

ABSTRACT
The generation of multiple fates from a uniform cell population via self-organisation is a recurring feature in development and regeneration. However, for most self-organising systems, we have little understanding of the processes that allow cells to become different. One of the clearest examples of developmental self-organisation is shown by Dictyostelium, with cells segregating into two major fates, stalk and spore, within multicellular aggregates. To characterise the gene expression decisions that underlie this cell fate bifurcation, we carried out single cell transcriptomics on Dictyostelium aggregates. Our data show the transition of progenitors into prespore and prestalk cells occurs via distinct developmental intermediates. Few cells were captured switching between states, with minimal overlap in fate marker expression between cell types, suggesting states are discrete and transitions rapid. Surprisingly, fate-specific transcript dynamics were a small proportion of overall gene expression changes, with transcript divergence coinciding precisely with large-scale remodelling of the transcriptome shared by prestalk and prespore cells. These observations suggest the stepwise separation of cell identity is temporally coupled to global expression transitions common to both fates.
KEY WORDS: Single cell transcriptomics, Stochastic gene expression, Symmetry-breaking, Dictyostelium, Attractor state, MS2
Summary: A stochastic differentiation programme shows stepwise separation of cell fate, with initiation of cell type-specific gene expression coupled to global transcriptome changes shared by all cells.
INTRODUCTION
At its most rudimentary, a self-organising developmental system is one in which a single cell or unstructured population of equivalent cells is able to spontaneously derive a functionally differentiated tissue. These phenomena are widespread, with notable examples including the specification and organisation of the cell types in the early mammalian embryo (Xenopoulos et al., 2012) and the ability of single haematopoietic stem cells to give rise to diverse blood cell types upon transplantation (Becker et al., 1963). The potential for development to be driven by self-organisation is perhaps most viscerally illustrated by the ability of uncommitted stem cells, in relatively non-patterned culture environments, to generate organoids (Sasai, 2013; Sato and Clevers, 2013) and, more recently, structures resembling entire embryos (Beccari et al., 2018).
Understanding how equivalent cells can self-pattern to produce complex differentiated structures is not trivial. More ‘instructive’ forms of development, which are anchored by a limited number of regulators, allow a more conventional experimental narrative based upon standard perturbation approaches. In contrast, self-organising systems are by their very nature adaptive and responsive – how else could the correct proportions of cells, in the correct alignments and orientations, be generated without precise instruction? Adaptive systems will often rapidly accommodate to standard perturbation approaches, giving minimal or indirect insight into how cells make decisions.
These difficulties in describing the early events of cell fate choice in self-organising systems, without the experimental ‘landmark’ of an instructive inducer to orientate our understanding, can potentially be addressed using single cell transcriptomics. The ability to sample the transcriptomes of individual cells, within complex tissues, as they decide their fates, provides the opportunity to delineate the early features of symmetry breaking. Clarity of understanding can be further aided by using a system with few differentiated cell types; this reduces the number of cells that need to be sampled, which permits a greater depth of sequencing, allowing more comprehensive insight into the gene expression dynamics underlying cell fate choice.
Dictyostelium cells show one of the clearest examples of self-organisation during development. Upon starvation, cells initiate a programme of differentiation resulting in the generation of the two major cell fates: stalk and spore. After 6 h of starvation, single cells chemotax together to form a multicellular mound. Cells entering this mound are initially equivalent, before deciding over the next few hours whether to become stalk or spore progenitors (prestalk and prespore, respectively). The final developed structure, formed 24 h after the induction of differentiation, consists of a spore head suspended over the substrate by a thin cellular stalk. Prestalk and prespore markers have been identified (Brown and Firtel, 1999; Maeda et al., 2003; Maruo et al., 2004; Mehdy et al., 1983; Williams, 2006), and specific perturbations and intrinsic cell states can favour specific developmental choices. In particular, the choice between stalk and spore fates is influenced by a cell's position in the cell cycle at the onset of starvation (Gomer and Firtel, 1987; Gruenheit et al., 2018; Weijer et al., 1984), with cells dividing around the onset of starvation favouring the stalk fate. These intrinsic fate tendencies can be further modulated by a variety of different extracellular signals, such as cAMP and DIF (Brown and Firtel, 1999; Kay et al., 1999; Loomis, 2014) and the nutritional history of the cell (Thompson and Kay, 2000). The full developmental programme of Dictyostelium involves a complex series of gene expression changes corresponding to different phases of differentiation (Rosengarten et al., 2015). However, the initial gene expression transitions occuring in individual cells during cell fate separation have not been defined.
To characterise the changes in gene expression accompanying the progression from an equivalent population of cells through a bifurcation to two separate fates, we carried out single cell transcriptomics on the mound phase of Dictyostelium development. Our data reveal that cells entering the mound transition through distinct intermediate cell states with stalk or spore tendencies. Following these intermediates, cells fully express the classical prestalk markers or more strongly induce the prespore programme. Transitions between cell states are rapid and cell states appear separated, with little spillover in the expression of cell type-specific markers between fates. Surprisingly, most changes in transcript abundance occurring during fate separation were common to both spore and stalk, with almost step-like progression in global expression profiles occurring alongside the initiation of cell type-specific programmes.
RESULTS
To characterise the gene expression decisions occurring during cell fate choice in Dictyostelium, we carried out single cell RNA sequencing (scRNA-seq) on cells developed for 14 h. By this time of development, cells have completed aggregation into multicellular mounds (Fig. 1A), with cells within the mound beginning to express markers of the final cell fates, stalk and spore (Williams, 2006). The cells were developed on an agar substrate, which favours temporally heterogeneous progression through the multicellular phase of development. Under these conditions, the developmental state of cells will vary between just having completed aggregation through to tip formation, in preparation for the later stages of morphogenesis. To allow for sensitive detection of early indications of cellular decision-making, we captured transcriptomes at a high read depth (∼3×106 reads/cell) for 116 cells.
Fig. 1.

Cell type divergence in Dictyostelium aggregates. (A) Schematic of Dictyostelium development. Single cell transcriptomics was carried out on 116 cells, over three replicates, at the mound stage (outlined). (B) Patterns of correlation within lineage-specific genes. Correlation heatmaps, split into prespore and prestalk genes, are shown for 0, 3, 6 and 14 h timepoints. We selected cell type-specific genes from the data of Parikh et al. (2010) with |log2FC|>1, FDR<0.1 and an expression level of at least 100 normalised read counts, in at least one cell. (C) PCA of individual cell transcriptomes reveals distinct subpopulations at the mound stage. Shown here are the first two principal components. Each dot is a cell, colour-coded by developmental time.
The single cell transcriptomes show clear indications of cell fate divergence. Fig. 1B and Fig. S1A show correlation heatmaps for markers of stalk and spore fate, at different stages of development. The fate markers were extracted from transcriptomic data of prespore and prestalk cells separated by gradient centrifugation (Parikh et al., 2010). In data from the unicellular phase of development (0-6 h) (Antolovic et al., 2017) there was no clear segregation of heatmaps into stalk and spore clusters. In contrast, the 14 h heatmap shows widespread single cell correlations between spore genes, clear correlations between stalk genes and anti-correlations between genes of the two fates. Divergence between cells at 14 h is also observed using principal component analysis (PCA) (Fig. 1C). Cells from 0-6 h of development projected as single populations in PCA. In contrast, 14 h data were more dispersed, showing at least two separate clusters.
Analysis of known cell fate markers did not correspond to specific isolated clusters in this PCA analysis of multiple developmental timepoints (Fig. S1B). For a more unambiguous analysis of the structure of the 14 h populations, we considered the transcriptomic data in PCA space for 14 h data alone (Fig. 2).
Fig. 2.
