

Abstract
A prominent function of TGIF1 is suppression of transforming growth factor beta (TGF‐β) signaling, whose inactivation is deemed instrumental to the progression of pancreatic ductal adenocarcinoma (PDAC), as exemplified by the frequent loss of the tumor suppressor gene SMAD4 in this malignancy. Surprisingly, we found that genetic inactivation of Tgif1 in the context of oncogenic Kras, KrasG12D, culminated in the development of highly aggressive and metastatic PDAC despite de‐repressing TGF‐β signaling. Mechanistic experiments show that TGIF1 associates with Twist1 and inhibits Twist1 expression and activity, and this function is suppressed in the vast majority of human PDACs by KrasG12D/MAPK‐mediated TGIF1 phosphorylation. Ablating Twist1 in Kras G12D ;Tgif1 KO mice completely blunted PDAC formation, providing the proof‐of‐principle that TGIF1 restrains KrasG12D‐driven PDAC through its ability to antagonize Twist1. Collectively, these findings pinpoint TGIF1 as a potential tumor suppressor in PDAC and further suggest that sustained activation of TGF‐β signaling might act to accelerate PDAC progression rather than to suppress its initiation.
Keywords: oncogenic Kras, pancreatic ductal adenocarcinoma, TGF‐β signaling, TGIF1, Twist1
Subject Categories: Cancer, Transcription
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death and one of the most aggressive human malignancies (Hidalgo, 2010; Jemal et al, 2010). The vast majority of PDAC patients present with inoperable disease and rapidly succumb from a devastating illness characterized by a very rapid tumor dissemination and severe cachexia, leading to treatment intolerance and general organ dysfunction (Hidalgo, 2010; Jemal et al, 2010; Stathis & Moore, 2010). Advanced PDAC has a cumulative 5‐year survival rate of < 5% (Kern et al, 2011; Maitra & Hruban, 2008).
Pancreatic ductal adenocarcinoma evolves through a series of pre‐neoplastic lesions, termed pancreatic intraepithelial neoplasia (PanIN), accompanied by recurrent genetic alterations, the earliest and most ubiquitous of which activating mutations in the KRAS proto‐oncogene, affecting more than 90% of PDAC patients (Almoguera et al, 1988; Hezel et al, 2006). Given their high incidence at very early stage PanIN, perturbations in KRAS are considered as key genetic determinants in PDAC initiation. The accumulation of additional inactivating mutations in other tumor suppressor genes (e.g., p16INK4A, TP53, and SMAD4) occurs at high frequency in late‐stage PanIN, and these alterations are deemed essential for tumor progression and metastasis (Hezel et al, 2006; Iacobuzio‐Donahue, 2012).
Considerable efforts have been made during the last two decades to generate animal models that faithfully recapitulate the natural history of human PDAC. For instance, mice with pancreas‐specific expression of KrasG12D develop PanINs that can gradually progress to invasive PDAC, providing evidence for oncogenic KRAS as the major driver in PDAC (Hingorani et al, 2003; Tuveson et al, 2004). In contrast, engineering p16Ink4a, Trp53, or Smad4 inactivating mutations into pancreas failed to induce pancreatic neoplasia per se, and expression of KrasG12D appeared critical for their malignant properties (Aguirre et al, 2003; Bardeesy et al, 2006a,b; Hingorani et al, 2005). Along these lines, concomitant KrasG12D expression and inactivation of p16Ink4a or Trp53 accelerate the development of PDAC with clinical and histological features that closely recapitulate key aspects of the human disease, such as the highly reactive desmoplastic stroma and aggressive metastatic behaviors (Aguirre et al, 2003; Bardeesy et al, 2006a; Hingorani et al, 2005). Likewise, concomitant KrasG12D expression and Smad4 inactivation result in rapid development of highly invasive PDAC tumors resembling intraductal papillary mucinous neoplasia (IPMN), a precursor to PDAC in humans (Bardeesy et al, 2006b; Whittle et al, 2015). Intriguingly, these IPMN/PDAC tumors retain a well‐differentiated ductal structure and manifest an attenuated metastatic potential, creating a conundrum of how Smad4 could enhance malignant conversion of PDAC while simultaneously compromising its metastatic potential. Owing to its prominent function in TGF‐β signaling, these seemingly distinct effects of Smad4 have been attributed to a potential bimodal role of TGF‐β signaling that typically manifests during cancer initiation and progression in a number of human malignancies (Massague, 2008). However, since these genetic studies relied exclusively upon suppression of TGF‐β/Smad signaling (Bardeesy et al, 2006b; Whittle et al, 2015), it remains unclear whether activation of the TGF‐β pathway is indeed needed to sustain the metastatic phenotype, and, if so, whether this entails an intact Smad signaling. Such information would produce insights into the so‐called divergent actions of TGF‐β signaling and also ascertain whether targeting this pathway is prudent for cancer therapeutics when both tumor‐suppressive and promoting features might co‐exist.
TGF‐β initiates responses by contacting two types of transmembrane Ser/Thr kinases called type I (TβRI) and type II (TβRII) receptors, promoting phosphorylation and activation of TβRI by the TβRII kinase (Attisano & Wrana, 2002; Massague et al, 2005). In the canonical pathway, the activated TβRI propagates signals by phosphorylating intracellular downstream effectors, Smad2 and Smad3 (Smad2/3), which then complex with Smad4 and translocate to the nucleus to regulate expression of TGF‐β target genes through cooperative interaction with general transcriptional coactivators or corepressors (Massague et al, 2005). The ability of TGF‐β pathway to regulate vast arrays of target genes enables the pathway to impact diverse cellular processes including proliferation, apoptosis, differentiation, and migration (Feng & Derynck, 2005; Massague, 2008).
Because of its critical roles in cell fate determination, TGF‐β signaling is subject to many levels of positive and negative regulation, targeting both receptors and intracellular mediators. For example, TGF‐β signaling can be limited by TG‐interacting factor 1 (TGIF1), a nuclear protein originally thought to repress Smad transcriptional activity by binding to Smad2 and Smad4 on promoters of target genes and recruiting corepressor complex containing histone deacetylases (HDAC; Wotton et al, 1999). However, recent studies from our laboratory have revealed that TGIF1 can also interfere with early phases of TGF‐β signaling, presumably by restricting access of Smad2 for phosphorylation by TβRI (Ettahar et al, 2013; Seo et al, 2006). Congruently, Tgif1 inactivation either in vitro or in vivo is sufficient for Smad2/3 phosphorylation and attendant integration of the TGF‐β transcriptional program. As such, exploring the role of TGIF1 in PDAC could provide new insights into how TGF‐β signaling influences PDAC behaviors, in particular whether TGF‐β's actions on proliferation and metastasis depend on its ability to impact cell proliferation, alter processes that orchestrate the malignant conversion and metastasis or both.
In this study, we combined several orthogonal approaches and models to demonstrate that TGIF1 functions as a potential tumor suppressor in PDAC driven by KrasG12D. We found that although Tgif1 inactivation in the pancreatic epithelium culminated in hyperactivation of TGF‐β signaling, it enabled rapid development of highly aggressive and metastatic PDAC. We provide molecular and genetic evidence that TGIF1 functions to suppress PDAC progression by antagonizing the pro‐malignant transcription factor Twist1. Collectively, these findings unveil an unanticipated tumor suppressor role for TGIF1 in PDAC, therefore offering an unprecedented framework for further exploring the concept in which TGF‐β cytostatic signaling fulfills biphasic role during PDAC initiation and progression.
Results
TGIF1 is dispensable for normal pancreatic development
Mice with global homozygous deletion of Tgif1 (Tgif1 −/−) have been shown to display a variety of subtle phenotypes, including runting and intestinal inflammation (Bartholin et al, 2008; Hneino et al, 2012; Taniguchi et al, 2012). However, further targeted comprehensive analysis of these mice failed to reveal any overt pathology in the pancreatic function, as evidenced by the normal fasting blood glucose levels, gain of body weight, and unperturbed pancreatic parenchymal architecture (Fig EV1A–C). To confirm these observations, we generated mice with pancreas‐specific deletion of Tgif1 (Tgif1 KO) by crossing Tgif1 fl/fl mice with Pdx1‐Cre mice, which express Cre recombinase in all pancreatic progenitor cells beginning during early development at E8.5 (Gu et al, 2003; Shen & Walsh, 2005). Tgif1 KO mice were born at the Mendelian ratio, showed no evidence of any gross anatomic or physiological abnormalities, and had normal body weight and life expectancy (Fig EV1D, see also Fig 1C). Immunoblotting analysis confirmed the loss of TGIF1 expression in pancreatic tissue from Tgif1 KO mice (Fig EV1E). As anticipated (Ettahar et al, 2013; Seo et al, 2006), both immunoblotting and immunofluorescence showed a marked increase in Smad2 phosphorylation in Tgif1 KO mice relative to wild‐type littermates (Figs 1A and EV1E). Despite elevated TGF‐β/Smad signaling, Tgif1 KO mice pancreata were indistinguishable from those of wild‐type mice in overall histology, as assessed by staining with hematoxylin and eosin (H&E) or immunohistochemistry (IHC) using antibodies to Amylase (acinar marker) or Cytokeratin 19 (CK19; ductal marker; Fig 1B). Likewise, there were no changes in the expression of pancreatic endocrine markers, including Insulin (β‐cells) and Glucagon (α‐cells), which is consistent with the normal fasting blood glucose (Fig EV1F and G). Finally, none of the Tgif1 KO mice developed pancreatic neoplasms during an observation period of 18 months (Fig 1C–E). Thus, the apparently healthy pancreas in both Tgif1 −/− and Tgif1 KO mice indicated that constitutive activation of TGF‐β/Smad signaling throughout embryonic development and postnatal life was insufficient to perturb pancreas homeostasis or promote sporadic pancreatic cancer development.
Figure EV1. Tgif1 deletion does not affect pancreas function or histology (related to Fig 1).
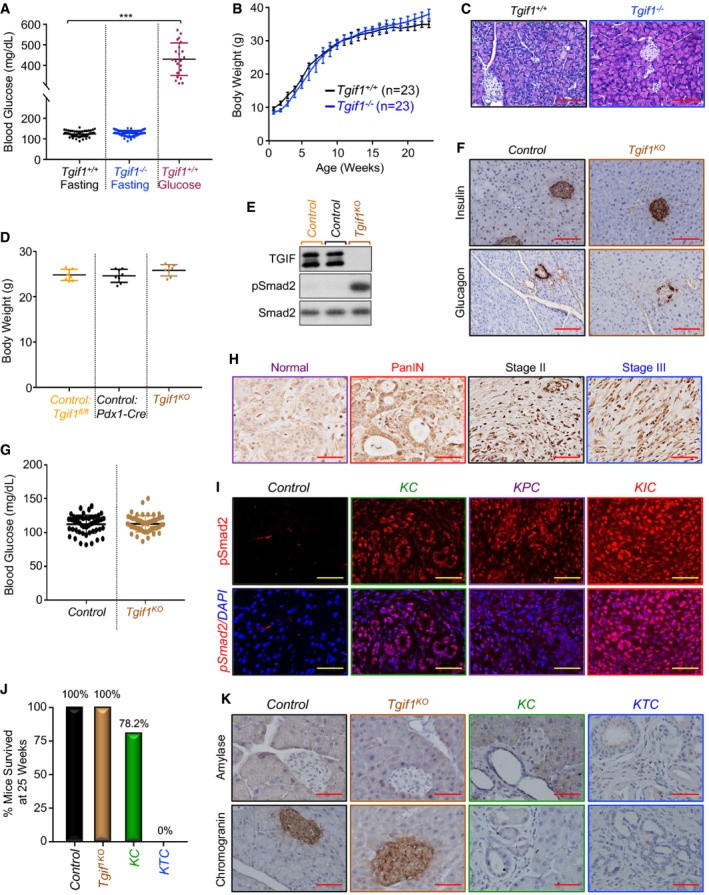
- Tgif1 +/+ or Tgif1 −/− mice were fasted for 6 h and blood glucose levels were measured using the ReliON method. Mice injected with glucose at 2 g/kg body weight were used as controls. ***P < 0.001; based on a two‐tailed Student's test.
- Body weight of Tgif1 +/+ or Tgif1 −/− mice was recorded every week for 25 weeks.
- FFPE pancreatic sections from Tgif1 +/+ or Tgif1 −/− mice were stained with H&E. Representative pictures at 20× are shown (n = 23). Scale bars, 200 μM.
