
2.3. Instrumentation and chromatographic conditions
The LC/UV measurements were performed on an Dionex Ultimate 3000 LC system equipped with a pump, autosampler, column compartment, and diode array detector. The LC/MS measurements were performed on an Agilent 1100 LC system equipped with a binary pump, degasser, autosampler, column compartment, and variable wavelength absorbance detector. The Agilent 1100 LC system was coupled to an Agilent 1956B quadrupole MS with electrospray ionization (ESI). Data acquisition and instrument control used Chromeleon software (Thermo Scientific) and ChemStation software (Agilent), respectively.
Separations were carried out on an octadecyl (C18) column (ACE 3 C18) manufactured by Advanced Chromatography Technologies (ACE, Aberdeen, Scotland) with the following characteristics: 15 cm length, 4.6 mm diameter, and 3 µm average particle diameter. The separation conditions were optimized with a flow rate 0.70 mL/min, column temperature of 25 °C, and the mobile phase program summarized in Table 1. The gradient was slightly modified for the LC/MS method because of partial co-elution between Rb2 and Rb3 using a different instrument from the LC/UV method. The injection volume was 2 µL. The LC/UV instrument was operated with an absorbance wavelength of 200 nm for all ginsenosides. The LC/MS instrument was operated in selected ion monitoring (SIM) mode according to the m/z ions for each ginsenoside with the most abundant peak signal in their mass spectra shown in Table 2, which includes additional identifying m/z ions. The mass spectra were collected using the following parameters: (1) capillary potential of 4.0 kV; (2) electrospray source temperature of 200 °C; (3) desolvation gas temperature of 250 °C; and (4) cone voltage of 70 V.
Table 1.
Mobile phase gradient for the LC/UV and LC/MS methods.
| LC/UV |
LC/MS |
||||
|---|---|---|---|---|---|
| Time | H2O (%) | ACN (%) | Time | H2O/0.1% FA (%) | ACN (%) |
| −5.0 | 78 | 22 | 0.0 | 78 | 22 |
| 0.0 | 78 | 22 | 10.0 | 78 | 22 |
| 10.0 | 78 | 22 | 38.0 | 42 | 58 |
| 29.0 | 58 | 42 | 45.0 | 0 | 100 |
| 59.0 | 0 | 100 | 60.0 | 0 | 100 |
| 60.0 | 0 | 100 | 65.0 | 78 | 22 |
| 60.1 | 78 | 22 | 70.0 | 78 | 22 |
Table 2.
Peak distribution in the negative ion mode mass spectra.
| Ginsenoside | Molecular Formula |
Molecular Mass (g/mol) |
m/z a |
|---|---|---|---|
| Rb1 | C54H92O23 | 1109.31 | 1107.5, 1108.5, 1109.5, 1153.5, 1154.5 |
| Rb2 | C53H90O22 | 1079.29 | 829.5, 1077.4, 1078.4, 1079.4, 1123.5, 1191.4, 1192.4 |
| Rb3 | C53H90O22 | 1079.29 | 1077.5, 1078.5, 1079.5, 1123.5, 1124.5, 1125.5, 1191.3 |
| Rc | C53H90O22 | 1079.29 | 829.4, 830.4, 1077.5, 1078.5, 1079.5, 1123.5, 1124.5, 1191.6 |
| Rd | C48H82O18 | 947.17 | 945.6, 991.7, 992.7, 1059.5, 1060.5 |
| Re | C48H82O18 | 947.17 | 991.7, 992.7, 993.7, 1059.5 |
| Rf | C42H72O14 | 801.03 | 799.3, 800.3, 845.5, 846.5, 847.5, 913.4 |
| Rg1 | C42H72O14 | 801.03 | 845.4, 846.4 |
| Rg2 | C42H72O13 | 785.03 | 829.5, 830.5, 831.5, 897 |
| Rg3 | C42H72O13 | 785.03 | 783.4, 829.4, 830.4, 831.4, 897.3 |
| Rh1 | C36H62O9 | 638.89 | 683.4, 684.4 |
| Rh2 | C36H62O8 | 622.89 | 667.4, 668.4, 669.4 |
All m/z peaks had relative abundance values greater than 10%.
The underlined m/z represents the most abundant peak signal in the mass spectra
3. Results and Discussion
In a previous study MacCrehan and White determined eight ginsenosides (Rg1, Re, Rf, Rg2, Rb1, Rc, Rb2, and Rd) in extracts of a powdered Panax ginseng rhizomes via LC/UV and LC/MS using a 3 µm ACE 3 C18 LC column and a ACN/H2O mobile phase gradient [15]. The ginsenosides were separated with a flow rate of 0.75 mL/min, column temperature of 23 °C, and mobile phase gradient with the following conditions: isocratic elution for 18 % ACN for 18 min, increased to 33 % ACN over 38 min, and held for 9 min. Similar approaches have been reported that require lengthy separation times [17,18].
