

Abstract
Bok (Bcl-2-related ovarian killer) is a Bcl-2 family member that, because of its predicted structural homology to Bax and Bak, has been proposed to be a pro-apoptotic protein. In this study, we demonstrate that Bok is highly expressed in neurons of the mouse brain but that bok was not required for staurosporine-, proteasome inhibition-, or excitotoxicity-induced apoptosis of cultured cortical neurons. On the contrary, we found that bok-deficient neurons were more sensitive to oxygen/glucose deprivation-induced injury in vitro and seizure-induced neuronal injury in vivo. Deletion of bok also increased staurosporine-, excitotoxicity-, and oxygen/glucose deprivation-induced cell death in bax-deficient neurons. Single-cell imaging demonstrated that bok-deficient neurons failed to maintain their neuronal Ca2+ homeostasis in response to an excitotoxic stimulus; this was accompanied by a prolonged deregulation of mitochondrial bioenergetics. bok deficiency led to a specific reduction in neuronal Mcl-1 protein levels, and deregulation of both mitochondrial bioenergetics and Ca2+ homeostasis was rescued by Mcl-1 overexpression. Detailed analysis of cell death pathways demonstrated the activation of poly ADP-ribose polymerase-dependent cell death in bok-deficient neurons. Collectively, our data demonstrate that Bok acts as a neuroprotective factor rather than a pro-death effector during Ca2+- and seizure-induced neuronal injury in vitro and in vivo.
SIGNIFICANCE STATEMENT Bcl-2 proteins are essential regulators of the mitochondrial apoptosis pathway. The Bcl-2 protein Bok is highly expressed in the CNS. Because of its sequence similarity to Bax and Bak, Bok has long been considered part of the pro-apoptotic Bax-like subfamily, but no studies have yet been performed in neurons to test this hypothesis. Our study provides important new insights into the functional role of Bok during neuronal apoptosis and specifically in the setting of Ca2+- and seizure-mediated neuronal injury. We show that Bok controls neuronal Ca2+ homeostasis and bioenergetics and, contrary to previous assumptions, exerts neuroprotective activities in vitro and in vivo. Our results demonstrate that Bok cannot be placed unambiguously into the Bax-like Bcl-2 subfamily of pro-apoptotic proteins.
Keywords: Bax, Bcl-2 family proteins, calcium, cell death, excitotoxicity, glutamate
Introduction
The Bcl-2 family of proteins are essential mediators of the intrinsic (mitochondrial) pathway of apoptosis by controlling mitochondrial outer membrane integrity (Tait and Green, 2010, Czabotar et al., 2014). The structural and functional characteristics of Bcl-2 proteins separate this family into anti-apoptotic and pro-apoptotic proteins. The latter are further divided into Bcl-2 homology 3 (BH3)-only and Bax-like proteins (Czabotar et al., 2014). Bcl-2 family proteins contain between one and four BH conserved domains, essential for mediating homodimeric or heterodimeric interactions among Bcl-2 family proteins (Czabotar et al., 2014). Anti-apoptotic Bcl-2 proteins, such as Bcl-2, Bcl-xL, Bcl-w, and Mcl-1, have four BH domains, with BH4 granting their anti-apoptotic activity (Youle and Strasser, 2008). BH3-only proteins, such as Bid, Bim, and Puma, possess only the BH3 domain, which is essential for apoptosis initiation (Giam et al., 2008). Members of the Bax-like subfamily (Bax, Bak, and Bok) are characterized by their BH1, BH2, and BH3 domains. Bax and Bak are inhibited by anti-apoptotic Bcl-2 proteins, activated by BH3-only proteins, and form “release channels” in the mitochondrial outer membrane as part of their role in mitochondrial permeabilization (Hsu et al., 1997b; Lovell et al., 2008). Most BH3-only proteins are not constitutively expressed in neurons and need to be transcriptionally or post-translationally activated in response to cell death signaling (Ward et al., 2004; Engel et al., 2011). In contrast, anti-apoptotic Bcl-2 proteins and Bax-like proteins are widely expressed in the nervous system (Anilkumar and Prehn, 2014). Their expression peaks during neuronal development; however, many of these proteins retain high levels of expression in the adult nervous system. Indeed, in addition to their role in controlling the intrinsic apoptosis pathway, Bcl-2 family proteins have essential “daytime” functions (Kilbride and Prehn, 2013). These include the regulation of mitochondrial fusion and fission (Autret and Martin, 2010), neuronal Ca2+ homeostasis (Pinton et al., 2000; Chen et al., 2004; Oakes et al., 2005; D'Orsi et al., 2015), and bioenergetics (Alavian et al., 2011; D'Orsi et al., 2015).
Bok (Bcl-2-related ovarian killer) has extensive amino-acid sequence similarity (70–80% homology) to Bax and Bak and a similar organization of BH domains (Hsu et al., 1997a); for this reason, Bok has long been considered part of the Bax-like pro-apoptotic subfamily (Inohara et al., 1998; Bartholomeusz et al., 2006; Rodriguez et al., 2006). Indeed, the yeast two-hybrid screening method identified Bok as a strong interactor of certain anti-apoptotic proteins, including Mcl-1, BHRF1, and Bfl-1 (Hsu et al., 1997a, Inohara et al., 1998); however, unlike Bax and Bak, interactions of Bok with Bcl-2, Bcl-xL, and Bcl-w were not detectable (Hsu et al., 1997a; Inohara et al., 1998). Similar to other Bcl-2 proteins, Bok localizes to the mitochondria, ER, and nuclear envelope, although the atypical C-terminal transmembrane region of Bok seems to lead its localization more toward the ER and Golgi than the mitochondria (Echeverry et al., 2013). At the ER, Bok binds to inositol 1,4,5-trisphosphate receptor proteins (IP3Rs) and protects these against proteolytic cleavage (Schulman et al., 2013). Bok overexpression in mammalian cells leads to cytochrome-c release, caspase-3 activation, and nuclear fragmentation, thereby inducing apoptosis (Igaki et al., 2000; Zhang et al., 2000; Yakovlev et al., 2004; Bartholomeusz et al., 2006). Interestingly, this role of Bok mostly requires the presence of Bax and Bak, suggesting that Bok activities may be distinct from those of Bax-like proteins (Ke et al., 2012; Echeverry et al., 2013). Bok is expressed in the cerebral cortex and highly enriched in the CA3 subfield of the hippocampus (Lein et al., 2004; Newrzella et al., 2007; Bonner et al., 2010), which are areas of the brain vulnerable to ischemic and seizure-induced injury. The significance of this is unknown because the potential physiological or pathophysiological role of Bok in the CNS has yet to be explored. Because of its high expression in central neurons, homology to Bax, and suggested involvement in Ca2+ regulation, our aim in this study was to investigate the role of bok in neuronal apoptosis and Ca2+- and seizure-induced neuronal injury.
Materials and Methods
Materials
Fetal bovine serum, horse serum, minimal essential medium, B27 supplemented Neurobasal medium, tetramethylrhodamine methyl ester (TMRM), and Fluo-4 AM came from BioSciences. All other chemicals, including NMDA, MK-801, staurosporine (STS), epoxomicin, and thapsigargin, came in analytical grade purity from Sigma-Aldrich. Calpeptin, Necrostatin-1 (Nec-1), and DPQ (3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone) were purchased from ENZO Life Sciences. zVAD (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]) was purchased from Bachem.
Gene targeted mice
The generation and genotyping of bax−/− mice has been described previously (D'Orsi et al., 2012, 2015). Several pairs of heterozygous breeder pairs of bax-deficient mice were obtained from The Jackson Laboratory and maintained in house. The bax−/− mice were originally generated on a mixed C57BL/6x129SV genetic background, using 129SV-derived embryonic stem (ES) cells, but had been backcrossed for >12 generations onto the C57BL/6 background. bok−/− mice generated by gene targeting using C57BL/6-derived Bruce4 ES cells (Ke et al., 2012) were bred as a homozygous knock-out colony. To generate bax/bok-double-deficient mice, bax+/− mice were crossed with bok−/− mice to produce mice heterozygous for both alleles (bax+/−/bok+/−). The double heterozygous offspring with a deleted bok allele were crossed with bok−/− mice to produce bax+/−/bok−/− mice, which were then intercrossed to generate mice double knock-out bax−/−/bok−/−. As controls, double heterozygous bax+/−/bok+/− mice were intercrossed to breed bax+/−/bok+/+ mice, which were then intercrossed to generate knock-out bax−/−/bok+/+ mice. As controls, wild-type (WT), bax−/−-deficient, and bok−/−deficient mice were used for all experiments. All mouse strains were backcrossed for >12 generations on an inbred C57BL/6 background. WT, heterozygote, and gene-deficient mice were generated and maintained in-house in the Royal College of Surgeons in Ireland (RCSI) Biological Research Facility.
The genotype of bax−/−, bok−/−, and bax+/−/bok−/− and bax+/−/bok+/+ mice was confirmed by PCR. DNA was extracted from tail snips using the High Pure PCR Template Preparation kit (Roche). Genotyping was performed using three specific primers as follows: 5′GTTGACCAGAGTGGCGTAGG3′ (common), 5′GAGCTGATCAGAACCATCATG3′ (WT allele-specific), and 5′CCGCTTCCATTGCTCAGCGG3′ (mutant allele-specific) for bax; 5′CGGGTTTGAATGGAAGGGTC3′ (common forward primer), in combination with two reverse primers, 5′TGTTCCCATGGTGCTACATCC3′ and 5′GAGCTAGCTAGCTATGTGTG3′ for bok. All animal work was performed with ethics approval and under licenses granted by the Health Products Regulatory Authority in accordance with European Communities Council Directive (86/609/EEC), and procedures were reviewed and approved by the RCSI Research Ethics Committee.
Preparation of mouse neocortical neurons
Primary cultures of cortical neurons were prepared at embryonic days 16–18 (D'Orsi et al., 2012, 2015). To isolate cortical neurons, hysterectomies of the uterus of pregnant female mice were performed after killing mice by cervical dislocation. The cerebral cortices were isolated from each embryo and pooled in a dissection medium on ice (PBS with 0.25% glucose and 0.3% bovine serum albumin). Tissue was incubated with 0.25% trypsin–EDTA at 37°C for 15 min. After incubation, trypsinization was stopped by the addition of fresh plating medium (minimal essential medium containing 5% fetal bovine serum, 5% horse serum, 100 U/ml penicillin/streptomycin, 0.5 mm l-glutamine, and 0.6% d-glucose). Neurons were then dissociated by gentle pipetting and after centrifugation (1500 rpm, 3 min), and medium containing trypsin was aspirated. Neocortical neurons were resuspended in plating medium, plated at 2 × 105 cells/cm2 on poly-d-lysine-coated plates (final concentration of 5 μg/ml) and then incubated at 37°C and 5% CO2. The plating medium was exchanged with 50% feeding medium (Neurobasal containing 100 U/ml penicillin/streptomycin, 2% B27, and 0.5 mm l-glutamine) and 50% plating medium with additional mitotic inhibitor cytosine arabinofuranoside (600 nm). Two days later, medium was again exchanged with complete feeding medium. All experiments were performed on 8–11 d in vitro (DIV).