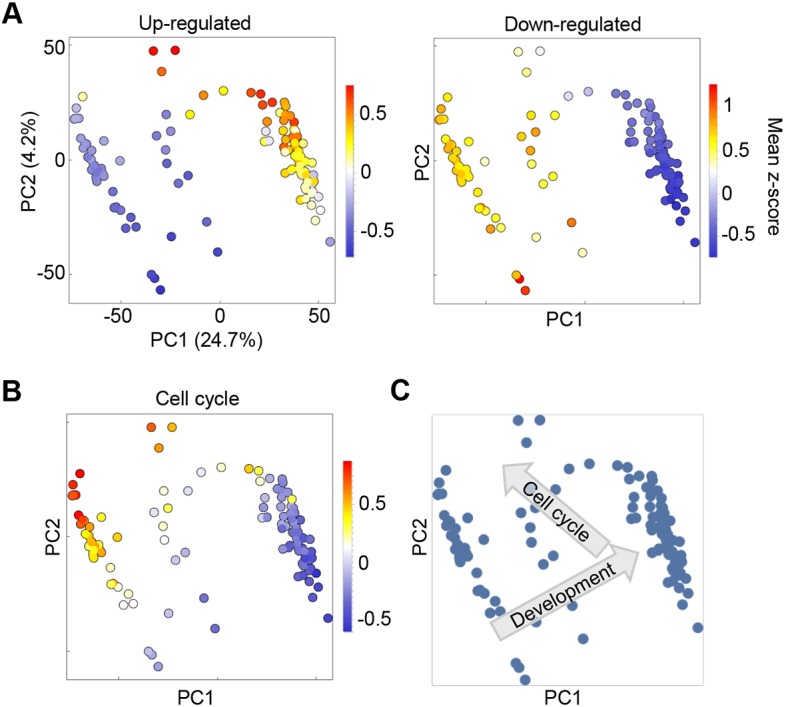
Heterogeneity in developmental timing and cell cycle within Dictyostelium mounds. (A) PCA of single cell transcriptomes reveals that developmental timing is highly heterogeneous in mounds. Shown here are the first two principal components. Each dot is a cell, coloured by the average expression value (z-score) of a set of genes that are upregulated (left panel) or downregulated during the mound stage (right panel). The gene set was taken from Rosengarten et al. (2015). PC1, and to a certain extent PC2, contains information about developmental advancement. (B) Same as A, but cells are coloured by the average expression value of a set of cell cycle-related genes. Differences in cell cycle-related gene expression are also captured by PC1 and PC2, which suggests that cell cycle advancement is not independent of developmental stage. (C) Vectors of developmental progression and the cell cycle state of cells in PC2/PC1 space.
At least three populations were detected for the two major components, PC1 and PC2. Differences in gene expression caused by heterogeneity in developmental progression are captured predominantly by PC1 (Fig. 2A). We compared 14 h single cell transcriptomes with population transcriptomic data (Rosengarten et al., 2015) for transcripts that change over the 4 h of the mound phase. Genes upregulated during this window are enriched in cells with high PC1 scores. Genes downregulated during this window are enriched in the cells with low PC1 scores. Consistent with this interpretation, gene ontology (GO) analysis of genes with high PC1 loadings (transcripts enriched in the cells with high PC1 values) reveals processes linked to multicellular development (Fig. S2A). In contrast, negative loadings (transcripts enriched in cells with strong negative PC1 values) are enriched in gene expression-related GO terms, consistent with data showing cells strongly reduced global transcription during development (Mangiarotti et al., 1981).
Differences in expression of cell cycle components are captured by both PC1 and PC2 (Fig. 2B). When PCA plots are labelled according to the expression of a curated set of 162 genes with annotated cell cycle functions (Table S1), enrichment of their expression occurs at low PC1 and high PC2 scores. Cell cycle gene enrichment in this part of PC1-PC2 space is independent of whether genes are linked to S or M phase (Fig. S2B), suggesting the absence of one of the gap phases, consistent with a number of reports indicating Dictyostelium lacks G1 (Muramoto and Chubb, 2008; Zimmerman and Weijer, 1993). Variability in cell cycle position and developmental timing may not be completely independent, with a wave of expression of many cell cycle genes occurring in the mound (Rosengarten et al., 2015).
Separation of cell fate progenitors
The first two principal components effectively show developmental progression, and an early wave of gene expression of cell cycle components in the mound (Fig. 2C). However, distinctions between cells with different fates only become apparent in higher order principal components.
Plots of PC3 against PC1 show at least three distinct clusters of cells (Fig. 3A). With PC1 as a proxy for developmental time, we observed one compact early population. This population became more dispersed later in development, resolving into two late populations, one with high PC3 scores and another with low PC3 scores. Genes with high positive loadings in PC3 are strongly enriched in prespore genes (Fig. 3B). Genes with negative loadings in PC3 are overwhelmingly enriched in prestalk markers (Fig. 3C). These data imply the presence, within the mound phase, of a single progenitor population, which becomes more diverse, before ultimately generating distinct prespore and prestalk cells.
Fig. 3.
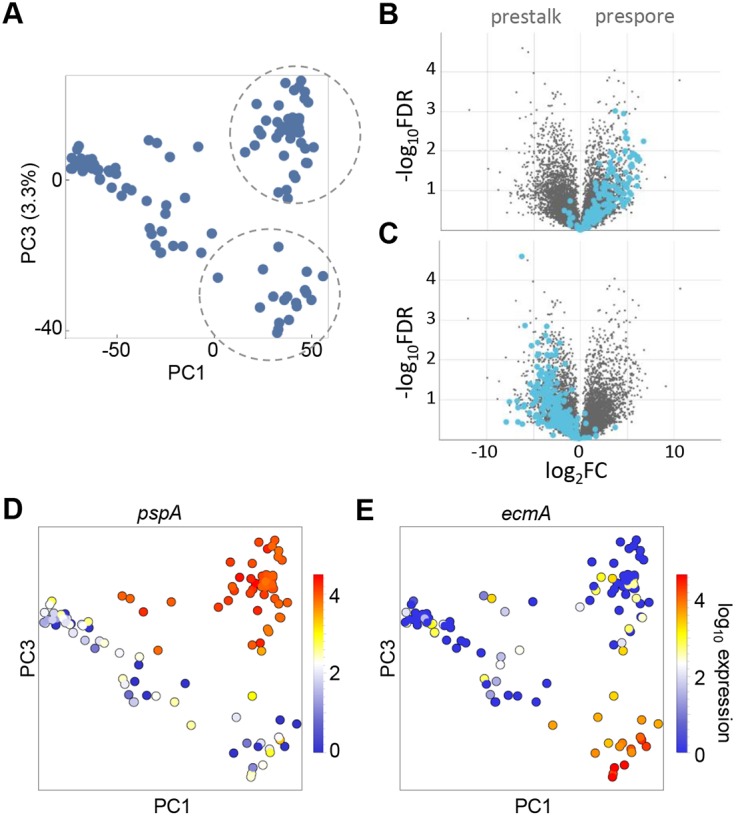
Identification of prespore and prestalk populations in late mounds. (A) The identification of cell type-specific subpopulations in mounds requires analysis of higher order principal components. Each dot is a cell shown in the PC3 versus PC1 space, with PC1 used as a proxy for developmental time. Distinct clusters separate at high values of PC1 (encircled). (B) Genes with high positive loadings in PC3 are strongly enriched for prespore markers, as shown in the volcano plot. (C) Genes with negative PC3 loadings are enriched in prestalk markers. Volcano plots labelled using dictyExpress (Stajdohar et al., 2017). (D,E) Expression of standard cell type-specific markers pspA and ecmA validates the clusters that are separated in PC3 as prespore and prestalk. Each dot is a cell coloured by the level of expression of pspA or ecmA. The prespore marker pspA is enriched in the population that was identified as prespore in B, and a few cells from earlier in development (D). The prestalk marker, ecmA, is enriched in the population that was identified as prestalk in C (E).