To minimize measurement biases during certification, a new method was developed to achieve rapid baseline separation of the eight ginsenosides without the need of ultra-high pressure conditions. The LC/UV chromatograms obtained for the eight ginsenosides are shown in Fig. S3 using a flow rate of 1.0 mL/min, column temperature of 25 °C, and the mobile phase gradient programs listed in Table S1. All ginsenosides were baseline resolved using gradient A: linear gradient from 20 % ACN to 23 % ACN over 10 min with a subsequent linear gradient to 58 % ACN over 19 min. The separations in Fig. S3 clearly show the sensitive nature of the ginsenoside separation to small changes in the mobile phase composition. The retention behavior of four additional ginsenosides (Rb3, Rg3, Rh1, and Rh2) standards were investigated for use as potential internal standards.
3.1. Liquid Chromatography/Absorbance Detection
An optimized separation of a 12 component ginsenoside mixture is shown in Fig. 1 using the mobile phase gradient listed in Table 1. The flow rate, column temperature, and absorbance wavelength were held constant at 0.7 mL/min, 25 °C, and 200 nm, respectively. Using these conditions, all the ginsenoside components present in the MIGS-1 and SRM 3389 calibration solutions were baseline resolved as illustrated in Fig. 1. The complete separation of these ginsenosides was required for the LC/UV certification of SRM 3389, which used MIGS-1 as a control. Several key parameters including flow rate, initial mobile phase composition, and column temperature are discussed below. Ginsenoside Rg3 and Rh2 were not included in these studies because they are significantly retained and are less suitable for use as internal standards.
Fig. 1.
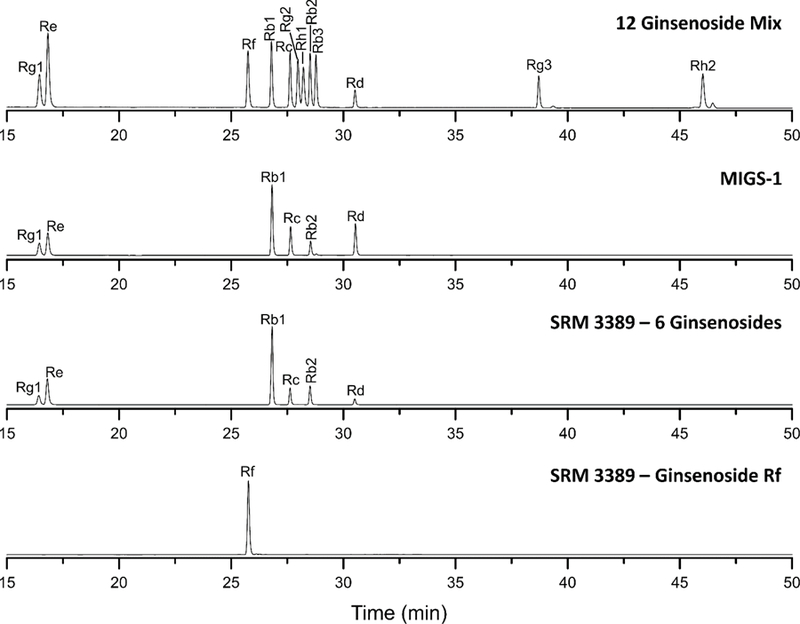
LC separations of the 12 ginsenoside mixture, MIGS-1, SRM 3389 Ginsenoside Rb1, Rb2, Rc, Rd, Re, and Rg1 and SRM 3389 Ginsenoside Rf.
3.1.1. Flow rate
In LC, the mobile phase flow rate influences separation efficiency and detection sensitivity. Increased flow produces narrower peaks and higher signal response from the detector. However, separation efficiency is reduced at high linear velocities since mass transfer is limited (i.e., the C term of the van Deemter equation becomes significant). At very low linear velocities, diffusion can decrease separation efficiency, although in practice this condition is rarely significant in modern LC. The most appropriate flow rate is typically based on the dimensions of the column and pressure limitation of the LC system to provide the highest efficiency possible while obtaining good chromatographic resolution.
Column selectivity is normally attributed to properties of the stationary phase and mobile phase composition and is not affected by the flow rate. For this reason, it was surprising to observe a significant difference in the retention behavior of two ginsenosides at different flow rates as illustrated in Fig. 2. As the flow rate increased from 0.6 mL/min to 1.0 mL/min, the relative retention of Rg2 and Rh1 unexpectedly changed. The ginsenosides have similar molecular structures, but differ in the number of carbohydrate units attached at the same hydroxyl position (Fig. S2). The origin of the selectivity differences for Rg2 and Rh1 is not immediately apparent; however, for gradient elution, changes in flow affect the residence time of the solute under changing environmental conditions. Thus, the changes in selectivity are probably related to changes in the modifier composition experienced by the solute even though the same gradient conditions are employed. The best chromatographic resolution was obtained with a flow rate of 0.7 mL/min.
Fig. 2.
