

ABSTRACT
Cyclin-dependent kinase 9 (CDK9) is critical for RNA Polymerase II (Pol II) transcription initiation, elongation, and termination in several key biological processes including development, differentiation, and cell fate responses. A broad range of diseases are characterized by CDK9 malfunction, illustrating its importance in maintaining transcriptional homeostasis in basal- and signal-regulated conditions. Here we provide a historical recount of CDK9 discovery and the current models suggesting CDK9 is a central hub necessary for proper execution of different steps in the transcription cycle. Finally, we discuss the current therapeutic strategies to treat CDK9 malfunction in several disease states.
Abbreviations: CDK: Cyclin-dependent kinase; Pol II: RNA Polymerase II; PIC: Pre-initiation Complex; TFIIH: Transcription Factor-II H; snoRNA: small nucleolar RNA; CycT: CyclinT1/T2; P-TEFb: Positive Transcription Elongation Factor Complex; snRNP: small nuclear ribonucleo-protein; HEXIM: Hexamethylene Bis-acetamide-inducible Protein 1/2; LARP7: La-related Protein 7; MePCE: Methylphosphate Capping Enzyme; HIV: human immunodeficiency virus; TAT: trans-activator of transcription; TAR: Trans-activation response element; Hsp70: Heat Shock Protein 70; Hsp90/Cdc37: Hsp90- Hsp90 co-chaperone Cdc37; DSIF: DRB Sensitivity Inducing Factor; NELF: Negative Elongation Factor; CPSF: cleavage and polyadenylation-specific factor; CSTF: cleavage-stimulatory factor; eRNA: enhancer RNA; BRD4: Bromodomain-containing protein 4; JMJD6: Jumonji C-domain-containing protein 6; SEC: Super Elongation Complex; ELL: eleven-nineteen Lys-rich leukemia; ENL: eleven-nineteen leukemia; MLL: mixed lineage leukemia; BEC: BRD4-containing Elongation Complex; SEC-L2/L3: SEC-like complexes; KAP1: Kruppel-associated box-protein 1; KEC: KAP1-7SK Elongation Complex; DRB: Dichloro-1-ß-D-Ribofuranosylbenzimidazole; H2Bub1: H2B mono-ubiquitination; KM: KM05382; PP1: Protein Phosphatase 1; CDK9i: CDK9 inhibitor; SHAPE: Selective 2’-hydroxyl acylation analyzed by primer extension; TE: Typical enhancer; SE : Super enhancer.
KEWORDS: Enhancer, CDK9, P-TEFb, 7SK, RNA polymerase II, Transcription, Pausing, Elongation, Cancer, HIV, Disease
Introduction
Several Ser/Thr cyclin dependent kinases (CDK) are regulators of diverse sets of processes in the cell, including transcription [1]. There are several transcriptional CDKs including CDK7, CDK8, CDK9, CDK12, CDK13, CDK18, and CDK19. CDK8 and CDK19 are mutually exclusive subunits of the Mediator complex and help to establish the pre-initiation complex (PIC), while CDK7 is part of the Transcription Factor-II H (TFIIH) complex necessary for the release of Pol II from the PIC [2,3]. Following PIC formation and promoter clearance, transition into productive elongation requires the activity of CDK9 and CDK12 or CDK13. While loss of CDK12/13 in human cells selectively affects DNA damage response and small nucleolar RNA (snoRNA) genes [4,5], CDK9 appears to be necessary for the global regulation of gene transcription under both basal and stimulated conditions [6–14]. Furthermore, CDK9 localizes to sites of active transcription throughout the nucleus [15,16]. Originally defined as a key factor for Pol II transcription elongation, CDK9 has recently emerged as a central regulatory hub at several stages of the transcription cycle [8,11,17–22]. Here we discuss the recent advances in our understanding of how CDK9 is normally regulated, how its kinase activity signals for different steps of the transcription cycle, and how mis-localization of CDK9 to chromatin promotes aberrant transcriptional programs leading to disease states.