In support of this view, the expression of classical markers of the different fates shows appropriate distributions when overlaid on PC3/PC1 space. The prespore marker pspA (Early et al., 1988) is strongly expressed in all the high PC3 cluster cells (Fig. 3D). Other prespore markers, such as the spore coat gene cotB, and dmtA, which encodes an enzyme that is required for the biogenesis of the stalk inducer DIF-1, are also enriched in the high PC3 population (Fig. S3A). We term this population ‘prespore’. The prestalk marker ecmA (Early et al., 1988) is strongly expressed in the low PC3 cluster (Fig. 3E). We term this population ‘prestalk’.
Defining early symmetry-breaking intermediates
The pspA gene is not exclusive to the distinct prespore cluster, as it shows clear expression in a distinct subset of the cells from an earlier developmental time (Fig. 3D). This distribution is also shared with the prespore markers cotB and dmtA (Fig. S3A). The early expression of prespore markers implies an early intermediate with prespore tendencies.
At a similar developmental time to these prespore intermediates, there were cells without strong prespore expression, with slightly negative PC3 scores, suggestive of an early prestalk intermediate population. However, these cells displayed no strong early expression of the prestalk marker ecmA (Fig. 3E). Does this imply that there is no intermediate prestalk population, or alternatively, do prestalk intermediates exist before ecmA induction? Earlier work using ecmA-lacZ promoter fusion transgenes, in combination with an antibody against the prestalk marker CP2 (encoded by cprB) (Clay et al., 1995), showed that CP2 protein could be detected before β-galactosidase staining during development. This suggested developmental progression within prestalk cells, consistent with the possibility of a distinct intermediate state. Our data allow us to test this possibility further, using transcriptome-wide analysis of native transcripts.
Analysis of the fourth principal component indicates the presence of a prestalk intermediate. In plots of PC4 versus PC1, an intermediate-stage population segregates with high PC4 scores (Fig. 4A, circled), with prespore and prestalk cells showing low or negative scores (Fig. S4A). Is this high PC4 cluster an early prestalk intermediate? Analysis of the positive loadings of different genes for PC4 initially suggested no bias to either stalk or spore lineage (Fig. S4B,C). However, we reasoned that, with the prestalk population having low or negative PC4 scores (Fig. S4A) and the candidate early prestalk population having high PC4 scores, any prestalk genes that are common to both populations would not contribute strongly to PC4. In other words, the mature prestalk population may be hiding the features of a candidate early prestalk population in the analysis. To evaluate the enrichments in this early population, we masked the mature prestalk population from the PCA. In the PC3/PC1 space of this reduced dataset, the candidate early population was again clearly separated from all other cells (Fig. 4B, circled). Genes with strong positive loadings for this PC3 were enriched in prestalk markers (Fig. 4C, Table S2). Genes with negative loadings showed a slight prespore bias (Fig. 4D). The enriched genes in Table S2 do not include cprB; however, cprE and cprG are present, which are 57% and 60% identical to cprB, and so may contribute to antibody staining against CP2. Carrying out the opposite masking process, with prespore cells excluded, did not reveal a prespore tendency in this candidate early population (Fig. S4D-F). These data indicate the presence of an early prestalk intermediate, with ecmA induction a feature of a more mature prestalk population.
Fig. 4.

Identification of an early intermediate in prestalk differentiation. (A) Identification of a population (encircled) of intermediate developmental progression with high values of PC4. PC1 is used as a proxy for developmental time. (B) PCA on the mound population after masking of prestalk cells from the analysis, with the same population encircled as in A. (C) After masking the stalk population, genes with high positive loadings in PC3 are enriched in prestalk markers, as shown by the volcano plot. (D) After masking the stalk population, genes with negative loadings in PC3 are enriched in prespore markers. (E) Visualising cell fate separation in the first four principal components. Individual cells are coloured based on their cell-type: unspecified early mound (blue), early prestalk (purple), early prespore (orange), prespore (red) and prestalk (green).
Overall, this analysis reveals that cells transit through multiple developmental intermediates during the ∼4 h mound phase (Fig. 4E) and suggests a view of developmental progression within the mound in which cells initially exist as a single population (blue). Cells then progress into early prespore and prestalk cell states (orange and purple, respectively) before maturation to more differentiated progenitors (green and red).
Inferring mechanisms of cell sorting and specification
During the mound phase, prestalk and prespore cells segregate, with prestalk cells translocating to the top of the mound, forming the tip. This migration has been proposed to result from several processes (Loomis, 2015), including chemotaxis to cAMP pulses emanating from the tip, differential adhesion and differential motility. To gain insight into the contributions of these different processes, we studied the expression of markers of chemotaxis, adhesion and motility in the different subpopulations in mounds (Fig. S3).
Well-studied genes involved in chemotaxis to cAMP show enrichments in expression in prestalk cells (Fig. S3B). Examples are carA, pdsA and acaA, which encode a cAMP receptor, cAMP phosphodiesterase and adenylyl cyclase, respectively. Adhesion genes tgrB1 and tgrC1 show no bias in expression between cell types (Fig. S3C); however, csbA and, to a certain extent, csaA, show enrichment in prestalk cells (Fig. S3D). Motility-associated genes also show prestalk enrichment, including myosin II (mhcA and mlcR) (Fig. S3E) and almost all the 17 genes of the act8 (actin) gene family (Fig. S3F). These data suggest the segregation of prestalk and prespore cells may have contributions from differences between cell types in chemotaxis, motility and adhesion. Contributions from multiple processes in cell sorting may contribute to the inability of some strong mutations in regulators of chemotaxis (Wang and Kuspa, 1997) and adhesion (Wong et al., 2002) to completely disrupt cell sorting.
The markers that identify the early prestalk population have varied expression characteristics (Fig. 5A and Fig. S3G). Tpp1F (V4-7) is one of the few genes that is exclusive to the early prestalk cells. Others, such as guaD and hacl1, show moderate induction in other cells earlier and later in development, with abcF2 expressed in most cells, and only slightly enriched in early prestalk cells. CryS, which encodes a peroxisomal protein, is strongly expressed in early prestalk cells, and continues through to the mature prestalk population. These diverse expression characteristics, forming an amalgam of genes with slight to moderate enrichments, may be a feature of intermediate and perhaps incompletely specified progenitors.
Fig. 5.

Characterisation of the early prestalk population. (A) Selected markers of the early prestalk population. Projection of cells in PC4/PC1 space, with each cell coloured by the expression level of one of the indicated genes. Each gene shows enrichment in the cluster with high PC4 values, with some genes showing detectable expression in other cell states. (B,C) Preferential enrichment of markers in A to the prestalk regions of multicellular structures, visualised using knock-in protein fusions. The fast-folding fluorescent protein mNeonGreen was fused to C-termini, by targeting to endogenous loci. Unless stated otherwise, each image is representative of at least three independent experiments [the frequency of occurrence of the pattern shown (per structure) is stated in each panel]. Inset for AbcF2-Neon (B) is a tipped mound (4/5 structures from two independent experiments). Tpp1F-Neon could not be detected. Arrowheads mark the anterior prestalk region, although additional prestalk cells (ALCs) are also scattered in the main slug body. An inclined view of early stage culminants of GuaD-Neon cells (C), showing localisation to basal disc and upper and lower cup structures (arrowheads). Scale bars: 50 µm; 20 µm in AbcF2 inset.
