

ABSTRACT
The mammalian embryo’s caudal lateral epiblast (CLE) harbours bipotent progenitors, called neural mesodermal progenitors (NMPs), that contribute to the spinal cord and the paraxial mesoderm throughout axial elongation. Here, we performed a single cell analysis of different in vitro NMP populations produced either from embryonic stem cells (ESCs) or epiblast stem cells (EpiSCs) and compared them with E8.25 CLE mouse embryos. In our analysis of this region, our findings challenge the notion that NMPs can be defined by the exclusive co-expression of Sox2 and T at mRNA level. We analyse the in vitro NMP-like populations using a purpose-built support vector machine (SVM) based on the embryo CLE and use it as a classification model to compare the in vivo and in vitro populations. Our results show that NMP differentiation from ESCs leads to heterogeneous progenitor populations with few NMP-like cells, as defined by the SVM algorithm, whereas starting with EpiSCs yields a high proportion of cells with the embryo NMP signature. We find that the population from which the Epi-NMPs are derived in culture contains a node-like population, which suggests that this population probably maintains the expression of T in vitro and thereby a source of NMPs. In conclusion, differentiation of EpiSCs into NMPs reproduces events in vivo and suggests a sequence of events for the emergence of the NMP population.
KEY WORDS: Neuromesodermal progenitors, Single cells, Transcription
Summary: Single cell analysis of mouse PSC in vitro-derived populations, using support vector machine-based comparison with embryo-derived cells, uncovers the origin and structure of neuro-mesodermal progenitors.
INTRODUCTION
In mammalian embryos, the trunk consists of the endoderm, the spinal cord and the derivatives of different kinds of mesoderm (axial, paraxial, intermediate and lateral plate). Much of our current understanding regarding the development of this body region has focused on two progenitor cell populations: the node, that will give rise to the axial mesoderm (Beddington, 1982; McGrew et al., 2008; Tam and Beddington, 1987) and the neural mesodermal progenitors (NMPs), a bipotent stem cell population that contributes to the spinal cord and the paraxial mesoderm (PXM) (Henrique et al., 2015; Selleck and Stern, 1991; Wilson et al., 2009). Both populations are closely related within the anterior region of the caudal epiblast (CE) in the embryo (Wymeersch et al., 2016). This association persists for as long as the node is visible, between embryonic day (E)7.5 and E9.0 (Fig. 1, Fig. S1; Wymeersch et al., 2016; Wymeersch et al., 2019; Yamanaka et al., 2007). It is not clear when the NMPs arise but their association with the node suggests that they might emerge at the same time, around E7.5, from a multipotent population (Edri et al., 2019); the NMP population must then proliferate to sustain the axial extension process. Absence of the node results in severe axial truncations (Ang and Rossant, 1994; Davidson and Tam, 2000; Weinstein et al., 1994), suggesting a relationship between the node and the establishment and maintenance of the NMPs. However, little is known about these interactions.
Fig. 1.

Organization and gene expression patterns in the E8.5 mouse embryo caudal region. Top, ventral view; bottom: lateral (a) and medial (b) views. The caudal region of the embryo is derived from the posterior epiblast of E7.5 (green in Fig. S1) when the primitive streak (pink) reaches the most distal region of the embryo and the node (purple) appears. This region proliferates and undergoes several morphogenetic events which lead to the organization visible at E8.5 and indicated in the figure. Bottom: marker genes that are expressed in each region are detailed. The sources for the outlines shown here can be found in Table S1 and (Edri et al., 2019).
The earliest identifiable NMPs emerge in the CE of E8.25 embryos distributed between the node streak border (NSB) and the caudal lateral epiblast (CLE) (Cambray and Wilson, 2007; Wymeersch et al., 2016, 2019). They are associated with the co-expression of T (Brachyury), Sox2 and NKx1-2 (Henrique et al., 2015; Steventon and Martinez Arias, 2017; Wilson et al., 2009). However, molecular analysis in embryos is limited, because of accessibility to primary material and the challenging temporal resolution. To circumvent these difficulties, over the last few years embryonic stem cells (ESCs) have emerged as a useful model for mammalian development. In the context of axial extension, it has been possible to generate NMPs in vitro from pluripotent stem cells (PSCs) (Edri et al., 2019; Gouti et al., 2014, 2017; Lippmann et al., 2015; Tsakiridis and Wilson, 2015; Turner et al., 2014). These studies provide large quantities of material and allow the study of details that are difficult to obtain in vivo, particularly the structure and the genetic profile of the NMP population. In these studies, it is important to establish the relationship between the in vitro and the in vivo populations. A recent study aiming to do this, using an ESC-based protocol, has established some features of an ESC-derived NMP population (Gouti et al., 2017).
Here, we perform a single cell analysis of different in vitro-derived populations, comparing them with those in the E8.25 embryo CLE (Ibarra-Soria et al., 2018; Pijuan-Sala et al., 2019), in which NMPs can be clearly observed (Cambray and Wilson, 2007; Wymeersch et al., 2016, 2019). We perform this analysis with a support vector machine (SVM) based on the reference CLE embryo data. We use the SVM as a classification model to analyse the different in vitro NMP-like populations and show that, whereas ESC-derived CLE-like populations are heterogeneous and contain few NMP-like cells, epiblast stem cell (EpiSC)-derived populations produce a high proportion of cells with the embryo NMP signature. Importantly, we find that Epi-CE, the population from which the Epi-NMPs are derived (Edri et al., 2019), contains a node-like population, and we show that this population can maintain the expression of T in vitro. Our results suggest a sequence of events for NMP emergence, which we discuss here.
RESULTS
To understand the complexity and identity of the cell populations that emerge when recapitulating NMPs in vitro and how they relate to the embryo CLE, we characterized these populations at a single cell level. We focused our study on the populations that we have previously described (Edri et al., 2019) and extracted mRNA from single cells of ES-NMP (Edri et al., 2019; Turner et al., 2014), Epi-CE and Epi-NMP (Edri et al., 2019), as well as of the T-expressing cells from the Epi-CE population (Epi-CE-T, see Materials and Methods). As a reference for the in vivo population, we used a gene expression dataset containing 7006 cells from E8.25 embryos (Ibarra-Soria et al., 2018; Pijuan-Sala et al., 2019). However, rather than using the complete dataset, we performed an in silico dissection of the caudal region of the embryo (Fig. 1). We selected cells that co-expressed Sox2 and T (putative NMPs) (Cambray and Wilson, 2007; Henrique et al., 2015; Koch et al., 2017; Tsakiridis et al., 2014; Wymeersch et al., 2016, 2019); cells that expressed Sox2 and Nkx1-2 but not T (preneural progenitors) (Henrique et al., 2015; Schubert et al., 1995); and cells that expressed T but not Sox2, Mixl1 or Bmp4, which represent mesodermal progenitors and exclude progenitors for the endoderm (Mixl1) and the allantois (Bmp4) (Dunty et al., 2014; Lawson et al., 1999; Robb et al., 2000; Wolfe and Downs, 2014). We refer to these three population as NMP, preNeuro and preMeso, respectively. The extraction process yielded 498 cells that represented the caudal region of the embryo (108 NMP cells, 133 preNeuro cells and 257 preMeso cells).
In vitro-derived populations reflect temporally overlapping embryonic populations
As a first step in our analysis we performed batch correction analysis between the embryo and the in vitro population datasets, based on the detection of mutual nearest neighbours (MNNs) in the high-dimensional expression space (Haghverdi et al., 2018; Figs S2-S3).
For the batch-corrected data we implemented the Seurat package (Butler et al., 2018; Stuart et al., 2018 preprint) to observe how the cells clustered together (Materials and Methods). Between two and eight clusters were tested and coloured according to the conditions and clusters, following the projected cells in tSNE plots (Fig. 2A,B). Seven clusters were chosen for the downstream analysis. The marker genes that distinguish between clusters are shown in Fig. S4. The tSNE plots in Fig. 2A,B allowed us to obtain a first approximation of the transcriptional complexity of the different samples. There is an overlap between the different NMP-like populations and also with the cells from the embryo in the dimensionally reduced gene space (Fig. 2B).
Fig. 2.

Visualization of the samples and their gene expression along the seven clusters. (A,B) tSNE plots coloured by seven detected clusters (A) and the sample names (B). Quantification of cell composition of clusters from different samples (B, right). (C) Expression of chosen marker genes of pluripotent state, CE (E7.5), CLE, neural, mesoderm and the node along the seven clusters. The genes are ordered according to hierarchical clustering.
Fig. 2B shows how each cluster relates to the different samples. Cluster 3 is composed mainly from Epi-CE-T, cluster 5 from ES-NMP and cluster 6 from E8.25 and Epi-CE-T. Using the major reference genes of the CLE gene expression signature (Figs 1, 2C and Fig. S5), we observed a spread in the markers expressed by the different populations which can be used to determine their identity. Cluster 5 contains cells that express pluripotent markers [Nanog, Rex1 (also known as Zfp42), Sox2, Esrrb, Fgf4], whereas cluster 3 exhibits cells with node identity (T, Foxa2, Nog, Chrd, Shh) and cluster 6 cells contains cells that express mesodermal markers (Tbx6, Cited1, Msx1 and Msx2), the range of Hox genes (Hox1-Hox9) and CE markers (Wnt3a, Fgf8, Cdx2, Cdx4, Cyp26a1). Clusters 0-2 are composed of cells belonging to the ES-NMP, Epi-CE, Epi-CE-T and Epi-NMP population, and exhibit some similarity to the gene expression profile of cluster 6.