Regulation of CDK9 kinase activity
Two CDK9 isoforms [short (CDK9-S: 42-kDa) and long (CDK9-L: 55-kDa)] (Figure 1(a)) are encoded by the same gene and are transcribed by two different promoters located more than 500-bp apart from each other. Both isoforms share exons 2–7, but exon 1 is longer in the 55-kDa isoform thus containing an additional 117 amino acids N-terminal stretch. The two CDK9 isoforms are expressed in cells at different levels with the short one being more abundant [23] (Figure 1(a)). The majority of the short CDK9 isoform appears to localize throughout the nucleoplasm and a minor fraction to the cytoplasm, while the long isoform is contained mostly in the nucleolus [23]. While CDK9-L has been implicated in regulating apoptosis and DNA repair, CDK9-S is the best studied and is implicated in global transcriptional regulation [12,13]. However, it is worth noting these results were obtained under ectopic expression of epitope-tagged constructs. Therefore, the normal subcellular distribution, cell type/tissue expression patterns and precise functions, especially of the long isoform, remain poorly understood. Given most of our understanding of CDK9 function on transcriptional regulation derives from studies with the short isoform, this review will focus on this canonical CDK9 isoform.
Figure 1.
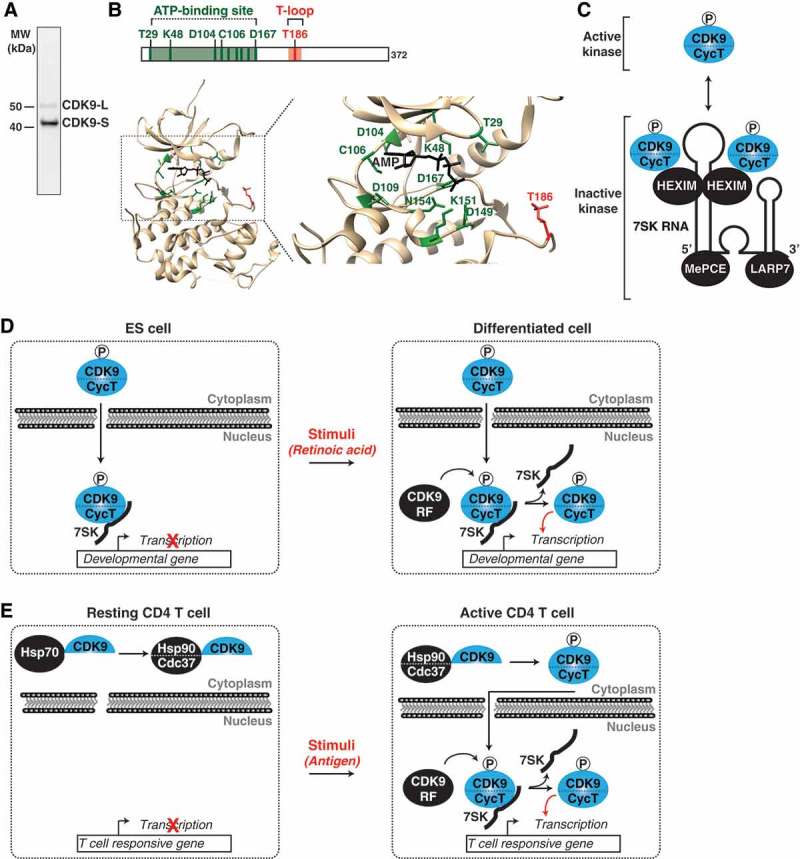
CDK9 activity is tightly regulated through incorporation into several regulatory complexes. (a) CDK9 Western Blot. CDK9 exists in two isoforms, a 55-kDa long form and the best characterized, 42-kDa short form. CDK9 probed in HCT-116 cells with anti-CDK9 (D-7, sc-13130). (b) CDK9 Crystal Structure. Structure of CDK9 showing the ATP binding site (green) and T186 in the T-loop (red). Note that the structure is of CDK9 complexed with CycT [27] but CycT was omitted for simplicity. CDK9 has not been crystallized in the apo form. (c) CDK9 kinase activity is regulated through incorporation into the 7SK snRNP complex. Phosphorylation of CDK9’s T-loop (T186) promotes its interaction with HEXIM and incorporation into the 7SK snRNP. The 7SK RNA/snRNP model depicted is based on Selective 2'-hydroxyl acylation analyzed by primer extension (SHAPE) data from the Price lab [35]. (d) ES cells utilize the 7SK snRNP for CDK9 kinase regulation. In ES cells, CDK9 is inactivated through incorporation into the 7SK snRNP (left). Upon ES stimulation (e.g., retinoic acid), CDK9 release factors (CDK9 RF) promote the release of CDK9, resulting in kinase activation for target gene transcription and cell differentiation (right). (e) Resting CD4 T cells prevent CDK9/CycT assembly. In resting T cells, CDK9 is sequentially incorporated into the Hsp70 and the co-chaperone Hsp90/CDC37 complex in the cytoplasm, where it awaits assembly with CycT (left). Upon T cell activation (e.g., antigen, Toll-like receptor signaling), CDK9 is released from the co-chaperone complex, assembled with newly-synthesized CycT and delivered to T cell responsive genes for transcription activation (right).