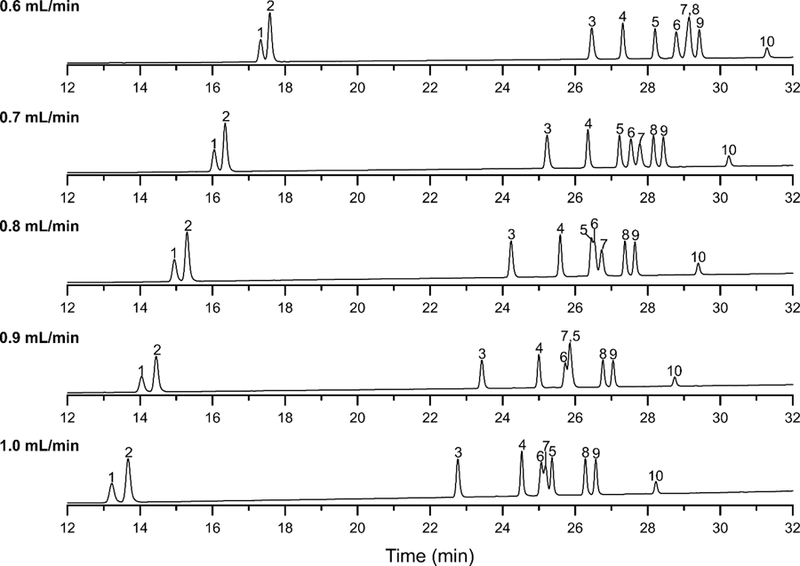
LC separations of the ginsenoside mixture on the ACE 3 C18 column using different mobile phase flow rates and the mobile phase gradient shown in Table 1. The column temperature and initial mobile phase composition was 25 °C and 22 % ACN, respectively. Peak identification is the following: (1) Rg1, (2) Re, (3) Rf, (4) Rb1, (5) Rc, (6) Rg2, (7) Rh1, (8) Rb2, (9) Rb3, and (10) Rd.
3.1.2. Initial Mobile Phase Composition
The separation of the 12 ginsenoside mixture was sensitive to the initial mobile phase composition of the gradient program as shown in Fig. 3. To evaluate the influence of mobile phase composition and selectivity, the initial conditions were varied from 19 to 23 % (v/v) ACN for 10 mins and followed by a linear gradient to 58 % ACN over 19 mins. The flow rate and column temperature were held constant at 0.7 mL/min and 25 °C, respectively. In the case of ginsenoside Rg1 and Re, better separations were obtained with increased levels of ACN at reduced retention. Separations of ginsenoside Rf, Rb1, Rc, Rb2, Rb3, Rd, Rg3, and Rh2 were influenced less by small changes in the mobile phase composition. However, retention of Rg2 and Rh1 was reduced relative to other ginsenosides, as the percentage of ACN increased. The best chromatographic resolution was obtained with an initial mobile phase composition of 22 % (v/v) ACN.
Fig. 3.
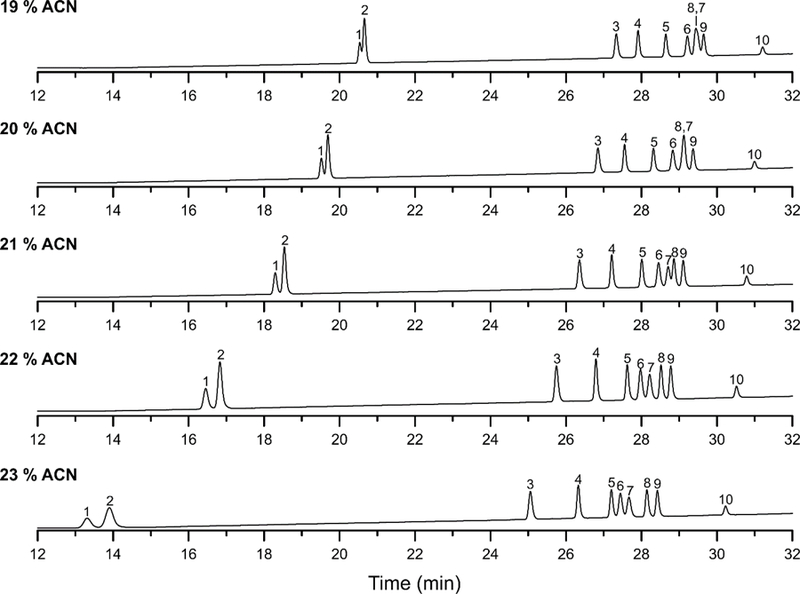
LC separations of the ginsenoside mixture on the ACE 3 C18 column using different initial isocratic conditions and linear gradient to 58 % ACN in 19 mins. The column temperature and mobile phase flow rate was 25 °C and 0.7 mL/min, respectively. Peak identification is the following: (1) Rg1, (2) Re, (3) Rf, (4) Rb1, (5) Rc, (6) Rg2, (7) Rh1, (8) Rb2, (9) Rb3, and (10) Rd.
3.1.3. Column Temperature
The effect of column temperature on the separation of the ginsenoside mixture is shown in Fig. 4. Separations were carried out over the temperature interval 15 °C to 35 °C, with a constant flow rate and initial mobile phase composition of 0.70 mL/min and 22 % (v/v) ACN, respectively. The chromatographic resolution of Rg1 and Re decreased as the temperature increased. Unexpectedly, the retention of the ginsenosides increased with increases in the column temperature, but Rg2 and Rh1 were affected to a lesser degree. The best chromatographic resolution was obtained with a column temperature of 25 °C.