Analysis of the gene expression patterns associated with each cluster (Fig. 2C and Fig. S5), revealed the heterogeneity of these populations, particularly in the ES-NMP sample, in which we can find cells with a mixed signature of pluripotency (Nanog, Rex1, Sox2, Esrrb, Fgf4), primed epiblast (Fgf5, Otx2 and Cdh1), a later epiblast population that expresses some CLE and NMP markers as well as cells with a neural identity and others with mixed mesodermal characteristic. Overlapping with the last population, we noticed a group of cells with mixed potential expressing Mixl1 and Fgf17 together with Evx1, Hoxb9, Oct4 (also known as Pou5f1) and Wnt genes, which might represent the posterior primitive streak population that will give rise to mesendodermal tissue (Dunty et al., 2014; Kojima et al., 2014; Robb et al., 2000; Wolfe and Downs, 2014). The heterogeneity of the ES-NMPs confirms the conclusion from our previous ensemble study (Edri et al., 2019) that differentiation in the absence of FGF leads to a highly heterogeneous and asynchronous population with some, but few, NMPs.
The Epi-NMP population is enriched in cells with expression profiles clearly associated with the E8.25-E8.5 embryo – expression of Cyp26a1 and Cdh2 – and an absence of Otx2, Oct4, Cdh1 and Fst, all of which are associated with earlier stages of the embryo (E7.5) (Fig. 2C and compare gene expression of E8.25 CLE embryo with Epi-NMP in Fig. S5). In vitro, Epi-NMPs are derived from Epi-CEs (Materials and Methods and Edri et al., 2019) which can explain how the expression of the different genes indicates a progression in the developmental stage from Epi-CE to Epi-NMP (early epiblast markers in Epi-CE versus CLE markers in Epi-NMP, Fig. S5). We also observed that Epi-NMP, but not Epi-CE, contains a few cells differentiated into mesoderm, as highlighted by the expression of Tbx6, Meox1 and Aldh1a2 (Fig. S5). Most surprisingly, we noted that the Epi-CE population, but not Epi-NMP, contained cells co-expressing genes that are associated with the node e.g. Nodal, Foxa2, Ccno, Chrd, Nog and Shh (Fig. S5). A similar population can also be found in the Epi-CE-T and suggests the presence of node-like cells in the Epi-CE population. These cells are reduced in the Epi-NMP population, following the characteristics of the E8.5 CLE (Fig. 1).
The above observations provide support for our conjecture that that Epi-CE and Epi-NMP correspond to temporally consecutive populations in the embryo, which probably reflect a spectrum between E7.5 [emergence of the node (Davidson and Tam, 2000), Epi-CE] and E8.25-E8.5 (Epi-NMP), when NMPs are clearly discernible (Wymeersch et al., 2016; Wymeersch et al., 2019). The temporal sequence can also be observed in the pattern of Hox genes expression, as the Epi-NMP population expresses more posterior Hox genes than the Epi-CE (Fig. S5).
The NMP landscape in the E8.25 embryo
To interpret the in vitro-derived cell populations, we used the caudal cells dissected in silico from the E8.25 embryo to build an SVM pipeline that would enable us to map the NMP-like cells to the in vivo CLE. As a first step, we attempted to identify phenotypically distinct populations amidst the three pools of cells that we defined based on their pattern of T, Sox2 and Nkx1-2 expression (Fig. 3A). After processing the single cell data for both the embryo and the in vitro samples, we found a total of 14,822 genes that can be used for the analysis (Materials and Methods). To provide identifier genes associated with the CLE region, we based our gene selection on the report from Koch et al. (2017), in which the authors perform an ensemble analysis of the caudal region of the E8.5 embryo based on the levels of Sox2 and T. This work identified 1402 genes that, together, provide specific signatures for five distinct subpopulations in the caudal end of the embryo: Group 1, axial elongation and trunk development; Group 2, early mesoderm; Group 3, later (committed) mesoderm; Group 4, early neural; and Group 5, later (committed) neural (Koch et al., 2017 and Table S3).
Fig. 3.

Building SVM based on E8.25 embryo data. (A) A total of 498 cells representing the CLE and NSB from three E8.25 embryos were dissected in silico and subjected to an unsupervised clustering approach (SC3 R package; Kiselev et al., 2017; Materials and Methods). This yielded four clusters and their marker genes: (1) genes associated with NMPs (pink); (2) mainly mesodermal genes (green); (3) genes associated with neural fate, mainly spinal cord (dark yellow); (4) genes associated with endoderm, mesoderm and extra-embryonic tissue (peach) (Table S1). Venn diagram shows the criteria for the in silico dissection: cells co-expressing Sox2 and T are NMP candidates; cells co-expressing Sox2 and Nkx1-2 but not T are neural progenitors (PreNeuro); cells co-expressing T but not Sox2, Mixl1 or Bmp4 are mesodermal progenitors (PreMeso), excluding progenitors for the endoderm (Mixl1) and the allantois (Bmp4). (B) Leave one out SVM workflow: an iterative process in which each cell is trained and tested (Materials and Methods).
In this study, the marker genes of Group 1 are significant in cells that are positive for Sox2 and T and are hence defined as putative NMPs. Moreover, these cells also have significant expression of marker genes which are upregulated in cells that are defined as early mesoderm (Group 2) and early neural (Group 4). We used these 1402 genes and added 69 genes that were expressed in the decision-making region of the embryo according to the literature (Table S1 and Edri et al., 2019), yielding 1471 genes, which were reduced to 1342 after the removal of genes with a mean expression of zero (Table S2). These 1342 genes were used to cluster the embryo data using an SC3 R package (Bioconductor; Kiselev et al., 2017), an algorithm based on k-means clustering (Materials and Methods).
The analysis yielded an optimal number of four clusters in the E8.25 cells (Fig. 3A, Materials and Methods) and 96 marker genes that act as discriminating identifiers of the clusters (Table S3). The top ten marker genes associated with each cluster are visualized in Fig. 3A. Having allocated cells to the four clusters based on their gene expression, we looked to see how each of the three functional groups (NMP candidates, preNeuro and preMeso) that compose the CLE region occupies each of the clusters.
Cluster 1 is a mixed cluster, composed of the three cell categories: NMP candidates, preMeso and preNeuro (Fig. 3A and Table S3); 71% of its 28 marker genes are part of the NMP profile gathered from the literature, including Cdx4, Nkx1-2, Fgf8 and Fgf17 (Fig. 3A, Table S3 and Koch et al., 2017). Cluster 2 is mainly composed from cells defined as preMeso, and the most highly expressed genes in this cluster exhibit a mesodermal affiliation [lateral plate mesoderm (LPM), intermediate mesoderm (IM), PXM and somites; see Table S1], with 91% of the 23 marker genes being mesodermal according to Koch et al. (2017) (Fig. 3A and Table S3). Cluster 3 is constructed mostly from preNeuro cells and has a neural identity characterized by genes related to the spinal cord and the nervous system: 85% of the 13 marker genes of cluster 3 are defined as neural based on Koch et al. (2017) (Fig. 3A and Table S3). Finally, cluster 4 is mostly composed of preMeso cells and, as defined in Koch et al. (2017), 34% of the 32 marker genes match to Group 3 (LPM and IM), but with additional genes affiliated to endoderm and IM (Tables S1 and S3).
Our clustering suggests that cluster 1 has an NMP signature, as it highlights genes such as Nkx1-2, Cdx1-4, Fgf8, Grsf1, Epha5 and Cystm1, which are all associated with NMPs (Cambray and Wilson, 2007; Edri et al., 2019; Gouti et al., 2014, 2017; Henrique et al., 2015; Koch et al., 2017; Wymeersch et al., 2016, 2019). Furthermore, it suggests that, rather than being a population of bipotent cells that is characterized mainly by the co-expression of Sox2 and T at the mRNA level, which makes only 29% of cluster 1, the ensemble appears to contain some pre-mesodermal (38%) and pre-neural (33%) cells. This analysis thus raises a question about the differences between the preMeso and preNeuro cells in cluster 1 in comparison with those that are found in clusters 2 and 3. One probable explanation is that cluster 1 encompasses very early neural and mesodermal cells, embedded in the NMP region of the mouse embryo, whereas the other clusters contain committed cells, similar to what was found in Koch et al. (2017). Indeed, cluster 1 includes genes that have been previously linked to the NMP profile together with genes that have neural or mesodermal characteristics. Based on the work of Koch et al. (2017), out of the 28 marker genes defining cluster 1, two genes (Ptk7 and Fgf8) are linked to Group 1 (axial elongation and trunk development), 15 genes (Epha5, Nkx1-2, Cdx2, Cdx4, Cystm1, Acot7, Stmn2, Fgf17, Lhpp, Mgst1, Lix1, Hoxc4, Ccnjl, Sp8 and Oat) are linked to Group 4 (early neural) and the rest of the genes are either expressed in the embryo CLE at around E8.5 (Grsf1, Cdx1, Hoxb9, Hoxc9, Wnt5b), or exhibit neural (Hes3, Ncam1, Pmaip1) or mesodermal (Evx1, Hes7, Foxb1, which also express in the neural plate) progenitor characteristics (see Table S1 for references).
Having identified an organization based on gene expression for the E8.25 CLE embryo the next step was to build an SVM classifier that would learn the gene profile of the four different clusters found in the embryo data. After testing its performance and its stability on the embryo (Fig. 3B, Materials and Methods), the SVM was used to assign cells of the in vitro populations to the four classes (clusters) based on their gene expression.
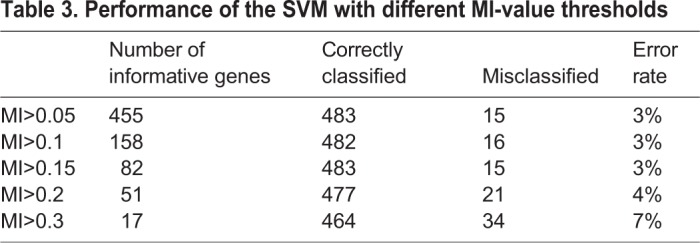
To build a robust and accurate classifier selecting the input features (genes) that the SVM needs to learn was an important task. Hence, we first wanted to identify the informative genes associated with the four clusters. To do this, and to avoid the underrepresentation of genes that were not previously linked to the NMPs, we used the whole set of qualified genes (14,822). We selected the genes by computing the mutual information (MI) measure between the genes and the four clusters (see Table 3 in Materials and Methods), which resulted in 82 informative genes (Table S4) that were used as input features to the SVM. The feature selection process leads to a classifier that, by reading the expression of these 82 genes, can correctly classify 97% of the input cells (Fig. 3B, Table 3). Of the 82 informative genes, 60% are identical to the 96 marker genes of the four clusters, whereas the remaining 40% include genes such as T, Hoxc8, Hoxb8 and Cdkn1c, which are expressed in the embryo CLE at E8.25-E8.5.