Mouse mixed glial cell preparation and isolation of primary microglia and astrocytes
Mixed cortical cell isolation for microglia and astrocytes was performed using cortices of postnatal day 0 (P0) to P2 C57BL/6 WT pups. The cerebral cortices were isolated from each pup and incubated with 0.25% trypsin–EDTA at 37°C for 10 min. After this incubation, trypsinization was stopped with the addition of DMEM–F-12/l-glutamine (Gibco) medium containing 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. Mixed cells were then dissociated by gentle pipetting, strained through a 40 μm nylon cell strainer (Falcon; BD Biosciences Discovery Labware), and centrifuged at 300 × g for 6 min. Cells were resuspended in fresh medium, plated at a density of approximately four cortices per T75 flask, and then incubated at 37°C and 5% CO2. One day after plating, medium was exchanged with fresh medium containing cytokines 10 ng/ml macrophage colony-stimulating factor (R&D Systems) and 20 ng/ml granulocyte-macrophage colony-stimulating factor (R&D Systems) to promote microglial proliferation (Suzumura et al., 1990). After 7–8 d, when astrocytes were confluent and overlaying microglia, the cells were isolated using a shaking method, consisting of placing the T75 flasks on a plate shaker (900 rpm, 20 min). The media containing the detached and floating microglia was immediately collected for Western blot analysis, whereas the remaining attached cells were trypsinized, replated, and cultured for 12–14 d in the absence of cytokines to obtain astrocyte cultures (Schildge et al., 2013). The murine BV2-like microglial cell line was cultured in RPMI (Lonza) with 100 U/ml penicillin/streptomycin, 2 mm l-glutamine, and 10% fetal bovine serum and incubated at 37°C in a humidified atmosphere with 5% CO2.
Cell death assay and determination of neuronal injury
Cortical neurons on 24-well plates exposed to NMDA-induced excitotoxic injury (100 μm NMDA for 5 min or 300 μm NMDA for 60 min; each with the addition of 10 μm glycine) or STS-induced (100 nm) or epoxomicin-induced (50 nm) cell death were then stained live with 1 μg/ml Hoechst 33258 (Sigma) and 5 μm propidium iodide (PI) (Sigma) dissolved in culture medium 24 h after treatment. In separate experiments, cortical neurons were exposed to NMDA in the presence and absence of Calpeptin (20 μm), Nec-1 (50 μm), zVAD (100 μm), or DPQ (100 μm), after which they were allowed to recover for 24 h. Neuronal injury was assessed using an Eclipse TE 300 inverted microscope (Nikon) with 20×, 0.43 numerical aperture (NA) phase-contrast objective using the appropriate filter set for Hoechst and PI and using a charge-coupled device camera (SPOT RT SE 6; Diagnostic Instruments), as described previously (D'Orsi et al., 2012). All experiments were performed at least three times with independent cultures, and, for each time point, images of nuclei were captured in three subfields containing ∼300–400 neurons each and repeated in triplicate. The number of PI-positive cells was expressed as a percentage of total cells in the field. Resultant images were processed using NIH ImageJ.
Oxygen/glucose deprivation in mouse neocortical neurons
WT, bok−/−, bax−/−/bok−/−, and bax−/−/bok+/+ cortical neurons, cultured on 24-well plates, were rinsed in a prewarmed glucose-free medium and then transferred to a hypoxic chamber (COY Lab Products). The hypoxic chamber had an atmosphere comprising 1.5% O2, 5% CO2, and 85% N2, and the temperature was maintained at 37°C. Neurons were then incubated with oxygen/glucose deprivation (OGD) medium preincubated in the hypoxia chamber for 1 h before use. The OGD medium consisted of glucose-free DMEM containing 100 U/ml penicillin/streptomycin and 0.5 mm l-glutamine and freshly supplemented with B27. After 90 min of OGD, medium was removed and conditioned medium replaced. Thereafter, cells were placed in normoxic conditions (21% O2 and 5% CO2) and allowed to recover for 24 h. Control cortical neurons were exposed to DMEM, as above, supplemented with 15 mm glucose, and kept in normoxic conditions (D'Orsi et al., 2015). Neuronal injury was assessed 24 h after OGD treatment, as described in the above paragraph for cell death assay and determination of neuronal injury.
Time-lapse live cell imaging
Primary neocortical neurons on Willco dishes (Willco Wells) were coloaded with the calcium dye Fluo-4 AM (3 μm) and the membrane-permeant cationic fluorescent probe TMRM (20 nm) for 30 min at 37°C (in the dark) in experimental buffer containing the following (in mm): 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 HEPES, 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, and 15 glucose, pH 7.4. Cells were washed and bathed in 2 ml of experimental buffer containing TMRM and a thin layer of mineral oil was added to prevent evaporation. Neurons were placed on the stage of an LSM 510 Meta confocal microscope equipped with a 63×, 1.4 NA oil-immersion objective and a thermostatically regulated chamber (Carl Zeiss Jena). After 30 min equilibration time, neurons were exposed to 100 μm NMDA plus 10 μm glycine for 5 min; MK-801 (5 μm) was added to terminate NMDA receptor activation as required (D'Orsi et al., 2012). TMRM was excited at 543 nm, and the emission was collected with a 560 nm long-pass filter. Fluo-4 was excited at 488 nm, and the emission was collected through a 505–550 nm barrier filter. Images were captured every 30 s during NMDA excitation and every 5 min during the rest of the experiments. For the Mcl-1 overexpression single-cell experiments, neurons were cotransfected with a vector expressing Mcl-1 (MC200829; OriGene; Anilkumar et al., 2013) and a plasmid expressing enhanced CFP (ECFP-C1; BD Biosciences Clontech) or, for control neurons, transfected with the CFP plasmid only. Neocortical neurons were transfected at 6 DIV using Lipofectamine 2000 (Invitrogen). Two days after transfection, neurons were coloaded with Fluo-4 AM (3 μm) and TMRM (20 nm) in experimental buffer and placed on the stage of an LSM 7.10 confocal microscope equipped with a 63×, 1.4 NA oil-immersion objective and a thermostatically regulated chamber set at 37°C (Carl Zeiss). After a baseline equilibration time, NMDA (100 μm/5 min) dissolved in experimental buffer was added to the medium. TMRM was excited at 561 nm, and the emission was collected by a 575 nm long-pass filter. Fluo-4 was excited at 488 nm, and the emission was collected through a 505–550 nm barrier filter. All microscope settings including laser intensity and scan time were kept constant for the whole set of experiments. Control experiments were performed and showed that phototoxicity had a negligible effect. All images were processed and analyzed using MetaMorph Software version 7.5 (Universal Imaging), and the data presented were normalized to the baseline.
Western blotting
Preparation of cell lysates from cortical neurons and mouse tissues and Western blotting was performed as described previously (Reimertz et al., 2001). The resulting blots were probed with the following: a rabbit monoclonal Bok antibody (clone 1–5) at 1:250 (Echeverry et al., 2013); a rabbit polyclonal Bax antibody at 1:1000 (ab7977; Abcam); a rabbit polyclonal Mcl-1 antibody diluted 1:250 (600-401-394; Rockland); a mouse monoclonal Bcl-xL antibody at 1:250 (clone H-5, sc8392; Santa Cruz Biotechnology); a rabbit polyclonal Bcl-w antibody at 1:500 (AAP-050C; Stressgen); a mouse monoclonal Bcl-2 antibody (5K140, sc-70411; Santa Cruz Biotechnology); a mouse monoclonal supernatant NR2A glutamate receptor antibody (clone N327/95; NeuroMab) diluted 1:10; a mouse monoclonal supernatant NR2B glutamate receptor antibody (clone N59/20; NeuroMab) diluted 1:10; a purified mouse monoclonal NR1 glutamate receptor antibody (clone N308/48; NeuroMab) diluted 1:10; a mouse monoclonal supernatant GluA2/GluR2 antibody (clone L21/32; NeuroMab) diluted 1:10; a mouse monoclonal β-actin antibody (clone DM 1A; Sigma) diluted 1:5000; and a mouse monoclonal GAPDH antibody (clone 6C5, ab8245; Abcam). Horseradish peroxidase-conjugated secondary antibodies diluted 1:10,000 (Pierce) were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and imaged using a FujiFilm LAS-3000 imaging system (Fuji).
Poly ADP-ribose polymerase activity assay
To assay cellular poly ADP-ribose polymerase (PARP) activity, cortical neurons, cultured on a 96-well flat-bottom plate, were exposed to NMDA-induced excitotoxic injury (100 μm NMDA for 5 min) in the presence and absence of DPQ (100 μm). Neurons were then allowed to recover for selected time points (2, 4, and 8 h), after which they were washed twice with 1× PBS and lysed in 100 μl of Cell Extraction PARP buffer on ice with periodic mixing for 30 min. PARP activity was assayed in cortical neurons extracts using the HT PARP/Apoptosis Assay according to the instructions of the manufacturer (4684-096-K; Trevigen). The HT PARP/Apoptosis Assay is an ELISA that semiquantitatively detects PAR deposited onto immobilized histone proteins in a 96-well format. Lysates (30 μg/well) were added in triplicates to the wells containing PARP buffer and PARP substrate mixture, followed by incubation at room temperature for 30 min. An anti-PAR monoclonal antibody, goat anti-mouse IgG–HRP conjugate, and HRP substrate (materials supplied) were used to generate a colorimetric signal. Absorbance was detected at 450 nm. The background reading was subtracted from the readings of the samples, and PARP activity was calculated using the standard curve obtained from readings of the PARP standards.
Status epilepticus
Mouse seizure model.
WT (n = 14) and bok−/− (n = 18) mice were anesthetized and placed in a stereotaxic frame. Three partial craniectomies were performed to affix cortical skull-mounted EEG electrodes (Plastics One) to record cortical EEG using a Grass Comet digital EEG. A guide cannula was affixed for intra-amygdala targeting, and the skull assembly was fixed in place with dental cement. After baseline EEG was obtained, kainic acid (KA; 0.3 μg in 0.2 μl of 1× PBS; Sigma) was microinjected into the basolateral amygdala. Nonseizure control mice received 0.2 μl of intra-amygdala vehicle (1× PBS). Forty minutes later, mice received intraperitoneal lorazepam (6 mg/kg) as described previously (Engel et al., 2012). Mice were then saline perfused under deep anesthesia to remove intravascular blood components, and brains were flash frozen whole in 2-methylbutane at 30°C for histopathology.
Quantification of electroencephalography.
Digitized EEG recordings were analyzed using automated software as described previously (Engel et al., 2012). EEG analysis was performed by uploading EEG into Labchart7 software (ADInstruments) to calculate frequency and amplitude of the EEG signal.
Histological analysis.
Measurement of acute cell death was assessed by Fluoro-Jade B (FJB; Millipore) staining as described previously (Engel et al., 2012). FJB is a polyanionic fluorescein derivative that specifically stains degenerating neurons (Schmued and Hopkins, 2000). Hippocampal FJB-positive neurons were the average of two adjacent sections for the entire hippocampus or the CA3 dorsal or CA3 ventral subregions of each genotype.
Statistical analysis
Data are given as means ± SEM. Data were analyzed using one-way ANOVA, followed by Tukey's post hoc test or Student's t test for two-group comparison. p values <0.05 were considered to be statistically significant. When significant, exact p values were stated in the figure legends.