- Body weight of control or Tgif1 KO mice was measured at the age of 25 weeks.
- Total lysates from pancreas of control or Tgif1 KO mice were pooled and analyzed by immunoblotting using antibodies to TGIF1, pSmad2, and Smad2 used as control (n = 6).
- FFPE pancreatic sections from control or Tgif1 KO mice were immunostained with antibodies to Insulin or Glucagon and revealed by IHC. Representative pictures at 20× are shown (n = 30). Scale bars, 200 μM.
- Fasting blood glucose of control or Tgif1 KO mice was measured as described in (A).
- Expression of TGF‐β1 in tissue microarrays of human PDAC samples was analyzed by IHC. Representative pictures at 40× are shown. Scale bars, 100 μM.
- FFPE pancreatic sections from control, KC, KPC or KIC mice were immunostained with anti‐pSmad2 antibody and revealed by IF and DAPI. Representative pictures at 40× are shown (n = 7–30). Scale bars, 100 μM.
- Tumor‐free survival of control, Tgif1 KO, KC and KTC mice at 25 weeks of age, time at which 100% of KTC mice succumbed to PDAC.
- FFPE pancreatic sections from control, Tgif1 KO, KC or KTC mice were immunostained with antibodies to chromogranin A or Amylase and revealed by IHC. Representative pictures at 40× are shown (n = 30). Scale bars, 100 μM.
Figure 1. Tgif1 inactivation deepens KrasG12D‐driven PDAC .
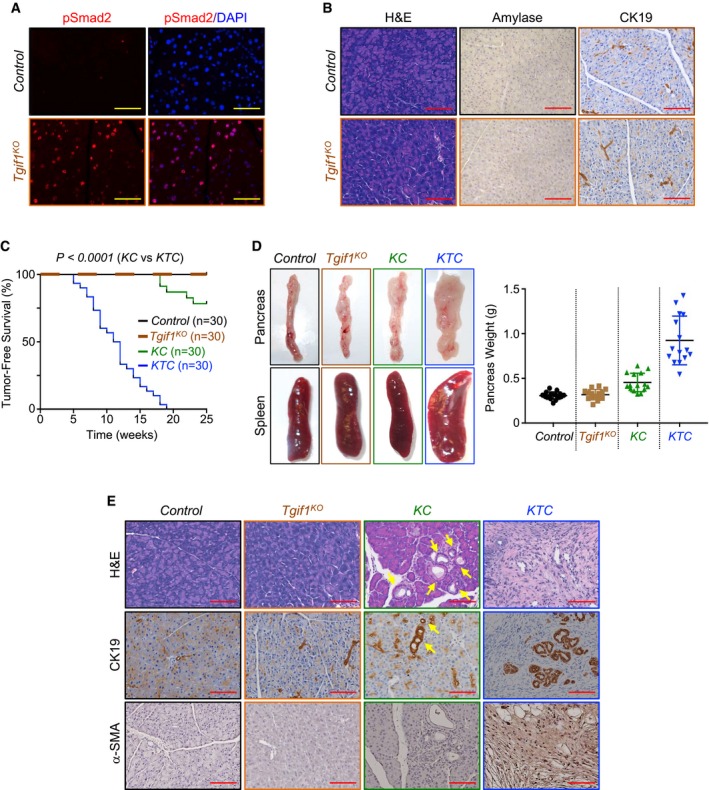
- Formalin‐fixed paraffin‐embedded (FFPE) sections from control or Tgif1 KO mice were immunostained with anti‐pSmad2 antibody and revealed by immunofluorescence (IF) and DAPI. Representative pictures at 40× are shown (n = 30). Scale bars, 100 μM.
- FFPE pancreatic sections from control or Tgif1 KO mice were stained with hematoxylin and eosin (H&E) or immunostained with antibodies to CK19 or Amylase and revealed by IHC. Representative pictures at 20× are shown (n = 30). Scale bars, 200 μM.
- Kaplan–Meier survival analysis of control, Tgif1 KO, KC, and KTC mice. A regular mosaic two‐color line was used to discriminate between control and Tgif1 KO mice.
- Pictures of whole pancreas and spleen tissues from control, Tgif1 KO, KC, or KTC mice (left). Weight of pancreas was measured and presented as dot plot (n = 30).
- FFPE pancreatic sections from control, Tgif1 KO, KC, or KTC were stained with H&E or subjected to IHC analysis using antibodies to CK19 or α‐SMA. Yellow arrows indicate PanINs in KC mice. Representative pictures at 20× are shown (n = 30). Scale bars, 200 μM.
Tgif1 inactivation accelerates KrasG12D‐driven PDAC
Based on PDAC mouse models with either TβRII or Smad4 deletion, it has been postulated that inactivation of TGF‐β signaling facilitates PDAC progression (Bardeesy et al, 2006b; Ijichi et al, 2006). Nevertheless, other studies have reported that TGF‐β signaling is highly active during late stages of PDAC (Bardeesy et al, 2006b; Friess et al, 1993; Wagner et al, 1999). We independently confirmed this observation by assessing TGF‐β1 expression in a human PDAC tissue microarray (Fig EV1H). We also detected increased Smad2 phosphorylation in PDAC tumors (Fig EV1I) from three well‐characterized mouse models of PDAC, e.g., Kras G12D ;Pdx1‐Cre (KC), Kras G12D ;Trp53 R172H/+ ;Pdx1‐Cre (KPC), and Kras G12D ;p16Ink4a −/− ;Pdx1‐Cre (KIC; Bardeesy et al, 2006a,b; Hingorani et al, 2005). The fact that Tgif1 inactivation leads to de‐repression of TGF‐β signaling provided us with a unique platform to delineate the exact role of TGF‐β signaling in PDAC under gain‐of‐function conditions instead of loss‐of‐function conditions, as performed in previous genetic studies (Bardeesy et al, 2006b; Ijichi et al, 2006). Thus, we set out to conduct comparative experiments using mice with KrasG12D alone (KC) or in combination with Tgif1 conditional deletion (termed thereafter as KTC mice). Consistent with previous findings (Hingorani et al, 2003; Tuveson et al, 2004), most KC mice showed uniformly good health through age of 20 weeks, and thereafter, a subset of these mice succumbed to PDAC (Fig 1C). Perhaps surprisingly, combining KrasG12D expression with Tgif1 deletion resulted in a dramatic acceleration of the onset of PDAC. Notably, at 8 weeks of age, the majority of KTC mice began to demonstrate jaundice and abdominal distension due to ascites and the tumor itself (Fig 1D). Kaplan–Meier analysis showed a dramatic decrease in median survival of KTC mice as compared to KC mice (Fig 1C). Noteworthy, all KTC mice died within 19 weeks, whereas more than 78% of KC survived within this time period (Fig EV1J). Gross inspection at necropsy revealed that KTC tumors were irregularly shaped with white appearance (Fig 1D), typical characteristic of PDAC (Hingorani et al, 2003). Moreover, splenomegaly was frequently observed in these mice (Fig 1D). At this stage, pancreas of KC mice displayed almost normal appearance (Fig 1D). Microscopic examination showed that KTC tumors displayed uniformly poorly differentiated architecture, which occupied the entire pancreas, resulting in almost complete loss of normal pancreatic tissue (Fig 1E). They stained positive for the ductal marker CK19 and were negative for Amylase and Chromogranin A (Figs 1E and EV1K), consistent with a PDAC phenotype. The tumors also displayed a marked desmoplastic stroma, as assessed by the strong expression of alpha smooth muscle actin (α‐SMA; Fig 1E), a feature frequently seen in human PDAC (Feig et al, 2012; Ozdemir et al, 2014). Of note, age‐matched KC mice developed PanINs that were confined within large areas of normal pancreas exhibiting well‐organized acinar architectures (Fig 1E), as reported previously (Bardeesy et al, 2006a,b; Hingorani et al, 2003; Ijichi et al, 2006). These data strongly suggest that loss of TGIF1 function might promote the development and/or progression of PDAC driven by KrasG12D.
Tgif1 inactivation drives PDAC metastasis
The accelerated progression of PDAC in KTC mice promoted us to conduct more detailed pathological analyses to assess the kinetics of PDAC formation and progression under TGIF1 deficiency conditions. KC mice have been reported to develop focal PanINs as early as 4 weeks, but most of the lesions do not progress into malignant PDAC over the next 40 weeks (Bardeesy et al, 2006a,b; Hingorani et al, 2003; Ijichi et al, 2006). Histological analysis at 4 weeks of age showed that KTC mice pancreata displayed very few multifocal isolated PanINs and acinar‐ductal metaplasia (ADM) lesions embedded within abundant normal pancreatic areas, similar to what observed in age‐matched KC mice (Appendix Fig S1A). At 8 weeks of age, most of KTC mice displayed abundant high‐grade PanINs and full PDAC lesions, whereas their age‐matched KC counterparts still have only focal low‐grade PanINs (Appendix Fig S1A). Consistently, PanINs in KTC mice displayed increased proliferation rate compared to PanINs in KC mice, as determined by BrdU incorporation (Appendix Fig S1A). The penetrance of tumor appearance across mice of the KTC genotype was 100% at the age of 18 weeks (Figs 1C and EV1J). Collectively, these findings illustrate that Tgif1 inactivation accelerates KrasG12D‐driven PDAC formation.
Apart from KTC mice that were sacrificed as they reached terminal morbidity due to heavy tumor burden, the remaining mice (63%) that survived beyond 8 weeks developed PDAC with both local invasion and distant macroscopic metastases into the liver (Fig 2A). Further histopathological analysis of primary PDAC tumors by H&E or immunofluorescence (IF) using anti‐Vimentin antibody showed massive invasion of lymph node, a phenotype never seen in age‐matched KC mice (Fig 2B). To provide further evidence that Tgif1 inactivation promotes PDAC metastasis, we generated KC and KTC mice harboring a Cre‐inducible luciferase (Cheung et al, 2008) to monitor PDAC metastatic spread in live animals. Relative to age‐matched KC Luc mice, which showed only weak local bioluminescence signal, 5 out of 6 KTC Luc mice displayed disseminated PDAC, affecting both lung and liver (Fig 2C), two sites associated with human PDAC (Stathis & Moore, 2010). None of the six mice developed metastasis to the brain (Fig 2C). H&E analysis confirmed the presence of metastatic lesions in liver and lung in KTC Luc mice but not in KC Luc mice (Fig 2B). As anticipated, both liver and lung of Tgif1 KO mice showed normal appearance (Appendix Fig S1B). Thus, Tgif1 inactivation appeared to accelerate the metastatic behaviors of PDAC driven by KrasG12D.
Figure 2. Tgif1 inactivation promotes PDAC metastasis.
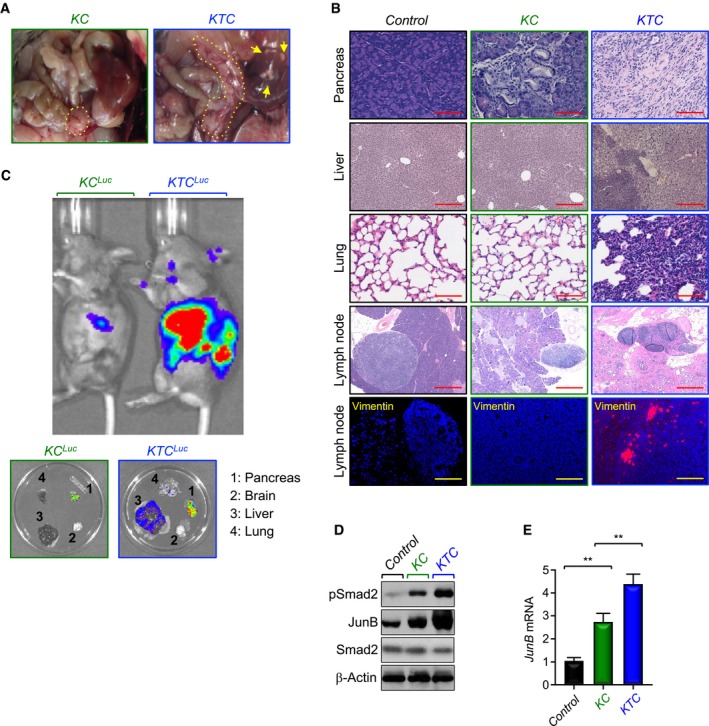
- Representative pictures of dissected abdominal region of KC and KTC mice. Yellow arrows indicate metastatic nodules in liver. Yellow dotted lines show the pancreas.