Table 3.
Performance of the SVM with different MI-value thresholds
A comparison between the in vitro and in vivo cell populations
We used the SVM that was established from the embryo data to explore the structure and nature of the in vitro populations. To do this, we first needed to ensure that the input cells from the in vitro populations did not contain cells with gene expression patterns on which the SVM had not been trained, as we only want to test the cells with similarity to the E8.25 caudal region (Fig. 3A and Step 1 in Fig. 4A). Similarly to the in silico dissection of the CLE from the embryo cells, we selected cells co-expressing Sox2 and T, cells expressing Sox2 and Nkx1-2 but not T, and cells expressing T but not Sox2, Mixl1 or Bmp4. This step resulted in filtering out a higher number of cells from the ES-NMP condition (45%) in comparison with the other conditions (∼30%), consistent with the previously noted heterogeneity. Feeding the remaining CLE-like cells to the classifier with the expression of the 82 informative genes, which are the features on which the SVM had been trained and needs to perform the classification task, resulted in the assignment of probabilities for each cell to be classified to each of the four classes (Fig. 4A). As the true classification of the in vitro cells is not known, and as there might be some hidden classes in the in vitro populations that were not trained using the embryo data, only the cells with a minimum probability of 0.8 are assigned to the class with the highest probability among the four classes and proceeded to the next step (see the probability plot under Step 6 in Fig. 4A: probability of 0.8 is indicated by the red line, see also Materials and Methods). In this step the highest filter of cells (50%) was observed in the ES-NMP condition compared with the others (∼30-35%), suggesting that this condition produces a high quantity of cells that do not correspond to the E8.25 embryo CLE. The last step (Step 7, Fig. 4A) was to summarize the distribution of the cells of each sample across the four classes. Most of the qualified cells (Fig. 4A, Table of Step 6) from ES-NMP (84%) and Epi-NMP (73%) were allocated to class 1 (Step 7, Fig. 4A), which is associated with the NMP signature. On the other hand, more than 90% of the qualified cells (Fig. 4A, Table of Step 6) from Epi-CE (91%) and Epi-CE-T (97%) were allocated to class 4 (Step 7, Fig. 4A), which is characterized by the expression of mesodermal and endodermal genes. Class 2 and class 3, which have mesodermal and neural differentiation characteristics, did not attract many cells from the different samples, suggesting that the in vitro cells, passed through this pipeline, are not very differentiated.
Fig. 4.

Classifying the in vitro cells using the SVM trained on the embryo data. (A) Workflow of the classification of the in vitro cells (for details see text and Materials and Methods). (B) Average expression of the 96 marker genes found in the embryo in the in vitro samples classified to the four classes. Rows of the expression heatmap are hierarchy clustered. Blue-red colour bar indicates the gene expression.
Fig. 4B shows the average expression of the 96 marker genes of the four clusters in the in vitro cell populations. This result emphasizes that the same classes from different samples clustered together, which displays the similarity of the cells from different conditions that assign to the same class. In addition, it shows that the in vitro cells exhibit the expression of marker genes for the four classes found in the embryo, demonstrating that the SVM pipeline detects the in vitro cells in agreement with the learned embryo cells.
A node-like population induced in vitro
The finding that Epi-CE and Epi-CE-T were allocated mainly to class 4, and that Epi-CE is the origin of Epi-NMP (Materials and Methods; Edri et al., 2019), led us to investigate further the identity of cluster 4. As a first step, we arranged all the qualified cells from the SVM pipeline (Fig. 4A, Table of Step 6) into pseudotime ordering using TSCAN (Bioconductor R package, version 1.16.0; Materials and Methods). This analysis revealed that class 4 cells (red cells in Fig. 5A) are split into two pseudotime ranges, with class 1 cells (blue cells in Fig. 5A) forming a bridge between these two classes. This result lends support to the possibility that Epi-NMP cells (mainly classified to class 1) are derived from Epi-CE (class 4, mainly composed from Epi-CE and Epi-CE-T). It also highlights the existence of two different populations in Epi-CE. When exploring the highly expressed genes that define the two pseudotime ranges of class 4 (Fig. 5A, Materials and Methods and Table S5), we observed that the later range is defined by genes that are associated with rapidly dividing cells, whereas the early one does not show this enrichment (Fig. 5A). This observation suggests the existence of a group of cells in a phase of large expansion in class 4. Similar results were obtained by pseudotime ordering the cells using Monocle (Bioconductor R package, version 2.10.1; Qiu et al., 2017a,b; Trapnell et al., 2014; Fig. S7), where class 4 is divided to two groups: an early one that contains Epi-CE-T cells and a later one that is mainly composed of Epi-CE. Class 1, which is composed of Epi-NMP and ES-NMP, is a later population in the pseudotime range, in comparison with class 4 (Epi-CE conditions). This result indicates that class 1 is derived from class 4, which is true in culture (Epi-NMP derived from Epi-CE) and in the embryo: the CE will harbour the NMP in a later state.
Fig. 5.

Class 4 contains node-like cells. (A) Pseudotemporal order of the in vitro cells that were classified to the four classes. Class 4 is divided to two pseudotime ranges: the later range of highly expressed genes contains 55% of cycling genes, whereas the early range does not contain any cycling genes (Materials and Methods). (B) E8.5 mouse embryo node: illustration of a sagittal view of the embryo shows the expression of T (red) in the NSB (Tsakiridis et al., 2014). Posterior view of the embryo exhibits the expression of Nodal (blue) in the node and in the LPM (Shiratori and Hamada, 2006) and its left (L) right (R) asymmetry. A transverse section (A′A′) reveals the pit and crown cells of the node, PXM, LPM, endoderm and the prospective floor plate. The expression of Nodal and Foxa2 is indicated in blue and green, respectively. The pit cells co-express T and Foxa2 and the crown cells express Nodal and T (Davidson and Tam, 2000; Jeong and Epstein, 2003; Lee and Anderson, 2008; Shiratori and Hamada, 2006). (C) The distribution of the node-like cells among the four classes: a significantly higher number of the node-like cells are found in class 4 in comparison with the other classes. ***P<0.001 (calculated empirical P-value; see Materials and Methods for details). (D) Gene expression heatmap of chosen node genes in class 4. The genes are hierarchically clustered and the cells are ordered in accordance with the decreasing expression of Oct4 (Pou5f1). Gene expression, which is defined as log2(CPM+1) (Materials and Methods), is indicated by the blue-red colour bar.
The presence of endodermal and mesodermal markers in class 4 is surprising, as it suggests the existence of a cell type in the embryo caudal region that would be associated with these germ layers. One structure that could fit this criterion is the node (Blum et al., 2007; Lee and Anderson, 2008; Martinez Arias and Steventon, 2018), a structure that appears at E7.5, contains the progenitors of the axial mesoderm (Beddington, 1982; McGrew et al., 2008; Tam and Beddington, 1987) and has been associated with the NMPs (Albors and Storey, 2016; Garriock et al., 2015; Henrique et al., 2015; Wymeersch et al., 2016). Thus, we considered the possibility that class 4 contains node cells.
At a very coarse level, the node can be identified as cells expressing combinations of three genes; Foxa2, Nodal and T (Fig. 5B; Davidson and Tam, 2000; Jeong and Epstein, 2003; Lee and Anderson, 2008; Shiratori and Hamada, 2006). Applying this coarse definition, we detected node-like cells in our in vitro samples with a very high representation in class 4 (Fig. 5C). The allocation of a node identity to cells in class 4 is not a bias of the sample size, as a statistical test controlling the size of the classes yielded that class 4 has the highest proportion of node-like cells (calculated empirical P<0.001; see Materials and Methods for details). To further test this coarse identification of node-like cells, we gathered a list of additional genes that are associated with the structure and function of the node, e.g. Shh, Ccno and Chrd (Davidson and Tam, 2000; Funk et al., 2015; Jeong and Epstein, 2003; Lee and Anderson, 2008; Shiratori and Hamada, 2006; Tam and Behringer, 1997), and tested for their expression in class 4 (Fig. 5D).
Having identified node-like cells in our in vitro populations, we thought we could use the dynamic changes in this region of the embryo to stage our in vitro populations. For example, at the time of its appearance the node expresses Oct4 and Otx2; however, by E8.0-E8.5 the expression of these genes have disappeared from the node (Cajal et al., 2012; Downs, 2008). The expression of Oct4 is particularly diagnostic for this transition. Ordering node-like cells of class 4 (Fig. 5D) from high to low Oct4 expression reveals additional patterns of gene expression that confirm the presence of a node-like population in the in vitro class 4 associated with Oct4 expression. Cells with decreasing levels of Oct4 display increasing levels of genes associated with the node: Foxa2, Bmp7, noggin, Chrd, Slit and, significantly, Shh (Fig. 5D; Davidson and Tam, 2000). Within the cells expressing low or no Oct4, we observe a further division based on Sox2 expression: although all cells express node genes, some of them express Sox2 and some do not. The ventral-most region of the neural plate is called the floor plate (Fig. 5B) and shares many of the pattern of gene expression of the node (Jeong and Epstein, 2003b; Wood and Episkopou, 1999). These results suggest that our experiment not only yields node-like cells (Sox2 negative) but also floor plate precursors (Sox2 positive).
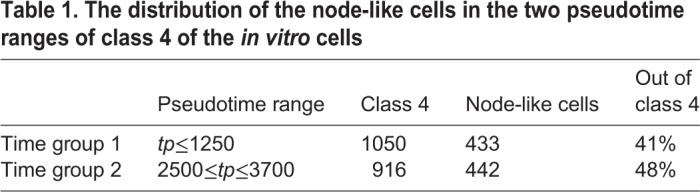
Moreover, we checked the proportion of the node-like cells in the two pseudotime ranges of class 4, as shown in Table 1, however no difference was found. This result supports our hypothesis that these two populations are very similar, however one of them represents an amplifying population, whereas the second one is more stable in terms of size.