Analysis of knock-in reporter cell lines of these markers is consistent with this ‘amalgam’ view (Fig. 5B). We inserted a sequence encoding the fast-folding fluorescent protein mNeonGreen (Neon) (Tunnacliffe et al., 2018) into the 3′ end of the coding sequences of abcF2, cryS, guaD, hacl1 and tpp1F. The expression of Tpp1F-Neon was too weak to detect. AbcF2 showed enrichment at the anterior prestalk region of slugs (arrowheads) after stronger enrichment in tipped mounds (inset). CryS-Neon also showed anterior localisation with a slight enrichment in the collar region and some cells showing a tendency to be shed from the rear of migrating slugs. Whether shedding is a tendency of prestalk cells, or some additional feature of cells with high CryS expression, is currently unclear. In contrast, Hacl1-Neon expression was low in the anterior zone, with moderate expression in the prespore zone and rare strong expressers throughout the slug. Cells expressing high levels of GuaD-Neon showed a distinctive scattered localisation in the posterior zone. These are likely to be prestalk cells, with images of the culmination step showing high GuaD-Neon cells consistently enriched in basal disc and lower cup structures, with the majority of structures also showing localisation to the upper cup (Fig. 5C, arrowheads).
This apparent complexity of the early prestalk population may also relate to the known diversity of mature prestalk types. In addition to the well-known anterior slug prestalk cells, ∼10% of the cells in the posterior spore zone also have pre-stalk marker expression – the so-called anterior-like cells (ALCs) (Sternfeld and David, 1982). Other cell types can also be identified in the slug, such as the immune-like sentinel cells (Chen et al., 2007). Sentinel cells are rare, ∼1% of the slug, and so may not leave a clear signature in our dataset. However, based on the abundance of ALCs, they should be apparent. To test this possibility, we evaluated the expression of markers previously shown to have some specificity to ALCs. Expression of the ampA gene (Casademunt et al., 2002) is not clearly segregated within the mature prestalk population into expressers and non-expressers (Fig. S4G), suggesting that, at the mound stage, there is no major restriction of ampA expression to a prestalk subset such as ALCs. In contrast, expression of six class 1 sigN genes (Vicente et al., 2008) shows clear enrichment in around half of the prestalk cells (Fig. S4H), suggesting these cells may correspond to the ALC population.
Sharp genome-wide expression transitions are shared between fates
To generate a more in-depth view of the transcriptome changes occurring in the mound, we carried out hierarchical clustering on single cell transcript counts. This revealed two separate branches, with one showing a significant side branch (Fig. 6A). These branches correspond exactly to the groupings and temporal ordering identified by PCA as ‘early’, ‘intermediate’ or ‘late’, with only three outlier cells. The top cluster corresponds to the late cells, and consists of both the prespore (red) and prestalk (green) populations. Within this cluster, the prespore and prestalk cells segregate into two separate groups, with the fate-specific genes visible as darker red patches (outlined by rectangles 1 and 2). In addition, although markers common to both fates are known (Mehdy et al., 1983), our transcriptome-wide data show a remarkable overlap in the transcript composition of prestalk and prespore cells. The fate-specific genes represent only a small fraction of the transcriptome changes shared by both fates.
Fig. 6.

Sharp genome-wide expression transitions shared between cell fates. (A) Hierarchical clustering of single cell transcriptomics data. The rows of the heatmap represent cells, with the ends of the clustergram branches coloured in the same way as in Fig. 4E – early mound (blue), early prestalk (purple), early prespore (orange), prestalk (green) and prespore (red). The columns of the heatmap represent 5229 genes. Clusters of cells correspond to the cell groups identified by PCA. The lower cluster in the plot represents the early mound cells (blue). The middle cluster contains the two intermediate populations (purple and orange), both still transcribing genes that are typical of early cells and starting to express genes that are typical of mature prestalk and prespore cells. Prestalk and prespore cells (green and red, respectively) segregate into two separate groups, with fate-specific gene clusters outlined by boxes (1, prespore specific; 2, prestalk specific). (B) High transcriptome entropy of the intermediate cells corresponds to the observation in A that transcript expression is more uniformly distributed across the transcriptome of the intermediate subpopulations (outlined by boxes 3 and 4). For comparison, 0, 3 and 6 h timepoints are in grey. (C) Evidence for discontinuous developmental progression in the mound. Pseudo-3D projection of cells in PC4/PC3/PC1 space shows separation of cell clusters.
Moving down the plot, the next cluster is the intermediate cells, which consists of the early prespore (orange) and early prestalk cells (purple). The early prespore cells show expression of the prespore genes. In contrast, the prestalk-specific genes defined in mature prestalk cells are not strongly induced in early prestalk cells, as earlier typified by ecmA, which is not an early marker. Therefore, although we identified early prestalk markers such as cryS and abcF2, at this intermediate stage the early prestalk cells are characterised more by the absence of prespore markers, rather than the presence of a mature prestalk programme. Other differences between the two fates can be detected in this intermediate population (rectangles 3 and 4). The bulk of the transcriptome shows similar changes for both fates during the transition to the late stage; however, many of these changes are apparent in the early prespore cells, but not yet clear in the early prestalk. This is illustrated in the left region of the plot, with early prestalk cells retarded in their developmental progression compared with early prespore cells. Overall, both intermediate populations displayed features of both early and late cells. This apparent disorder is summarised by the transcript entropy statistic (Martinez and Reyes-Valdes, 2008), which is elevated for intermediate cells (Fig. 6B).
The lower cluster in the plot (blue) is the early progenitor pool, with cells showing a markedly different transcriptome to the late cells. GO terms related to translation and post-transcriptional control are particularly enriched (Fig. S5A). Unlike the late and intermediate cells, very little transcriptional heterogeneity was detectable in this pool. One exception is a small cluster of ∼60 transcripts in the centre of the heatmap (Fig. S5A). The majority of these transcripts are retrotransposons, primarily DIRS-1 elements. A slight enrichment of prestalk markers in the early progenitor pool was suggested by analysis of genes with strong negative PC1 loadings (Fig. S5B,C). It is unclear whether this effect is biologically meaningful, as PC1 is heavily dominated by developmental time, hence the genes with negative loadings are downregulated through the mound phase (Fig. S5D), and will be well below their expression maxima.
Putting aside specific classes of genes, two general features emerge from the hierarchical clustering. Firstly, the cell groupings are separated by large-scale genome-wide differences in transcript content (Fig. 6A). Separation of the cell groups is also clearly visible in the pseudo-3D PCA plot in Fig. 6C. This separation implies there were few cells caught between states, consistent with a scenario in which transitions between different clusters are rapid (Moris et al., 2016). Secondly, although the signatures of different cell fates are detectable in our data, these signatures correspond to a relatively small proportion of the genome. Cells initiate and maintain cell fate tendencies during large-scale global changes in transcript content that are shared by both cell types. These global changes coincide with the transitions to more mature fate-specific expression profiles.
How distinct are the prespore and prestalk fates? Although two independent unbiased methods (PCA and hierarchical clustering) imply bunching of cells into discrete states, markers such as pspA and ecmA show some promiscuity, with low but detectable expression outside their primary cell type (see Fig. 3D,E). To quantify the extent of overlap, read counts for pspA and ecmA expression were plotted for each cell, with cells ordered by decreasing pspA expression (Fig. 7A). The prespore and early prespore cells, marked in red and orange respectively, are strongly enriched for pspA expression, compared with other cells in the mound. However, most cells express pspA to some extent, including several cells with high ecmA expression. Conversely, several prespore and early prespore cells showed some expression of ecmA. A similar overlap was observed between cotB and ecmA transcripts (Fig. S6A).
Fig. 7.