- FFPE sections of pancreas, liver, lungs, or lymph nodes from control, KC, or KTC mice were stained with H&E or immunostained with anti‐Vimentin antibody and revealed by IF. Representative pictures at 4× (Lymph node, H&E), 10× (Liver, H&E), and 20× (Pancreas, H&E; Lung, H&E; Lymph node, IF‐Vimentin) are shown (n = 18–30). Scale bars, 1,000 μM (Lymph node, H&E), 400 μM (Liver, H&E), and 200 μM (Pancreas, H&E; Lung, H&E; Lymph node, IF‐Vimentin).
- Tumor volume and metastasis in live KC Luc and KTC Luc mice were analyzed by IVIS bioluminescence. Following imaging live animals, pancreas, brain, liver, and lung were dissected and analyzed again by IVIS bioluminescence (n = 6, 5 out of 6 mice developed metastasis to liver and lung).
- Total cell lysates from pancreas of control, KC, or KTC mice were pooled and analyzed by immunoblotting using antibodies to pSmad2, Smad2, or JunB (n = 6).
- Expression of JunB in pancreas from control, KC, or KTC mice (same as in D) was analyzed by qRT–PCR (n = 6). Data are expressed as mean ± SEM. **P < 0.01; based on a two‐tailed Student's test.
Next, we conducted comparative experiments to assess TGF‐β signaling in PDAC from KTC and KC mice. As gauged by IHC, KTC mice displayed more pronounced pSmad2 abundance compared to KC mice irrespective of whether the lesions correspond to PanINs or PDAC (Appendix Fig S1C). Consistently, KTC mice also displayed increased staining of JunB (Appendix Fig S1C), a well‐characterized TGF‐β target gene (Sundqvist et al, 2018). Similar results were obtained when Smad2 phosphorylation and JunB expression were analyzed by immunoblotting and/or qRT–PCR (Fig 2D and E). Collectively, these data reveal that TGF‐β signaling is hyperactive in KTC mice during PDAC progression, therefore raising the possibility that activation of this pathway may predominately serve to accelerate the malignant conversion of PDAC rather than to fulfill a checkpoint mechanism to constrain tumor initiation.
TGIF1 represses Twist1 expression
Accumulating evidence supports a role of Twist1 in KrasG12D‐driven tumorigenesis and metastasis in a variety of organ systems, including pancreas (Hong et al, 2011; Lee & Bar‐Sagi, 2010). To gain mechanistic insight into how TGIF1 deficiency promotes PDAC development and progression, we sought to explore further an interaction between TGIF1 and Twist1 that we identified in a yeast two‐hybrid system screening (Ettahar et al, 2013). Using transfected HEK293T cells, we confirmed that TGIF1 displayed strong affinity for Twist1 in mammalian cells (Appendix Fig S2A). Endogenous TGIF1 and Twist1 also exhibit strong affinity for each other in the human PDAC cell line MIAPaCa‐2 (Fig 3A). We also detected an endogenous interaction between TGIF1 and Twist1 in pancreatic extracts from wild‐type mice, but not from Tgif1 KO mice, used as a negative control (Fig 3B). Of note, the TGIF1‐Twist1 interaction does not require binding of TGIF1 and/or Twist1 to chromatin, as treatment of samples with DNase I had little or no effect on the assembly of TGIF1‐Twist1 complex (Fig 3C). We also found that treatment of Panc‐1 cells with TGF‐β up to 8 h had little or no effect on the TGIF1‐Twist1 interaction despite eliciting strong activation of this pathway (Fig 3D and Appendix Fig S2C), suggesting that TGIF1 might interact with Twist1 in a manner independent of TGF‐β signaling.
Figure 3. TGIF1 associates with and inhibits Twist1 expression.
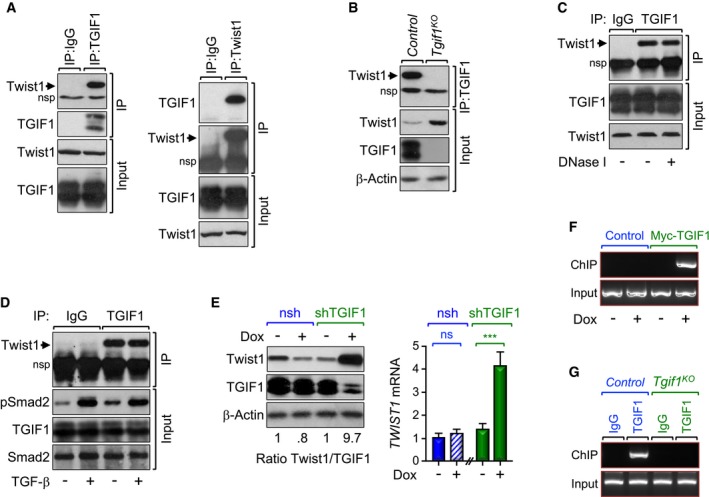
-
A–CProtein extracts from MIAPaCa‐2 cells (left) and control or Tgif1 KO mice (right) were immunoprecipitated using normal IgG, anti‐TGIF1, or anti‐Twist1 antibody and analyzed by immunoblotting using antibodies to Twist1 or TGIF1 (A–C). In (C), cell lysates were treated with DNase I before coimmunopreciptation. To control for loading, total cell lysates were analyzed by immunoblotting using antibodies to Twist1, TGIF1, or β‐Actin.
-
CPanc‐1 cells were treated with TGF‐β for 30 min, and protein extracts were immunoprecipitated with anti‐TGIF1 antibody and analyzed by immunoblotting using anti‐Twist1 antibody. To control for activation of TGF‐β signaling, total cell lysates were analyzed by immunoblotting using anti‐pSmad2 antibody.
-
DPL45 stably expressing Dox‐inducible control or shRNA targeting TGIF1 were treated with vehicle or Dox for 48 h and analyzed for Twist1 expression by immunoblotting or qRT–PCR (n = 6). In left panel, the intensity of the bands was assessed by densitometry, and the result was presented as a ratio of Twist1/TGIF1. In right panel, data are expressed as mean ± SEM. ***P < 0.001; ns, not significant; based on a two‐tailed Student's test.
-
EPL45 stably expressing Dox‐inducible TGIF1 were treated with vehicle or Dox for 48 h and analyzed for binding of TGIF1 to the Twist1 promoter by ChIP and agarose gel.
-
FPancreatic chromatin from control or Tgif1 KO mice was analyzed for binding of TGIF1 to the Twist1 promoter by ChIP and agarose gel.
Intriguingly, we noticed during the analysis of the TGIF1‐Twist1 interaction that Tgif1 inactivation was associated with increased abundance of the Twist1 protein (Fig 3B). Corroborating this finding, overexpression of TGIF1 in the human PDAC cell line PL45 stably expressing a doxycycline‐inducible TGIF1 decreased the abundance of endogenous Twist1 protein and mRNA (Appendix Fig S2C and D). Conversely, endogenous Twist1 protein and mRNA expression was markedly increased upon depletion of TGIF1 in PL45 expressing a Dox‐inducible TGIF1 shRNA (Fig 3E), hinting at the possibility that TGIF1 might repress Twist1 gene expression.
Several lines of evidence support a model in which TGIF1 functions as a direct transcriptional repressor of Twist1. First, overexpression of TGIF1 in MIAPaCa‐2 cells elicited a marked decrease in the activity of a luciferase reporter driven by the Twist1 promoter (TGTLuc; Appendix Fig S2E). Second, overexpression of a TGIF1 mutant (TGIF1∆), which is defective in its ability to recruit the general transcription corepressor HDAC1 (Wotton et al, 1999), had little or no effect on TGTLuc activity (Appendix Fig S2E). Third, the Twist1 promoter contains a conserved canonical TGIF1 binding site (TGCTGCTGTCAC; Bertolino et al, 1995), whose mutation rendered the TGTLuc reporter insensitive to TGIF1 (Appendix Fig S2E). Fourth, an oligonucleotide pull‐down assay using purified HA‐TGIF1 confirmed the ability of TGIF1 to bind to the Twist1 promoter (Appendix Fig S2F). Substitution of TG for AA in the oligonucleotide blunted this binding (Appendix Fig S2F), attesting to the specificity of this protein–DNA interaction. Fifth, chromatin immunoprecipitation (ChIP) assays using PL45‐Dox‐TGIF1 cells showed a robust binding of TGIF1 to the TWIST1 promoter (Fig 3F and Appendix Fig S2G). Finally, we detected strong binding of endogenous TGIF1 to the Twist1 promoter in wild‐type mice, but not in Tgif1 KO mice, used as a negative control (Fig 3G and Appendix Fig S2H).
TGIF1 antagonizes Twist1 transcriptional activity
Next, using PL45‐Dox‐TGIF1 cells expressing a Tam‐inducible variant of Twist1 (Twist1ER; Mani et al, 2008), we found that Twist1 was able to induce its own expression, and this effect was almost completely blocked by TGIF1 expression (Fig 4A). We noted that Tam stimulation increased Twist1ER expression, which could probably be due to increased stability of Twist1ER following its translocation to the nucleus. Although the exact underlying mechanism is presently unknown, the observation that Twist1 induces its own expression together with the previous biochemical data showing a physical interaction between TGIF1 and Twist1 prompted us to investigate whether TGIF1 could interact with Twist1 on the Twist1 promoter and represses Twist1 auto‐transcriptional activity. An in silico analysis identified a Twist1‐binding sequence (E‐box) located close to the TGIF1‐binding sequence in the Twist1 promoter. This Twist1‐binding sequence is functional, as overexpression of Twist1 activated the wild‐type TGTLuc reporter, but had no effect on a mutant lacking the Twist1 consensus sequence (Fig 4B). Interestingly, overexpression of TGIF1 blocked Twist1‐induced expression from its own promoter (Fig 4B), highlighting an ability of TGIF1 to suppress Twist1 auto‐transcriptional activity. To investigate whether TGIF1 and Twist1 could bind independently or cooperatively to the Twist1 promoter, we performed ChIP assays using chromatin from mice with pancreas‐specific deletion of Tgif1 (Tgif1 KO) or Twist1 (Twist1 KO). We found that deleting Tgif1 did not influence binding of Twist1 to the Twist1 promoter, and vice versa (Appendix Fig S3A and B). Collectively, these findings provide strong evidence that TGIF1 binds to and represses Twist1 auto‐transcriptional activity.
Figure 4. TGIF1 inhibits Twist1 transcriptional activity.
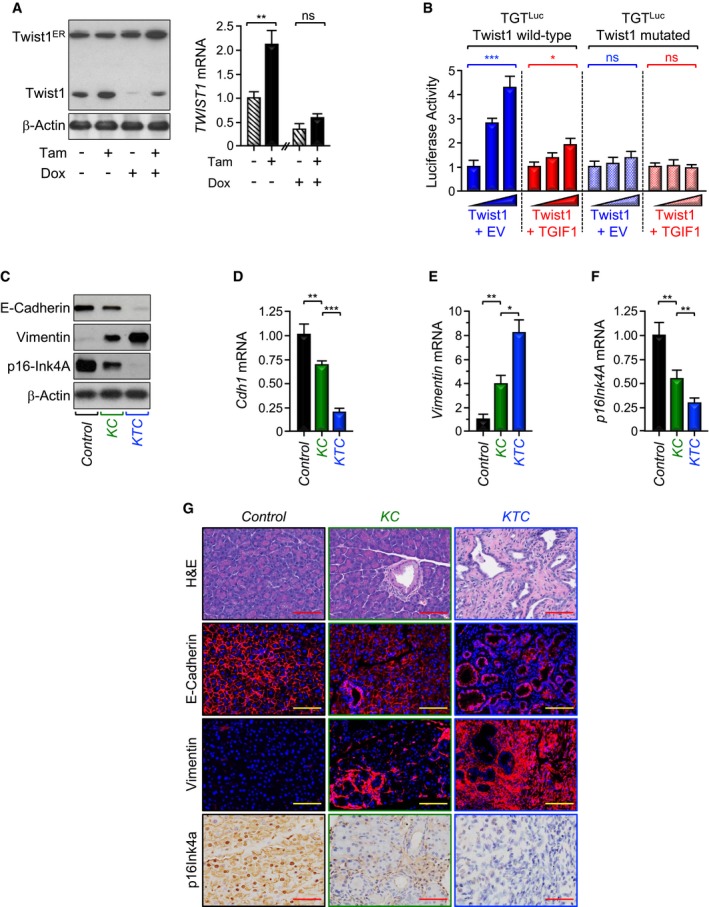
-
APL45 stably expressing both Dox‐inducible TGIF1 and Tam‐inducible Twist1 (Twist1ER) were treated with Tam and/or Dox for 48 h and analyzed for Twist1 expression by immunoblotting or qRT–PCR (n = 6).