Table 1.
The distribution of the node-like cells in the two pseudotime ranges of class 4 of the in vitro cells
In the in silico E8.25 embryo CLE, we found 38 node-like cells (Fig. 6A,B), 30 of which were mapped to the embryo class 4 cells (Fig. 6A). When comparing the node-like cells in class 4 of the embryo with those of the in vitro cells, some notable differences become apparent (Fig. 6B), for example Oct4, which is off in the embryo cells. As Epi-NMPs express very few node genes (Step 7, Fig. 4A, and Fig. 6C) and no Oct4, this supports our previous assertion that it has the closest relationship to the E8.25 CLE region, that the node-like cells are lost in the transition between Epi-CE and Epi-NMP (Fig. 6C) and that the Epi-CE cells represent a developmentally earlier cell state than the Epi-NMPs.
Fig. 6.

Node cells are needed to maintain the NMPs. (A) Distribution of the node cells among the four classes in the embryo. (B) Expression of chosen node genes in the embryo class 4. Genes and cells are hierarchically clustered. Gene expression, which is defined as log2(CPM+1), is indicated by the blue-red colour bar. (C) Proportion of node-like cells in the Epi-CE and Epi-NMP samples. (D) YFP-positive cells of Epi-CE Nodal sample composed of Nodal::YFP cells and RFP-positive cells of Epi-NMP RFP sample composed of Ubiquitin::Tomato cells were used to make Epi-meso2 mixture (Materials and Methods). This mixture was grown for 4 days then the cells were sorted based on their RFP fluorescence: RFP-positive cells (EM2-RFP+4d) and RFP-negative cells (EM2-RFP-4d). EM2-RFP4d is the control. The sorted cells and the control sample were quantified for their mRNA of a chosen set of genes using RT-qPCR. (E) Expression heatmap of 11 genes, obtained using RT-qPCR, in cells grown in the three conditions indicated in Fig. 6D. The normalized expression of each gene to the housekeeping gene Ppia was scaled between 0 and 1 across the different conditions. Gene expression is indicated by the blue-red colour bar.
An in vitro functional test of the in vitro induced node-like population
Previously, we have shown that the Epi-NMP population has a limited but clear self-renewing ability in culture when exposed to FGF and Chiron (Edri et al., 2019). These cells maintain T and Sox2 expression for at least two passages (Epi-NMP, Epi-meso2, Epi-meso3); however, over time the levels of NMP markers go down and the cells exhibit a slow increase in the expression of differentiation genes associated with neural fates (Edri et al., 2019). In the embryo, the self-renewing population also decreases with time, and this is associated with the disappearance of the node (Steventon and Martinez Arias, 2017; Wymeersch et al., 2016). Thus, we considered that in our in vitro system, the loss of T might be associated with the loss of node-like cells. To test this, we added node-like cells from Epi-CE to Epi-NMP and passaged the mixed sample to make Epi-meso2, then we checked whether the addition of node-like cells could maintain the levels of T expression (Fig. 6D).
We used a Ubiquitin::tomato cell line as a source of NMP-like cells and a Nodal::YFP cell line as a source of node cells. Both were cultured to produce Epi-CE: Epi-CE RFP (from the Ubiquitin::tomato cell line) and Epi-CE Nodal (from the Nodal::YFP cell line). The Epi-CE RFP were further grown to make Epi-NMP (Epi-NMP RFP). After two days of culturing Epi-NMP RFP, we plated a mixture that equally consisted of Epi-NMP RFP-positive cells and Epi-CE Nodal-positive YFP cells (Fig. 6D, Fig. S8A,B, Materials and Methods). The mixture (Epi-meso2) was cultured for 4 days (Fig. 6D) until cells were sorted into RFP-positive (EM2-RFP+4d, contains only the Ubiquitin::tomato cells) and RFP-negative (EM2-RFP–4d, contains Nodal::YFP cells and might contain Ubiquitin::tomato cells that did not express RFP; Fig. S8C and Materials and Methods). These populations of cells were compared with the EM2-RFP4d, which are only Epi-NMP RFP cells cultured for 4 days to make Epi-meso2 (Fig. 6D, Materials and Methods).
Addition of node-like cells to the Epi-NMP population elevates the level of T and Foxa2, maintains the expression of Cdx2 and Nkx1-2 and decreases the level of the neural fate markers Sox2 and Sox1 (Fig. 6E and Fig. S9, EM2-RFP4d versus EM2-RFP+4d). In addition, there is not much difference in the expression of Tbx6, Hoxc6, Fgf8 and Cyp26a1 when comparing those genes between EM2-RFP4d and EM2-RFP+4d.
This result, aligned with what we have previously shown (Edri et al., 2019), suggests that node-like cells are necessary to maintain the relative levels of Sox2 and T and buffer the tendency that the Epi-NMPs have towards the neural fate when passaging them in culture.
DISCUSSION
Over the last few years, ESCs have emerged as a useful experimental system to study mammalian development. Although they are no substitute for the embryo, they have some advantages when addressing processes that happen early in development, when material and experimental accessibility are scarce. However, their validation as an experimental system depends on testing how the events observed in culture relate to those taking place in the embryo. Here, we have used mouse PSCs to analyse, at the single cell level, the origin and structure of NMPs, a bipotent population that is thought to give rise to the spinal cord and the paraxial mesoderm. As an important reference for our study, we have used a single cell dataset from E8.25 embryos, the stage at which NMPs are first distinguishable.
Analysis of PSC-derived NMPs suggests that different protocols produce heterogeneous populations in terms of gene expression (Edri et al., 2019). To gain insights into these heterogeneities and their origins, we have performed a single cell transcriptomics analysis of the different populations. As a reference, we have used data from E8.25 embryos out of which we have dissected in silico the CLE/NSB region based on T, Sox2 and Nkx1-2 expression patterns, as cells that express these genes are often identified as NMPs. Our results suggest that, by this stage, these cells are distinct from those in the pluripotent epiblast. The transition between the states appears to be associated with the loss of expression of Cdh1, Oct4, Fst and Otx2 and the gain of expression of Cdh2 and Cyp26a1 among others (Fig. 1 and Fig. S1). Our results contrast with those of a recent study which allocated expression of Cdh1 and Oct4 to NMPs at E8.5 (Gouti et al., 2017). Analysis of published gene expression patterns (Fig. 1, Fig. S1 and Table S1) supports our conclusions that these markers are associated with the pluripotent epiblast. It might be that changes in the transcription of these genes happens abruptly at ∼E8.25 and that there is a difficulty in staging the embryos. The transition from pluripotent epiblast to the bipotent cells in the CLE/NSB region can be detected in our in vitro samples, as represented by the transition from Epi-CE to Epi-NMP (Fig. 2 and Fig. S5; see also Edri et al., 2019).
As a reference for the in vitro-derived populations, we used a clustering algorithm on existing datasets (Ibarra-Soria et al., 2018; Pijuan-Sala et al., 2019) to classify populations in the embryo: class 1 with NMP signature; class 2 with mesodermal signature; class 3 with neural signature and class 4 with extra-embryonic, endoderm and IM signature. Class 1 contains cells co-expressing Sox2 and T and cells in a pre-neural and pre-mesodermal state, i.e. not all of them co-express exclusively Sox2 and T. This observation emphasizes the notion that the co-expression of Sox2 and T alone is not a valid definition, or at least it is not an absolute structural condition, for NMPs (see also Edri et al., 2019). It also raises the possibility that an NMP population is not only a collection of Sox2 and T co-expressing cells (Gouti et al., 2014, 2017; Turner et al., 2014), but includes a heterogeneous population of mesodermal and neural poised and early differentiated cells. This situation is reminiscent of many stem and progenitor cell populations and suggests that, as in some of those cases (Huang, 2009; Moris et al., 2016; Pina et al., 2012), these different cell populations are in dynamic equilibrium. A suggestion has been made that differentiation from the T and Sox2 co-expressing population is a stochastic event biased by cell signalling (Gouti et al., 2017); our results support that observation but also suggest that the NMP population includes differentiation-poised cells.
The structure of the E8.25 caudal region inferred from our analysis was used as a reference for the study of the in vitro-derived populations. To do this, we used the four classes derived from the embryo data to build an SVM classification model that allowed us to allocate cells from the different protocols to our reference. We found that the ESC-based protocol contained few cells allocated to the E8.25 embryo CLE, but that the EpiSC samples are enriched in this population. Furthermore, we found that Epi-NMP cells, which are derived from Epi-CE (Materials and Methods), contained the most E8.25 CLE-like cells (>70% of the selected cells, Fig. 4A), and most of them map to class 1. Furthermore, we find many E8.25 CLE-like cells in the Epi-CE population (>60% of the selected cells, Fig. 4A) but, in contrast with Epi-NMPs, these cells predominantly map to class 4. Interestingly, very few cells of the Epi-CE descendant, Epi-NMP, map to class 4. A detailed analysis of class 4 reveals that it has a large representation of node-like cells and, interestingly, cells of the floor plate. The floor plate in the embryo shares many features with the node and its main derivative, the notochord. This allocation is confirmed by the identification of node-like cells in the embryo reference data.
The representation of cells from two different sequentially induced in vitro populations to one embryonic stage is, at first sight, surprising; however, we believe that there is an explanation. The CLE at E8.25 is derived from an earlier caudal region, at E7.5, the most prominent feature of which is the node, which is maintained until E9.0. Thus, at E8.25 the embryo has a signature of an early stage in the node. The representation of node cells in Epi-CE, but not much in its Epi-NMP progeny, suggests that, in adherent culture the conditions are not conducive to maintenance of the node. What we find interesting, given the relationship between Epi-CE and Epi-NMP, is the presence of NMP-like cells in the Epi-NMP population. This led us to speculate that, in the embryo, there might be a very close relationship between the emergence of the node and of the NMPs, something that has been suggested before (Albors and Storey, 2016; Garriock et al., 2015; Henrique et al., 2015; Wymeersch et al., 2016, 2019).