Quantifying spillover of fate-specific markers. (A) scRNA-seq detects co-expression of pspA and ecmA in mound cells. Cells are ordered by the level of pspA expression, with their pspA and ecmA expression levels shown in parallel (as light grey and dark green bars, respectively). For reference, pspA expression in prespore and early prespore cells is marked in red and orange, respectively, and ecmA expression in prestalk cells is marked in green. (B) Single cell transcript counts for pspA and ecmA in disaggregated cells from early mounds (232 cells), late (tipped) mounds (200 cells) and slugs (227 cells). Transcript counts were determined using smFISH. Data shown are from one representative of three independent experiments. The AX2 cell line was used to facilitate greater temporal homogeneity in development. (C) Live imaging of pspA transcription in intact slugs. 128 MS2 repeats were inserted into the pspA gene. Nascent transcripts were detected by expression of MCP-GFP. The inset shows a close-up of the boundary between prespore and prestalk regions, showing no detectable nascent transcripts in the prestalk region. Images are representative of 12 slugs, from two independent experiments on two independent pspA-MS2 cell lines. Scale bars: 20 µm; 10 µm in inset.
The single cell transcriptomics protocol involves amplification steps, which can affect read counts of genes in a non-linear manner. To more quantitatively evaluate the extent of overlap between spore and stalk gene expression, we used single molecule RNA fluorescent in situ hybridisation (smFISH) to count pspA and ecmA transcripts in single cells, over multiple phases of multicellular development. As with the transcriptomic data, expression of the two genes overlapped in single cells; however, in all but 1-2% of cells, strong expressers of one gene usually only had weak (<5 molecules) expression of the other gene, over all developmental times tested (Fig. 7B). Similar observations were also made when comparing single cell smFISH counts of ecmA with those of the alternative prespore marker, cotB (Fig. S6B). In these smFISH measurements for pspA, ecmA and cotB, we observed few nuclear spots corresponding to nascent RNA transcription. We reasoned this was because the smFISH was carried out on disaggregated cells, which lack their normal spatial and signalling context. We therefore adapted the smFISH protocol for use on intact aggregates. As the overlap between ecmA and pspA was consistent over multiple developmental stages, for clear spatial discrimination of different transcriptional states we used slugs. An example of a slug hybridised to fluorescent probes for both pspA and ecmA is shown in Fig. S7. In these intact structures, nascent RNA foci were abundant, for both genes. The slug shows a very clear boundary between prespore and prestalk regions, with no clear examples of cells with nuclear foci (nascent transcripts) of both genes. No cytoplasmic or nuclear staining could be detected in the prestalk region for pspA and only a single nascent transcript signal (arrow in Fig. S7) was observed in the prespore region for ecmA.
These observations on fixed aggregates were confirmed by live imaging of transcription. We generated cells with MS2 repeats (Tantale et al., 2016) inserted into the pspA gene. Nascent transcripts were detected by expression of MCP-GFP (Chubb et al., 2006) in these cells. Fig. 7C and Movie 1 show the prespore-prestalk boundary of a slug that was derived from these cells. At the top of the image (prespore cells) strong pspA transcription is visible as fluorescent nuclear dots in many cells. PspA dots were absent from the prestalk region of the slug (bottom), which implies the gene is switched off or transcribed weakly. A similar conclusion emerged from imaging cotB-MS2 reporter slugs (Fig. S8). Nascent transcripts could be detected in the prestalk region; however, the dots were rare, and weak. As inferred by hierarchical clustering and PCA, these imaging-based observations argue against substantial overlap in the expression of cell type-specific genes between different fates. Indeed, our smFISH and live cell data caution against inference of so-called mixed lineage progenitors from transcriptomics data alone. Instead, our observations may reflect that individual cells, even after choosing a fate, show a low level of ‘inappropriate’ transcriptional firing, perhaps in response to what is likely to be a highly complex signalling environment.
DISCUSSION
To gain insight into how cells make decisions during development, we have used single cell approaches to define the temporal sequence of global gene expression transitions during the cell fate bifurcation of Dictyostelium. Our data reveal the separation of a relatively homogeneous population into two well-defined progenitor fates within ∼4 h. This developmental progression occurs via distinct intermediates. We observed few cells in transit between different temporal states, indicating the transitions are rapid, probably in the order of minutes. Despite the rapid progression of cells through development, the mature progenitor states are relatively discrete, with little overlap in the expression of cell type-specific markers. During transitions between states, transcript dynamics were dominated by changes common to both fates, with cell-type specific expression a small proportion of the overall transcriptome remodelling.
The intermediate states were characterised by a relatively disordered transcriptome, with overlap of transcripts from both the initial population and the mature progenitors. This disorder was also apparent for markers of the early prestalk intermediate. Here, the enriched transcripts, and proteins, do not appear to be highly specific, unlike, for example, ecmA in the mature prestalk cells. This early prestalk state is characterised by a convergence of many expression profiles with a modest enrichment. This may imply that intermediate states are an approximation of the state to come and suggest a consensus model of decision-making, based upon the integration of many weak influences.
Decision-making based upon multiple small influences may be an emerging theme in developmental self-organisation, which likely extends well beyond Dictyostelium (Cannon et al., 2015; Chubb, 2017). Our data provide some clues as to what these influences are, most clearly the heterogeneity in the expression of cell cycle components in the initial pool of cells entering the mound. Cell cycle position can influence cell fate choice (Gomer and Firtel, 1987; Thompson and Kay, 2000; Weijer et al., 1984), with M and S-phase cells at the time of starvation biased towards the stalk fate (there is no G1). Given that the cells essentially stop dividing after starvation, and reside in G2 until the mound phase, it seems feasible that the cells enriched for cell cycle gene expression in the mound are those that did not divide at starvation and will be biased towards spore. Indeed, several studies have shown a broad wave of cell cycle progression occurs in prespore cells in the slug (Muramoto and Chubb, 2008; Zimmerman and Weijer, 1993). In extension, if cell cycle is contributing to fate choice, then when is it instructive to the fate-regulating machinery? Is it at the onset of starvation, in line with the early literature, or in the mound, as might be suggested from the present study? The abundance of cell cycle progression in the slug, compared with the clear strong activation of the prespore programme in the mound, implies that cycle progression lags behind prespore fate activation, despite prior activation of cell cycle genes.
The apparent lack of cells in our data that were caught between states suggests that the transition time between states is rapid. The mound phase lasts ∼4 h. Assuming the sampled cells are randomly distributed in developmental time through the mound phase, then the time gap between each cell would be ∼2 min. This seems unexpectedly fast, although this time gap will be greater if cells are distributed over multiple trajectories. In addition, temporal heterogeneity exists within clusters, which shrinks the effective jump in high dimensional expression space. Are there limits to the speed of switching? Switching will likely require concerted transcription and RNA turnover. Measurements of signal-induced mRNA degradation suggest a timescale of 5-10 min can ensure removal of the transcript for the cAMP receptor, cAR1 (Van Haastert et al., 1992). Based on transcription rates (Corrigan et al., 2016) and response times to developmental inducers (Stevense et al., 2010), it is feasible for a gene of ∼1 kb to express tens of transcripts within ∼5 min. Our analysis here suggests that even a handful of transcripts can be identified by sequencing methods, implying such a rapid transcriptome change could be detected. Rapid transition times between states suggest the continuous downward gradient of the landscape metaphor of developmental progression (Waddington, 1957) might instead be considered as a set of shallow gradients interspersed with steep sections.