-
BMIAPaCa‐2 cells were transfected with wild‐type or mutated (Twist1 binding site) Twist1 luciferase reporters (TGTLuc) together with increasing amounts of Twist1 expression vector in the presence of empty vector (EV) or TGIF1 expression vector. Luciferase activity was measured 48 h following transfection and normalized on the basis of co‐transfected Renilla luciferase (n = 6).
-
CTotal lysates from pancreas of control, KC, or KTC mice were pooled and analyzed by immunoblotting using antibodies to E‐Cadherin, Vimentin, p16Ink4A, and β‐Actin (n = 6).
-
D–FExpression of Cdh1, Vimentin, and p16Ink4A in pancreas from control, KC, or KTC mice (same as in C) was analyzed by qRT–PCR (n = 6).
-
GFFPE pancreatic sections from control, KC, or KTC mice were stained with H&E or immunostained with antibodies to E‐Cadherin, Vimentin, or p16Ink4a and revealed by IF or IHC. Representative pictures at 20× are shown (n = 30). Scale bars, 200 μM.
TGIF1 represses PDAC formation and progression by antagonizing Twist1
It is well established that Twist1 promotes cell invasion and metastasis by orchestrating the epithelial‐to‐mesenchymal transdifferentiation (EMT), which is associated with loss of the epithelial marker E‐Cadherin and gain of the mesenchymal marker Vimentin (Mani et al, 2008; Valastyan & Weinberg, 2011; Yang et al, 2004). Twist1 directly represses expression of the E‐Cadherin gene (Cdh1), thereby leading to disassembly of cell–cell junction and attendant cell migration. To provide further evidence that TGIF1 antagonizes Twist1 activity, we investigated whether TGIF1 could counteract Twist1‐mediated repression of E‐Cadherin. Using PL45‐dox‐TGIF1 cells stably expressing Twist1ER, we found that inducing Twist1 activity resulted in a significant decrease in E‐Cadherin protein expression, and this effect was blunted by inducing TGIF1 expression (Appendix Fig S3C). A similar conclusion could be drawn when CDH1 (gene encoding E‐Cadherin) mRNA expression was examined by qRT–PCR (Appendix Fig S3D). We also detected decreased expression of both E‐Cadherin protein and mRNA in KTC mice as compared to KC mice (Fig 4C and D). To substantiate these findings, we compared the extent of EMT between KTC and KC mice. Relative to KC mice, E‐Cadherin expression was decreased in KTC mice, concurring with increased expression of Vimentin (Fig 4C–E and G), indicative of Twist1 activation and the resulting EMT. These findings suggest that TGIF1 might function to oppose Twist1‐induced EMT, perhaps thereby inhibiting PDAC metastasis.
Besides its role in mediating EMT and cell invasion, Twist1 is deemed to contribute directly to malignant transformation owing to its ability to influence major tumor suppressor and oncogenic signaling pathways that are deregulated in most human cancers (Lee & Bar‐Sagi, 2010; Maestro et al, 1999; Piccinin et al, 2012; Shiota et al, 2008; Vichalkovski et al, 2010). In the context of PDAC, Twist1 has been shown to directly repress expression of p16Ink4A, thereby allowing KrasG12D to bypass cell senescence and thus to initiate tumorigenesis (Lee & Bar‐Sagi, 2010). To further substantiate the relationship between TGIF1 and Twist1, we extended our experiments to analyze expression of p16Ink4A. Similar to E‐Cadherin, activation of Twist1ER by Tamoxifen in PL45‐dox‐TGIF1‐Twist1ER cells reduced p16Ink4A expression, and this inhibitory effect was relieved upon inducing TGIF1 expression (Appendix Fig S3C and E). In vivo, KC mice displayed decreased expression of p16Ink4a when compared to control mice (Fig 4C, F, and G), which is consistent with previous studies (Lee & Bar‐Sagi, 2010). More importantly, relative to KC mice, KTC mice displayed a further decrease in p16Ink4A expression (Fig 4C, F, and G), lending further support to the hypothesis that TGIF1 might inhibit Twist1 oncogenic activity.
To investigate whether Twist1 could contribute to the PDAC phenotype driven by the combined Tgif1 deletion and KrasG12D expression, we generated mice with pancreas‐specific Twist1 deletion in the KTC background (referred to hereafter as KTWC mice). KTWC mice were born with Mendelian frequencies, and no phenotypic differences between KTWC mice and KTC or wild‐type littermates were observed. Remarkably, the vast majority of KTWC mice remained healthy and free of tumors, and none of them developed metastatic lesions into the liver during the entire observation period of 25 weeks (Fig 5A and B). In comparison, all KTC mice succumbed to invasive PDAC by 19 weeks, 63% of which had liver metastasis (Fig 5B). During the course of this study, we also generated KC mice deleted of Twist1 (KWC) and found that Twist1 inactivation completely blocked the PDAC phenotype driven by KrasG12D alone (Appendix Fig S4A–C). Noteworthy, Twist1 deletion prevented the decrease in expression of Cdh1 and p16Ink4A mRNA as well as the increase in expression of Vimentin mRNA in KTC mice (Fig 5C–E), and these were further reflected in their protein levels (Appendix Fig S4D), providing further evidence that Twist1 deletion can block both PDAC formation and progression in KTC mice. Collectively, these genetic experiments provide strong evidence that Twist1 plays an important role in PDAC progression driven by KrasG12D alone or in combination with Tgif1 inactivation, which is consistent with our mechanistic model in which TGIF1 functions to antagonize Twist1.
Figure 5. Twist1 inactivation blocks PDAC formation and metastasis in KTC mice.
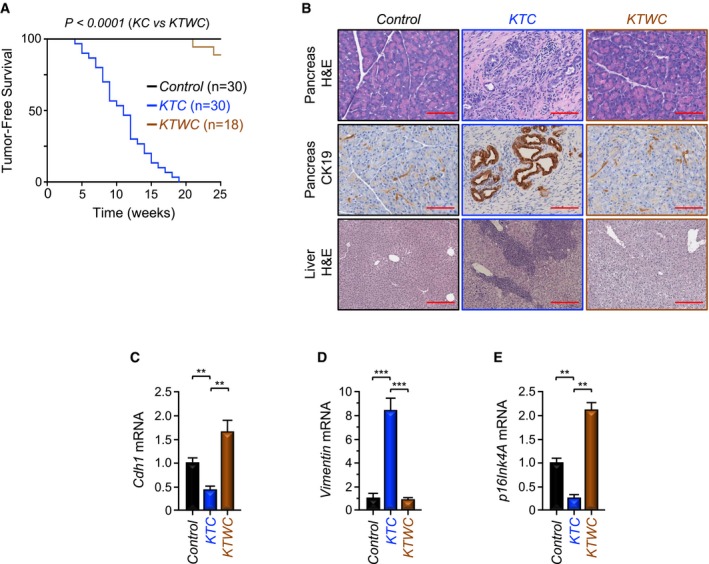
-
AKaplan–Meier survival analysis of control, KTC, and KTWC mice.
-
BFFPE sections of pancreas or liver from control, KTC, or KTWC mice were stained with H&E or immunostained with anti‐CK19 antibody and revealed by IHC. Representative pictures at 10× (Liver, H&E) or 20× (Pancreas, H&E; Pancreas, IHC‐CK19) are shown (n = 30). Scale bars, 400 μM (Liver, H&E) and 200 μM (Pancreas, H&E; Pancreas, IHC‐CK19).
-
C–EExpression of Cdh1, Vimentin, and p16Ink4A in pancreas from control, KTC, or KTWC mice was analyzed by qRT–PCR (n = 6). Data are expressed as mean ± SEM. **P < 0.01; ***P < 0.001; based on a two‐tailed Student's test.
TGIF1 phosphorylation in human PDAC disrupts its tumor suppressor function
To attest to the clinical relevance of our findings, we took advantage of previous studies from our and other laboratories reporting that activation of Ras/MAPK signaling can trigger TGIF1 phosphorylation at Thr235/Thr239 (Ferrand et al, 2007; Lo et al, 2001), though the impact of this phosphorylation remained unexplored. Noteworthy, TGIF1 migrates as a double band in SDS–PAGE, with the slower migrating band corresponding to the phosphorylated form (see Figs 3, 6 and 7, and EV1, Appendix Figs S2 and S5). To explore more deeply the impact of TGIF1 phosphorylation, we generated an antibody that specifically recognizes TGIF1 phosphorylated at Thr235/Thr239 (T235/239), as gauged by its inability to recognize a TGIF1 mutant in which Thr235/Thr239 were replaced by Ala (TGIF1.2TA) in immunoblotting assays using extracts from reconstituted Tgif1 −/− MEFs (Fig 6A). In an alternative control experiment, we found that TGIF1 was less phosphorylated in BxPC3 than MiaPaCa‐2 cells, which harbor wild‐type and oncogenic KRAS, respectively (Appendix Fig S5A). Using this antibody, we performed IHC experiments to assess phosphorylation of TGIF1 in two human PDAC tissue microarrays (TMAs) containing 80 and 103 samples, respectively. We detected a marked increase in TGIF1 phosphorylation in more than 90% of PDAC samples, and this increase was observed at any stage of tumorigenesis examined, although its extent tends to increase with PDAC progression (Fig 6B and C, and Appendix Fig S5B), which is in agreement with the literature that most of human PDAC tumors harbor constitutively active Kras signaling (Almoguera et al, 1988; Hezel et al, 2006). Interestingly, high levels of TGIF1 phosphorylation were associated with high levels of Twist1 expression, and vice versa (Fig 6B), further attesting to the relationship between TGIF1 and Twist1.
Figure 6. Increased TGIF1 phosphorylation in human PDAC .
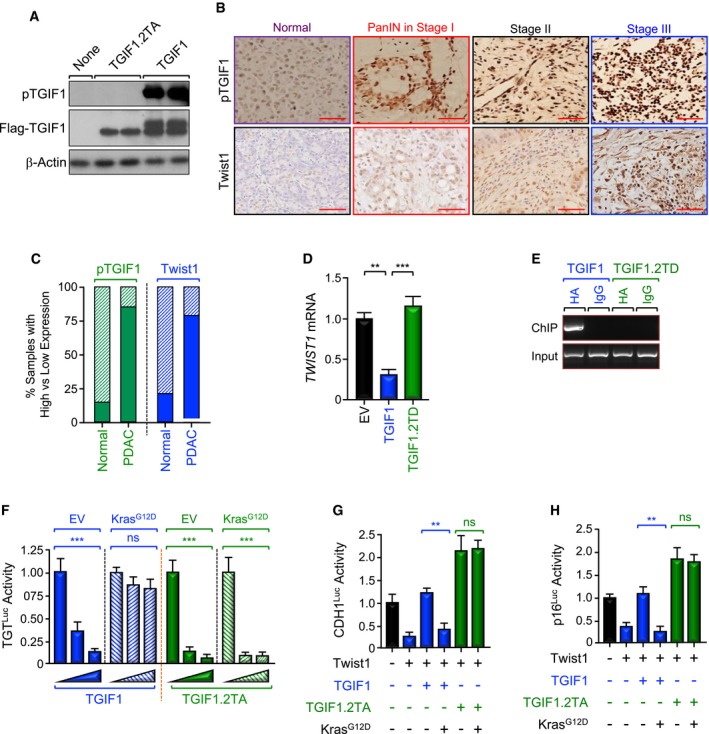
-
ASpecificity of anti‐phospho‐TGIF1 (pTGIF1) antibody. Cell extracts from Tgif1 −/− MEFs cells reconstituted with empty vector (EV), wild‐type (TGIF1) or non‐phosphorylated form (TGIF1.2TA) of Flag‐TGIF1 were immunoblotted with anti‐phospho‐TGIF1 or β‐Actin as a loading control.