Results
bok is not required for STS-, proteasome inhibition-, or excitotoxicity-induced apoptosis of cortical neurons in vitro
Bok has originally been reported to be expressed predominantly in the reproductive system (Hsu et al., 1997a). Western blotting analysis of several organs from adult C57BL/6 WT and bok-deficient mice revealed that Bok was widely but differentially expressed in mouse tissue. Bok protein levels were particularly abundant in brain, kidney, and spleen (Fig. 1A). Bok protein expression was also compared in protein lysates obtained from cultured mouse primary cortical neurons, primary astrocytes, and microglia, as well as microglial-like BV2 cells. Western blotting showed that Bok was highly expressed in cultured cortical neurons and astrocytes, with very low levels detected in microglia (Fig. 1B).
Figure 1.
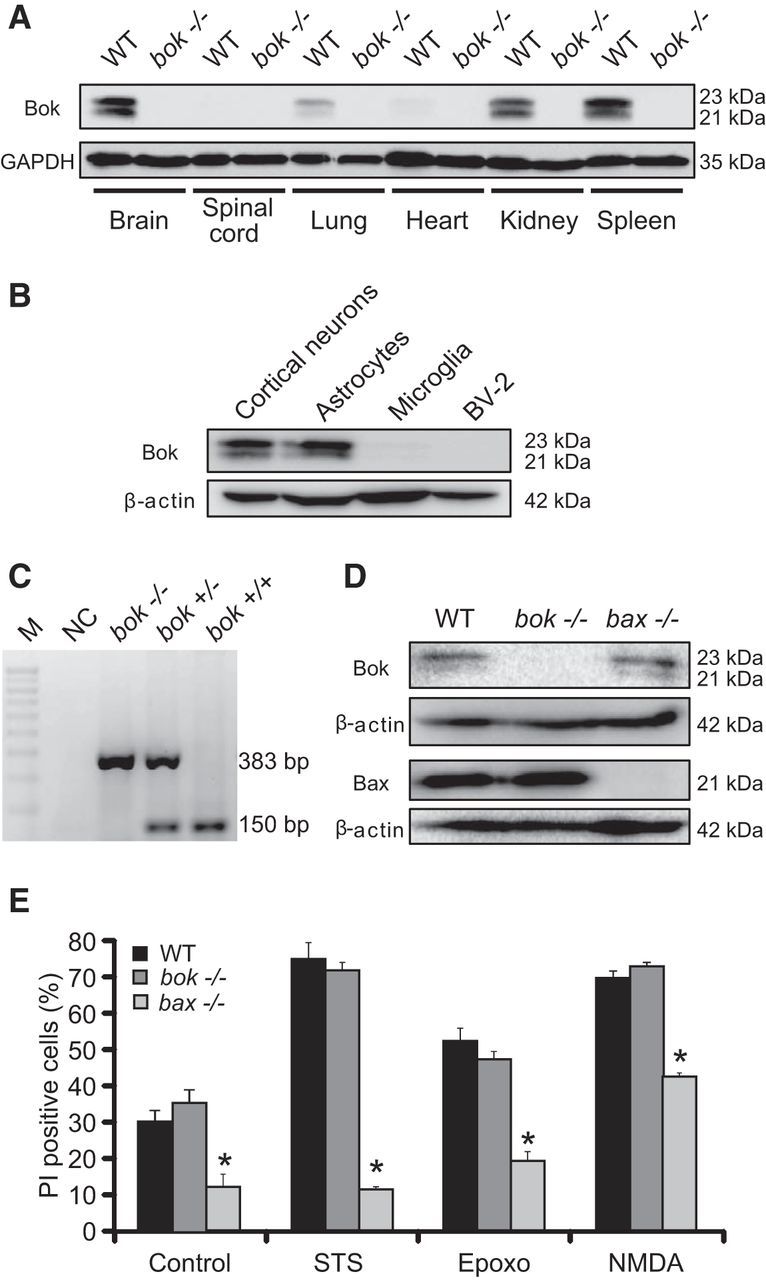
bok is not pro-apoptotic in response to several cell death stimuli in vitro. A, B, Western blot analysis comparing the levels of Bok in several organs from C57BL/6 WT and bok-deficient mice (A) and in cortical neurons, microglia, astrocytes, and BV2-like microglial cells (B). GAPDH and β-actin were used as loading controls. C, Representative standard PCR analysis of genomic DNA shows indicated strain and expected PCR band sizes during bok genotyping. M, 100 bp ladder; NC, negative control without the tail DNA template. D, Western blot analysis comparing the levels of Bok and Bax in WT, bok−/− and bax−/− cortical neurons. β-Actin was used as a loading control. Experiments were repeated three times with different preparations and with similar results. E, Cortical neurons from WT, bok−/−, and bax−/− mice were either treated with STS (100 nm) or epoxomicin (Epoxo; 50 nm) or exposed to 100 μm NMDA for 5 min, after which they were allowed to recover for 24 h. Controls were the pool of DMSO and sham conditions for 24 h and 5 min, respectively. Cell death was assessed by Hoechst 33258 (1 μg/ml) and PI (5 μm) staining. Three subfields containing 300–400 neurons each were captured, and n = 9 wells were analyzed per condition from three separate cultures. PI-positive nuclei were scored as dead neurons and expressed as a percentage of the total population. Data are means ± SEM. *p ≤ 0.05. p values are the following: p = 0.0005 (control), p = 0.0008 (STS), p = 0.0012 (Epoxo), p = 0.0012 (NMDA) versus WT cultures; p = 0.0001 (control), p = 0.0011 (STS), p = 0.0037 (Epoxo), p = 0.0004 (NMDA) versus bok−/− cultures; nonstatistical significance was observed in any treatment for WT versus bok−/− cultures (ANOVA, post hoc Tukey's test).
Having identified significant Bok protein levels in neurons, we aimed to explore a potential role of Bok in neuronal apoptosis and Ca2+-induced neuronal injury. We generated primary cortical neuron cultures from bok-deficient mice that were genotyped as described previously (Ke et al., 2012; Fig. 1C). The effect of bok deficiency was compared with that of bax deficiency, because Bax has been identified previously as a key regulator of neuronal apoptosis (Deckwerth et al., 1996; Vekrellis et al., 1997; Cregan et al., 1999; Putcha et al., 1999; D'Orsi et al., 2015). In contrast to non-neuronal cells, bax deficiency is often sufficient to protect against neuronal apoptosis, because mature neurons express a splice variant of Bak, N-Bak, that is anti-apoptotic (Sun et al., 2001). Western blot analysis confirmed deficiency of Bok and Bax protein in primary cortical neuron cultures (Fig. 1D). We exposed WT, bok-deficient, and bax-deficient cortical neurons to the apoptosis-inducing protein kinase inhibitor STS (Krohn et al., 1998) and the proteasome inhibitor epoxomicin (Tuffy et al., 2010). Neuronal injury was quantified 24 h after addition of STS or epoxomicin by PI uptake and Hoechst 33258 staining of nuclear chromatin. bok deficiency failed to afford protection against STS- and epoxomicin-induced apoptosis, whereas deletion of bax prevented neuronal apoptosis, as reported previously (Akhtar et al., 2006; Tuffy et al., 2010; Fig. 1E). We and others have observed previously that bax deficiency also provides neuroprotection against transient NMDA-induced excitotoxic cell death (Wang et al., 2004; D'Orsi et al., 2015). We found that cortical neurons exposed to NMDA (100 μm for 5 min) were protected by bax deficiency but not bok deficiency (Fig. 1E).
Deletion of bok increases OGD-induced neuronal injury in vitro and seizure-induced neuronal injury in vivo
Excitotoxic injury is a key component of ischemic/hypoxic and seizure-induced neuronal injury (Dirnagl et al., 1999; Ben-Ari and Cossart, 2000; Engel et al., 2011). To explore the effects of Bok in clinically relevant paradigms that involve excitotoxic injury, bok-deficient cultured cortical neurons were exposed to combined OGD for 90 min to simulate ischemic neuronal injury. Neurons were allowed to recover over a 24 h time period, after which neuronal injury was assessed by PI uptake and Hoechst 33258 staining. Surprisingly, bok deficiency potentiated rather than protected against OGD-induced neuronal injury when compared with WT cultures (Fig. 2A).
Figure 2.
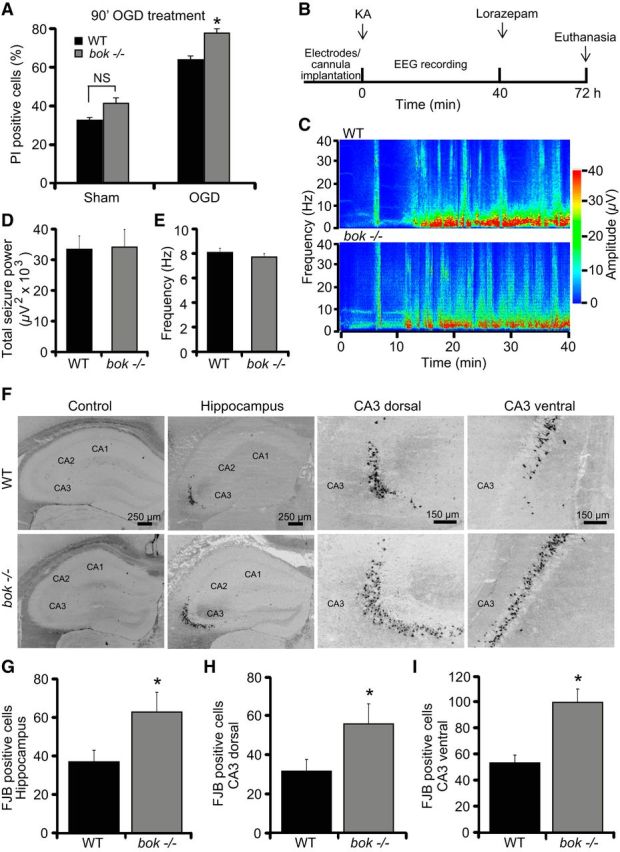
bok-deficient mice show increased cell death during OGD- and seizure-induced injury. A, Cortical neurons from WT and bok−/− mice were exposed to either OGD or sham conditions for 90 min and were allowed to recover for 24 h. Cell death was assessed by Hoechst and PI staining, and PI-positive nuclei were scored as dead neurons and expressed as a percentage of the total population. Three subfields containing 300–400 neurons each were captured, and n = 9 wells were analyzed per condition from three separate cultures. Means ± SEMs are shown. *p ≤ 0.05. p values are the following: p = 0.068 (Sham; NS), p = 0.041 (OGD) versus WT cultures (ANOVA, post hoc Tukey's test). B, Schematic showing experimental paradigm of status epilepticus (SE) and hippocampal damage after intra-amygdala KA in 8-week-old WT and bok−/− mice. KA (0.3 μg in 0.2 μl of 1× PBS) was injected into the amygdala of WT and bok−/− mice, followed by 40 min EEG recordings. After lorazepam (6 mg/kg) injection, mice were killed after 72 h for brain analysis. C, EEG heat map depicting typical amplitude frequency data during SE in WT and bok−/− mice and onset of continuous seizures after KA and lorazepam microinjection. D, E, WT (n = 14) and bok−/− (n = 18) mice semiquantitative analysis of EEG total seizure power and frequency in the SE model is shown. F–I, WT (n = 14) and bok−/− (n = 18) mice were subjected to SE or to vehicle injection (0.2 μl of 1× PBS) for 40 min. Mice were killed 72 h after status epilepticus, and 12 μm sections from each brain sample were collected at the level of the dorsal hippocampus. Neurodegeneration was assessed by FJB staining. Representative photomicrographs of vehicle-injected and KA-injected WT and bok−/− hippocampal sections and CA3 dorsal and CA3 ventral hippocampal fields stained with FJB are shown (F). Neuronal injury occurs exclusively in the CA3 hippocampal subfield after SE. Hippocampal FJB-positive neurons were the average of two adjacent sections for the entire hippocampus (G) and CA3 dorsal (H) and CA3 ventral (I) fields for WT (n = 15) and bok−/−(n = 17). Means ± SEMs are shown. *p ≤ 0.05. p values are the following: p = 0.047 (hippocampus), p = 0.024 (CA3 dorsal), p = 0.047 (CA3 ventral) versus SE-exposed WT mice (Student's t test).