Related to the landscape model is the view of a cell state as a high dimensional attractor or ‘potential well’ (Mojtahedi et al., 2016), with the attractor representing, at least in part, the configuration of the regulatory network. In this view, cells enter subsequent attractors (fates) when the initial attractor is destabilised – perhaps by some signal. In aligning our data to this view, we would add additional features, reminiscent of the ‘rugged’ epigenetic landscape used to describe the inefficient progression of cells during reprogramming (Lang et al., 2014). The progress of Dictyostelium cells towards stalk or spore occurs via intermediate states, so requires intermediate attractors. In addition, the conversion between different cell fates is concurrent with large-scale temporal changes in gene expression that are common to both fates. A potential realisation is that the initial attractor destabilises, to a second attractor field, with the intermediate prespore and prestalk cells existing in ‘furrows’ or ‘wrinkles’ in this field, rather than independent attractors in their own right. This intermediate stage is then itself destabilised, perhaps coupled to concerted changes in gene expression common to both fates, which leads to the cells entering the deeper wells of mature prespore and prestalk.
The major themes emerging from our study are the bunching of cells together into distinct states and the coupling of non-specific large-scale transcriptome changes to fate separation. Are these features to be found in other developmental contexts? The prevailing approach in the field is to emphasise large cell numbers rather than sequencing depth, and connectivity rather than discreteness, useful for describing complex mammalian tissues and organs, and inferring trajectories between intermediates. Our approach has been different, with the focus on sequencing depth rather than large numbers of cells, which may be better suited to embellishing the detailed transcriptomic features of a single cell fate bifurcation over a relatively narrow window of developmental time. Potential examples of global changes and discreteness during fate separation can be discerned in vertebrate haematopoiesis, in which expression of lineage-specific factors temporally overlaps, in single cells, the downregulation of ribosome protein transcripts (Athanasiadis et al., 2017) and terminal differentiation occurs alongside a sharp transcriptional switch (Tusi et al., 2018). The current broad expansion in the availability of single cell datasets provides a rich source of data for assessing the generality of these features of cell fate choice.
MATERIALS AND METHODS
Cell handling
For scRNA-seq, we used Dictyostelium AX3 cells expressing an H2B-mCherry fusion protein under the control of the endogenous rps30 promoter (Antolovic et al., 2017; Corrigan and Chubb, 2014). For targeting fluorescent reporters, we used AX3 cells (from Robert Insall, Beatson Institute, Glasgow, UK). For smFISH and live transcription imaging, we used the AX2 strain (from Rob Kay, Laboratory of Molecular Biology, Cambridge, UK), which shows highly synchronous development. Cell lines were authenticated using PCR and Southern blotting. Cells were maintained in Petri dishes in HL5 media (Formedium) under selection against bacterial contamination. Cells were not allowed to exceed confluence and were not used beyond 10 days of culture. For development, cells were washed in KK2 [20 mM KPO4 (pH 6.2)] then plated on KK2/1.5% agar in 35 mm Petri dishes with 1-5×106 cells. The dishes were incubated at 22°C for 14 h. For sequencing and smFISH, aggregates were disaggregated to single cells by taking them up in 1 ml KK2/10 mM EDTA and repeatedly passing the structures through a 20G needle. Selections for knock-in and transgene expression constructs used 10 µg/ml blasticidin S (Merck) and 20 µg/ml G418 (Invitrogen), respectively.
Single cell RNA-seq
Single cell transcriptomes of disaggregated mound cells were determined using the Fluidigm C1 system, with read quality assessment, mapping of 75 bp paired-end reads and read counting for each gene carried out as previously described (Antolovic et al., 2017). An average of 3 million reads were generated from each single cell library. We excluded seven cells based on the following criteria: total number of reads was lower than 5×105, percentage of mitochondrial reads was higher than 15%, or the data was missing a contiguous part of the transcriptome. Reads from ribosomal RNA contaminants were excluded. We retained a total of 116 cells, from three experimental replicates. Read counts of cells within each replica were normalised using the size factor that was calculated using the DESeq package (Anders and Huber, 2010). Read counts from early developmental timepoints (0, 3 and 6 h) were taken from Antolovic et al. (2017), incorporating replicas sequenced with only 75 bp paired-end reads for consistency.
Data analysis
Unless otherwise stated, data processing was carried out in Mathematica. For cell-type structure at different time points, we selected cell type-specific genes from published population transcriptomic data (Parikh et al., 2010) with, |log2FC|>1 FDR<0.1 and an expression level of >100 normalised read counts, in at least one cell in our dataset. Depending upon the time point, this gave 250-251 prespore markers and 298-309 prestalk markers. For more stringent analyses, we used an FDR<0.01. This gave a smaller set of genes: 45-46 prespore markers and 60-63 prestalk markers. For analysis of developmental progression within mounds, genes were selected from Rosengarten et al. (2015) as genes with |log2FC|>1 between the loose aggregate stage and tipped mound stage, and an expression level, in the Rosengarten dataset, of at least 100 reads per kilobase of transcript per million mapped reads (RPKM), at at least one timepoint. The cell cycle-related genes that were used are listed in Table S1. We curated this gene set based upon GO annotations, supplemented with homologues of well-described cell cycle genes from other systems. For characterisation of the subpopulations of cells that were identified by PCA, we ranked the genes by their contribution score, then used the top-ranked genes, the total contribution of which sums to 25% of the component's variance. GO enrichment analysis was performed using the PANTHER Classification System version 13.1. Hierarchical clustering was performed in MATLAB using 5229 genes that satisfied the following condition: their mean normalised read count was >10 and they are correlated with at least 10 other genes (r>0.5). The transcriptome entropy of cell j was defined as 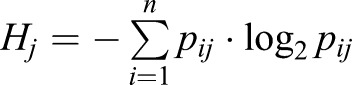 , with pij being the proportion of gene i’s read count in the cell and n being the total number of genes (Martinez and Reyes-Valdes, 2008).
, with pij being the proportion of gene i’s read count in the cell and n being the total number of genes (Martinez and Reyes-Valdes, 2008).
Cell line generation
For imaging fluorescent protein fusions of early prestalk markers, we targeted a cassette that contained a codon-optimised mNeonGreen gene (Paschke et al., 2018; Shaner et al., 2013; Tunnacliffe et al., 2018) upstream of, and in opposite orientation to, the blasticidin resistance marker (Faix et al., 2004) after the final protein-coding triplet of the endogenous cryS, tpp1F, abcF2, guaD and hacl1 genes. This operation allowed the tagged gene to utilise the terminator of the selection marker and placed GlySer between the native protein and the fluorescent tag.
For imaging nascent transcripts of pspA, we targeted a cassette containing 128 MS2 repeats (Tantale et al., 2016) upstream of the blasticidin resistance cassette at position 28 of the pspA coding sequence (the A of ATG is position 1). For imaging the transcription of cotB, we targeted the MS2-blasticidin to position 490 of the cotB genomic sequence, just downstream of the 5′ intron. Nascent RNA was detected by expression of MCP-GFP using a dual expression plasmid, which was a combination of the MCP-GFP expression vector (Corrigan et al., 2016) and PCP-TdTomato expression plasmid based upon the shuttle vector pDM344 (Veltman et al., 2009). The dual expression plasmid was generated by ligating an NgoMIV fragment containing the PCP-TdTomato expression cassette into the MCP-GFP vector.
Imaging gene expression
For smFISH, disaggregated cells were plated in two-well chambered coverslips and, after leaving for 10 min to settle, were processed as described (Raj et al., 2008). For intact aggregates, slugs were washed off the agar surface with KK2 and left to settle and adhere for 2-3 min on the coverglass. Fixation was extended to 30 min and permeabilisation was 2 h at room temperature followed by >4 h at 4°C. We used sets of 20mer probes from Stellaris, labelled with Quasar570 (ecmA) and Quasar670 (pspA and cotB) together with DAPI for nuclear labelling.