We find two interesting features of the possible relationship between these two populations. The first one is the observation that within the Epi-CE population there is a subpopulation in a high proliferative state. Second, there appears to be a relationship between a node population and the maintenance of the T and Sox2 expression ratio. These observations lead us to suggest that, in the embryo, the NMP population arises early in development, near the node, and that the node might play a role in its maintenance and amplification at that early stage. A need for amplification of the initial NMP pool could be explained by the size of the primordia relative to the size of the tissue that needs to be generated. It is not clear how the node would mediate this function, but an interaction between BMP and Nodal (Edri et al., 2019) might be important. A relationship between the node and axial elongation can be gauged from the effect of mutations in which the node is absent, which leads to a loss of T expression in the caudal region of the embryo and severe truncations (Ang and Rossant, 1994; Davidson and Tam, 2000; Weinstein et al., 1994). In this context, there might be an effect of Oct4 as we observe a clear transition in the behaviour of the in vitro populations depending on whether they express Oct4 (Epi-CE) or not (Epi-NMP). This transition might correspond to the proliferative amplification phase and the start of the differentiation phase of the NMPs. Oct4 might create a molecular context for Sox2; as long as both are expressed the cells in the epiblast are multipotent and, only when Oct4 is downregulated, Sox2 becomes engaged in neural differentiation. It will be interesting to test this hypothesis.
Our study highlights the value of comparing embryonic and in vitro-derived cell populations. This can not only provide useful information for the derivation of specific populations, but might also generate hypotheses and thus provide insights into normal development which might not be obtainable by classical genetic methods.
MATERIALS AND METHODS
ESC culture and routine cell culture
E14-Tg2A, Bra::GFP (Fehling et al., 2003), Nodal::YFP (Papanayotou et al., 2014) and Sox17::GFP Ubiquitin::Tomato (Niakan et al., 2010) mouse ESCs were cultured in tissue-culture plastic flasks coated with 0.1% gelatine in PBS (with calcium and magnesium), filled with GMEM (Gibco) supplemented with non-essential amino acids, sodium pyruvate, GlutaMAX™ (Gibco), β-mercaptoethanol, foetal bovine serum and LIF. Cell medium was changed daily and cells passaged every other day. The differentiation protocols are described below.
ES-NMP
E14-Tg2A cells were plated at a density of 4.44×103 cells/cm2 in a 0.1% gelatine-coated flask with a base medium of N2B27 (NDiff 227, Takara Bio) for 2 days. After 48 h, N2B27 was supplemented with 3 μM of CHIR99021 (Chiron 10 mM, Tocris Bioscience) for an additional 24 h.
EpiSCs
E14-Tg2A or Bra:GFP were grown in a culture flask coated with 0.5% plasma fibronectin (FC010, 1 mg/ml, Chemicon) in PBS (with calcium and magnesium) with N2B27 supplemented with 12 ng/ml FGF2 (R&D Systems, 50 µg/ml) and 25 ng/ml Activin A (Stem Cells Institute, University of Cambridge, 100 μg/ml), known as Epi-media, for at least four passages. These cells were considered to be EpiSCs, confirmed by seeding them in a colony assay at a density of 67 cells/cm2 in restricted medium [2i: N2B27 supplemented with 3 μM Chiron and 1 μM PD0325901 (PD03, Tocris Bioscience, 10 mM)], resulting in no growth of cells; this ensured that the cells were no longer in the naïve pluripotent state and they had moved on to the prime pluripotent state (data not shown).
Epi-CE and Epi-CE-T
EpiSCs were plated at a 5×104 cells/cm2 density in a 0.5% fibronectin pre-coated flask with Epi-media for the first day. On day 2, the concentration of FGF2 was increased to 20 ng/ml in the base medium of N2B27 and Activin A removed. On day 3, N2B27 was supplemented with 3 μM Chiron, which was added to the 20 ng/ml FGF2. After 72 h those cells were known as Epi-CE. This protocol is a variation of one that has been used to derive NMP-like cells from human ESCs (Lippmann et al., 2015). Epi-CE-T were cultured from the Bra:GFP cell line at the same way as Epi-CE, with the modification that after 72 h the cells were sorted for positive GFP cells only.
Epi- NMP
Epi-CE cells were detached from the culture flask using Accutase (BioLegend, 0.5 Mm) and seeded on a flask coated with 0.5% fibronectin at a density of 5×104 cells/cm2. The cells were grown for 2 days in N2B27 supplemented with 20 ng/ml FGF2 and 3 μM Chiron.
Single cells transcriptomic analysis
10x Genomics single cell transcriptomic service was used to sequence our four different samples. We loaded 8700 cells from each sample into the 10x Chromium system. The preparation of the libraries and the Illumina sequencing (HiSeq 4000) was carried out by Cambridge Genomic Services. Cell Ranger version 1.3.1 (10x Genomics) was used to process raw sequencing data and the Seurat R package (version 2.0; Butler et al., 2018; Macosko et al., 2015) was used to read the data from Cell Ranger to R and build the expression matrix. Gene expression was quantified using UMI counts. The final output was a matrix of genes versus cells, utilized for further analysis.
Embryo data
In this work, we used the published transcriptomic single cell data from three mouse embryos (females and males) at E8.25 (Ibarra-Soria et al., 2018; Pijuan-Sala et al., 2019) including their extra-embryonic tissues. These embryos were dissociated to single cells and processed on a 10x microfluidic chip. The resulting libraries were sequenced on an Illumina HiSeq 2500, providing 7006 cells out of which 4706 are male and 2300 are female.
Single cell data clean up and quality control
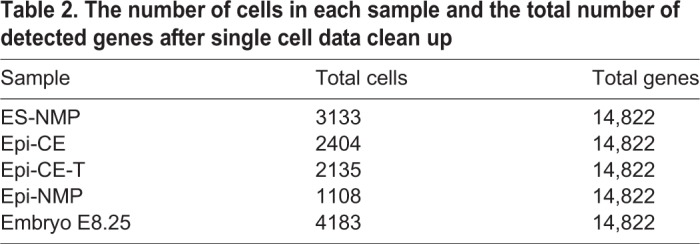
Using the Scater package in R (McCarthy et al., 2017), the expression matrix was cleaned according to the four following aspects: (1) UMI counts – drawing the histogram of the RNA UMI total counts per cell allowed us to set a threshold of above 8000 UMI counts in a cell, ensuring a sufficient sequencing depth for each cell; (2) detected genes – from the histogram of total detected genes in a cell we set a threshold of above 2500 unique genes in a cell, ensuring the reads are distributed across the transcriptome; (3) mitochondrial gene expression – plotting the percentage of mitochondrial gene counts in a cell versus the total detected genes in a cell allowed us to set a threshold of 20%, ensuring the cells to be further analysed are not likely to be dead or stressed; (4) gene filtering – undetectable genes were filtered out by setting a threshold of having at least two cells containing more than 1 UMI of a gene. The number of cells and total genes following the clean up are presented in Table 2.
Table 2.
The number of cells in each sample and the total number of detected genes after single cell data clean up
The UMI count normalization, which is necessary to make an accurate comparison of gene expression between samples, was carried out by first scaling the counts of each gene in a cell to the total counts in that cell per million counts (known as counts per million, CPM). Then the log2(CPM+1) was calculated for each gene, this is the normalized gene expression (the 1 was added to the CPM to keep zero counts as zero in the binary logarithm scale).
Seurat clustering
We used Seurat R package (version 2.3.4; Butler et al., 2018; Stuart et al., 2018preprint) for clustering, which is based on a community detection approach. This package calculates highly variable genes and focuses on them for downstream analysis. It calculates the average expression and dispersion for each gene, places these genes into bins, and then calculates a z-score for dispersion within each bin. This helps control for the relationship between variability and average expression.
Clustering the embryo cells
The dissection of CLE in silico from the whole mouse embryo was carried out by selecting cells that co-express Sox2 and T; cells that express Sox2 and Nkx1-2 but not T; and cells that express T but not Sox2, Mixl1 and Bmp4 (see text). Clustering the embryo CLE cells was guided using a selection of genes. The selection was made to focus on the caudal region of the embryo and, importantly, to avoid biases towards clustering results led by genes that are associated with different processes or regions; for example, the embryo data is a mixture of male and female embryos and, in this situation, Xist expression leads to clusters of female and males (S.E., unpublished observation). The genes that were selected for our analysis were 1402 genes reported by Koch et al., 2017 in a study of the NMPs and the caudal region of the embryo (Koch et al., 2017). Further genes were added to this list owing to their association with the CLE region of the E8.5 embryo (Edri et al., 2019), reaching a total of 1471 genes. From this list, genes with zero mean expression were removed, yielding a total of 1342 genes for analysis (Table S2). Clustering was performed using the Cell Consensus Clustering (SC3) package in R (Kiselev et al., 2017) with the following steps: (1) Gene filter – filtering genes that are either expressed in less than 6% of the cells (rare genes) or expressed in at least 94% of cells (ubiquitous genes). (2) Distance matrices calculations – distances between the cells are calculated using the Euclidean, Pearson and Spearman matrices. (3) Transformations – all distance matrices are then transformed using either principal component analysis or by calculating the eigenvectors of the associated Laplacian matrix. (4) k-means – k-means clustering is performed on the first set of eigenvectors of the transformed distance matrices. The number of clusters, k, is set by the user. (5) Consensus clustering – a binary similarity matrix is constructed for each individual clustering result from the corresponding cell labels obtained in the previous step: if two cells belong to the same cluster, their similarity is 1; otherwise the similarity is 0. A consensus matrix is calculated by averaging all similarity matrices of the individual clustering results. The resulting consensus matrix is clustered using hierarchical clustering.
The clustering of the embryo cells was carried out between k=2 and k=8. The consensus matrices for the different k are shown in Fig. S6. The averaged Silhouette width values for each clustering results between k=2 and k=8 are 0.8, 0.9, 0.85, 0.78, 0.77, 0.77 and 0.72, respectively.