-
B, CExpression of pTGIF1 and Twist1 in human tissue microarrays of human PDAC samples was analyzed by immunohistochemistry. Representative pictures at different stages (40×) are shown. The percentages of samples with high versus low expression of pTGIF1 and Twist1 in normal tissues versus PDAC tissues are shown. Scale bars, 100 μM.
-
D, EMIAPaCa‐2 cells were transfected with TGIF1 or TGIF1.2TD expression vectors and selected with G418, and resistance colonies were pooled (n = 6). Expression of Twist1 was examined by qRT–PCR (D). Binding of TGIF1 to the Twist1 promoter was examined by ChIP and agarose gel.
-
FBxPC3 cells were transfected with TGTLuc together with TGIF1 or TGIF1.2TA in the presence or absence of KrasG12D. Luciferase activity was measured 48 h following transfection and normalized on the basis of co‐transfected Renilla luciferase (n = 6).
-
G, HBxPC3 cells were transfected with CDH1Luc or p16Luc reporter together with TGIF1 or TGIF1.2TA in the presence or absence of KrasG12D, and luciferase activity was measured 48 h following transfection as described in (E) (n = 6).
Figure 7. TGIF1 phosphorylation in PDAC disrupts its anti‐tumor activity.
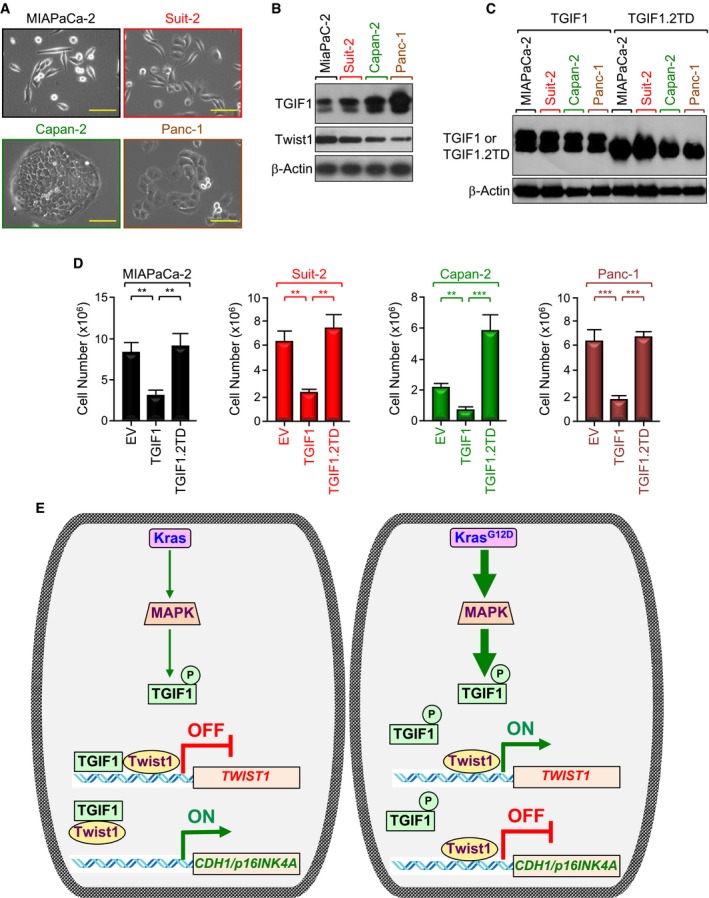
-
ARepresentative microscope pictures (20×) of MIAPaCa‐2, Suit‐2, Capan‐2, and Panc‐1 cells. Scale bars, 200 μM.
-
BExpression of TGIF1 and Twist1 in MIAPaCa‐2, Suit‐2, Capan‐2, and Panc‐1 cells was analyzed by immunoblotting.
-
C, DMIAPaCa‐2, Suit‐2, Capan‐2, and Panc‐1 cells were transfected with empty vector (EV), wild‐type (TGIF1), or phosphorylation‐mimic mutant (TGIF1.2TD) of TGIF1. Cell extracts were analyzed 48 h following transfection by immunoblotting using anti‐Flag antibody (C). Cells were selected with neomycin for 2–3 weeks and counted using an automatic cell counter (D) (n = 6). Data are expressed as mean ± SEM. **P < 0.01; ***P < 0.001; based on a two‐tailed Student's test.
-
EModel of suppression of TGIF1 function by KrasG12D/MPK‐mediated phosphorylation. In this model, acquisition of KrasG12D leads to constitutive activation of MAPK/ERK, which in turn phosphorylates TGIF1. Phosphorylation of TGIF1 disrupts its ability to inhibit Twist1 expression, thereby leading to suppression of CDH1 and p16INK4A expression and attendant PDAC formation and progression.
In light of this clinically relevant finding, we sought to investigate whether constitutive phosphorylation of TGIF1 could affect its ability to suppress PDAC. To probe this possibility, we first conducted experiments to analyze the effects of TGIF1 phosphorylation on the interaction of TGIF1 with Twist1 as well as on the expression of Twist1. Using a mammalian two‐hybrid approach, we observed a significant decrease in the TGIF1‐Twist1 interaction upon expression of KrasG12D in BxPC3 cells (Appendix Fig S5C). Under these experimental conditions, TGIF1.2TA (phosphorylation‐defective mutant) exhibited more affinity for Twist1, and its interaction with Twist1 was not affected by KrasG12D (Appendix Fig S5C). These results provide further evidence that TGIF1 and Twist1 form a physical complex, whose level appears to decline in the presence of oncogenic KRAS.
Next, we found that overexpressing a double mutant that mimics phosphorylation at T235/239 (TGIF1.2TD) completely blocked TGIF1‐induced repression of the TGTLuc reporter in MIAPaCa‐2 cells (Appendix Fig S5D). Congruently, TGIF1.2TD was neither able to bind to the Twist1 promoter, nor able to inhibit Twist1 expression in MIAPaCa‐2 cells (Fig 6D and E, and Appendix Fig S5E). In an alternative experimental approach, expressing KrasG12D dampened the ability of wild‐type TGIF1 to repress Twist1 transcription activity, but failed to do so in the presence of the phosphorylation‐defective mutant TGIF1.2TA (Fig 6F). A similar conclusion could be drawn when the expression of Twist1 target genes (e.g., CDH1, p16INK4A) was examined (Fig 6G and H). To substantiate the findings that TGIF1 phosphorylation disrupts its ability to repress Twist1 transcription, we extended our experiments to examine whether TGIF1 phosphorylation could also disrupt its anti‐tumor activity. To this end, we used four different human PDAC cell lines harboring constitutive active mutant KRAS (i.e., MIAPaCa‐2, KRASG12C; Suit‐2, KRASG12D; Capan‐2, KRASG12V; Panc‐1, KRASG12D). As shown in Fig 7A, Suit‐2 and MIAPaCa‐2 cells display a more mesenchymal phenotype, whereas Capan‐2 and Panc‐1 cells display a more epithelial phenotype. We also found that cells with high TGIF1 expression displayed low Twist1 expression, and vice versa (Fig 7B), making these cell lines suitable for our study. Remarkably, we found that although transfecting wild‐type TGIF1 had little or no toxicity 48 h after transfection, it almost completely blocked proliferation of all four cell lines after 2 to 3 weeks, depending on the cell type (Fig 7C and D, and Appendix Fig S6). Under these experimental conditions, overexpression of phosphorylation‐mimic mutant TGIF1.2TD was void of any significant effect on MIAPaCa‐2, Suit‐2, and Panc‐1 cell growth. Intriguingly, expression of TGIF1.2TD stimulated proliferation of Capan‐2 cells (Fig 7D), suggesting that TGIF1 phosphorylation might switch TGIF1 from an inhibitor to an activator of cell proliferation in this particular cell line. Of note, TGIF1.2TD was expressed at a level similar to that observed for wild‐type TGIF1 (Fig 7C). When taken together with our genetic experiments using KTC mice, these findings provide strong evidence that TGIF1 acts to restrain oncogenic Kras‐driven PDAC and further suggest that this TGIF1 tumor‐suppressive function might be inactivated by phosphorylation in human PDAC harboring constitutive Kras signaling.
Discussion
TGIF1 belongs to the superfamily of TALE homeodomain proteins, which control an array of important cellular processes, such as specification of developmental fate, proliferation, apoptosis, and differentiation (Burglin & Affolter, 2016; Wotton & Massague, 2001). Although mice with global Tgif1 deletion develop normally and are fertile, they show a large spectrum of subtle abnormalities, which were mainly attributed to de‐repression of TGF‐β/Smad signaling (Bartholin et al, 2006; Hneino et al, 2012; Mar & Hoodless, 2006; Taniguchi et al, 2012). This observation provided us with a unique opportunity to further interrogate the role of TGF‐β/Smad signaling during PDAC pathogenesis and progression under gain‐of‐function conditions, since most of studies available to date relied exclusively on loss‐of‐function approaches targeting either TβRII or Smad4 (Bardeesy et al, 2006b; Ijichi et al, 2006; Whittle et al, 2015). In this study, we show that pancreas‐specific deletion of Tgif1 had no discernible impact on pancreatic development or physiology despite hyperactivation of TGF‐β signaling. Quite intriguingly, Tgif1 inactivation in the context of oncogenic KrasG12D resulted in a dramatic acceleration of PDAC development and progression. From a mechanistic perspective, we demonstrate that TGIF1 functions as a direct transcriptional repressor of Twist1 (see model in Fig 7E). We went on to obtain genetic evidence demonstrating a requirement of Twist1 for the acquisition of the PDAC metastatic phenotype driven by the combined KrasG12D expression and Tgif1 inactivation. These results have several ramifications for our understanding of the molecular mechanisms underlying PDAC pathogenesis and progression, which could ultimately leverage current efforts to curb this lethal malignancy.
TGF‐β/Smad is well known to suppress normal epithelial growth, thereby functioning as an early tumor suppressor. This function has been extensively studied, as has TGF‐β's late‐stage role in metastasis (Massague, 2008). However, in the absence of genetic tools to explore intermediate stages of tumor progression, the prior speculation as to TGF‐β dual function during PDAC progression had been that loss of the TGF‐β cytostatic program might cause cells to escape from appropriate growth regulation, which would result in cell transformation. At later stages, other TGF‐β responses then purportedly prevail that are unrelated to TGF‐β cytostatic effects but which favor PDAC invasion and metastasis (Bardeesy et al, 2006b; David et al, 2016; Ijichi et al, 2006; Whittle et al, 2015). In further support to this notion, TGF‐β is highly expressed in human PDAC tumors, a feature correlating with poor prognosis (Bardeesy et al, 2006b; Friess et al, 1993; Wagner et al, 1999). Paradoxically, genetic deletion of either Smad4 or TβRII in a wild‐type background revealed no obvious effects on normal pancreatic development, demonstrating that inactivation of TGF‐β signaling per se is not sufficient to induce cell transformation (Bardeesy et al, 2006b; Ijichi et al, 2006). However, Smad4 or TβRII inactivation led to rapid progression of pancreatic tumors in the context of KrasG12D, though the tumors retained epithelial differentiation and manifest an attenuated metastatic potential, in line with the hypothesis that TGF‐β signaling might enhance malignant conversion and promote metastasis (Bardeesy et al, 2006b; Ijichi et al, 2006). So far, definitive evidence on whether TGF‐β signaling indeed promotes PDAC is still lacking. Remarkably, we found in this study that Tgif1 ablation in the pancreatic epithelium culminated in constitutive activation of TGF‐β signaling but failed to oppose KrasG12D‐driven PDAC, suggesting that activation of this signaling during early development (E8.5, stage in which Pdx1‐Cre is expressed) might not be sufficient to suppress PDAC tumor formation. Although the mechanistic basis for this unanticipated finding remains to be established experimentally, one would speculate that inactivation of Tgif1 might lead to activation of a bona fide oncogene(s) that enable KrasG12D to bypass the TGF‐β tumor‐suppressive barrier during PDAC development. Alternatively, Tgif1 inactivation might result in de‐repression of signaling pathways activated by other TGF‐β superfamily members, such as Activins and BMPs, which are known to signal through Smad proteins to enhance malignancy and promote cancer metastasis in a variety of human malignancies (Attisano & Wrana, 2000; Feng & Derynck, 2005; Massague, 2008; Massague et al, 2005). Under this latter scenario, sustained activation of Activin or BMP signaling should overcome the tumor‐suppressive function of TGF‐β signaling while eliciting at the same pro‐tumorigenic and pro‐metastatic effects. Finally, it is also conceivable that Tgif1 inactivation might affect other signaling pathways (i.e., Wnt, TNF‐α) that together with TGF‐β ensure a dynamic balance between pancreatic cell growth and differentiation to maintain homeostatic pancreatic physiology even in the presence of oncogenic insults (Demange et al, 2009; Zhang et al, 2015). As such, a thorough exploration of such potential mechanisms may provide valuable insights into the existence of tumor suppressor or oncogenic pathways that either enable or oppose TGF‐β bimodal functions during the course of PDAC pathogenesis and progression. Moreover, in light of the general notion that the TGF‐β bimodal function operates in a non‐tissue‐specific manner, it will be interesting to determine whether the molecular mechanisms we've unearthed here will extend to other human malignancies.