For smFISH on single cells, z-stacks were collected using a Nikon ECLIPSE Ti-E inverted microscope with 100×1.49NA TIRF objective and Andor iXon DU897E (EMCCD) camera in low-angle epifluorescence illumination mode. Raw images were deconvolved using classical maximum likelihood estimation (Huygens Essential, version 16.05, Scientific Volume Imaging). Transcript counting was performed using batch-processing workflows in FISHQuant (Mueller et al., 2013). For intact slugs, z-stacks were collected using a spinning disk confocal microscope (3i) with 63×1.4NA oil immersion objective and Prime 95B CMOS camera (Photometrics).
For live imaging, cells were developed on KK2/1.5% agar. Agar pads that contained multicellular structures were excised and inverted into a drop of silicone oil on a Delta TPG imaging dish (Bioptechs). Z-stacks were collected using the spinning disk with a 63× objective for transcription imaging and a 25× oil objective for imaging protein fusions.
Supplementary Material
Acknowledgements
Imaging was carried out at the Medical Research Council Laboratory for Molecular Cell Biology (MRC LMCB) Light Microscopy Facility. Sequencing was carried out at Barts and the London Genome Centre. We thank Lindsey Millward for assistance with molecular biology. The MRC LMCB University Unit at UCL receives MRC funding (MC_U12266B).
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: V.A., J.R.C.; Methodology: V.A., T.L., A.M., J.R.C.; Software: V.A., T.L.; Validation: V.A., T.L.; Formal analysis: V.A., T.L.; Investigation: V.A., T.L., A.M., J.R.C.; Resources: V.A., T.L., J.R.C.; Data curation: V.A.; Writing - original draft: V.A., J.R.C.; Writing - review & editing: V.A., T.L., A.M., J.R.C.; Visualization: V.A., T.L.; Supervision: J.R.C.; Project administration: V.A., J.R.C.; Funding acquisition: J.R.C.
Funding
Work was supported by Wellcome Trust Senior Fellowship 202867/Z/16/Z to J.R.C. Deposited in PMC for release after 6 months.
Data availability
All scRNA-seq data have been deposited in GEO under accession number GSE128974.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.173740.supplemental
References
- Anders S. and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolovic V., Miermont A., Corrigan A. M. and Chubb J. R. (2017). Generation of single-cell transcript variability by repression. Curr. Biol. 27, 1811-1817.e1813. 10.1016/j.cub.2017.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis E. I., Botthof J. G., Andres H., Ferreira L., Lio P. and Cvejic A. (2017). Single-cell RNA-sequencing uncovers transcriptional states and fate decisions in haematopoiesis. Nat. Commun. 8, 2045 10.1038/s41467-017-02305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccari L., Moris N., Girgin M., Turner D. A., Baillie-Johnson P., Cossy A.-C., Lutolf M. P., Duboule D. and Arias A. M. (2018). Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids. Nature 562, 272-276. 10.1038/s41586-018-0578-0 [DOI] [PubMed] [Google Scholar]
- Becker A. J., Mc C. E. and Till J. E. (1963). Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 197, 452-454. 10.1038/197452a0 [DOI] [PubMed] [Google Scholar]
- Brown J. M. and Firtel R. A. (1999). Regulation of cell-fate determination in Dictyostelium. Dev. Biol. 216, 426-441. 10.1006/dbio.1999.9485 [DOI] [PubMed] [Google Scholar]
- Cannon D., Corrigan A. M., Miermont A., McDonel P. and Chubb J. R. (2015). Multiple cell and population-level interactions with mouse embryonic stem cell heterogeneity. Development 142, 2840-2849. 10.1242/dev.120741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casademunt E., Varney T. R., Dolman J., Petty C. and Blumberg D. D. (2002). A gene encoding a novel anti-adhesive protein is expressed in growing cells and restricted to anterior-like cells during development of Dictyostelium. Differentiation 70, 23-35. 10.1046/j.1432-0436.2002.700103.x [DOI] [PubMed] [Google Scholar]
- Chen G., Zhuchenko O. and Kuspa A. (2007). Immune-like phagocyte activity in the social amoeba. Science 317, 678-681. 10.1126/science.1143991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb J. R. (2017). Symmetry breaking in development and stochastic gene expression. Wiley Interdiscip. Rev. Dev. Biol. 6, e284 10.1002/wdev.284 [DOI] [PubMed] [Google Scholar]
- Chubb J. R., Trcek T., Shenoy S. M. and Singer R. H. (2006). Transcriptional pulsing of a developmental gene. Curr. Biol. 16, 1018-1025. 10.1016/j.cub.2006.03.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay J. L., Ammann R. R. and Gomer R. H. (1995). Initial cell-type choice in a simple eukaryote: cell-autonomous or morphogen-gradient dependent? Dev. Biol. 172, 665-674. 10.1006/dbio.1995.8045 [DOI] [PubMed] [Google Scholar]
- Corrigan A. M. and Chubb J. R. (2014). Regulation of transcriptional bursting by a naturally oscillating signal. Curr. Biol. 24, 205-211. 10.1016/j.cub.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan A. M., Tunnacliffe E., Cannon D. and Chubb J. R. (2016). A continuum model of transcriptional bursting. eLife 5, e13051 10.7554/eLife.13051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early A., McRobbie S. J., Duffy K. T., Jermyn K. A., Tilly R., Ceccarelli A. and Williams J. G. (1988). Structural and functional characterization of genes encoding Dictyostelium prestalk and prespore cell-specific proteins. Dev. Genet. 9, 383-402. 10.1002/dvg.1020090419 [DOI] [PubMed] [Google Scholar]
- Faix J., Kreppel L., Shaulsky G., Schleicher M. and Kimmel A. R. (2004). A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system. Nucleic Acids Res. 32, e143 10.1093/nar/gnh136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomer R. H. and Firtel R. A. (1987). Cell-autonomous determination of cell-type choice in Dictyostelium development by cell-cycle phase. Science 237, 758-762. 10.1126/science.3039657 [DOI] [PubMed] [Google Scholar]
- Gruenheit N., Parkinson K., Brimson C. A., Kuwana S., Johnson E. J., Nagayama K., Llewellyn J., Salvidge W. M., Stewart B., Keller T. et al. (2018). Cell cycle heterogeneity can generate robust cell type proportioning. Dev. Cell 47, 494-508.e494. 10.1016/j.devcel.2018.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay R. R., Flatman P. and Thompson C. R. L. (1999). DIF signalling and cell fate. Semin. Cell Dev. Biol. 10, 577-585. 10.1006/scdb.1999.0341 [DOI] [PubMed] [Google Scholar]
- Lang A. H., Li H., Collins J. J. and Mehta P. (2014). Epigenetic landscapes explain partially reprogrammed cells and identify key reprogramming genes. PLoS Comput. Biol. 10, e1003734 10.1371/journal.pcbi.1003734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis W. F. (2014). Cell signaling during development of Dictyostelium. Dev. Biol. 391, 1-16. 10.1016/j.ydbio.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis W. F. (2015). Genetic control of morphogenesis in Dictyostelium. Dev. Biol. 402, 146-161. 10.1016/j.ydbio.2015.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M., Sakamoto H., Iranfar N., Fuller D., Maruo T., Ogihara S., Morio T., Urushihara H., Tanaka Y. and Loomis W. F. (2003). Changing patterns of gene expression in dictyostelium prestalk cell subtypes recognized by in situ hybridization with genes from microarray analyses. Eukaryot. Cell 2, 627-637. 10.1128/EC.2.3.627-637.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarotti G., Altruda F. and Lodish H. F. (1981). Rates of synthesis and degradation of ribosomal ribonucleic acid during differentiation of Dictyostelium discoideum. Mol. Cell. Biol. 1, 35-42. 10.1128/MCB.1.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O. and Reyes-Valdes M. H. (2008). Defining diversity, specialization, and gene specificity in transcriptomes through information theory. Proc. Natl. Acad. Sci. USA 105, 9709-9714. 10.1073/pnas.0803479105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruo T., Sakamoto H., Iranfar N., Fuller D., Morio T., Urushihara H., Tanaka Y., Maeda M. and Loomis W. F. (2004). Control of cell type proportioning in Dictyostelium discoideum by differentiation-inducing factor as determined by in situ hybridization. Eukaryot. Cell 3, 1241-1248. 10.1128/EC.3.5.1241-1248.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdy M. C., Ratner D. and Firtel R. A. (1983). Induction and modulation of cell-type-specific gene expression in Dictyostelium. Cell 32, 763-771. 10.1016/0092-8674(83)90062-4 [DOI] [PubMed] [Google Scholar]