In their active kinase state, CDKs form heterodimers with Cyclin proteins [1]. CDK9 is no exception to this rule, as it forms a heterodimer with either CyclinT1 or CyclinT2 (hereafter referred to as CycT) to form the Positive Transcription Elongation Factor Complex (P-TEFb, hereafter referred to as CDK9) [24,25]. Besides a role for CycT in controlling kinase activity, the current model suggests that CycT is also required for assembly of CDK9 and its substrates at sites of active transcription in the nucleus [16,26]. Interestingly, the CDK9-CycT heterodimer was found to be critical for localization of both CDK9 and Pol II to phase-separated droplets, which are thought to be sites of active transcription in living cells [16].
While the structure of CDK9 in the unbound state has not been solved, the atomic resolution of CDK9 bound to its cognate CycT has established a framework for understanding the molecular and chemical details of the mechanism of kinase activation and inhibition by anti-cancer drugs such as flavopiridol [27]. As any other kinase, CDK9 contains an ATP binding site (Figure 1(b), green) and an activating T-loop containing the key T186 residue (Figure 1(b), red). CDK9 activity and CycT interaction are governed by in cis auto-phosphorylation of T186 and three C-terminal phosphorylation sites [18,27–31].
While T-loop phosphorylation at S175 was reported to increase under signal-regulated conditions (T-cell activation) to potentially control CDK9 interaction with other transcription elongation complexes (see CDK9 Delivery to Genome Regulatory Regions section below), this modification does not impact kinase activity [30]. In addition to evidence for in cis auto-phosphorylation, both T186 and S175 can be phosphorylated by other kinases, such as CDK7 in basal conditions [18]. However, it remains unclear whether these molecular events are important for global or pathway-specific transcription in basal- and/or signal-regulated conditions in key biological contexts [18,27,29,30].
Proper regulation of CDK9 kinase activity is essential for the cell to maintain transcriptional homeostasis. One-way the cell has evolved to inactivate kinase activity is through its incorporation into the 7SK small nuclear ribonucleo-protein (snRNP) complex (Figure 1(c)), a mechanism that has been well characterized in vitro and in cancer cell lines [32–35]. Two CycT/CDK9 molecules directly interact with the kinase inhibitor Hexamethylene Bis-acetamide-inducible Protein 1/2 (HEXIM) in a 7SK RNA-dependent manner, which forms dimers to provide a scaffold to organize the snRNP (Figure 1(c)) [31,35]. In addition to CycT/CDK9-HEXIM, the La-related Protein 7 (LARP7) and 7SK snRNA Methylphosphate Capping Enzyme (MePCE) [37,38] are both required for 7SK RNA stability and overall snRNP complex integrity [36–40].
Interestingly, using biochemical approaches the Price lab found that CDK9 T-loop (T186) phosphorylation is a pre-requisite for kinase assembly with the 7SK snRNP through HEXIM interaction (Figure 1(c)) [31]. As expected, T-loop mutations prevented CDK9 from binding HEXIM and subsequent incorporation of the kinase into 7SK snRNP [31]. These studies solidified the model that physical interaction between T-loop phosphorylated CDK9 and HEXIM prevents kinase activation.