In attempts to delineate the mechanisms by which Tgif1 inactivation accelerates KrasG12D‐induced PDAC progression, we found that TGIF1 associates with and represses Twist1 transcriptional activity, thereby promoting expression of E‐Cadherin, which is known to suppress cell invasion and metastasis owing to its ability to maintain epithelium structure and integrity (Mani et al, 2008; Valastyan & Weinberg, 2011; Yang et al, 2004). The ability of TGIF1 to antagonize Twist1 transcriptional activity also promotes expression of the tumor suppressor gene p16Ink4A, which is inactivated in more than 90% of human PDAC (Hezel et al, 2006; Iacobuzio‐Donahue, 2012). Subsequent genetic experiments showed that Twist1 deletion was sufficient to reverse the aggressive PDAC phenotype driven by the simultaneous Tgif1 inactivation and KrasG12D expression. Although this model clearly underscored the relevance of TGIF1‐dependent suppression of Twist1 expression and activity to PDAC formation and progression, an important question remains as to whether this molecular route would unleash the TGF‐β‐driven tumor invasiveness and metastasis, and, if so, how it acts mechanistically. At present, we found that activation of TGF‐β signaling had little or no effect on the physical interaction between TGIF1 and Twist1, raising the possibility that Tgif1 inactivation might lead to activation of Twist1 independently of TGF‐β signaling. Based on our in vivo findings, one would speculate that Tgif1 inactivation might impact at least two distinct networks (i.e., TGF‐β, Twist1) that perhaps converge together to deepen PDAC proliferative and metastatic behaviors. While we cannot exclude the possibility that other pathways also contribute to the metastatic PDAC phenotype in KTC mice, our study provides tantalizing insights into mechanistic paradigms of PDAC pathogenesis and progression and thus offers a framework for further studies seeking alternative therapeutic options for this incurable malignancy. For instance, it would be highly informative to apply other integrative approaches, such as RNA sequencing, ChIP‐sequencing, proteomic, and lipodomic, to identify other genes and pathways that cooperate with Tgif1 inactivation to bolster the PDAC phenotype initiated by KrasG12D.
There is compelling evidence that Twist1 plays multiple roles during carcinogenesis, initially facilitating malignant transformation through suppression of key tumor suppressor pathways, and subsequently executing key aspects of cell movement, extravasion, and metastasis following tumor formation (Lee & Bar‐Sagi, 2010; Maestro et al, 1999; Piccinin et al, 2012; Shiota et al, 2008; Vichalkovski et al, 2010). Further bolstering those oncogenic functions, increased Twist1 expression correlates with increased risk of metastasis and poor prognosis in a number of malignancies, including PDAC (Lee & Bar‐Sagi, 2010; Valastyan & Weinberg, 2011). Of particular relevance, a landmark study from the Bar‐Sagi group has suggested that Twist1 functions to promote KrasG12D‐driven PDAC progression by repressing p16INK4A (Lee & Bar‐Sagi, 2010). Our present study reinforces and extends this finding and provides another layer of the complexity of PDAC by demonstrating that TGIF1 functions upstream of Twist1 to regulate p16INKA expression. Interestingly, Twist1 ablation resulted in complete abrogation of PDAC development and metastasis in KTC mice, providing the proof‐of‐principle experiments that Twist1 might not only function to execute cell invasion and metastasis once the PDAC tumor has developed, but also facilitate the initial malignant transformation following oncogenic Kras acquisition. Although these findings seem to confirm earlier observations from the Bar‐Sagi group, another recent study has revealed that Twist1 deletion was ineffective at suppressing PDAC driven by the combined expression of Kras G12D and gain‐of‐function mutant p53, and, therefore, questioning the contribution of Twist1 to PDAC development (Zhang et al, 2015). In light of our present study, this discrepancy could be attributed to the genetic alterations in question, as TGIF1 seems to act mainly through repression of Twist1, whereas mutant p53 likely elicits its malignant effects through very complex mechanisms, often involving multiple genes and signaling pathways. It should be noted that our recent published studies have shown that pancreas‐specific deletion of Twist1 elicited either no effect or complete suppression of PDAC, depending on the genetic background (i.e., p16Ink4A deletion versus Trp53 deletion; Parajuli et al, 2018). These observations appear to reconcile all previously incongruent data regarding Twist1 in the context of PDAC and further shed important insights into the complex roles of this pro‐malignant transcription factor in PDAC, and perhaps in other human malignancies.
Perhaps the most impressive finding in this study is the massive increase in TGIF1 phosphorylation in human PDAC, which occurs in more than 90% of samples irrespective of the stages of cancer examined. Interestingly, pharmacological inhibition of MEK/ERK signaling has been shown to suppress PDAC formation in KC mice, providing direct evidence that constitutive Kras/MEK/ERK plays a key role in PDAC pathogenesis (Collins et al, 2014). Since MAPK/ERK phosphorylates TGIF1 (Ferrand et al, 2007; Lo et al, 2001), the high occurrence of TGIF1 phosphorylation fits well with the general notion that the vast majority of human PDAC tumors harbor constitutively active Kras signaling (Almoguera et al, 1988; Hezel et al, 2006). Subsequent functional studies showed that TGIF1 phosphorylation completely disrupted its ability to repress Twist1 expression. Likewise, TGIF1 phosphorylation also disrupted its ability to inhibit proliferation of human PDAC cell lines, raising the provocative possibility that TGIF1 tumor‐suppressive function might be inactivated by phosphorylation in human PDAC displaying constitutive Kras signaling. Quite intriguingly, we found that expression of the phospho‐mimic mutant TGIF1.2TD cells stimulated proliferation of Capan‐2 cells, providing an initial hint that phosphorylation might switch TGIF1 from an inhibitor to a promoter of PDAC cell growth at least under specific circumstances. It would be appealing in future studies to examine whether TGIF1 phosphorylation also contributes to its oncogenic functions in breast cancer and acute promyelocytic leukemia described in our previous studies (Prunier et al, 2015; Zhang et al, 2015). Likewise, since TGF‐β can also activate the MAPK/ERK signaling pathway, it would also be appealing to determine whether increased abundance of TGF‐β at late stages of PDAC could cooperate with KrasG12D to inactivate TGIF1 tumor suppressor function. As such, our identification of TGIF1 as a potential tumor suppressor in PDAC that connects oncogenic Kras to Twist1 provides an unprecedented platform for future identification of potential targets amenable to therapeutic intervention in PDAC, and possibly in other malignancies with frequent oncogenic Kras alterations, such as colorectal and lung cancers (Lau & Haigis, 2009).
Materials and Methods
Plasmids
pCMV5‐2xHA‐TGIF1, pCMV5‐2xHA‐TGIF1∆148‐177, pBICEP‐CMV2‐3xFlag‐TGIF1, pMT‐Gal4‐TGIF1, pcDNA5‐TO‐6xMyc‐TGIF1, pTRIPZ‐sh.TGIF1, pBABE‐puro‐mTwist1, pcDNA6‐TR, pMT (Gal4), pVP (VP16), pBICEP‐CMV2‐3xFlag and pG5E1b‐Luc were previously described (Faresse et al, 2008; Pessah et al, 2001; Prunier et al, 2015; Seo et al, 2006; Zhang et al, 2015). pWZL‐Blast‐Twist1ER and pBABE‐puro‐mTwist1 were obtained from Dr. Robert Weinberg (Addgene #18799 and #1783, respectively). The Twist1 luciferase reporter (TGTLuc) was kindly provided by Dr. Lu‐Hai Wang (Cheng et al, 2008). The CDH1Luc reported was kindly provided by Dr. Jing Yang (Yang et al, 2004).
Introduction of mutations into the Twist1 binding site (CAGCTG) or the TGIF1 binding site (TGCTGTCACA) in the TGTLuc reporter was performed by PCR using the QuickChange Site‐Directed Mutagenesis kit according to the manufacturer's instructions (Stratagene). The TGIF1 phospho‐mimic and phospho‐defective mutations were introduced into the pBICEP‐CMV2‐3xFlag‐TGIF1 or pMT‐Gal4‐TGIF1 using QuickChange Site‐Directed Mutagenesis.
To generate the expression vector encoding Flag‐Twist1, Twist1 cDNA was obtained by PCR using pBABE‐puro‐mTwist1 as template and cloned into pBICEP‐CMV2‐3xFlag. To generate the p16Luc reporter, genomic DNA (1,200 bp) upstream of the translation start site was amplified by the Genomic‐GC PCR amplification kit (BD Biosciences) using genomic DNA from HMLE cells as previously described (Parajuli et al, 2018). All cloned DNA fragments and their corresponding mutants were checked by sequencing. The sequence of all primers used for cloning is available upon request.
To produce lentiviruses encoding shRNAs in pTRIPZ (doxycycline‐inducible), the expression vector was transfected into HEK293T cells along with the packaging mix and lentiviruses were purified following the manufacturer's guidelines (Thermo‐scientific).
Antibodies
Anti‐Flag M2 and anti‐α‐Smooth Muscle Actin (Sigma‐Aldrich); anti‐HA and anti‐c‐Myc 9E10 (Roche); anti‐TGIF1, anti‐Twist1, and α‐Amylase (Santa Cruz Biotechnology); anti‐phophso‐Smad2‐S465/S467, anti‐Smad2, anti‐BrdU, anti‐E‐Cadherin, anti‐Glucagon, anti‐Insulin, anti‐JunB, and anti‐Vimentin (Cell Signaling Technology); and anti‐TGIF1, anti‐Twist1, anti‐CDK2A/p16INK4A, anti‐Chromogranin, and anti‐Cytokeratin 19 (Abcam). Polyclonal anti‐phospho‐TGIF1 antibody was generated using a phosphopeptide encompassing Thr235 and Thr239 (performed by Primm Biotech). Goat anti‐rabbit or goat anti‐mouse secondary antibodies conjugated to horseradish peroxidase were purchased from Cell signaling. The specificity of antibodies against TGIF1 and Twist1 was confirmed using extracts of pancreatic tissue deleted of Tgif1 or Twist1. All other antibodies were extensively characterized in other studies.
Cell lines and culture
All cell lines used in this study were obtained from American Type Culture Collection. They were cultured in Dulbecco's modified Eagle's medium (DMEM) with or without phenol red and supplemented with 10% fetal calf serum (FCS) and antibiotics. MIAPaCa‐2, Suit‐2, Capan‐2, and Panc‐1 cells were chosen to study the role of TGIF1 phosphorylation in PDAC because they harbor oncogenic KRAS, which activates MAPK/ERK that in turn phosphorylates TGIF1. BxPC3 cells were also chosen to study the role of TGIF1 phosphorylation in the absence or presence of KarsG12D because they harbor the wild‐type KRAS gene.
Generation of stable cell lines
To generate PL45‐Dox‐Myc‐TGIF1, cells were first transfected with pcDNA6‐TR (Invitrogen) and selected with blasticidin. Then, cells expressing the Tet‐transactivator were transfected with pcDNA5‐TO‐6xMyc‐TGIF1 and selected with hygromycin, and colonies expressing the transgenes (20 colonies) were identified by Western blotting and pooled. For inducible depletion of TGIF1, cells were infected with pTRIPZ‐sh.TGIF1 and selected with puromycin. Then, resistant colonies were pooled and expanded as single populations. For Twist1ER expression, PL45‐Dox‐Myc‐TGIF1 were transfected with pWZL‐Blast‐Twist1ER together with the empty vector pBabe‐puro and selected with puromycin and resistant positive colonies (identified by immunoblotting using anti‐Twist1 antibody) were pooled and expanded as single populations.