- Mojtahedi M., Skupin A., Zhou J., Castaño I. G., Leong-Quong R. Y. Y., Chang H., Trachana K., Giuliani A. and Huang S. (2016). Cell fate decision as high-dimensional critical state transition. PLoS Biol. 14, e2000640 10.1371/journal.pbio.2000640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moris N., Pina C. and Arias A. M. (2016). Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 17, 693-703. 10.1038/nrg.2016.98 [DOI] [PubMed] [Google Scholar]
- Mueller F., Senecal A., Tantale K., Marie-Nelly H., Ly N., Collin O., Basyuk E., Bertrand E., Darzacq X. and Zimmer C. (2013). FISH-quant: automatic counting of transcripts in 3D FISH images. Nat. Methods 10, 277-278. 10.1038/nmeth.2406 [DOI] [PubMed] [Google Scholar]
- Muramoto T. and Chubb J. R. (2008). Live imaging of the Dictyostelium cell cycle reveals widespread S phase during development, a G2 bias in spore differentiation and a premitotic checkpoint. Development 135, 1647-1657. 10.1242/dev.020115 [DOI] [PubMed] [Google Scholar]
- Parikh A., Miranda E. R., Katoh-Kurasawa M., Fuller D., Rot G., Zagar L., Curk T., Sucgang R., Chen R., Zupan B. et al. (2010). Conserved developmental transcriptomes in evolutionarily divergent species. Genome Biol. 11, R35 10.1186/gb-2010-11-3-r35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschke P., Knecht D. A., Silale A., Traynor D., Williams T. D., Thomason P. A., Insall R. H., Chubb J. R., Kay R. R. and Veltman D. M. (2018). Rapid and efficient genetic engineering of both wild type and axenic strains of Dictyostelium discoideum. PLoS ONE 13, e0196809 10.1371/journal.pone.0196809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., van den Bogaard P., Rifkin S. A., van Oudenaarden A. and Tyagi S. (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877-879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengarten R. D., Santhanam B., Fuller D., Katoh-Kurasawa M., Loomis W. F., Zupan B. and Shaulsky G. (2015). Leaps and lulls in the developmental transcriptome of Dictyostelium discoideum. BMC Genomics 16, 294 10.1186/s12864-015-1491-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai Y. (2013). Cytosystems dynamics in self-organization of tissue architecture. Nature 493, 318-326. 10.1038/nature11859 [DOI] [PubMed] [Google Scholar]
- Sato T. and Clevers H. (2013). Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190-1194. 10.1126/science.1234852 [DOI] [PubMed] [Google Scholar]
- Shaner N. C., Lambert G. G., Chammas A., Ni Y., Cranfill P. J., Baird M. A., Sell B. R., Allen J. R., Day R. N., Israelsson M. et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407-409. 10.1038/nmeth.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajdohar M., Rosengarten R. D., Kokosar J., Jeran L., Blenkus D., Shaulsky G. and Zupan B. (2017). dictyExpress: a web-based platform for sequence data management and analytics in Dictyostelium and beyond. BMC Bioinformatics 18, 291 10.1186/s12859-017-1706-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternfeld J. and David C. N. (1982). Fate and regulation of anterior-like cells in Dictyostelium slugs. Dev. Biol. 93, 111-118. 10.1016/0012-1606(82)90244-5 [DOI] [PubMed] [Google Scholar]
- Stevense M., Muramoto T., Muller I. and Chubb J. R. (2010). Digital nature of the immediate-early transcriptional response. Development 137, 579-584. 10.1242/dev.043836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantale K., Mueller F., Kozulic-Pirher A., Lesne A., Victor J.-M., Robert M.-C., Capozi S., Chouaib R., Backer V., Mateos-Langerak J. et al. (2016). A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat. Commun. 7, 12248 10.1038/ncomms12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. R. and Kay R. R. (2000). Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Dev. Biol. 227, 56-64. 10.1006/dbio.2000.9877 [DOI] [PubMed] [Google Scholar]
- Tunnacliffe E., Corrigan A. M. and Chubb J. R. (2018). Promoter-mediated diversification of transcriptional bursting dynamics following gene duplication. Proc. Natl. Acad. Sci. USA 115, 8364-8369. 10.1073/pnas.1800943115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusi B. K., Wolock S. L., Weinreb C., Hwang Y., Hidalgo D., Zilionis R., Waisman A., Huh J. R., Klein A. M. and Socolovsky M. (2018). Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54-60. 10.1038/nature25741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haastert P. J., Wang M., Bominaar A. A., Devreotes P. N. and Schaap P. (1992). cAMP-induced desensitization of surface cAMP receptors in Dictyostelium: different second messengers mediate receptor phosphorylation, loss of ligand binding, degradation of receptor, and reduction of receptor mRNA levels. Mol. Biol. Cell 3, 603-612. 10.1091/mbc.3.6.603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman D. M., Akar G., Bosgraaf L. and Van Haastert P. J. M. (2009). A new set of small, extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid 61, 110-118. 10.1016/j.plasmid.2008.11.003 [DOI] [PubMed] [Google Scholar]
- Vicente J. J., Galardi-Castilla M., Escalante R. and Sastre L. (2008). Structural and functional studies of a family of Dictyostelium discoideum developmentally regulated, prestalk genes coding for small proteins. BMC Microbiol. 8, 1 10.1186/1471-2180-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington C. H. (1957). The Strategy of the Genes; a Discussion of Some Aspects of Theoretical Biology. London: Allen & Unwin. [Google Scholar]
- Wang B. and Kuspa A. (1997). Dictyostelium development in the absence of cAMP. Science 277, 251-254. 10.1126/science.277.5323.251 [DOI] [PubMed] [Google Scholar]
- Weijer C. J., Duschl G. and David C. N. (1984). Dependence of cell-type proportioning and sorting on cell cycle phase in Dictyostelium discoideum. J. Cell Sci. 70, 133-145. [DOI] [PubMed] [Google Scholar]
- Williams J. G. (2006). Transcriptional regulation of Dictyostelium pattern formation. EMBO Rep. 7, 694-698. 10.1038/sj.embor.7400714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E., Yang C., Wang J., Fuller D., Loomis W. F. and Siu C. H. (2002). Disruption of the gene encoding the cell adhesion molecule DdCAD-1 leads to aberrant cell sorting and cell-type proportioning during Dictyostelium development. Development 129, 3839-3850. [DOI] [PubMed] [Google Scholar]
- Xenopoulos P., Kang M. and Hadjantonakis A.-K. (2012). Cell lineage allocation within the inner cell mass of the mouse blastocyst. Results Probl. Cell Differ. 55, 185-202. 10.1007/978-3-642-30406-4_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman W. and Weijer C. J. (1993). Analysis of cell cycle progression during the development of Dictyostelium and its relationship to differentiation. Dev. Biol. 160, 178-185. 10.1006/dbio.1993.1296 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.