We next evaluated the effect of bok deficiency in a well characterized model of status epilepticus-induced neuronal injury in vivo (Engel et al., 2010; Murphy et al., 2010; Jimenez-Mateos et al., 2012). Status epilepticus was triggered by a unilateral microinjection of KA into the amygdala. In this model, the resulting seizure-induced cell death is mainly restricted to the ipsilateral hippocampal CA3 subfield (Murphy et al., 2010), in which Bok is known to be enriched (Lein et al., 2004, Newrzella et al., 2007). WT and bok-deficient mice were subjected to status epilepticus for 40 min, and, 72 h later, seizure-induced neuronal death was examined (Fig. 2B). Analysis of the total seizure power and frequency showed comparable high-amplitude and high-frequency spikes during (Fig. 2C–E) and after seizure [total power 2 h after lorazepam recording: 16.253 ± 5.3 and 10.96 ± 2.57 μV2 × 103 in WT (n = 7) and bok−/− (n = 5) mice, respectively; p = 0.44, Student's t test; frequency 2 h after lorazepam recording: 10.5 ± 1.34 and 10.49 ± 1.16 Hz in WT (n = 7) and bok−/− (n = 5) mice, respectively; p = 0.99, Student's t test] in both genotypes, excluding the possibility that any variation in damage may be attributable to altered susceptibility to KA or seizure severity. As expected, status epilepticus caused damage mostly to the ipsilateral CA3 subfield, whereas hippocampal neurons in the CA1 and CA2 subfields were essentially spared (Fig. 2F). Interestingly, mice lacking bok displayed more damage to the hippocampus compared with their WT control mice (Fig. 2F,G). This was most prominent in the CA3 region, as evidenced by the analysis of the neurodegenerative marker FJB in either the dorsal (Fig. 2F,H) or ventral (Fig. 2F,I) CA3 hippocampal subdomains. Together, these data suggest that Bok has a neuroprotective rather than pro-death role during OGD- and seizure-induced neuronal injury.
bok does not substitute for bax deficiency
Although the above experiments suggested that bok was not a mediator of neuronal apoptosis or Ca2+- and seizure-induced neuronal injury, the possibility remained that any bok requirement for cell death was masked by the simultaneous presence of bax. To address this hypothesis, we crossbred bax- and bok-deficient mice to generate bax/bok double-deficient mice. Again, bak-deficient mice were not included in this crossbreeding program because Bak is translationally repressed, and its splice variant, N-Bak, is anti-apoptotic in mature neurons (Sun et al., 2001; Jakobson et al., 2012). Indeed, several studies demonstrated that deletion of the bax gene is sufficient to confer protection against numerous apoptotic stimuli in cultured neurons in vitro, including neurotrophic factor deprivation (Deckwerth et al., 1996; Deshmukh and Johnson, 1998) and DNA damage-induced neuronal apoptosis (Xiang et al., 1998). Western blot analysis was performed to demonstrate bax/bok double-gene deficiency in cortical neurons (Fig. 3A) and to test whether bok deficiency influenced protein levels of other key Bcl-2 family proteins when bax is also deleted (Fig. 3B). bok deficiency did not affect the protein levels of the Bcl-2 family proteins, as determined by quantitative Western blot analysis, with the exception of Mcl-1, which was found to be significantly lower in bok-deficient neurons (Fig. 3B). Next, we focused our investigations on STS-, NMDA-, and OGD-induced neuronal injury. Exposure of bax/bok double-deficient cortical neurons to STS, a transient and brief NMDA exposure (100 μm/5 min), or a more severe NMDA exposure (300 μm/60 min) significantly increased neuronal death compared with the bax-deficient control neurons (Fig. 3C). bax/bok double-deficient cortical neurons exposed to OGD for 90 min also displayed significantly increased neuronal death compared with the bax-deficient control neurons (Fig. 3D), suggesting that bok did not substitute for bax in excitotoxic neuronal death and may exert protective effects in the absence of bax.
Figure 3.
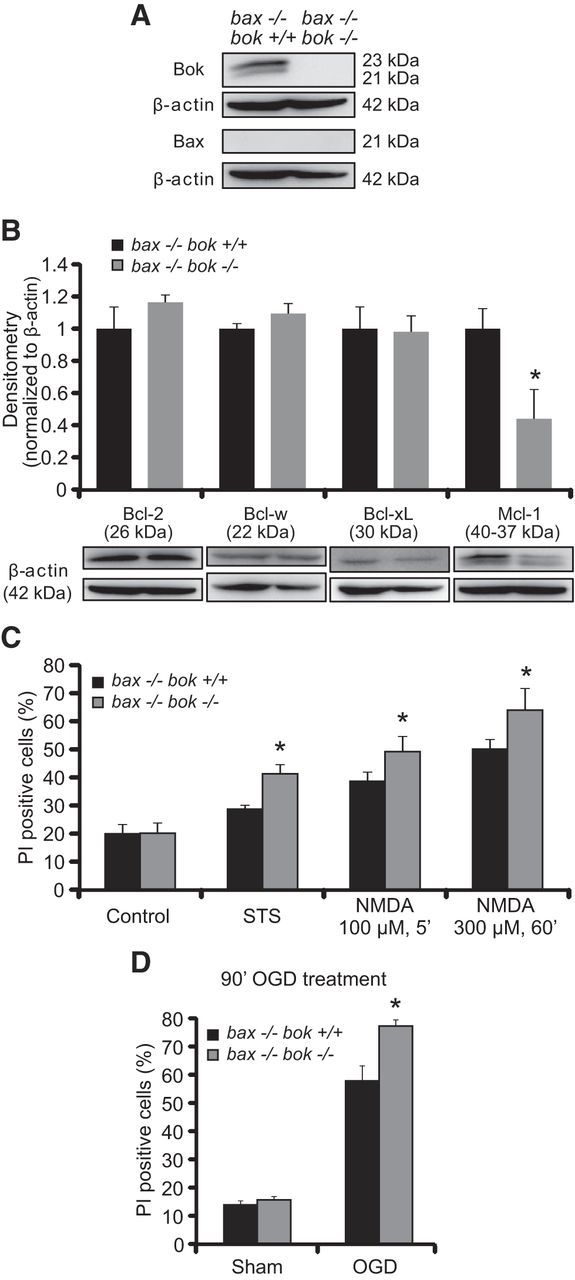
bok/bax double deficiency causes greater sensitivity to NMDA- and OGD-induced neuronal death. A, bax−/−/bok+/+ and bax−/−/bok−/− cortical neurons were used to confirm Bok and Bax protein absence in culture by Western blotting. β-Actin was used as a loading control. B, Western blot and densitometry analysis comparing the levels of several anti-apoptotic members of the Bcl-2 family proteins, including Bcl-2, Bcl-w, Bcl-xL, and Mcl-1 in bax−/−/bok+/+ and bax−/−/bok−/− cortical neurons. β-Actin was used as a loading control. Experiments were repeated three times with different preparations and with similar results. Means ± SEMs are shown. *p ≤ 0.05. p = 0.021 (Mcl-1) versus bax−/−/bok+/+ cultures (ANOVA, post hoc Tukey's test). C, D, Cortical neurons from bax−/−/bok+/+ and bax−/−/bok−/− mice were either treated with STS (100 nm) or exposed to the following: 100 μm NMDA for 5 min; 300 μm NMDA for 60 min; OGD for 90 min; or sham conditions for 90 min. Controls were the pool of DMSO and sham conditions for 24 h and 5 min, respectively. After treatments, cortical neurons were allowed to recover for 24 h. Cell death was assessed by Hoechst 33258 and PI staining, and PI-positive nuclei were scored as dead neurons and expressed as a percentage of the total population. Three subfields containing 300–400 neurons each were captured, and n = 9 wells were analyzed per condition from three separate cultures. Means ± SEMs are shown. *p ≤ 0.05. p values are the following: p = 0.0011 (STS), p = 0.0092 (NMDA at 100 μm, 5 min), p = 0.0053 (NMDA at 300 μm, 60 min), p = 0.018 (OGD) versus bax−/−/bok+/+ cultures (ANOVA, post hoc Tukey's test).
bok-deficient neurons show an early and prolonged deregulation of neuronal Ca2+ homeostasis and reduced mitochondrial energetics
To investigate in more detail the possible mechanisms of increased or potentially altered cell death induced by bok deficiency, we turned to the more controlled environment of excitotoxic injury (Ward et al., 2007; D'Orsi et al., 2012, 2015). Because excitotoxic injury is primarily mediated by glutamate receptor-mediated Ca2+ influx (Hardingham and Bading, 2010), we exposed cortical neurons from WT and bok-deficient mice to 100 μm NMDA for 5 min and monitored intracellular Ca2+ and mitochondrial membrane potential (Δψm) changes by confocal imaging. Cells were loaded using the fluorescent calcium indicator Fluo-4 AM and the membrane-permeant cationic fluorescent probe TMRM and monitored during and after NMDA exposure. Exposure of WT neurons to NMDA resulted in a significant increase in intracellular Ca2+; however, this returned to baseline levels 30 min after NMDA excitation (Fig. 4A,C). WT neurons maintained their intracellular Ca2+ homeostasis for up to 6 h (Fig. 4A,C,D), after which dying neurons underwent a delayed Ca2+ deregulation, followed by Δψm depolarization and plasma membrane permeabilization, as described previously (data not shown; Ward et al., 2007; D'Orsi et al., 2012, 2015). bok-deficient neurons, although displaying comparable baseline intracellular Ca2+ levels compared with their WT controls (7.324 ± 1.116 and 5.954 ± 0.678 arbitrary Fluo-4 fluorescence units in WT and bok−/− neurons, data from n = 178 and n = 247 neurons in 9 and 12 experiments, respectively; p = 0.2832, Student's t test), exhibited decreased Ca2+ levels at the point of NMDA exposure (Fig. 4B,C). Despite reduced Ca2+ levels during the NMDA exposure, bok-deficient neurons failed to recover their Ca2+ homeostasis after termination of the NMDA excitation (Fig. 4B–D). bok-deficient neurons displayed this prolonged Ca2+ dysregulation until neurons succumbed to cell death, as evidenced by complete neuronal TMRM loss (data not shown).