Yeast two‐hybrid screen
To identify novel TGIF1 partners, we performed a yeast two‐hybrid screening using a fragment of human TGIF1 (amino acid 1–192) as bait, as described in our previous study (Ettahar et al, 2013). Briefly, after PCR amplification, the TGIF1 fragment was cloned into pGBKT7 (Clontech) as a C‐terminal fusion to the GAL4 DNA‐binding domain (DNA‐BD; amino acids 1–147) and transformed into the Y2HGold Yeast strain (Clontech). For the prey, we used a universal human cDNA library cloned into pGADT7 (Clontech), a yeast two‐hybrid expression vector designed to express a prey protein fused to the GAL4 activation domain (AD; amino acids 768–881). The whole library was transformed into the Y187 Yeast strain (Clontech), and ten million independent yeast colonies were collected and pooled. The bait and prey strains were mated, and colonies were selected on a medium deficient for tryptophan and leucine and supplemented with X‐Gal. Positive colonies were confirmed on a medium lacking tryptophan, leucine, and histidine. The prey fragments of the positive colonies were sequenced, and the resulting sequences were used to interrogate GenBank databases. Twist1 was one of the preys that exhibit strong and specific binding to TGIF1.
Mice
Tgif1 global knockout (Tgif1 −/−), Tgif1.Loxp/Loxp (Tgif1 fl/fl), Twist1.Loxp/Loxp (Twist1 fl/fl), p16Ink4A‐Luciferase (p16 Luc), Loxp‐Stop‐Loxp‐Kras G12D (LSL.Kras G12D), CAG‐Loxp‐Stop‐Loxp‐Luciferase (LSL‐Luc), and Pdx1‐Cre were described previously (Parajuli et al, 2018; Zhang et al, 2015). Loxp‐Stop‐Loxp‐Trp53 R172H (LSL‐Trp53 R172H) mice were obtained from the NCI Mouse Repository.
The PDAC mouse models were generated through successive crossbreeding of Tgif1 fl/fl, Twist1 fl/fl, p16 Luc, Trp53 fl/fl, LSL‐Kras G12D, LSL‐Luc, and Pdx1‐Cre mice as appropriate. Their genotypes are as follows:
Tgif1 KO : Tgif1 fl/fl ;Pdx1‐Cre
Twist1 KO : Twist1 fl/fl ;Pdx1‐Cre
KC: LSL‐Kras G12D ;Pdx1‐Cre
KC Luc : LSL‐Kras G12D ;LSL‐Luc;Pdx1‐Cre
KTC Luc : LSL‐Kras G12D ;Tgif1 fl/fl ;LSL‐Luc;Pdx1‐Cre
KWC: LSL‐Kras G12D ;Twist1 fl/fl ;Pdx1‐Cre
KTWC: LSL‐Kras G12D ;Tgif1 fl/fl ;Twist1 fl/fl ;Pdx1‐Cre
KPC: LSL‐Kras G12D ;LSL‐Trp53 R172H ;Pdx1‐Cre
KIC: LSL‐Kras G12D ;p16 Luc ;Pdx1‐Cre
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Mississippi Medical Center. All mice were maintained on a mixed C57BL/6 and FVB/N genetic background. Mice were maintained in 12‐h light:dark cycles (6:00 am to 6:00 pm) at 22°C and fed a standard rodent chow diet. Formation of PDAC in all mice enrolled in the study was confirmed using pancreatic tissue sections stained with hematoxylin and eosin (H&E) or immunostained with an antibody to the ductal marker Cytokeratin 19 (see below). For in vivo imaging, mice were anaesthetized by isoflurane inhalation and imaged after injection of D‐luciferin (Perkin Elmer) using a Xenogen IVIS Spectrum (Caliper Life Sciences, Hopkinton, MA). Tumor metastasis was assessed by bioluminescence imaging of dissected tissues and confirmed by H&E staining. For the study involving blood glucose levels, mice were fasted for 6 h, injected with vehicle or 2 g/kg body weight glucose (Sigma‐Aldrich), and blood drained from the tail was quantified by the ReLiOn system (ReLiOn). To measure cell proliferation in vivo, mice were injected with 100 mg/kg body weight BrdU (Sigma‐Aldrich) and euthanized 48 h following injection. To take pancreas pictures inside the abdomen, the orientation of the tissue was preserved in order to avoid altering the liver (principal site of PDAC metastasis), which is located in close proximity to the pancreas (see pictures in Fig 2A). To take pancreas pictures after tissue collection, the excised pancreas was unrolled before the photo acquisition in order to show both the head and tail of the pancreas.
Immunoblotting and coimmunopreciptation
Cell extracts were prepared in lysis buffer containing 50 mM Tris HCl (pH 8.0), 120 mM NaCl, 5 mM EDTA, 1% Igepal, protease inhibitors (Roche Diagnostics), and phosphatase inhibitors (Calbiochem). Protein concentrations were determined using the Bradford reagent (Sigma‐Aldrich), and samples were denatured using SDS sample buffer (Invitrogen). For coimmunoprecipitation, cell extracts were cleared by pre‐incubation with Sepharose‐coupled protein G and centrifugation. Then, cell lysates were incubated with the appropriate antibody for 2 h in the absence or presence of bovine DNase I (Sigma‐Aldrich), followed by adsorption to Sepharose‐coupled protein G for 1 h. Immune complexes were washed five times with lysis buffer. For both coimmunoprecipitation and direct immunoblotting, samples were boiled in denaturation sample buffer, loaded into a NuPAGE Bis‐Tris Gel (Invitrogen) and separated by electrophoreses at 200 V. The gels were then transferred onto a nitrocellulose membrane (BioRad) by a wet transfer system (BioRad) and blocked by incubation with 5% dry milk in TBST (TBS with 0.2% Tween20). Membranes were probed with the primary antibody for 2 h at room temperature in the blocking buffer, washed with TBST, and incubated with the peroxidase‐conjugated secondary antibody. Enhanced chemiluminescence (ECL) Western blotting substrates (Pierce) were used for visualization of the results. Primary antibodies against the following antigens were used at specified dilutions: Twist1, 1:500; pSmad2, 1:1,000; Smad2, 1:1,000; TGIF1 1:2,000; pTGIF1, 1:5,000; E‐Cadherin, 1:1,000; Vimentin, 1:1,000; p16Ink4A, 1:200; Flag, 1:2,000; HA, 1:5,000; Myc, 1:1,000; and β‐Actin, 1:5,000.
Immunofluorescence and histology
Tissue samples were fixed in 10% formalin and embedded in paraffin. Human tissue microarrays (TMA) were obtained from US Biomax, Inc. Tissue sections were deparaffinized with xylene and rehydrated in a graded series of ethanol. Antigen retrieval was performed for 30 min at high temperature in citrate buffer. Then, slides were blocked and incubated overnight with anti‐BrdU (1:100), anti‐JunB (1:100), anti‐E‐Cadherin (1:100), anti‐pSmad2 (1:100), anti‐Vimentin (1:100), anti‐p16Ink4a (1:100), anti‐Twist1 (1:100), anti‐pTGIF1 (1:50), or IgG‐matched isotype control antibody (negative control) at 4°C. Finally, slides were incubated with the secondary antibodies conjugated to Alexa‐Fluor®568 or Alexa‐Fluor®448 and co‐stained with DAPI. Slides were viewed on a fluorescence microscope. For quantification of human TMA, samples were scored 0 (undetectable), 1 (low), 2 (medium), and 3 (high) under a microscope in a blinded manner.
For pancreatic tissue histology, paraffin sections were stained with hematoxylin and eosin (H&E) using standard techniques. For immunohistochemistry, formalin‐fixed paraffin‐embedded sections were immunostained with anti‐Amylase (1:100), anti‐α‐Smooth muscle actin (1:500), anti‐Chromogranin (1:100), anti‐Cytokeratin 19 (1:100), anti‐Glucagon (1:200), anti‐Insulin (1:400), anti‐pTGIF1 (1:100), anti‐p16Ink4a (1:100), or anti‐JunB (1:100), followed by incubation with secondary antibody conjugated with peroxidase and revealed by DAB using standard techniques.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP assay kit following the manufacturer's instructions (Millipore). Briefly, chromatin was extracted from tissue or cell samples, sonicated, and immunoprecipitated with antibodies against TGIF1, Twist1, or IgG‐matched isotype control. PCR was run on the chromatin, and the products were analyzed on a 2% agarose gel. Relative DNA binding was determined by quantitative PCR (qPCR) as described previously (Parajuli et al, 2018; Zhang et al, 2015). The following primers were used:
Primers for human ChIP
TWIST1, TGIF1 site‐For 5′‐CAATCCCAAATCGGCCCCAC‐3′
TWIST1, TGIF1 site‐Rev 5′‐CGGAGGAGACTGTCCTGGCC‐3′
GAPDH‐For 5′CGGGATTGTCTGCCCTAATTAT‐3′
GAPDH‐Rev 5′GCACGGAAGGTCACGATGT‐3′
Primers for mouse ChIP
Twist1, TGIF1 site‐For 5′‐GGCCCGGAGAACTCCGAGGG‐3′
Twist1, TGIF1 site‐Rev 5′‐CTGTCTGGGTCGCTGTTGCAG‐3′
Twist1, Twist1 site‐For 5′‐CCGCTTGCAATCAACCAACATG‐3′
Twist1, Twist1 site‐Rev 5′‐CCTTGAGAACATTTCATGTCC‐3′
Gapdh‐For 5′‐ATCCACGACGGACACATTGG‐3′
Gapdh‐Rev 5′‐TGGTGCTGCCAAGGCTGTGG‐3′
Real‐time PCR (qRT–PCR)
Total RNA was extracted from frozen mice tissue samples or cultured cells using TRIzol, purified with QIAGEN RNeasy minicolumns, and reverse transcribed using a High‐Capacity cDNA Reverse Transcription kit (Applied Biosystems). The resulting cDNA was analyzed by qRT–PCR. Briefly, 25 ng cDNA and 150 nmol of each primer were mixed with SYBR GreenER qPCR SuperMix (Invitrogen). Reactions were performed in the 96‐well format using an ABI PRISM 7900HT instrument (Applied Biosystems). Relative mRNA levels were calculated using the comparative CT method and normalized to Gapdh mRNA.
Primers used for mouse samples:
Twist1‐For 5′‐CTCGGACAAGCTGAGCAAGA‐3′
Twist1‐Rev 5′‐GCAGGACCTGGTACAGGAAG‐3′
JunB‐For 5′‐TCACGACGACTCTTACGCAG‐3′
JunB‐Rev 5′‐CCTTGAGACCCCGATAGGGA‐3′
Cdh1‐For 5′‐GTCTCCTCATGGCTTTGC‐3′
Cdh1‐Rev 5′‐CTTTAGATGCCGCTTCAC‐3′
p16Ink4A‐For 5′‐GAACTCTTTCGGTCGTACCC‐3′
p16Ink4A‐Rev 5′‐CGAATCTGCACCGTAGTTGA‐3′
Gapdh‐For 5′‐CACCATCTTCCAGGAGCGAG‐3′
Gapdh‐Rev 5′‐CACCATCTTCCAGGAGCGAG‐3′
Primers used for human cell samples:
TWIST1‐For 5′‐CGGGAGTCCGCAGTCTTA‐3′
TWIST1‐Rev 5′‐GCTTGAGGGTCTGAATCTTG‐3′
CDH1‐For 5′‐CTGAGAACGAGGCTAACG‐3′
CDH1‐Rev 5′‐TTCACATCCAGCACATCC‐3′
p16INK4A‐For 5′‐CCCAACGCACCGAATAGTTA‐3′
p16INK4A‐Rev 5′‐ACCAGCGTGTCCAGGAAG‐3′
GAPDH‐For 5′‐CGCTGAGTACGTGGAGTC‐3′
GAPDH‐Rev 5′‐GCAGGAGGCATTGCTGATGA‐3′
Note that the primers used to amplify human TWIST1 in PL45 cells do not recognize exogenous mouse Twist1 expressed from the pWZL‐Blast‐Twist1ER expression vector, thus enabling us to discriminate between endogenous human TWIST1 and mouse Twist1ER.