The silhouette is a quantitative measure that represents the consensus matrix diagonally. An average silhouette width, which is calculated as the weighted average between the silhouette values of each cluster, varies from 0 to 1, and the closer it is to 1 the better the clustering is for that value of k. From the consensus matrices on Fig. S6 and from the averaged silhouette width in Table 3, we estimated that the optimal number of clusters could be k=3 or k=4. For k=3, the three clusters are: a mixed cluster – containing cells from all the three categories; NMP candidates, preNeuro and preMeso; and the two other, mainly composed from preMeso cells (Fig. S6). For k=4 the clusters are: a mixed cluster composed from all the three cell categories; two others which are mainly composed of cells with a mesodermal identity; and a fourth one which is mainly constructed from neural-oriented cells (Fig. S6). We decided to continue to downstream analysis with k=4 because of the appearance of a clear neural cluster along with mesodermal clusters. K=4 ensures a representation for all the three cell categories: NMP candidates, mesodermal and neural cells.
Marker genes
Using the SC3 package in R (Kiselev et al., 2017), 96 marker genes were identified for the four obtained clusters (see Table S3). Marker genes are defined as genes that are highly expressed in only one of the clusters and can lead to the segregation of one cluster from the rest. The marker genes were found according to the following steps (Kiselev et al., 2017): constructing a binary classifier for each gene based on comparing the mean expression values across the clusters; calculating the classifier prediction by comparing the gene expression ranks across clusters; quantifying the accuracy of the prediction by calculating for each gene the area under the receiver operating characteristic (ROC) curve (true positive rate versus false positive rate); calculating the P-value for each gene using the Wilcoxon signed rank test and comparing the gene ranks in the cluster with the highest mean expression with all others; setting a threshold for the area under the ROC curve and the P-value to determine the marker genes.
The genes with the area under the ROC curve >0.65 and with the P-value<0.01 are defined as marker genes. The top 10 marker genes of each cluster are visualized in Fig. 3A.
Mutual information between genes and classes
After identifying the four different clusters in the in silico CLE embryo data, the downstream analysis was constructed using the whole set of qualified genes (14,822) rather than with the genes restricted to CLE (1342). This step was performed to avoid an underrepresentation of genes that were not previously linked to the NMPs. However, there is a need for dimensionality reduction to elucidate the data and to feasibly reduce computer calculation time. Here, similar to the work of Vanitha et al. (2015), we used an MI technique (Battiti, 1994) to select the informative genes related to the four clusters. The steps of computing the MI between the clusters (denoted as Y) and genes (denoted as X) start with calculating the cluster's entropy:
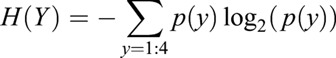 |
(1) |
where p(y) is the probability of each cluster y=1, 2, 3, 4, which is computed based on the distribution of the four clusters in the embryo data. We then discretized the gene expression values into ten bins and calculate the conditional entropy H(Y|X) as follows:
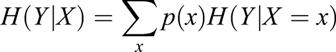 |
(2) |
where p(x) is the probability of the discretized expression values of a gene across the cell population and H(Y|X=x) is the cluster's entropy given a specific gene expression value, calculated following Eqn 1. Finally, we computed the MI between the clusters and each gene according to the below equation:
| (3) |
setting a threshold of the MI of all the genes and selecting the informative genes above this value to train the SVM.
Testing different values of MI between the genes and the clusters in which there are genes with MI above these values determined which genes were selected as input features for building the SVM (Table 3). The gene selection step helped to remove many irrelevant genes, which improved the classification accuracy. As can be seen in Table 3, setting a higher threshold to the MI value led to a lower number of informative genes that were fed to the classifier and that influenced its performance. Using an MI threshold above 0.15 led to 82 useful genes (Table S4) without damaging the classifier performance.
Multiclass SVM
In machine learning, SVM is a supervised learning model used either for classification or regression analysis, introduced in 1992 by Boser, Guyon and Vapnik (Boser et al., 1992). Given labelled training data, an SVM classifies it by finding the best hyperplane that separates all the data points of one class from the other class. The best hyperplane for an SVM means the one with the largest margin between the two classes. The support vectors are the data points that are on the margins of the separating hyperplane. New data points are then mapped into the same space and predicted to belong to a specific class based on which side of the hyperplane they fall. It often happens that the sets to discriminate are not linearly separable in a finite dimensional space. In that case a kernel function is used to map the original finite dimensional space into a much higher dimensional space, making the separation easier in that space. The selection of an appropriate kernel function is important, as it defines the space in which the training set will be classified. Exploration of the different kernel functions is described in the PhD thesis of S.E. (Edri, 2019); here, we show the result of the linear kernel function.
The classification problem encountered in this work is a multiclass classification rather than a binary classification. To solve this problem, the dominant approach is to reduce the single multiclass problem into multiple binary classification problems. Using the R package e1071 (version 1.6.8), the ‘one-against-one’ approach was selected in which n(n−1)/2 binary classifiers are trained, where n is the number of classes (in this work, n=4); the appropriate class is assigned by the majority output of a voting scheme.
We chose SVM in this work as a classifier owing to its high accuracy and its ability to deal with high dimensional data, as has been previously proven in large-scale image classification and gene expression data (Abdullah et al., 2011; Jiang et al., 2007; Lin et al., 2011; Vanitha et al., 2015).
To train the SVM and test its performance, a leave one out cross validation (LOOCV) method was used. In this method, the train data is N−1 cells, where N is the total number of cells in the embryo data (498 cells) and the remaining Nth cell is used for testing the model, and the same is repeated N times such that each cell is tested, classified and contributing to the model performance (Fig. 3B). The informative genes that passed the MI threshold were used as input features to the SVM. The LOOCV method makes the best use of the available data, especially when the number of samples is small (498 cells), and avoids the problem of random selection (Ben-Dor et al., 2000).
Predicting the class of the in vitro cells
Predicting the class of the in vitro cells involved selecting the CLE cells in the same way as for the embryo data, selecting the same informative genes that were used to build the SVM on the embryo data, and inserting the expression matrix of the in vitro cells as an input to the SVM. The output is the probability of each cell to be assigned to any of the four trained clusters. The dominant class that the cell was assigned in agreement with the maximum probability out of the four probabilities was then chosen (see the plot under Step 6 in Fig. 4A). As the true classification of the cells is not known, and as there might be some hidden classes in the in vitro data that were not trained using the embryo data, a harsh constrain needs to be taken: only the cells with minimum probability of 0.8 to be assigned to the dominant class are proceeded to the next step (see the probability plot under Step 6 in Fig. 4A: probability of 0.8 is indicated by the red line. The classification results are that only the qualified cells from the previous step are assigned to any of the four classes.
Pseudotime analysis
The cells from the in vitro samples (ES-NMP, Epi-CE, Epi-CE-T and Epi-NMP) that were classified to the four classes (the qualified output cells from the SVM pipeline) went through a pseudotemporal cell ordering. For pseudotime reconstruction of single cell RNA-seq data there are not a lot of available tools that have been systematically tested and have easily accessible software. Moreover, in this work we are analysing a heterogeneous cell population of different conditions rather than cells from a time course experiment, hence the supervised pseudotime reconstruction approaches are not applicable and one should rely on unsupervised methods. We decided to use TSCAN, the Bioconductor R package (version 1.16.0), as it has demonstrated reliable unsupervised pseudotime reconstruction results compared with alternative methods.
TSCAN first clusters the cells, then it builds a minimum spanning tree to connect the clusters. The branch of this tree that connects the largest number of clusters is the main branch, which is used to determine the pseudotime order of the cells. This algorithm does not detect starting or ending points, and previous biological information is needed to understand the start of the pseudotime order. The pseudotime order might represent the underlying developmental trajectory.
Defining the highly expressed genes in the two pseudotime ranges of class 4
The cells in class 4 were split into two groups based on their pseudotime order: tp ≤ 1250; 2500 ≤ tp ≤ 3700. We then identified the differentially expressed genes between the two groups using the two-sided Wilcoxon rank sum test. The P-value was corrected using the ‘BY’ method of Benjamini and Yekutieli (2001). This method controls the false discovery rate and the proportion of false discoveries among the rejected hypotheses. We detected 4569 differentially expressed genes by setting the adjusted P-value to ≤ 0.01. The mean expression of the 4569 genes across the cells in each group and the log2-fold between the mean expression of the two groups was calculated and the highly expressed genes in each group were defined as the genes with log2-fold above 1, resulting in 24 genes in the early pseudotime range and 178 genes in the later range (Table S5). Using the ccRemover R package (version 1.0.4; Barron and Li, 2016) each gene from the identified highly expressed genes could be identified as a cycling gene; 55% of the highly expressed genes in the later pseudotime range group are defined as cycling genes, whereas the cells in the earlier range do not show this enrichment (no cycling genes).
Statistical test for controlling the sample size
The numbers of in vitro cells classified to each of the four clusters were: class 1, 1141 cells; class 2, 264 cells; class 3, 70 cells; class 4, 2036 cells. Class 4 is approximately twice the size of class 1, and the node-like cells were assigned almost exclusively to class 4. Hence, one might think that the different size of the classes might bias the finding of the node-like cells in class 4. The statistical test that was designed in this case was to control for the size of the classes: 570 cells (half of class 1) were randomly selected from each of class 1 and class 4, and the null hypothesis is that there is no difference in the number of the node-like cells between class 1 and class 4. This step was repeated 1000 times, with the result that, in 1000 cases, class 4 contained more node cells than class 1, meaning that the calculated P-value<0.001 and the null hypothesis was rejected.