Figure 4.
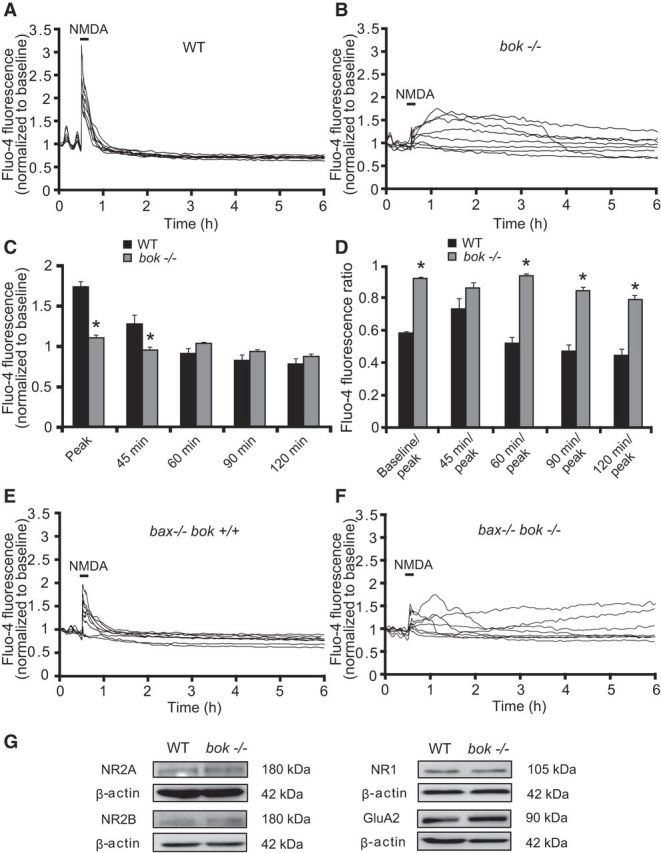
bok-deficient neurons show early and prolonged Ca2+ deregulation. WT, bok−/−, bax−/−/bok+/+, and bax−/−/bok−/− cortical neurons cultured separately on Willco dishes were preloaded with TMRM (20 nm) and Fluo-4 AM (3 μm) for 30 min at 37°C before being monitored by a confocal microscope (LSM 510). Neurons were exposed to 100 μm NMDA for 5 min, after which alterations in Δψm and intracellular Ca2+ were monitored in single cells over a 24 h period. A, B, Representative traces of NMDA-treated WT and bok−/− cortical neurons depicting the extent of peak Ca2+ influx at point of stimulation (100 μm/5 min NMDA) and maintained calcium homeostasis (A) or early and prolonged calcium deregulation (B) after the initial excitotoxic stimulus. C, D, Analysis of the Fluo-4 AM fluorescence at the indicated time points (C) and the relationship between the indicated time points and peak initial Fluo-4 AM fluorescence at NMDA exposure point (D) of WT (n = 178) and bok−/− (n = 246) cortical neurons from n = 9 and n = 12 separate cultures, respectively, are quantified. Data are means ± SEMs. *p ≤ 0.05. p values are the following: p = 0.00004 (Peak), p = 0.0415 (45 min), p = 0.00016 (Baseline/peak), p = 0.0705 (45 min/peak; NS), p = 0.00034 (60 min/peak), p = 0.00006 (90 min/peak), p = 0.00016 (120 min/peak) versus WT cultures (ANOVA, post hoc Tukey's test). E, F, Representative traces of NMDA-treated bax−/−/bok+/+ and bax−/−/bok−/− cortical neurons depicting the extent of peak Ca2+ influx at the point of stimulation (100 μm/5 min NMDA) and maintained calcium homeostasis (E) or partial calcium deregulation (F) after the initial excitotoxic stimulus. G, Western blot analysis comparing the levels of NMDA receptors NR2A, NR2B, and NR1 and the AMPA receptor GluA2 in WT and bok−/− neurons. β-Actin was used as a loading control. Experiments were repeated three times with three separate cultures with similar results.
Similar effects of bok deficiency on neuronal Ca2+ handling were also observed in a bax-deficient background. In agreement with our previous report, bax-deficient neurons displayed reduced neuronal Ca2+ levels during NMDA excitation (D'Orsi et al., 2015). bax/bok double-deficient neurons also showed decreased Ca2+ transients during the period of NMDA excitation (Fig. 4E,F). After NMDA stimulation, intracellular Ca2+ promptly returned to baseline levels in bax-deficient/bok-proficient neurons, whereas bax/bok double-deficient neurons displayed a Ca2+ dysregulation after excitation (Fig. 4E,F). Although Ca2+ deregulation was attenuated when compared with the previous experiments, quantification of Ca2+ levels in bax-deficient/bok-proficient versus bax/bok double-deficient neurons revealed significant differences after termination of the NMDA exposure (baseline/peak, 0.893 and 0.953 for bax−/−/bok+/+ and bax−/−/bok−/−, n = 121 and n = 144 neurons in 10 and 8 experiments, respectively; p = 0.045, Student's t test) and after 60 min (60 min/peak, 0.813 ± 0.072 and 0.894 ± 0.022 for bax−/−/bok+/+ and bax−/−/bok−/−, n = 121 and n = 144 neurons in 10 and 8 experiments, respectively; p = 0.027, Student's t test).
Western blotting confirmed that bok deficiency did not alter protein levels of the NMDA receptor subunits NR2A, NR2B, and NR1 or the AMPA receptor GluA2 subunit (Fig. 4G).
In parallel, we also monitored mitochondrial membrane potential dynamics using TMRM in WT and bok-deficient cortical neurons. WT and bok-deficient neurons showed similar baseline TMRM fluorescence levels (Fig. 5D). NMDA excitation induced a similar loss of TMRM fluorescence in WT and bok−/− neurons (Fig. 5A,B,E); however, whereas WT neurons exhibited a significant Δψm repolarization/hyperpolarization within 1–2 h (Fig. 5A,E), bok-deficient neurons displayed no recovery to Δψm, as evidenced by persisting low levels of TMRM fluorescence (Fig. 5B,E). These alterations in mitochondrial energetics were paralleled by morphological changes, such as swelling of neuronal somata and nuclear pyknosis, as evidenced by differential interface contrast (DIC) images (Fig. 5C). After additional analysis of the TMRM fluorescence, we identified that neurons tolerant to transient NMDA excitation had a significantly higher TMRM fluorescence than neurons that underwent cell death in either WT and bok-deficient neurons (Fig. 5F). However, the extent of the hyperpolarization of Δψm after NMDA excitation was more closely associated with neuronal survival in WT rather than bok-deficient neurons (Fig. 5F).
Figure 5.
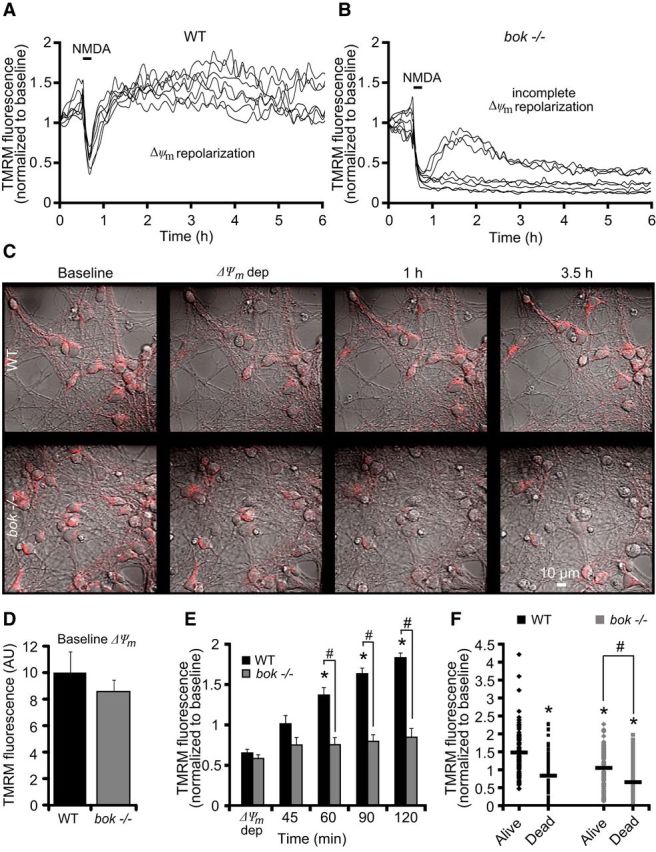
bok-deficient neurons fail to recover their mitochondrial membrane potential in response to NMDA. WT and bok−/− cortical neurons, preloaded with TMRM (20 nm) and Fluo-4AM (3 μm), were exposed to NMDA (100 μm/5 min NMDA) and monitored by confocal microscopy (LSM 510). A, B, Representative TMRM traces measuring alterations in mitochondrial membrane potential (Δψm) of NMDA-treated WT and bok−/− cortical neurons. WT neurons showed a Δψm repolarization after the initial NMDA excitation (A). bok−/− neurons showed an incomplete Δψm repolarization to basal levels even at later times after excitation (B). C, TMRM fluorescence in WT and bok−/− cortical neurons plated on Willco dishes was monitored over time (before, during, and after NMDA excitation). DIC and TMRM fluorescent images were chosen at selected time points (baseline, Δψm depolarization, 1 h, and 3.5 h) from a representative experiment. D, E, Analysis of baseline levels of Δψm (D) and average of TMRM fluorescence in WT (n = 178) and bok−/− (n = 246) neurons during and after NMDA excitation are represented. A significant increase in the whole-cell TMRM fluorescence of the WT neurons was identified within a 1–2 h period after NMDA excitation. In contrast, bok gene deletion did not produce a considerable repolarization of the Δψm. Means ± SEMs are shown from at least three independent experiments for each genotype. *p ≤ 0.05. p values are the following: p = 0.015 (60 min), p = 0.007 (90 min), p = 0.00008 (120 min) versus Δψm WT cultures (ANOVA, post hoc Tukey's test). #p ≤ 0.05. p values are the following: p = 0.0055 (60 min), p = 0.00002 (90 min), p = 0.00004 (120 min) versus WT cultures (ANOVA, post hoc Tukey's test). F, Whole-cell TMRM fluorescence in NMDA-treated WT (n = 178) and bok−/− (n = 246) neurons at the 120 min time point dividing neurons undergoing cell death from neurons that survive for >24 h after NMDA excitation. *p ≤ 0.05. p values are the following: p = 3.11E-13 (dead, WT), p = 0.0000001 (alive, bok−/−), p = 2.59E-13 (dead, bok−/−) versus alive neuron WT cultures (ANOVA, post hoc Tukey's test). #p ≤ 0.05. p = 1.16E-07 (dead, bok−/−) versus alive neuron bok−/− cultures (ANOVA, post hoc Tukey's test).
mcl-1 overexpression rescues deregulation of mitochondrial energetics in bok-deficient neurons
We demonstrated that bok deficiency induced a prominent reduction in Mcl-1 protein levels in bax/bok double-deficient neurons (Fig. 3B), an effect that we also observed in bok-deficient neurons when compared with WT neurons (data not shown). Moreover, Mcl-1 has been shown previously to modulate mitochondrial bioenergetics and normalize Ca2+ handling and to improve bioenergetics (Perciavalle et al., 2012; Anilkumar et al., 2013). Therefore, we next explored whether Mcl-1 overexpression rescued the defects in Ca2+ handling and mitochondrial bioenergetics in the bok-deficient cortical neurons. To address this, bok-deficient neurons were transfected with a plasmid expressing mcl-1 and cotransfected with an ECFP-expressing plasmid (bok −/− + mcl-1) or only transfected with an empty vector (bok −/− + CFP). Analysis of bok-deficient neurons transfected with the mcl-1-overexpressing plasmid revealed significantly higher cytosolic Ca2+ levels in response to the NMDA challenge compared with the control transfected bok-deficient neurons at the point of NMDA exposure (Fig. 6A,B,E). Furthermore, quantification of the individual Δψm changes showed that bok-deficient, mcl-1-transfected neurons displayed significant recovery of Δψm dynamics after NMDA-induced depolarization, as evidenced by increasing levels of TMRM fluorescence compared with the control transfected bok-deficient neurons (Fig. 6C,D,F).