While the majority of 7SK-bound CDK9 is nuclear, a small fraction appears to be located in the cytoplasm [41]. However, some controversy still exists in the field regarding 7SK snRNP complex subcellular distribution. While 7SK RNA is predominantly, if not exclusively, nuclear, this data is derived from RNA fluorescence in situ hybridization in which the probes used could, potentially, only detect the nuclear fraction of 7SK RNA and 7SK snRNP complex due to probe accessibility [42]. Similarly, various studies using immunofluorescence to detect the protein components of the snRNP have provided conflictive results (nuclear vs nuclear-cytoplasmic) potentially due to antibodies recognizing different 7SK component forms [30,41–43]. Thus, rigorous studies examining the localization of the complex (and not its components in isolation) would be required to precisely address this conundrum and to further evaluate functional roles for the nuclear and cytoplasmic 7SK snRNP pools.
Besides its role in controlling basal transcription, CDK9 functions as a hub for transducing environmental signaling into transcriptional outputs. In embryonic stem (ES) cells, stimulus-dependent activation of CDK9 is necessary for transcriptional activation of target genes to induce cell differentiation (Figure 1(d)) [10,44–46]. In the absence of any given stimuli, developmental genes are restricted by kinase inactivation through the 7SK snRNP complex. As such, CDK9 must be released from this state through the action of CDK9 release factors (CDK9 RF), which are enzymes/factors depositing or removing post-translation modifications on various 7SK snRNP subunits (CycT, CDK9, and HEXIM) or through direct interactions with the 7SK snRNP complex to promote kinase eviction. Once released, CDK9 becomes activated to phosphorylate its substrates (such as Pol II) at target promoters thereby inducing gene activation to facilitate cell differentiation in response to specific stimuli (Figure 1(d)). These mechanisms will be thoroughly discussed below.
Because CDK9 T-loop phosphorylation is required for its incorporation into the 7SK snRNP complex, it came as no surprise that dephosphorylation of the kinase T-loop by several phosphatases could promote release from the 7SK snRNP in the nucleus [47,48]. The first description from the Zhou lab revealed that PP2B and PP1α dephosphorylate the CDK9 T-loop and that this mechanism appears to be a key example for signal-regulated transcription [47]. Additionally, two members of the PPM/PP2C family of metal-dependent phosphatases (PPM1A and PPM1G) have also been shown to promote CDK9 T-loop dephosphorylation [28,48,49]. While the physiologic involvement of PPM1A on CDK9 function remains to be clarified, PPM1G functions under signal-regulated conditions (e.g., NF-κB induction stimulus) and during activation of the human immunodeficiency virus (HIV) transcriptional program [28,49]. Interestingly, not only does PPM1G promote the release of CDK9 from the 7SK snRNP by T-loop dephosphorylation, it is also capable of directly interacting with 7SK RNA and HEXIM to block the reassembly of released CDK9 back into the snRNP to maintain transcription elongation programs under signal-regulated conditions [28,49]. Collectively, given the large number of Ser/Thr phosphatases in the cell, the variety of cell types, and activating stimuli, as well as the complexity of gene-/pathway-specific transcriptional regulation, future systematic studies will be required to hone our understanding on the roles of these and other phosphatases in the control of CDK9-mediated transcriptional regulation in normal and disease states.
Phosphorylation is not the only post-translational modification purposed to facilitate CDK9 release from the 7SK snRNP complex. The HIV trans-activator of transcription (Tat) protein was recently shown to recruit the UBE2O ubiquitin ligase to cytoplasmic and nuclear 7SK snRNP pools, leading to the non-degradative ubiquitination of HEXIM [41]. This ubiquitination event promotes release of HEXIM from the 7SK snRNP and its redistribution from the nucleus to the cytoplasm thereby promoting an enrichment of free, active CDK9 in the nucleus for HIV gene activation [41]. Besides ubiquitination, factor acetylation has also been implicated in CDK9 activation [50]. In vitro assays suggest that the histone acetyl transferase p300 acetylates CycT at various sites, promoting its dissociation from HEXIM, and facilitating CDK9-mediated transcription activation in cells [50].