Fig. 4.
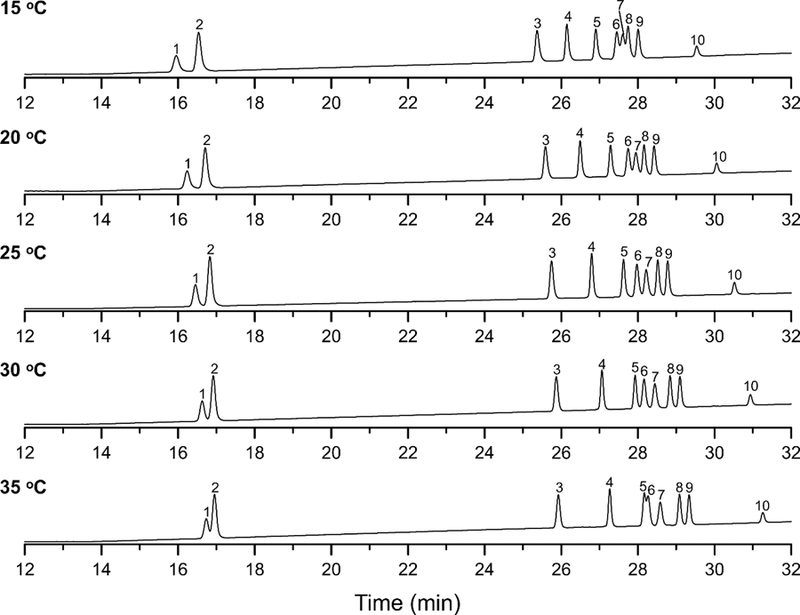
LC separations of the ginsenoside mixture on the ACE 3 C18 column using different column temperatures and the mobile phase gradient shown in Table 1. The mobile phase flow rate and initial composition was 0.7 mL/min and 22 % ACN, respectively. Peak identification is the following: (1) Rg1, (2) Re, (3) Rf, (4) Rb1, (5) Rc, (6) Rg2, (7) Rh1, (8) Rb2, (9) Rb3, and (10) Rd.
3.2. Liquid Chromatography/Mass Spectrometry Detection
3.2.1. Mass Spectra of 12 Ginsenosides
In LC/MS, the choice between positive and negative ion modes for ESI-MS has been shown to play a significant role in the detection sensitivity of ginsenosides [19, 25, 27, 32]. Based on previous work by Miao et al. [33], negative ion mode was shown to provide higher sensitivity, but positive mode provided more structural information. Negative mode was chosen in the current studies because the collection of structural information was not a priority. The ESI mass spectra of the 12 ginsenosides are shown in Figs. S4 – S6 and the peak distribution (≥ 10 %) is summarized in Table 2. The maximum signal abundance was observed for the formation of the FA adduct ion, which is represented by the singly charged ion [M – 2H + HCO2H]− except for ginsenosides Rc and Rb1. Doubly charged ion, [M – 2H]2-, was observed in the mass spectra of ginsenosides Rc, Rd, Re, Rf, Rb1, Rb2, Rb3, Rc, Rg2, and Rg3. The focus was to determine the appropriate ion to use in the selected ion monitoring (SIM) mode for the LC/MS certification measurements. The ion peaks with the maximum signal abundance reported in Table 2 were chosen for these studies except for ginsenoside Rc (m/z 1123.5).
3.2.2. LC/MS Separation of 12 Ginsenosides
Based on LC/UV method development, an ACE 3 C18 column (150 mm x 4.6 mm) with 3 µm particles was selected for all LC/MS studies. The optimized separation conditions from the initial LC/UV study are as follows: 0.7 mL/min flow rate, 25 °C column temperature, absorbance wavelength of 200 nm, and the mobile phase gradient summarized in Table 1. The LC/MS chromatograms in SIM mode for the 12 ginsenosides using these conditions are shown in Fig. S7. Under these conditions on a different instrument, Rb2 and Rb3 partially co-eluted in the m/z 1123.5 ion chromatogram. The 10 remaining ginsenosides are baseline resolved in their respective ion chromatograms. The baseline separation of these two ginsenosides are critical to the certification measurements of SRM 3389. For this reason, the separation conditions for the LC/MS measurements were slightly adjusted to achieve a baseline separation for Rb2 and Rb3 (see Table 1). The SIM chromatograms obtained with the modified separation conditions are shown in Fig. 5.
Fig. 5.