Figure 6.
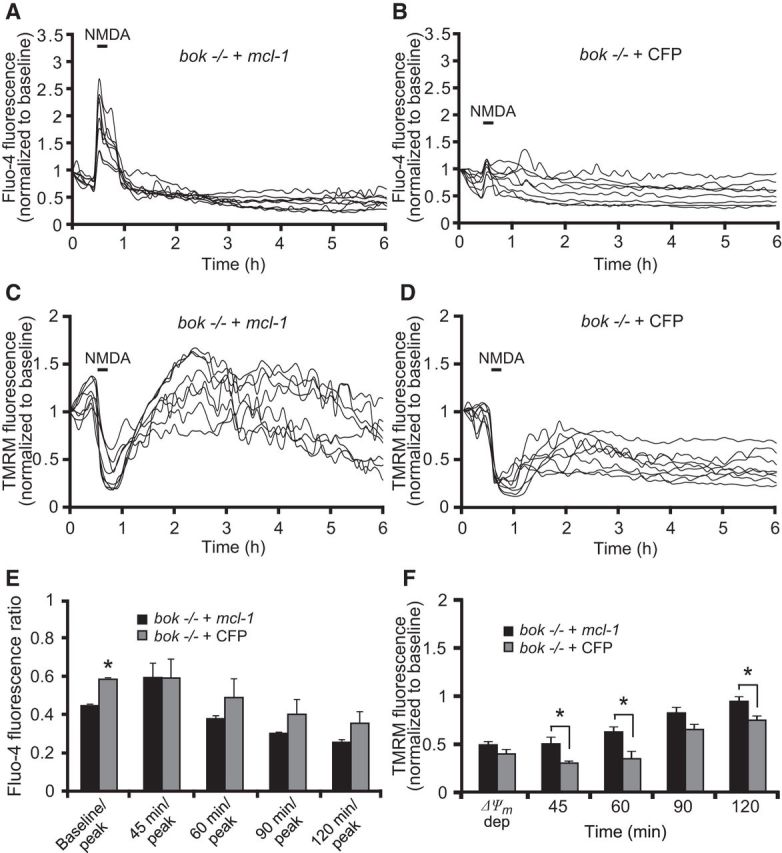
Mcl-1 overexpression rescues the mitochondrial phenotype in bok-deficient cortical neurons. bok−/− cortical neurons, transfected with an Mcl-1-overexpressing plasmid or a control empty vector (ECFP) for 48 h and coloaded with TMRM (20 nm) and Fluo-4 AM (3 μm), were exposed to NMDA (100 μm/5 min NMDA) and monitored by confocal microscopy (LSM 710). A, B, Representative Fluo4-AM traces measuring alterations in intracellular Ca2+ influx at the point of stimulation (100 μm/5 min NMDA) and after the initial excitotoxic stimulus. C, D, Representative TMRM traces measuring alterations in Δψm of NMDA-treated bok−/− cortical neurons transfected with Mcl-1-overexpressing plasmid (C) or empty plasmid (D). E, Analysis of the relationship of Fluo-4 fluorescence at the indicated time points compared with peak Fluo-4 AM fluorescence at NMDA exposure. bok−/− cortical neurons transfected with Mcl-1 overexpressing plasmid: n = 56 neurons from n = 6 separate cultures. Empty vector: n = 45 neurons from n = 5 separate cultures. Data are shown as means ± SEMs. *p ≤ 0.05. p values are the following: p = 0.012 (peak), p = 0.029 (Baseline/peak) versus overexpressing Mcl-1 bok−/− cultures (ANOVA, post hoc Tukey's test). F, Average of TMRM fluorescence in bok−/− cortical neurons transfected with Mcl-1-overexpressing plasmid (n = 56; from n = 6 separate cultures) or control vector (n = 45; from n = 5 separate cultures) during and after NMDA excitation. Means ± SEMs are shown. *p ≤ 0.05. p values are the following: p = 0.0035 (45 min), p = 0.0026 (60 min), p = 0.038 (120 min) versus overexpressing Mcl-1 bok−/− cultures (ANOVA, post hoc Tukey's test).
bok-deficient cortical neurons activate PARP-dependent cell death pathways
To investigate whether deficiency in bok promoted alternative cell death pathways in neurons exposed to NMDA (100 μm/5 min), we treated WT and bok-deficient cortical neurons with the selective calpain inhibitor Calpeptin (20 μm), the caspase inhibitor zVAD (100 μm), the necroptosis inhibitor Nec-1 (50 μm), and the selective PARP inhibitor DPQ (100 μm). We previously demonstrated that calpains are able to substitute for caspases in the execution of excitotoxic apoptosis (Lankiewicz et al., 2000; D'Orsi et al., 2012). Indeed, treatment with Calpeptin exerted significant neuroprotection in WT cortical neurons (Fig. 6A,B), whereas treatment with DPQ, zVAD, or Nec-1 showed no protection (Fig. 6A,B). Interestingly, whereas Calpeptin failed to protect bok-deficient neurons from excitotoxic injury (Fig. 6A,B), the PARP inhibitor DPQ provided significant neuroprotection, suggesting that deficiency in bok promoted the activation of PARP-dependent cell death pathways during NMDA excitotoxicity. Analysis of PARP activity in WT and bok-deficient neurons, using an ELISA that quantitatively determined the incorporation of poly ADP-ribose onto histone proteins, demonstrated that bok-deficient neurons had a significantly elevated PARP activity during excitotoxic injury when compared with WT neurons that was sensitive to DPQ (Fig. 7C). Previous studies demonstrated that PARP activation triggers strong nuclear pyknosis (Yu et al., 2002; Wang et al., 2004). Indeed, we found that nuclear pyknosis in response to NMDA was also blocked by DPQ in bok-deficient neurons (Fig. 6A).
Figure 7.
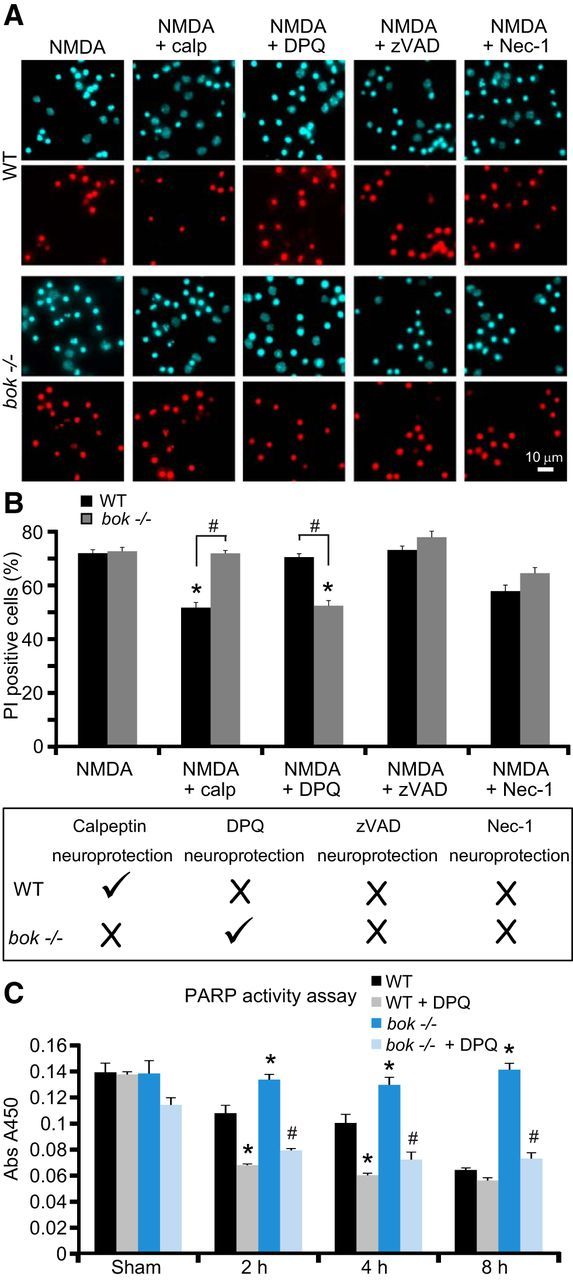
PARP inhibition provides significant neuroprotection in bok-deficient cortical neurons. A, B, Representative images of Hoechst-stained (in blue) and PI-stained (in red) neurons from WT and bok−/− mice exposed to 100 μm NMDA for 5 min in the presence and absence of Calpeptin (20 μm), DPQ (100 μm), zVAD (100 μm), or Nec-1 (50 μm). Images were taken 24 h after treatment. Cell death was assessed by Hoechst 33258 and PI staining (B, top). Three subfields containing 300–400 neurons each were captured, and n = 9 wells were analyzed per condition from three separate cultures. PI-positive nuclei were scored as dead neurons and expressed as a percentage of the total population. Data are means ± SEMs. *p ≤ 0.05. p values are the following: p = 0.0038 (NMDA + calp), p = 0.0053 (NMDA + DPQ) versus NMDA-treated WT cultures; p = 0.0075 (NMDA + DPQ) versus NMDA-treated bok−/− cultures. #p ≤ 0.05. p values are the following: p = 0.0032 (NMDA + calp), p = 0.0084 (NMDA + DPQ) between genotypes (ANOVA, post hoc Tukey's test). Schematic showing experimental results from the cell death assay (B, bottom). WT cortical neurons are sensitive to calpain inhibition, whereas bok-deficient neurons are sensitive to PARP inhibition. C, PARP activity measured as the amount of ribosylation on histone-coated plates in WT and bok−/− cortical neurons extracts exposed to 100 μm NMDA for 5 min in the presence and absence of DPQ (100 μm) and allowed to recover as indicated. Each point represents the mean value from triplicate experiments. Means ± SDs are shown. *p ≤ 0.05. p values are the following: p = 0.0037 (2 h, bok−/−), p = 0.0046 (4 h, bok−/−), p = 0.0001 (8 h, bok−/−), p = 0.0004 (2 h, WT + DPQ), p = 0.0005 (4 h, WT + DPQ) versus WT control extracts. #p ≤ 0.05. p values are the following: p = 0.0001 (2 h, bok−/− + DPQ), p = 0.0003 (4 h, bok−/− + DPQ), p = 0.0001 (8 h, bok−/− + DPQ) versus bok−/− extracts (ANOVA, post hoc Tukey's test).