DNA pull‐down assay
Purified HA‐TGIF1 prepared from HEK293T cells by immunoprecipitation was incubated with 1 μg of biotinylated double‐stranded oligonucleotides and 10 mg Poly(deoxyinosine‐deoxycytidine; Amersham Biosciences) in binding buffer [20 mmol/l Tris (pH 7.5) and 150 mmol/l NaCl] for 16 h. DNA‐bound proteins were collected with streptavidin–agarose beads, washed with binding buffer, and analyzed by Western blotting. Sense strand oligonucleotides were 5′ biotinylated, and antisense oligonucleotides were unbiotinylated. The following oligonucleotides pairs were used:
Wild‐type sense: 5′‐CTGTTGCCATTGCTGCTGTCACAGCCACTCCGG‐3′
Wild‐type antisense: 5′‐CCGGAGTGGCTGTGAGAGCAGCAATGGCAACAG‐3′
Mutated sense: 5′‐CTGTTGCCATTGCTGCCACTGTAGCCACTCCGG‐3′
Mutated antisense: 5′‐CCGGAGTGGCTACAGTGGCAGCAATGGCAACAG‐3′
Luciferase reporter assays and mammalian two‐hybrid system
For gene reporter assays, MIAPaCa‐2 cells were plated in 6‐well plates and transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen). The pRL‐SV40 plasmid (Promega) was co‐transfected as a normalization control. Cells were incubated for 24 h with the transfection mixtures and allowed to recover for another 24 h before measuring luciferase activity using the Dual‐Luciferase Reporter Assay System. Firefly Luciferase activity was normalized on the basis of Renilla luciferase expressed from the pRL‐SV40 plasmid.
The mammalian two‐hybrid system was performed as described previously (Prunier et al, 2015; Zhang et al, 2015). Briefly, BxPC3 cells were transfected with pG5E1b‐Luc together with Gal4‐TGIF1 mutants, VP16‐Twist1, KrasG12D, and pRL‐SV40, as indicated in the figure. After 48 h, cells were assessed for luciferase activity as described above. In all experiments, data are expressed as mean ± SEM of six samples from a representative experiment performed at least three times.
Statistical analysis
For every experiment, sample size was determined empirically (preliminary experiments were performed) to ensure that the desired statistical power could be achieved. The values are expressed as mean ± SEM. The error bars (SEM) shown for all results were derived from biological replicates, not technical replicates. Significant differences between two groups were evaluated using a two‐tailed, unpaired t‐test, which was found to be appropriate for the statistics, as the sample groups displayed a normal distribution and comparable variance. Statistical significance of survival differences was determined by log‐rank test.
Author contributions
PP and PS were responsible for the experimental design and data analysis. PP, PS, SE, and SO were responsible for mice breeding, genotyping, and harvesting and preparation of tissues for biochemical, molecular, and histological analyses. OF, ZW, and CP contributed to the in vitro experiments. PP, PS, ZW, MSR, LL, and KX contributed to all PDAC experiments, including survival and histological analysis. MSR, KX, and AA wrote the article with inputs from other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Drs. Xu X. and Sharpless N. for providing Twist1 fl/fl and p16Inkk4a‐Luc, LSL.Kras G12D and Pdx1.Cre mice, respectively. We thank Dr. Wang LH for providing the Twist1 luciferase reporter (TGTLuc). We thank Atfi's laboratory members (Asrar Ahmad, Gopalakrishnan Ramakrishnan, Yadong Wang) for help with genotyping analyses, plasmids, luciferase assay, and immunoblotting. Special thanks go to the UMC Animal Imaging and Histology Cores. We thank Dr. Jennifer Koblinski for critical reading of the manuscript. This work was supported by grants from the NIH (R01AR059070 and R01CA210911), Department of Defense (PR162051), and Association pour la Recherche sur le Cancer (R11120DD) to A.A.
The EMBO Journal (2019) 38: e101067
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA (2003) Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17: 3112–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M (1988) Most human carcinomas of the exocrine pancreas contain mutant c‐K‐ras genes. Cell 53: 549–554 [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL (2000) Smads as transcriptional co‐modulators. Curr Opin Cell Biol 12: 235–243 [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL (2002) Signal transduction by the TGF‐beta superfamily. Science 296: 1646–1647 [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U et al (2006a) Both p16(Ink4a) and the p19(Arf)‐p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA 103: 5947–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D et al (2006b) Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20: 3130–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D (2006) TGIF inhibits retinoid signaling. Mol Cell Biol 26: 990–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholin L, Melhuish TA, Powers SE, Goddard‐Leon S, Treilleux I, Sutherland AE, Wotton D (2008) Maternal Tgif is required for vascularization of the embryonic placenta. Dev Biol 319: 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino E, Reimund B, Wildt‐Perinic D, Clerc RG (1995) A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid‐responsive motif. J Biol Chem 270: 31178–31188 [DOI] [PubMed] [Google Scholar]
- Burglin TR, Affolter M (2016) Homeodomain proteins: an update. Chromosoma 125: 497–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283: 14665–14673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AF, Dupage MJ, Dong HK, Chen J, Jacks T (2008) Regulated expression of a tumor‐associated antigen reveals multiple levels of T‐cell tolerance in a mouse model of lung cancer. Cancer Res 68: 9459–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Yan W, Sebolt‐Leopold JS, Pasca di Magliano M (2014) MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 146: 822–834 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio‐Donahue CA, Massague J (2016) TGF‐beta tumor suppression through a lethal EMT. Cell 164: 1015–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demange C, Ferrand N, Prunier C, Bourgeade MF, Atfi A (2009) A model of partnership co‐opted by the homeodomain protein TGIF and the Itch/AIP4 ubiquitin ligase for effective execution of TNF‐alpha cytotoxicity. Mol Cell 36: 1073–1085 [DOI] [PubMed] [Google Scholar]
- Ettahar A, Ferrigno O, Zhang MZ, Ohnishi M, Ferrand N, Prunier C, Levy L, Bourgeade MF, Bieche I, Romero DG et al (2013) Identification of PHRF1 as a tumor suppressor that promotes the TGF‐beta cytostatic program through selective release of TGIF‐driven PML inactivation. Cell Rep 4: 530–541 [DOI] [PubMed] [Google Scholar]
- Faresse N, Colland F, Ferrand N, Prunier C, Bourgeade MF, Atfi A (2008) Identification of PCTA, a TGIF antagonist that promotes PML function in TGF‐beta signalling. EMBO J 27: 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA (2012) The pancreas cancer microenvironment. Clin Cancer Res 18: 4266–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R (2005) Specificity and versatility in tgf‐beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693 [DOI] [PubMed] [Google Scholar]
- Ferrand N, Demange C, Prunier C, Seo SR, Atfi A (2007) A mechanism for mutational inactivation of the homeodomain protein TGIF in holoprosencephaly. FASEB J 21: 488–496 [DOI] [PubMed] [Google Scholar]
- Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI, Korc M (1993) Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 105: 1846–1856 [DOI] [PubMed] [Google Scholar]
- Gu G, Brown JR, Melton DA (2003) Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mech Dev 120: 35–43 [DOI] [PubMed] [Google Scholar]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA (2006) Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 20: 1218–1249 [DOI] [PubMed] [Google Scholar]
- Hidalgo M (2010) Pancreatic cancer. N Engl J Med 362: 1605–1617 [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA et al (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450 [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483 [DOI] [PubMed] [Google Scholar]
- Hneino M, Francois A, Buard V, Tarlet G, Abderrahmani R, Blirando K, Hoodless PA, Benderitter M, Milliat F (2012) The TGF‐beta/Smad repressor TG‐interacting factor 1 (TGIF1) plays a role in radiation‐induced intestinal injury independently of a Smad signaling pathway. PLoS ONE 7: e35672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J (2011) Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res 71: 3980–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio‐Donahue CA (2012) Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 61: 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, Moses HL (2006) Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas‐specific blockade of transforming growth factor‐beta signaling in cooperation with active Kras expression. Genes Dev 20: 3147–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277–300 [DOI] [PubMed] [Google Scholar]
- Kern SE, Shi C, Hruban RH (2011) The complexity of pancreatic ductal cancers and multidimensional strategies for therapeutic targeting. J Pathol 223: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Haigis KM (2009) Non‐redundancy within the RAS oncogene family: insights into mutational disparities in cancer. Mol Cells 28: 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, Bar‐Sagi D (2010) Oncogenic KRas suppresses inflammation‐associated senescence of pancreatic ductal cells. Cancer Cell 18: 448–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RS, Wotton D, Massague J (2001) Epidermal growth factor signaling via Ras controls the Smad transcriptional co‐repressor TGIF. EMBO J 20: 128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ (1999) Twist is a potential oncogene that inhibits apoptosis. Genes Dev 13: 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Hruban RH (2008) Pancreatic cancer. Annu Rev Pathol 3: 157–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar L, Hoodless PA (2006) Embryonic fibroblasts from mice lacking Tgif were defective in cell cycling. Mol Cell Biol 26: 4302–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19: 2783–2810 [DOI] [PubMed] [Google Scholar]
- Massague J (2008) TGFbeta in cancer. Cell 134: 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva‐Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV et al, et al (2014) Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli P, Kumar S, Loumaye A, Singh P, Eragamreddy S, Nguyen TL, Ozkan S, Razzaque MS, Prunier C, Thissen JP et al (2018) Twist1 activation in muscle progenitor cells causes muscle loss akin to cancer cachexia. Dev Cell 45: 712–725 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah M, Prunier C, Marais J, Ferrand N, Mazars A, Lallemand F, Gauthier JM, Atfi A (2001) c‐Jun interacts with the corepressor TG‐interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc Natl Acad Sci USA 98: 6198–6203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, Zanatta L, Pivetta F, Grizzo A, Sonego M et al (2012) A “twist box” code of p53 inactivation: twist box: p53 interaction promotes p53 degradation. Cancer Cell 22: 404–415 [DOI] [PubMed] [Google Scholar]
- Prunier C, Zhang MZ, Kumar S, Levy L, Ferrigno O, Tzivion G, Atfi A (2015) Disruption of the PHRF1 tumor suppressor network by PML‐RARalpha drives acute promyelocytic leukemia pathogenesis. Cell Rep 10: 883–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SR, Ferrand N, Faresse N, Prunier C, Abecassis L, Pessah M, Bourgeade MF, Atfi A (2006) Nuclear retention of the tumor suppressor cPML by the homeodomain protein TGIF restricts TGF‐beta signaling. Mol Cell 23: 547–559 [DOI] [PubMed] [Google Scholar]
- Shen J, Walsh CA (2005) Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol 25: 3639–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M, Izumi H, Onitsuka T, Miyamoto N, Kashiwagi E, Kidani A, Hirano G, Takahashi M, Naito S, Kohno K (2008) Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 27: 5543–5553 [DOI] [PubMed] [Google Scholar]
- Stathis A, Moore MJ (2010) Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol 7: 163–172 [DOI] [PubMed] [Google Scholar]
- Sundqvist A, Morikawa M, Ren J, Vasilaki E, Kawasaki N, Kobayashi M, Koinuma D, Aburatani H, Miyazono K, Heldin CH et al (2018) JUNB governs a feed‐forward network of TGFbeta signaling that aggravates breast cancer invasion. Nucleic Acids Res 46: 1180–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Anderson AE, Sutherland AE, Wotton D (2012) Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway. PLoS Genet 8: e1002524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D et al (2004) Endogenous oncogenic K‐ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387 [DOI] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147: 275–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichalkovski A, Gresko E, Hess D, Restuccia DF, Hemmings BA (2010) PKB/AKT phosphorylation of the transcription factor Twist‐1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene 29: 3554–3565 [DOI] [PubMed] [Google Scholar]
- Wagner M, Kleeff J, Friess H, Buchler MW, Korc M (1999) Enhanced expression of the type II transforming growth factor‐beta receptor is associated with decreased survival in human pancreatic cancer. Pancreas 19: 370–376 [DOI] [PubMed] [Google Scholar]
- Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, Goggins M, Hruban RH, Chang AE et al (2015) RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell 161: 1345–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massague J (1999) A Smad transcriptional corepressor. Cell 97: 29–39 [DOI] [PubMed] [Google Scholar]
- Wotton D, Massague J (2001) Smad transcriptional corepressors in TGF beta family signaling. Curr Top Microbiol Immunol 254: 145–164 [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939 [DOI] [PubMed] [Google Scholar]
- Zhang MZ, Ferrigno O, Wang Z, Ohnishi M, Prunier C, Levy L, Razzaque M, Horne WC, Romero D, Tzivion G et al (2015) TGIF governs a feed‐forward network that empowers Wnt signaling to drive mammary tumorigenesis. Cancer Cell 27: 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File