Culturing Nodal-YFP cells and ubiquitous-tomato cells
Nodal::YFP and Sox17::GFP Ubiquitin::Tomato cells were cultured under the Epi-CE protocol [Epi-CE Nodal and Epi-CE RFP (for red fluorescent protein), respectively]. The Epi-CE RFP were further grown to make Epi-NMP (Epi-NMP RFP). After two days of culturing Epi-NMP RFP, we plated a mixture that consists of 50% Epi-NMP RFP-positive cells and 50% Epi-CE Nodal YFP-positive cells, at a total density of 5×104 cells/cm2 (Fig. S8A,B). After sorting, the cells might be in stress, so we decided to culture the mixture for 4 days and not for the normal period of 2 days to let the cells recover. The mixture was grown in N2B27 supplemented with 20 ng/ml FGF2 and 3μM Chiron to make Epi-meso2 (EM2), until sorting the cells to RFP-positive (EM2-RFP+4d, contains only the Ubiquitin::Tomato cells) and RFP-negative (EM2-RFP-4d, contains Nodal::YFP cells and might contain Ubiquitin::Tomato cells that did not express RFP, see Fig. S8C). This population of cells was compared with the EM2-RFP4d (Epi-NMP RFP cells plated in a flask and cultured for 4 days in N2B27 supplemented with 20 ng/ml FGF2 and 3 μM Chiron to make Epi-meso2). Total RNA was isolated from the three samples (EM2-RFP4d, EM2-RFP+4d and EM2-RFP-4d) using TRIzol (Invitrogen/Thermo Fisher Scientific). First-strand cDNA synthesis was performed using the Superscript III system (Invitrogen) and the quantification of double-stranded DNA was obtained using specific primers (see Table S6) using QuantiFast SYBR Green PCR Master Mix (Qiagen) and the standard cycler program (Qiagen RotorGene Q). The qPCR was carried out in technical triplicates. Expression values were normalized against the housekeeping gene Ppia. To calculate the normalized gene expression values we identified the Ct (threshold cycle) for each gene (technical triplicates) and calculated the expression values (2−Ct). We then calculated the mean and s.d. for each gene from the triplicate expression values, and divided the mean and s.d. of each gene by the expression value of Ppia. The gene expression across the different conditions was scaled between 0 and 1.
Cell sorting
Epi-CE Nodal cells were sorted according to their YFP-positive fluorescence in a MoFlo sorter (Beckman Coulter) using a 488 nm laser with an emission filter of 530/40 (Fig. S8A). Epi-NMP RFP cells were sorted according to their RFP-positive fluorescence using a 647 nm laser with an emission filter of 610/20 (Fig. S8B). Cells were collected, counted and replated in N2B27 supplemented with 20 ng/ml FGF2 and 3 μM Chiron medium to make the 50% Epi-CE Nodal YFP-positive/Epi-NMP RFP-positive mixture of cells, as described above. After 4 days, the mixture was sorted to RFP-positive and -negative cells in the MoFlo sorter using the same laser and filter sets mentioned above (Fig. S8C).
Supplementary Material
Acknowledgements
We thank Bertie Gottgens for sharing the embryo data before publication, Meritxell Vinyoles for helping in experimental design and insightful discussion, and James Briscoe, Valerie Wilson and Ben Steventon for insightful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.E., A.M.A.; Methodology: S.E., P.H., W.J.; Software: S.E.; Validation: S.E., W.J.; Formal analysis: S.E., P.H.; Investigation: S.E., P.H.; Resources: W.J., A.M.A.; Data curation: S.E., W.J.; Writing - original draft: S.E., A.M.A.; Writing - review & editing: S.E., A.M.A; Visualization: S.E.; Supervision: A.M.A.; Project administration: A.M.A.; Funding acquisition: A.M.A.
Funding
This work was supported by the Cambridge Commonwealth, European and International Trust and Cambridge Philosophical Society scholarships and an AJA Karten Trust (the Anglo-Jewish Association Trust and the Ian Karten Charitable Trust) award to S.E., a Wellcome Trust Clinical PhD Fellowship (103392/Z/13/Z and 103392/Z/13/A) to W.J., and a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and Biotechnology and Biological Sciences Research Council project grants (BB/M023370/1 and BB/P003184/1) to A.M.A. Deposited in PMC for release after 6 months.
Data availability
Single cell RNA-seq data data have been deposited in Gene Expression Omnibus under accession number GSE132504.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.180190.supplemental
References
- Abdullah N., Ngah U. K. and Aziz S. A. (2011). Image classification of brain MRI using support vector machine. In 2011 IEEE International Conference on Imaging Systems and Techniques, pp. 242-247. IEEE; 10.1109/IST.2011.5962185 [DOI] [Google Scholar]
- Albors A. R. and Storey K. G. (2016). Mapping body-building potential. eLife 5, e14830 10.7554/eLife.14830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang S.-L. and Rossant J. (1994). HNF-3β is essential for node and notochord formation in mouse development. Cell 78, 561-574. 10.1016/0092-8674(94)90522-3 [DOI] [PubMed] [Google Scholar]
- Barron M. and Li J. (2016). Identifying and removing the cell-cycle effect from single-cell RNA-Sequencing data. Sci. Rep. 6, 33892 10.1038/srep33892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiti R. (1994). Using mutual information for selecting features in supervised neural net learning. IEEE Trans. Neural Networks 5, 537-550. 10.1109/72.298224 [DOI] [PubMed] [Google Scholar]
- Beddington R. S. P. (1982). An autoradio graphic analysis of tissue potency in different regions of the embryonic ectoderm during gastrulation in the mouse. Embryol. Exp. Morph 69, 265-285. [PubMed] [Google Scholar]
- Ben-Dor A., Bruhn L., Friedman N., Nachman I., Schummer M. and Yakhini Z. (2000). Tissue classification with gene expression profiles. J. Comput. Biol. 7, 559-583. 10.1089/106652700750050943 [DOI] [PubMed] [Google Scholar]
- Benjamini Y. and Yekutieli D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 29, 1165-1188. 10.1214/aos/1013699998 [DOI] [Google Scholar]
- Blum M., Andre P., Muders K., Schweickert A., Fischer A., Bitzer E., Bogusch S., Beyer T., van Straaten H. W. M. and Viebahn C. (2007). Ciliation and gene expression distinguish between node and posterior notochord in the mammalian embryo. Differentiation 75, 133-146. 10.1111/j.1432-0436.2006.00124.x [DOI] [PubMed] [Google Scholar]
- Boser B. E., Guyon I. M. and Vapnik V. N. (1992). A training algorithm for optimal margin classifiers. In COLT ’92 Proceedings of the fifth annual workshop on Computational learning theory, pp. 144-152. ACM Press. [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E. and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411-420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal M., Lawson K. A., Hill B., Moreau A., Rao J., Ross A., Collignon J., Camus A., Simeone A. and Levi G. (2012). Clonal and molecular analysis of the prospective anterior neural boundary in the mouse embryo. Development 139, 423-436. 10.1242/dev.075499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambray N. and Wilson V. (2007). Two distinct sources for a population of maturing axial progenitors. Development 134, 2829-2840. 10.1242/dev.02877 [DOI] [PubMed] [Google Scholar]
- Davidson B. P. and Tam P. P. L. (2000). The node of the mouse embryo. Curr. Biol. 10, R617-R619. 10.1016/S0960-9822(00)00675-8 [DOI] [PubMed] [Google Scholar]
- Downs K. M. (2008). Systematic localization of Oct-3/4 to the gastrulating mouse conceptus suggests manifold roles in mammalian development. Dev. Dyn. 237, 464-475. 10.1002/dvdy.21438 [DOI] [PubMed] [Google Scholar]
- Dunty W. C., Kennedy M. W. L., Chalamalasetty R. B., Campbell K. and Yamaguchi T. P. (2014). Transcriptional profiling of Wnt3a mutants identifies Sp transcription factors as essential effectors of the Wnt/β-catenin pathway in neuromesodermal stem cells. PLoS ONE 9, e87018 10.1371/journal.pone.0087018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edri S. (2019). Date with Destiny: Genetic and epigenetic factors in cell fate decisions in populations of multipotent stem cells. PhD thesis, University of Cambridge, UK: 10.17863/CAM.35678 [DOI] [Google Scholar]
- Edri S., Hayward P., Baillie-Johnson P., Steventon B. J. and Arias A. M. (2019). An Epiblast stem cell-derived multipotent progenitor population for axial extension. Development 146, dev.168187 10.1242/dev.168187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehling H. J., Lacaud G., Kubo A., Kennedy M., Robertson S., Keller G. and Kouskoff V. (2003). Tracking mesoderm induction and its specification to the hemangioblast during embryonic stem cell differentiation. Development 130, 4217-4227. 10.1242/dev.00589 [DOI] [PubMed] [Google Scholar]
- Funk M. C., Bera A. N., Menchen T., Kuales G., Thriene K., Lienkamp S. S., Dengjel J., Omran H., Frank M. and Arnold S. J. (2015). Cyclin O (Ccno) functions during deuterosome-mediated centriole amplification of multiciliated cells. EMBO J. 34, 1078-1089. 10.15252/embj.201490805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriock R. J., Chalamalasetty R. B., Kennedy M. W., Canizales L. C., Lewandoski M. and Yamaguchi T. P. (2015). Lineage tracing of neuromesodermal progenitors reveals novel Wnt-dependent roles in trunk progenitor cell maintenance and differentiation. Development 142, 1628-1638. 10.1242/dev.111922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M., Tsakiridis A., Wymeersch F. J., Huang Y., Kleinjung J., Wilson V. and Briscoe J. (2014). In vitro generation of neuromesodermal progenitors reveals distinct roles for Wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol. 12, e1001937 10.1371/journal.pbio.1001937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M., Delile J., Stamataki D., Wymeersch F. J., Huang Y., Kleinjung J., Wilson V. and Briscoe J. (2017). A gene regulatory network balances neural and mesoderm specification during vertebrate trunk development. Dev. Cell 41, 243-261.e7. 10.1016/j.devcel.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A. T. L., Morgan M. D. and Marioni J. C. (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421-427. 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrique D., Abranches E., Verrier L. and Storey K. G. (2015). Neuromesodermal progenitors and the making of the spinal cord. Development 142, 2864-2875. 10.1242/dev.119768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. (2009). Non-genetic heterogeneity of cells in development: more than just noise. Development 136, 3853-3862. 10.1242/dev.035139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Soria X., Jawaid W., Pijuan-Sala B., Ladopoulos V., Scialdone A., Jörg D. J., Tyser R. C. V., Calero-Nieto F. J., Mulas C., Nichols J. et al. (2018). Defining murine organogenesis at single-cell resolution reveals a role for the leukotriene pathway in regulating blood progenitor formation. Nat. Cell Biol. 20, 127 10.1038/s41556-017-0013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y. and Epstein D. J. (2003). Distinct regulators of Shh transcription in the floor plate and notochord indicate separate origins for these tissues in the mouse node. Development 130, 3891-3902. 10.1242/dev.00590 [DOI] [PubMed] [Google Scholar]