Discussion
In this study, we set out to explore the role of bok in neuronal apoptosis, specifically in the setting of Ca2+- and seizure-induced neuronal cell death. Although Bok is highly expressed in neurons of the mouse brain, we found that deletion of bok failed to protect cortical neurons against several apoptosis-inducing stimuli. On the contrary, using models of OGD- and seizure-induced neuronal injury in vitro and in vivo, we demonstrated that bok-deficient neurons showed significantly increased neuronal injury. In the absence of bax, deletion of bok also increased STS-, excitotoxicity-, and OGD-induced cell death. Additional single-cell imaging experiments revealed that bok deficiency decreased neuronal Ca2+ homeostasis and mitochondrial energetics during excitotoxic injury through a downregulation of Mcl-1 protein levels, suggesting that the combined presence of Bok and Mcl-1 is required for the maintenance of mitochondrial energetics. In contrast to WT neurons that were sensitive to calpain inhibition, bok-deficient neurons maintained high levels of PARP activity and were sensitive to PARP inhibition, demonstrating that physiological Bok levels suppress the activation of PARP-dependent cell death pathways.
Based on its high amino acid sequence similarity, arrangement of BH domains, and findings from overexpression studies (Hsu et al., 1997a; Inohara et al., 1998; Yakovlev et al., 2004; Bartholomeusz et al., 2006; Rodriguez et al., 2006; Ke et al., 2012), Bok has been grouped into the pro-apoptotic Bax-like family of Bcl-2 proteins. Our data obtained from in vitro and in vivo studies in neurons suggest that Bok cannot be easily categorized into any of the current Bcl-2 family subgroups. Previous studies have already shown that bax deficiency in mature neurons or bax/bak double deficiency in other cell types is sufficient to completely block apoptosis in response to multiple pro-apoptotic stimuli (Deckwerth et al., 1996; Miller et al., 1997; Vekrellis et al., 1997; Xiang et al., 1998; Putcha et al., 1999; Wei et al., 2001; Cregan et al., 2002; D'Sa et al., 2003; Lindsten et al., 2003). These findings indirectly suggested that Bok is likely not a significant gatekeeper in the mitochondrial apoptosis pathway. Previous studies have also reported that mutated Bok (D76A) prevents Bok dimerization in HEK293T cells without affecting its killing capabilities, which suggests that Bok, unlike Bax and Bak, may induce cell death as a monomer when overexpressed (Echeverry et al., 2013). Although overexpression studies have revealed a pro-apoptotic effect of Bok (which primarily requires the presence of Bax and Bak; Echeverry et al., 2013), gene knock-out studies have so far provided little evidence for a pro-apoptotic role of Bok. Similar to our results obtained in bok-deficient neurons, bok-deficient hematopoietic cells have been shown to respond normally to several apoptosis-inducing stimuli (Ke et al., 2012; Echeverry et al., 2013). This was confirmed in a recent report in non-neuronal cells (Carpio et al., 2015), which determined that Bok was not required for STS-, etoposide-, and UV-induced apoptosis. However, the latter study demonstrated that bok-deficient mouse embryonic fibroblasts were selectively protected against ER stress-induced apoptosis. Because of its accumulation and unique functions at the ER (Echeverry et al., 2013; Schulman et al., 2013), it is possible that the activities of Bok during ER stress are not directly related to Bcl-2-dependent cell death signaling. Conversely, recent studies by Echeverry and coworkers did not observe reduced, but rather enhanced, ER stress-induced apoptosis in bok-deficient fibroblasts, mast cells, and myeloid progenitor cells (Echeverry et al., 2013; Fernandez-Marrero et al., 2016).
bok-deficient mice and bax/bok and bak/bok double-deficient mice also develop normally and show no evidence of morphological or functional abnormalities (Ke et al., 2013). This contrasts strongly with the effects of bax and bak double deletion (Wei et al., 2001). In bax/bok double-deficient cortical neurons, we found that Bok did not substitute for Bax in inducing neuronal death in response to excitotoxicity- and OGD-induced neuronal injury. Of note, no compensatory upregulation of Bax protein levels in the absence of bok have been observed by Western blotting (Fig. 1). Interestingly, we also found that bok deficiency increased rather than decreased Ca2+-mediated excitotoxicity-, OGD-, and seizure-induced neuronal death in vitro and in vivo (Figs. 2, 3). Collectively, these data suggest that Bok cannot be placed unambiguously into the Bax-like Bcl-2 subfamily of proteins. Interestingly, a previous study also demonstrated a protective rather than pro-death role of bax in newborn mice infected with Sindbis virus (Lewis et al., 1999).
With regards to the in vivo component of this study, the focal-onset seizures induced by intra-amygdala KA are considered a clinically relevant model of status epilepticus, a neurological emergency associated with high mortality and morbidity attributable to brain damage (Shorvon, 2011). Hippocampal damage has been examined previously in mice lacking multi-BH domain Bcl-2 family members, including Bak (Fannjiang et al., 2003), Bad (Giménez-Cassina et al., 2012), Mcl-1 (Mori et al., 2004), and Bcl-w (Murphy et al., 2007). Bak in particular has been shown to also exert protective activities during seizure-induced injury, but the interpretation of those previous reports are complicated by altered electrographic seizures in the gene-deficient mice (Fannjiang et al., 2003; Murphy et al., 2007; Giménez-Cassina et al., 2012) and the need to use heterozygous mice (Mori et al., 2004). Our quantitative EEG analysis in bok-deficient mice excludes the observed phenotype being attributable to altered seizure susceptibility.
Another key finding of our experiments is that bok deficiency impaired intracellular neuronal Ca2+ handling during NMDA excitotoxicity (Fig. 4). Specifically, bok-deficient cortical neurons, similar to bax-deficient neurons (D'Orsi et al., 2015), displayed reduced intracellular Ca2+ transients during NMDA exposure. However, diverging from neurons lacking bax, which maintained their Ca2+ homeostasis for longer periods and underwent delayed calcium deregulation at much lower frequency than WT control neurons (D'Orsi et al., 2015), bok-deficient neurons exhibited an early and prolonged Ca2+ dysregulation after NMDA excitation (Fig. 4). In addition to ATP-dependent Ca2+ extrusion into the extracellular space, neurons also restore their intracellular Ca2+ homeostasis through mitochondrial and ER Ca2+ uptake (Hajnóczky et al., 1999; Ward et al., 2005; Rizzuto et al., 2012). Bok has been suggested to play an important role in regulating Ca2+ fluxes between the cytosol and the ER, and potentially Ca2+ fluxes between the ER and mitochondria (Hajnóczky et al., 1999; Chipuk et al., 2010; Esterberg et al., 2014). However, we noted no differences in IP3R2 protein levels and in the IP3R2 proteolytic cleavage in WT compared with bok-deficient neurons (data not shown). Rather, we found that bok deficiency resulted in reduced neuronal Mcl-1 protein levels, as seen by Western blot analysis (Fig. 3). It has been reported previously that Bok binds tightly to Mcl-1 (Hsu et al., 1997a; Inohara et al., 1998). Indeed, Mcl-1 is primarily regulated at the post-transcriptional level and has a short protein half-life, attributed partly to proteasome-mediated degradation (Wang et al., 1999; Wei et al., 2001; Nijhawan et al., 2003). Therefore, it is possible that liberation from Bok binding releases Mcl-1 for degradation and that reduced Mcl-1 protein levels in the bok-deficient neurons contributed to the sensitization to OGD- or seizure-induced injury. Mcl-1 overexpression has been shown to protect neurons against excitotoxic/ischemic hypoxic injury (Anilkumar et al., 2013). Deletion of mcl-1 results in increased neuronal death (Mori et al., 2004; Arbour et al., 2008; Malone et al., 2012) and autophagy activation under energetic stress conditions, indicating that Mcl-1 is vital for neuronal development and also in the adult CNS (Germain et al., 2011). In addition, we have shown previously that Mcl-1 overexpression increases neuronal Ca2+ handling and improves mitochondrial bioenergetics during NMDA excitation (Anilkumar et al., 2013). We here demonstrate that mcl-1 overexpression rescued mitochondrial bioenergetics defects observed in the bok-deficient phenotype (Fig. 6), suggesting that Mcl-1 mediates the effects of Bok on mitochondrial bioenergetics and that the combined presence of Bok and Mcl-1 is required for neuronal Ca2+ handling during excitotoxicity.
Interestingly, mcl-1 mRNA is alternatively spliced, a process that leads to the expression of two different Mcl-1 proteins that reside in the mitochondrial outer membrane and mitochondrial matrix (Mcl-1MOM and Mcl-1matrix), respectively. The latter variant, Mcl-1matrix, has been proposed to improve mitochondrial bioenergetics (Perciavalle et al., 2012). The reduced Δψm observed in bok-deficient cortical neurons indicated the existence of such a mitochondrial defect. A reduced Δψm will result in reduced mitochondrial Ca2+ uptake and pose a significant burden on cellular energetics. Furthermore, our study indicated that neurons lacking bok maintained high levels of PARP activity during excitotoxic injury and were protected from excitotoxic injury when pretreated with the PARP inhibitor DPQ (Fig. 7). During excitotoxicity, intracellular Ca2+ overloading induces PARP activation, which depletes NAD+, resulting in a neuronal energy imbalance and further causing ATP consumption and neuronal death (Ying et al., 2001). Hence, whereas WT neurons undergo cell death in response to transient NMDA excitation by undergoing a transcription- and Bax-dependent excitotoxic apoptosis after the re-establishment of mitochondrial bioenergetics (Concannon et al., 2010; D'Orsi et al., 2012), bok-deficient neurons do not re-establish their energetics after transient NMDA exposure and undergo PARP-dependent cell death associated with cell swelling and strong nuclear pyknosis. Collectively, these data indicate that bok deletion disrupts neuronal Ca2+ homeostasis and mitochondrial bioenergetics in response to NMDA excitation, leading to the enhanced sensitivity of bok-deficient neurons to Ca2+- and PARP-mediated neuronal injury.
In conclusion, our data redefine the role of the Bcl-2 family protein Bok as a cell death regulator in neurons. We show that neuronal Bok suppresses PARP-dependent cell death pathways and exerts protective activities during OGD-induced neuronal injury in vitro and seizure-induced neuronal injury in vivo. Our data also demonstrate that Bok plays a key role in the regulation of neuronal Ca2+ homeostasis and mitochondrial energetics and delineates a key role for Mcl-1 depletion in this process. Therefore, our study highlights the emerging role of Bcl-2 proteins in the control of neuronal “daytime” functions.
Footnotes
This work was generously supported by Science Foundation Ireland Grants 08/IN.1/B1949, 13/IA/1881, and 14/JP-ND/B3077 (J.H.M.P.) and Health Research Board Grant HRA_POR/2011/41 (T.E. and D.C.H.). We thank Prof. Andreas Strasser (WEHI, Melbourne, Australia) for the gift of bok-deficient mice, Dr. Janis Noonan for proofreading this manuscript, and Ina Woods for excellent technical support.
The authors declare no conflict of interest.