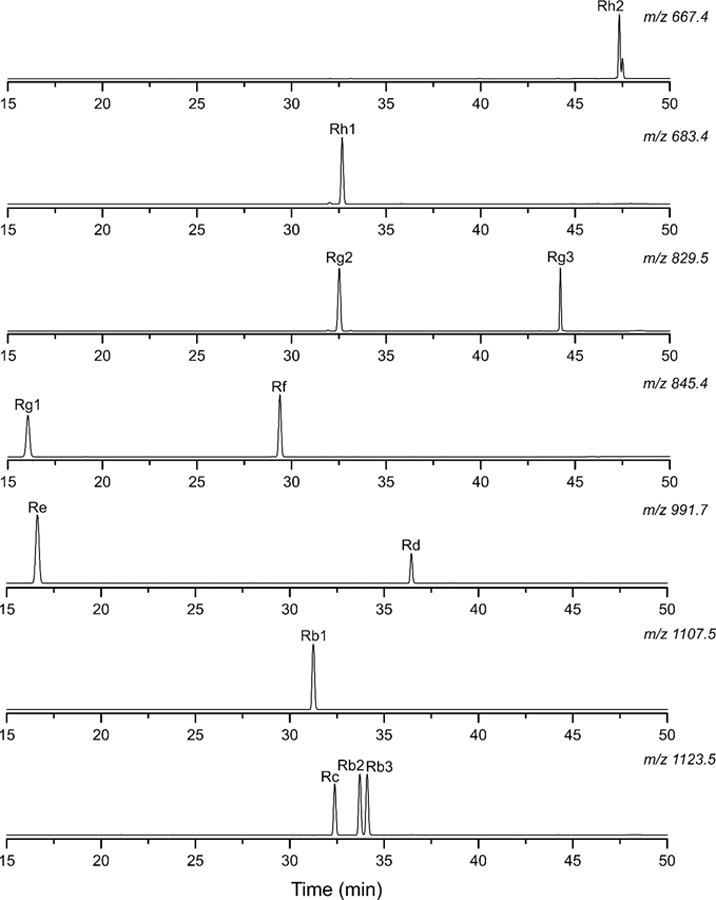
LC/MS chromatograms in SIM mode for a 12 component ginsenoside mixture using the optimized separation conditions summarized in Table 1. The flow rate was 0.7 mL/min and the column temperature was 25 °C.
3.3. Certification of SRM 3389
SRM 3389 Ginsenosides Calibration Solution consists of two different ampouled solutions: (1) Ginsenosides Rb1, Rb2, Rc, Rd, Re, and Rg1 and (2) Ginsenoside Rf. A summary of the certification measurements is reported in Table 3. The mass fraction values reported for both LC methods are similar to the initial gravimetry values from the preparation of SRM 3389. The MIGS-1 calibration solution (NRC Canada) was used as a control for the measurements. The LC/UV and LC/MS methods provided similar mass fraction values to the reported values in the Certificate of Analysis (COA) for the MIGS-1 solution. The mass fraction values obtained via the new LC/UV and LC/MS methods were statistically equivalent for SRM 3389 and MIGS-1 within a confidence interval of 95 % [34]. Detailed descriptions of the certification measurements are discussed below.
Table 3.
Summary of the certification results for the SRM 3389 and MIGS-1 solutions.
| Gravimetry |
LC/UVa |
LC/MSc |
|||
|---|---|---|---|---|---|
| Ginsenoside | Mass Fraction (mg/g) |
Mass Fractionb (mg/g) |
RSD (%) |
Mass Fractionb (mg/g) |
RSD (%) |
| SRM 3389 | |||||
| Rg1 | 0.51 | 0.518 ± 0.013 | 2.53 | 0.530 ± 0.023 | 4.27 |
| Re | 1.53 | 1.55 ± 0.039 | 2.52 | 1.54 ± 0.095 | 6.13 |
| Rf | 1.27 | 1.31 ± 0.039 | 3.02 | 1.31 ± 0.138 | 10.59 |
| Rb1 | 3.81 | 3.80 ± 0.097 | 2.55 | 3.86 ± 0.345 | 8.92 |
| Rc | 0.76 | 0.761 ± 0.019 | 2.55 | 0.750 ± 0.059 | 7.81 |
| Rb2 | 0.89 | 0.903 ± 0.023 | 2.54 | 0.877 ± 0.078 | 8.85 |
| Rd | 0.25 | 0.251 ± 0.008 | 3.18 | 0.246 ± 0.025 | 8.68 |
| MIGS-1: Control | |||||
| Rg1 | 0.258 ± 0.005 | 0.254 ± 0.005 | 1.90 | 0.257 ± 0.015 | 5.67 |
| Re | 0.506 ± 0.002 | 0.504 ± 0.009 | 1.86 | 0.525 ± 0.028 | 5.40 |
| Rf | - | - | - | - | - |
| Rb1 | 1.249 ± 0.016 | 1.24 ± 0.024 | 1.96 | 1.35 ± 0.114 | 8.42 |
| Rc | 0.505 ± 0.005 | 0.489 ± 0.010 | 1.98 | 0.497 ± 0.044 | 8.89 |
| Rb2 | 0.252 ± 0.002 | 0.254 ± 0.005 | 2.02 | 0.261 ± 0.017 | 6.39 |
| Rd | 0.507 ± 0.006 | 0.489 ± 0.010 | 1.91 | 0.504 ± 0.055 | 10.92 |
LC/UV values quantified using an external calibration approach.
The mass fraction values are the average ± standard deviation of ten measurements for each calibrant, candidate SRM 3389, and MIGS-1 control solutions. In the cases of LC/MS, the candidate SRM 3389 was measured nine times.
LC/MS values quantified using ginsenoside Rh1 for the internal standard calibration approach.