- Jiang Y., Li Z., Zhang L. and Sun P. (2007). An improved SVM classifier for medical image classification. In Rough Sets and Intelligent Systems Paradigms, pp. 764-773. Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Kiselev V. Y., Kirschner K., Schaub M. T., Andrews T., Yiu A., Chandra T., Natarajan K. N., Reik W., Barahona M., Green A. R. et al. (2017). SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483-486. 10.1038/nmeth.4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F., Scholze M., Wittler L., Schifferl D., Sudheer S., Grote P., Timmermann B., Macura K. and Herrmann B. G. (2017). Antagonistic activities of Sox2 and brachyury control the fate choice of neuro-mesodermal progenitors. Dev. Cell 42, 514-526.e7. 10.1016/j.devcel.2017.07.021 [DOI] [PubMed] [Google Scholar]
- Kojima Y., Kaufman-Francis K., Studdert J. B., Steiner K. A., Power M. D., Loebel D. A. F., Jones V., Hor A., de Alencastro G., Logan G. J. et al. (2014). The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell 14, 107-120. 10.1016/j.stem.2013.09.014 [DOI] [PubMed] [Google Scholar]
- Lawson K. A., Dunn N. R., Roelen B. A., Zeinstra L. M., Davis A. M., Wright C. V. E., Korving J. P. W. F. M. and Hogan B. L. M. (1999). Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 13, 424-436. 10.1101/gad.13.4.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. D. and Anderson K. V. (2008). Morphogenesis of the node and notochord: the cellular basis for the establishment and maintenance of left-right asymmetry in the mouse. Dev. Dyn. 237, 3464-3476. 10.1002/dvdy.21598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Lv F., Zhu S., Yang M., Cour T., Yu K., Cao L. and Huang T. (2011). Large-scale image classification: Fast feature extraction and SVM training. In CVPR 2011, pp. 1689-1696. IEEE; 10.1109/CVPR.2011.5995477 [DOI] [Google Scholar]
- Lippmann E. S., Williams C. E., Ruhl D. A., Estevez-Silva M. C., Chapman E. R., Coon J. J. and Ashton R. S. (2015). Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Rep. 4, 632-644. 10.1016/j.stemcr.2015.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M. et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202-1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Arias A. and Steventon B. (2018). On the nature and function of organizers. Development 145, dev159525 10.1242/dev.159525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D. J., Campbell K. R., Lun A. T. L. and Wills Q. F. (2017). Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179-1186. 10.1093/bioinformatics/btw777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew M. J., Sherman A., Lillico S. G., Ellard F. M., Radcliffe P. A., Gilhooley H. J., Mitrophanous K. A., Cambray N., Wilson V. and Sang H. (2008). Localised axial progenitor cell populations in the avian tail bud are not committed to a posterior Hox identity. Development 135, 2289-2299. 10.1242/dev.022020 [DOI] [PubMed] [Google Scholar]
- Moris N., Pina C. and Arias A. M. (2016). Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 17, 693-703. 10.1038/nrg.2016.98 [DOI] [PubMed] [Google Scholar]
- Niakan K. K., Ji H., Maehr R., Vokes S. A., Rodolfa K. T., Sherwood R. I., Yamaki M., Dimos J. T., Chen A. E., Melton D. A. et al. (2010). Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes Dev. 24, 312-326. 10.1101/gad.1833510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanayotou C., Benhaddou A., Camus A., Perea-Gomez A., Jouneau A., Mezger V., Langa F., Ott S., Sabéran-Djoneidi D. and Collignon J. (2014). A novel nodal enhancer dependent on pluripotency factors and Smad2/3 signaling conditions a regulatory switch during epiblast maturation. PLoS Biol. 12, e1001890 10.1371/journal.pbio.1001890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B., Griffiths J. A., Guibentif C., Hiscock T. W., Jawaid W., Calero-Nieto F. J., Mulas C., Ibarra-Soria X., Tyser R. C. V., Ho D. L. L. et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490-495. 10.1038/s41586-019-0933-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C., Fugazza C., Tipping A. J., Brown J., Soneji S., Teles J., Peterson C. and Enver T. (2012). Inferring rules of lineage commitment in haematopoiesis. Nat. Cell Biol. 14, 287-294. 10.1038/ncb2442 [DOI] [PubMed] [Google Scholar]
- Qiu X., Hill A., Packer J., Lin D., Ma Y.-A. and Trapnell C. (2017a). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309-315. 10.1038/nmeth.4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H. A. and Trapnell C. (2017b). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979-982. 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L., Hartley L., Begley C. G., Brodnicki T. C., Copeland N. G., Gilbert D. J., Jenkins N. A. and Elefanty A. G. (2000). Cloning, expression analysis, and chromosomal localization of murine and human homologues of aXenopus Mix gene. Dev. Dyn. 219, 497-504. 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1070>3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- Schubert F. R., Fainsod A., Gruenbaum Y. and Gruss P. (1995). Expression of the novel murine homeobox gene Sax-1 in the developing nervous system. Mech. Dev. 51, 99-114. 10.1016/0925-4773(95)00358-8 [DOI] [PubMed] [Google Scholar]
- Selleck M. A. and Stern C. D. (1991). Fate mapping and cell lineage analysis of Hensen's node in the chick embryo. Development 112, 615-626. [DOI] [PubMed] [Google Scholar]
- Shiratori H. and Hamada H. (2006). The left-right axis in the mouse: from origin to morphology. Development 133, 2095-2104. 10.1242/dev.02384 [DOI] [PubMed] [Google Scholar]
- Steventon B. and Martinez Arias A. (2017). Evo-engineering and the cellular and molecular origins of the vertebrate spinal cord. Dev. Biol. 432, 3-13. 10.1016/j.ydbio.2017.01.021 [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Stoeckius M., Smibert P. and Satija R. (2018). Comprehensive integration of single cell data. bioRxiv, 460147 10.1101/460147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam P. P. L. and Beddington R. S. P. (1987). The formation of mesodermal tissues in the mouse embryo during gastrulation and early organogenesis. Development 99, 109-126. [DOI] [PubMed] [Google Scholar]
- Tam P. P. L. and Behringer R. R. (1997). Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 68, 3-25. 10.1016/S0925-4773(97)00123-8 [DOI] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N. J., Livak K. J., Mikkelsen T. S. and Rinn J. L. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridis A. and Wilson V. (2015). Assessing the bipotency of in vitro-derived neuromesodermal progenitors. F1000Research 4, 100 10.12688/f1000research.6345.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridis A., Huang Y., Blin G., Skylaki S., Wymeersch F., Osorno R., Economou C., Karagianni E., Zhao S., Lowell S. et al. (2014). Distinct Wnt-driven primitive streak-like populations reflect in vivo lineage precursors. Development 141, 1209-1221. 10.1242/dev.101014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D. A., Hayward P. C., Baillie-Johnson P., Rué P., Broome R., Faunes F. and Martinez Arias A. (2014). Wnt/β-catenin and FGF signalling direct the specification and maintenance of a neuromesodermal axial progenitor in ensembles of mouse embryonic stem cells. Development 141, 4243-4253. 10.1242/dev.112979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanitha C. D. A., Devaraj D. and Venkatesulu M. (2015). Gene expression data classification using support vector machine and mutual information-based gene selection. Procedia Comput. Sci. 47, 13-21. 10.1016/j.procs.2015.03.178 [DOI] [Google Scholar]
- Weinreb C., Wolock S. and Klein A. M. (2017). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246-1248. 10.1093/bioinformatics/btx7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein D. C., Ruiz i Altaba A., Chen W. S., Hoodless P., Prezioso V. R., Jessell T. M. and Darnell J. E. (1994). The winged-helix transcription factor HNF-3β is required for notochord development in the mouse embryo. Cell 78, 575-588. 10.1016/0092-8674(94)90523-1 [DOI] [PubMed] [Google Scholar]
- Wilson V., Olivera-Martinez I. and Storey K. G. (2009). Stem cells, signals and vertebrate body axis extension. Development 136, 1591-1604. 10.1242/dev.021246 [DOI] [PubMed] [Google Scholar]
- Wolfe A. D. and Downs K. M. (2014). Mixl1 localizes to putative axial stem cell reservoirs and their posterior descendants in the mouse embryo. Gene Expr. Patterns 15, 8-20. 10.1016/j.gep.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood H. B. and Episkopou V. (1999). Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech. Dev. 86, 197-201. 10.1016/S0925-4773(99)00116-1 [DOI] [PubMed] [Google Scholar]
- Wymeersch F. J., Huang Y., Blin G., Cambray N., Wilkie R., Wong F. C. K. and Wilson V. (2016). Position-dependent plasticity of distinct progenitor types in the primitive streak. eLife 5, e10042 10.7554/eLife.10042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymeersch F. J., Skylaki S., Huang Y., Watson J. A., Economou C., Marek-Johnston C., Tomlinson S. R. and Wilson V. (2019). Transcriptionally dynamic progenitor populations organised around a stable niche drive axial patterning. Development 146, dev.168161 10.1242/dev.168161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka Y., Tamplin O. J., Beckers A., Gossler A. and Rossant J. (2007). Live imaging and genetic analysis of mouse notochord formation reveals regional morphogenetic mechanisms. Dev. Cell 13, 884-896. 10.1016/j.devcel.2007.10.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.