References
- Akhtar RS, Geng Y, Klocke BJ, Roth KA. Neural precursor cells possess multiple p53-dependent apoptotic pathways. Cell Death Differ. 2006;13:1727–1739. doi: 10.1038/sj.cdd.4401879. [DOI] [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar U, Prehn JH. Anti-apoptotic BCL-2 family proteins in acute neural injury. Front Cell Neurosci. 2014;8:281. doi: 10.3389/fncel.2014.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar U, Weisová P, Düssmann H, Concannon CG, König HG, Prehn JH. AMP-activated protein kinase (AMPK)-induced preconditioning in primary cortical neurons involves activation of MCL-1. J Neurochem. 2013;124:721–734. doi: 10.1111/jnc.12108. [DOI] [PubMed] [Google Scholar]
- Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC, Kelly MA, MacKenzie AE, Park DS, Opferman JT, Slack RS. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28:6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autret A, Martin SJ. Bcl-2 family proteins and mitochondrial fission/fusion dynamics. Cell Mol Life Sci. 2010;67:1599–1606. doi: 10.1007/s00018-010-0286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeusz G, Wu Y, Ali Seyed M, Xia W, Kwong KY, Hortobagyi G, Hung MC. Nuclear translocation of the pro-apoptotic Bcl-2 family member Bok induces apoptosis. Mol Carcinog. 2006;45:73–83. doi: 10.1002/mc.20156. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/S0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Bonner HP, Concannon CG, Bonner C, Woods I, Ward MW, Prehn JH. Differential expression patterns of Puma and Hsp70 following proteasomal stress in the hippocampus are key determinants of neuronal vulnerability. J Neurochem. 2010;114:606–616. doi: 10.1111/j.1471-4159.2010.06790.x. [DOI] [PubMed] [Google Scholar]
- Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2015;112:7201–7206. doi: 10.1073/pnas.1421063112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon CG, Tuffy LP, Weisová P, Bonner HP, Dávila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/S0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/S0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- D'Orsi B, Bonner H, Tuffy LP, Düssmann H, Woods I, Courtney MJ, Ward MW, Prehn JH. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci. 2012;32:1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orsi B, Kilbride SM, Chen G, Perez Alvarez S, Bonner HP, Pfeiffer S, Plesnila N, Engel T, Henshall DC, Düssmann H, Prehn JH. Bax regulates neuronal Ca2+ homeostasis. J Neurosci. 2015;35:1706–1722. doi: 10.1523/JNEUROSCI.2453-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Sa C, Klocke BJ, Cecconi F, Lindsten T, Thompson CB, Korsmeyer SJ, Flavell RA, Roth KA. Caspase regulation of genotoxin-induced neural precursor cell death. J Neurosci Res. 2003;74:435–445. doi: 10.1002/jnr.10738. [DOI] [PubMed] [Google Scholar]
- Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, Kaufmann T. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 2013;20:785–799. doi: 10.1038/cdd.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Caballero-Caballero A, Schindler CK, Plesnila N, Strasser A, Prehn JH, Henshall DC. BH3-only protein Bid is dispensable for seizure-induced neuronal death and the associated nuclear accumulation of apoptosis-inducing factor. J Neurochem. 2010;115:92–101. doi: 10.1111/j.1471-4159.2010.06909.x. [DOI] [PubMed] [Google Scholar]
- Engel T, Plesnila N, Prehn JH, Henshall DC. In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:1196–1210. doi: 10.1038/jcbfm.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012;26:1616–1628. doi: 10.1096/fj.11-196089. [DOI] [PubMed] [Google Scholar]
- Esterberg R, Hailey DW, Rubel EW, Raible DW. ER-mitochondrial calcium flow underlies vulnerability of mechanosensory hair cells to damage. J Neurosci. 2014;34:9703–9719. doi: 10.1523/JNEUROSCI.0281-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Dike S, Sappington AL, Kerr DA, Jonas EA, Kaczmarek LK, Hardwick JM. BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell. 2003;4:575–585. doi: 10.1016/S1534-5807(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marrero Y, Ke F, Echeverry N, Bouillet P, Bachmann D, Strasser A, Kaufmann T. Is BOK required for apoptosis induced by endoplasmic reticulum stress? Proc Natl Acad Sci U S A. 2016;113:E492–E493. doi: 10.1073/pnas.1516347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene. 2008;27(Suppl 1):S128–S136. doi: 10.1038/onc.2009.50. [DOI] [PubMed] [Google Scholar]
- Giménez-Cassina A, Martínez-Francois JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74:719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+ J Biol Chem. 1999;274:14157–14162. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A. 1997a;94:12401–12406. doi: 10.1073/pnas.94.23.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997b;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanuka H, Inohara N, Sawamoto K, Núñez G, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci U S A. 2000;97:662–667. doi: 10.1073/pnas.97.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Ekhterae D, Garcia I, Carrio R, Merino J, Merry A, Chen S, Núñez G. Mtd, a novel Bcl-2 family member activates apoptosis in the absence of heterodimerization with Bcl-2 and Bcl-XL. J Biol Chem. 1998;273:8705–8710. doi: 10.1074/jbc.273.15.8705. [DOI] [PubMed] [Google Scholar]
- Jakobson M, Lintulahti A, Arumäe U. mRNA for N-Bak, a neuron-specific BH3-only splice isoform of Bak, escapes nonsense-mediated decay and is translationally repressed in the neurons. Cell Death Dis. 2012;3:e269. doi: 10.1038/cddis.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, Sano T, O'Tuathaigh C, Waddington JL, Prenter S, Delanty N, Farrell MA, O'Brien DF, Conroy RM, Stallings RL, DeFelipe J, Henshall DC. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med. 2012;18:1087–1094. doi: 10.1038/nm.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, Kaufmann T. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 2012;19:915–925. doi: 10.1038/cdd.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, Voss AK. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013;4:e650. doi: 10.1038/cddis.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbride SM, Prehn JH. Central roles of apoptotic proteins in mitochondrial function. Oncogene. 2013;32:2703–2711. doi: 10.1038/onc.2012.348. [DOI] [PubMed] [Google Scholar]
- Krohn AJ, Preis E, Prehn JH. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998;18:8186–8197. doi: 10.1523/JNEUROSCI.18-20-08186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Lein ES, Zhao X, Gage FH. Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci. 2004;24:3879–3889. doi: 10.1523/JNEUROSCI.4710-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med. 1999;5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003;23:11112–11119. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Malone CD, Hasan SM, Roome RB, Xiong J, Furlong M, Opferman JT, Vanderluit JL. Mcl-1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci. 2012;49:439–447. doi: 10.1016/j.mcn.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Burgess DL, Gefrides LA, Foreman PJ, Opferman JT, Korsmeyer SJ, Cavalheiro EA, Naffah-Mazzacoratti MG, Noebels JL. Expression of apoptosis inhibitor protein Mcl1 linked to neuroprotection in CNS neurons. Cell Death Differ. 2004;11:1223–1233. doi: 10.1038/sj.cdd.4401483. [DOI] [PubMed] [Google Scholar]
- Murphy BM, Engel T, Paucard A, Hatazaki S, Mouri G, Tanaka K, Tuffy LP, Jimenez-Mateos EM, Woods I, Dunleavy M, Bonner HP, Meller R, Simon RP, Strasser A, Prehn JH, Henshall DC. Contrasting patterns of Bim induction and neuroprotection in Bim-deficient mice between hippocampus and neocortex after status epilepticus. Cell Death Differ. 2010;17:459–468. doi: 10.1038/cdd.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B, Dunleavy M, Shinoda S, Schindler C, Meller R, Bellver-Estelles C, Hatazaki S, Dicker P, Yamamoto A, Koegel I, Chu X, Wang W, Xiong Z, Prehn J, Simon R, Henshall D. Bcl-w protects hippocampus during experimental status epilepticus. Am J Pathol. 2007;171:1258–1268. doi: 10.2353/ajpath.2007.070269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newrzella D, Pahlavan PS, Krüger C, Boehm C, Sorgenfrei O, Schröck H, Eisenhardt G, Bischoff N, Vogt G, Wafzig O, Rossner M, Maurer MH, Hiemisch H, Bach A, Kuschinsky W, Schneider A. The functional genome of CA1 and CA3 neurons under native conditions and in response to ischemia. BMC Genomics. 2007;8:370. doi: 10.1186/1471-2164-8-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Lankiewicz S, Poppe M, Prehn JH. Ca(2+)-induced inhibition of apoptosis in human SH-SY5Y neuroblastoma cells: degradation of apoptotic protease activating factor-1 (APAF-1) J Neurochem. 2001;78:1256–1266. doi: 10.1046/j.1471-4159.2001.00503.x. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rodriguez JM, Glozak MA, Ma Y, Cress WD. Bok, Bcl-2-related ovarian killer, is cell cycle-regulated and sensitizes to stress-induced apoptosis. J Biol Chem. 2006;281:22729–22735. doi: 10.1074/jbc.M604705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013;(71):50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/S0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schulman JJ, Wright FA, Kaufmann T, Wojcikiewicz RJ. The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J Biol Chem. 2013;288:25340–25349. doi: 10.1074/jbc.M113.496570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon S. The treatment of status epilepticus. Curr Opin Neurol. 2011;24:165–170. doi: 10.1097/WCO.0b013e3283446f31. [DOI] [PubMed] [Google Scholar]
- Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–16247. doi: 10.1074/jbc.M010419200. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Sawada M, Yamamoto H, Marunouchi T. Effects of colony stimulating factors on isolated microglia in vitro. J Neuroimmunol. 1990;30:111–120. doi: 10.1016/0165-5728(90)90094-4. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tuffy LP, Concannon CG, D'Orsi B, King MA, Woods I, Huber HJ, Ward MW, Prehn JH. Characterization of Puma-dependent and Puma-independent neuronal cell death pathways following prolonged proteasomal inhibition. Mol Cell Biol. 2010;30:5484–5501. doi: 10.1128/MCB.00575-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, McCarthy MJ, Watson A, Whitfield J, Rubin LL, Ham J. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development. 1997;124:1239–1249. doi: 10.1242/dev.124.6.1239. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/MCB.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW, Kögel D, Prehn JH. Neuronal apoptosis: BH3-only proteins the real killers? J Bioenerg Biomembr. 2004;36:295–298. doi: 10.1023/B:JOBB.0000041756.23918.11. [DOI] [PubMed] [Google Scholar]
- Ward MW, Kushnareva Y, Greenwood S, Connolly CN. Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J Neurochem. 2005;92:1081–1090. doi: 10.1111/j.1471-4159.2004.02928.x. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisová P, Düssmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Di Giovanni S, Wang G, Liu W, Stoica B, Faden AI. BOK and NOXA are essential mediators of p53-dependent apoptosis. J Biol Chem. 2004;279:28367–28374. doi: 10.1074/jbc.M313526200. [DOI] [PubMed] [Google Scholar]
- Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci U S A. 2001;98:12227–12232. doi: 10.1073/pnas.211202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhang H, Holzgreve W, De Geyter C. Evolutionarily conserved Bok proteins in the Bcl-2 family. FEBS Lett. 2000;480:311–313. doi: 10.1016/S0014-5793(00)01921-9. [DOI] [PubMed] [Google Scholar]