3.3.1. LC/UV Certification Measurements
Quantitation was based on external standard calibration method [34]. SRM 3389 and MIGS-1 solutions were analyzed by direct injection without sample manipulation, which makes the external calibration method suitable for LC/UV measurements. The main concern with external standard calibration measurements is the reproducibility of the injection volume. To minimize the injection volume error, the autosampler injection syringe was primed and the performance of the autosampler was verified prior to starting certification by evaluating multiple injections of a dilute solution of acetone. Acetone was selected to test the autosampler because of its short retention time (≈ 1.20 min) and absorbance at 200 nm. Multiple injections of acetone were made under isocratic conditions of 100 % ACN and a flow rate of 1.5 mL/min. The relative standard deviation (RSD) of these measurements was 0.48 % (n = 26).
The external standard calibration method in the present study used the linear regression relationship between the chromatographic peak area and the mass fraction (mg/g) of the ginsenoside standards. Three independently prepared calibrants were utilized, and each solution was determined ten times (see Fig. S8). In all cases, the correlation coefficients (R2) for the calibration curves were greater than or equal to 0.9989, demonstrating excellent linearity [34]. LC/UV measurements of the control MIGS-1 are in good agreement with the values reported in the COA and the measurements were under control. Good precision was obtained from the LC/UV measurements for SRM 3389 and MIGS-1 solutions, with RSD values of ≈ 3.0 % and ≈ 2.0 %, respectively.
3.3.2. LC/MS Certification Measurements.
LC/MS analyses were based on the internal standard calibration method [35], which is typically used for quantitative MS to account for variations in detection response and injection irreproducibility. Better accuracy often results when the internal standard exhibits chemical properties similar to the analyte. Ideally, an internal standard should offer similar detector response to measured analytes and be resolved from potential interferences [35]. Isotopically labeled internal standards are preferred for quantitation; however, no isotopically labeled ginsenosides were commercially available at the time of analysis. For this reason, five ginsenosides (Rb3, Rg2, Rg3, Rh1, and Rh2) were investigated for use as possible internal standards.
Rb3 was unsuitable because it was found to be present as an impurity in the Rb2 reference standard. Rg3 and Rh2 elute significantly later than the ginsenosides present in SRM 3389. Rg2 and Rh1 exhibit the traits desired of internal standards. Rh1 was selected for use in the certification of SRM 3389. A different ginsenoside can be selected when measuring ginsenosides in a natural product material such as root extract if Rh1 is present in the sample material. Small quantities of the calibrant solutions, MIGS-1 solutions, and SRM 3389 were mixed with equivalent quantities of the internal standard in most cases. The three calibrants were measured nine times and the calibration curves are shown in Fig. S9. The R2 values were greater than or equal to 0.9919 except for Rg1, which had an R2 value of 0.8965 [34]. Ginsenoside levels determined by LC/MS are in good agreement with the LC/UV measurements and the targeted mass fraction values (gravimetry and COA). Considering the internal standard was not isotopically labeled, the LC/MS method provided good precision for the mass fraction values of SRM 3389 and MIGS-1 with RSD values between 4.27 % (Rg1, SRM 3389) and 10.92 % (Rd, MIGS-1).
4. Conclusions
LC/UV and LC/MS methods were developed for the certification of a ginsenoside mixture reference material. The optimal flow rate, initial mobile phase composition, and column temperature were evaluated to provide the best separation: 0.7 mL/min, 22 % ACN, and 25 °C, respectively. In the case of Rg2 and Rh1, retention was strongly affected by these conditions. Mass spectra were collected for the 12 ginsenosides in negative ion mode to achieve less fragmentation and improve method selectivity. The FA adduct ion, [M – 2H + HCO2H]−, provided the highest signal abundance in most of the mass spectra. Both LC methods were used in the certification of SRM 3389 Ginsenoside Calibration Solution.
Supplementary Material
Acknowledgements
W. B. Wilson acknowledges financial support from the Office of Dietary Supplements at the National Institutes of Health (ODS-NIH). The authors would like to thank Catherine Rimmer from NIST and Joseph M. Betz from ODS-NIH for continue guidance throughout the project.
Footnotes
Publisher's Disclaimer: Disclaimer
Certain commercial equipment or materials are identified in this manuscript to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References
- [1].Li N, Liu B, Dluzen DE, Jin Y, Protective effects of ginsenoside rg2 against glutamate-induced neurotoxicity in pc12 cells, Journal of Ethnopharmacology, 111 (2007) 458–463. [DOI] [PubMed] [Google Scholar]
- [2].Liao B, Newmark H, Zhou R, Neuroprotective effects of ginseng total saponin and ginsenosides rb1 and rg1 on spinal cord neurons in vitro, Experimental Neurology, 173 (2002) 224–234. [DOI] [PubMed] [Google Scholar]
- [3].Tian J, Fu F, Geng M, Jiang Y, Yang J, Jiang W, Wang C, Liu K, Neuroprotective effect of 20(s)-ginsenoside rg3 on cerebral ischemia in rats, Neuroscience Letters, 374 (2005) 92–97. [DOI] [PubMed] [Google Scholar]
- [4].Ye R, Li N, Han J, Kong X, Cao R, Rao Z, Zhao G, Neuroprotective effects of ginsenoside rd against oxygen-glucose deprivation in cultured hippocampal neurons, Neuroscience Research, 64 (2009) 306–310. [DOI] [PubMed] [Google Scholar]
- [5].Kang K-S, Kang B-C, Lee B-J, Che J-H, Li G-X, Trosko JE, Lee Y-S, Preventive effect of epicatechin and ginsenoside rb2 on the inhibition of gap junctional intercellular communication by tpa and h2o2, Cancer Letters, 152 (2000) 97–106. [DOI] [PubMed] [Google Scholar]
- [6].Yue PYK, Wong DYL, Wu PK, Leung PY, Mak NK, Yeung HW, Liu L, Cai Z, Jiang Z-H, Fan TPD, Wong RNS, The angiosuppressive effects of 20(r)- ginsenoside rg3, Biochemical Pharmacology, 72 (2006) 437–445. [DOI] [PubMed] [Google Scholar]
- [7].Wang T, Yu X, Qu S, Xu H, Han B, Sui D, Effect of ginsenoside rb3 on myocardial injury and heart function impairment induced by isoproterenol in rats, European Journal of Pharmacology, 636 (2010) 121–125. [DOI] [PubMed] [Google Scholar]
- [8].Lee M-S, Hwang J-T, Kim S.-h., Yoon S, Kim M-S, Yang HJ, Kwon DY, Ginsenoside rc, an active component of panax ginseng, stimulates glucose uptake in c2c12 myotubes through an ampk-dependent mechanism, Journal of Ethnopharmacology, 127 (2010) 771–776. [DOI] [PubMed] [Google Scholar]
- [9].Mogil JS, Shin Y-H, McCleskey EW, Kim S-C, Nah S-Y, Ginsenoside rf, a trace component of ginseng root, produces antinociception in mice, Brain Research, 792 (1998) 218–228. [DOI] [PubMed] [Google Scholar]
- [10].Kang KS, Kim HY, Yamabe N, Yokozawa T, Stereospecificity in hydroxyl radical scavenging activities of four ginsenosides produced by heat processing, Bioorganic & Medicinal Chemistry Letters, 16 (2006) 5028–5031. [DOI] [PubMed] [Google Scholar]
- [11].Park YC, Lee CH, Kang HS, Kim K-W, Chung HT, Kim HD, Ginsenoside-rh1 and rh2 inhibit the induction of nitric oxide synthesis in murine peritoneal macrophages, IUBMB Life, 40 (1996) 751–757. [DOI] [PubMed] [Google Scholar]
- [12].Xie J-T, Shao Z-H, Vanden Hoek TL, Chang W-T, Li J, Mehendale S, Wang C-Z, Hsu C-W, Becker LB, Yin J-J, Yuan C-S, Antioxidant effects of ginsenoside re in cardiomyocytes, European Journal of Pharmacology, 532 (2006) 201–207. [DOI] [PubMed] [Google Scholar]
- [13].Kim ND, Kim EM, Kang KW, Cho MK, Choi SY, Kim SG, Ginsenoside rg3 inhibits phenylephrine-induced vascular contraction through induction of nitric oxide synthase, British Journal of Pharmacology, 140 (2003) 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee H-U, Bae E-A, Han MJ, Kim D-H, Hepatoprotective effect of 20(s)-ginsenosides rg3 and its metabolite 20(s)-ginsenoside rh2 on tert-butyl hydroperoxide-induced liver injury, Biological and Pharmaceutical Bulletin, 28 (2005) 1992–1994. [DOI] [PubMed] [Google Scholar]
- [15].MacCrehan WA, White CM, Simplified ultrasonically- and microwave-assisted solvent extractions for the determination of ginsenosides in powdered Panax ginseng rhizomes using liquid chromatography with UV absorbance or electrospray mass spectrometric detection, Anal. Bioanal. Chem 405 (2013) 4511–4522. [DOI] [PubMed] [Google Scholar]
- [16].Li WK, Fitzloff JF, Hplc determination of ginsenosides content in ginseng dietary supplements using ultraviolet detection, Journal of Liquid Chromatography & Related Technologies, 25 (2002) 2485–2500. [Google Scholar]
- [17].Liu Z, Xia J, Wang CZ, Zhang JQ, Ruan CC, Sun GZ, Yuan CS, Remarkable impact of acidic ginsenosides and organic acids on ginsenoside transformation from fresh ginseng to red ginseng, Journal of Agricultural and Food Chemistry, 64 (2016) 5389–5399. [DOI] [PubMed] [Google Scholar]
- [18].Ma L, Liu F, Zhong Z, Wan J, Comparative study on chemical components and anti-inflammatory effects of Panax notoginseng flower extract by water and methanol, J. Sep. Sci 40 (2017) 4730–4739. [DOI] [PubMed] [Google Scholar]
- [19].Stavrianidi A, Stekolshchikova E, Porotova A, Rodin I, Shpigun O, Combination of hplc–ms and qams as a new analytical approach for determination of saponins in ginseng containing products, Journal of Pharmaceutical and Biomedical Analysis, 132 (2017) 87–92. [DOI] [PubMed] [Google Scholar]
- [20].Quiming NS, Denola NL, Soliev AB, Saito Y, Jinno K, High performance liquid chromatographic separation and quantitative analysis of ginsenosides using a polyvinyl alcohol-bonded stationary phase, Chromatographia, 66 (2007) 5–11. [Google Scholar]
- [21].Lee GJ, Shin BK, Yu YH, Ahn J, Kwon SW, Park JH, Systematic development of a group quantification method using evaporative light scattering detector for relative quantification of ginsenosides in ginseng products, Journal of Pharmaceutical and Biomedical Analysis, 128 (2016) 158–165. [DOI] [PubMed] [Google Scholar]
- [22].Zhan SY, Guo WJ, Shao Q, Fan XH, Li Z, Cheng YY, A pharmacokinetic and pharmacodynamic study of drug-drug interaction between ginsenoside rg1, ginsenoside rb1 and schizandrin after intravenous administration to rats, Journal of Ethnopharmacology, 152 (2014) 333–339. [DOI] [PubMed] [Google Scholar]
- [23].Kwon H-J, Jeong J-S, Lee Y-M, Hong S-P, A reversed-phase high-performance liquid chromatography method with pulsed amperometric detection for the determination of glycosides, Journal of Chromatography A, 1185 (2008) 251–257. [DOI] [PubMed] [Google Scholar]
- [24].Cai Q, Yang Z, Chen N, Zhou X, Hong J, Selective capture and rapid identification of panax notoginseng metabolites in rat faeces by the integration of magnetic molecularly imprinted polymers and high-performance liquid chromatography coupled with orbitrap mass spectrometry, Journal of Chromatography A, 1455 (2016) 65–73. [DOI] [PubMed] [Google Scholar]
- [25].Gao JW, Qiu Y, Chen JM, Mu SX, Sun LX, Simultaneous determination of nineteen major active compounds in qiangshen tablet by uplc-esi-ms/ms, Journal of Pharmaceutical and Biomedical Analysis, 128 (2016) 519–527. [DOI] [PubMed] [Google Scholar]
- [26].Dai GL, Jiang ZT, Zhu LJ, Zhang Q, Zong Y, Liu SJ, Li CY, Ju WZ, Simultaneous determination of notoginsenoside r1 and ginsenoside re in rat plasma by ultra high performance liquid chromatography with tandem mass spectrometry and its application to a pharmacokinetic study, Journal of Separation Science, 39 (2016) 3368–3374. [DOI] [PubMed] [Google Scholar]
- [27].Liu J, Liu Y, Zhao L, Zhang ZH, Tang ZH, Profiling of ginsenosides in the two medicinal panax herbs based on ultra-performance liquid chromatography-electrospray ionization-mass spectrometry, Springerplus, 5 (2016) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang XQ, Ma RJ, Liu XJ, Jiang XH, Wang L, Simultaneous determination of ginsenoside rg(1), re and notoginsenoside r-1 in human plasma by lc-ms/ms and its application in a pharmacokinetic study in chinese volunteers, Biomedical Chromatography, 30 (2016) 1915–1921. [DOI] [PubMed] [Google Scholar]
- [29].Wang H, Zhang Y, Yang X, Zhao D, Wang Y, Rapid characterization of ginsenosides i nthe roots and rhizomes of Panax ginseng by UPLC-DAD-QTOF-MS/MS and simultaneousb determination of 19 ginsenosides by HPLC-ESI-MS, Journal of Ginseng Research, 40 (2016) 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rimmer CA, Putzbach K, Sharpless KE, Sander LC, Yen JH, Preparation and certification of Standard Reference Material 3278 tocopherols in edible oils, J. Agric. Food Chem, 60 (2012) 6794–6798. [DOI] [PubMed] [Google Scholar]
- [31].Sander LC, Bedner M, Tims MC, Yen JH, Duewer DL, Porter B, Christopher SJ, Day RD, Long SE, Molloy JL, Murphy KE, Lang BE, Lieberman R, Wood LJ, Payne MJ, Roman MC, Betz JM, NguyenPho A, Sharpless KE, Wise SA, Development and certification of green tea-containing standard reference materials, Anal. Bioanal. Chem 402 (2012) 473–487. [DOI] [PubMed] [Google Scholar]
- [32].Phillips MM, Case RJ, Rimmer CA, Sander LC, Sharpless KE, Wise SA, Yen JH, Deterimination of organic acids in Vaccinium bery standard reference materials, Anal. Bioanal. Chem 398 (2010) 425–434. [DOI] [PubMed] [Google Scholar]
- [33].Miao X-S, Metcalfe CD, Hao C, March RE, Electrospray ionization mass spectrometry of ginsenosides, Journal of Mass Spectrometry, 37 (2002) 495–506. [DOI] [PubMed] [Google Scholar]
- [34].Miller JN, Miller JC, Statistics and chemometrics for analytical chemistry, Pearson/Prentice Hall, 2005. [Google Scholar]
- [35].Cunico RL, Gooding KM, Wehr T, Basic hplc and ce of biomolecules, Bay Bioanalytical Laboratory, 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.