In addition to protein modification mechanisms, the Wysocka lab discovered that RNA modifying enzymes contribute to CDK9 activation from the 7SK snRNP complex. The DDX21 DEAD box helicase directly unwinds 7SK RNA to facilitate kinase release from the 7SK snRNP at promoters of ribosomal genes [51]. Similarly, the splicing factor SRSF2 promotes CDK9 activation through direct interaction with the 7SK snRNP at exonic recognition sequences potentially linking splicing dynamics with efficient transcription elongation [52]. However, the molecular details of the activation mechanisms remain unclear.
The substantial number of modifying enzymes that have been reported to function through various 7SK snRNP complex subunits to promote CDK9 release suggests that they may co-operate to facilitate kinase activation. However, we do not have a clear picture of which enzymes function together and whether an ordered recruitment is required to facilitate 7SK snRNP complex disassembly for transcription activation in various biological contexts. One could envision a model in which these enzymes are activated at select target genes, in a context-dependent manner, and/or in response to different regulatory stimuli, thus opening a wide array of potential scenarios to be discovered.
Another more recent, but less-understood, mechanism of CDK9 control is through down-regulation of its cognate CycT, reduction in CDK9 T-loop (T186) phosphorylation, and CDK9 sequestration into the cytoplasm as part of a chaperone complex (Figure 1(e)) [53,54]. Biochemical evidence suggests that CDK9 sequentially binds Heat Shock Protein 70 (Hsp70) and the Hsp90- Hsp90 co-chaperone Cdc37 (Hsp90/Cdc37) complex, thereby stabilizing CDK9 in the cytoplasm until assembly of the active CycT/CDK9 dimer (Figure 1(e)) [54]. Interestingly, resting primary CD4 T cells display low levels of CDK9 T-loop phosphorylation and low expression of CycT. Thus, CDK9 is primarily bound to the Hsp90/Cdc37 complex in the cytoplasm [30,54]. Upon T cell receptor (TCR) activation through antigen engagement, resting cells transition into the active state, in which levels of T186 phosphorylation, CycT, and HEXIM readily increase, thereby augmenting levels of 7SK snRNP complex formation and facilitating assembly at target gene promoters for transcription activation (Figure 1(e)) [53]. Thus, at difference to ES cells and cancer cells (which have higher transcriptional demand), resting T cells have low-to-undetectable levels of active CDK9 and 7SK snRNP formation, consistent with the lower transcription activity of the cell in the resting state. Collectively, although different mechanisms have been proposed to regulate CDK9 activity, all share common themes. To prevent spurious transcriptional activation, CDK9 activity must be suppressed either through incorporation into the 7SK snRNP complex or through decreased expression of its cognate CycT.
To become activated, CycT and CDK9 T-loop phosphorylation levels should increase to facilitate 7SK snRNP complex assembly, and then the kinase must be released from the 7SK snRNP through the action of several modifying enzymes, the “so-called” CDK9 RF, leading to a pathway of kinase activation through T-loop re-phosphorylation. While the observed phosphorylation-dephosphorylation-rephosphorylation cycle appears convoluted, it has biological implications to guarantee an ordered pathway of kinase activation to precisely regulate its function and thus avoid spurious gene activation programs.
Collectively, the multitude of factors that have been implicated in the regulation of CDK9 activity illustrate the importance of proper kinase regulation to the cell’s overall health.
CDK9 regulation of promoter transcription
The elongation step of transcription is a highly regulated process. Shortly after initiation, Pol II must escape into elongation through CDK7-mediated phosphorylation of the PIC [2,3]. Once released from the PIC, Pol II begins elongating but pauses at the promoter-proximal region ~ 20–50 bp downstream of the transcription start site (TSS) [14,55–58]. The establishment of paused Pol II is facilitated by nucleosomal barriers near the TSS and negative elongation factors [DRB Sensitivity Inducing Factor (DSIF) and Negative Elongation Factor (NELF)] (Figure 2(a), top) [17,56,59–64]. By regulating the precise spatio-temporal release of Pol II into productive elongation the cell ensures that both basal- and signal-regulated genes are expressed at the appropriate time.
Figure 2.
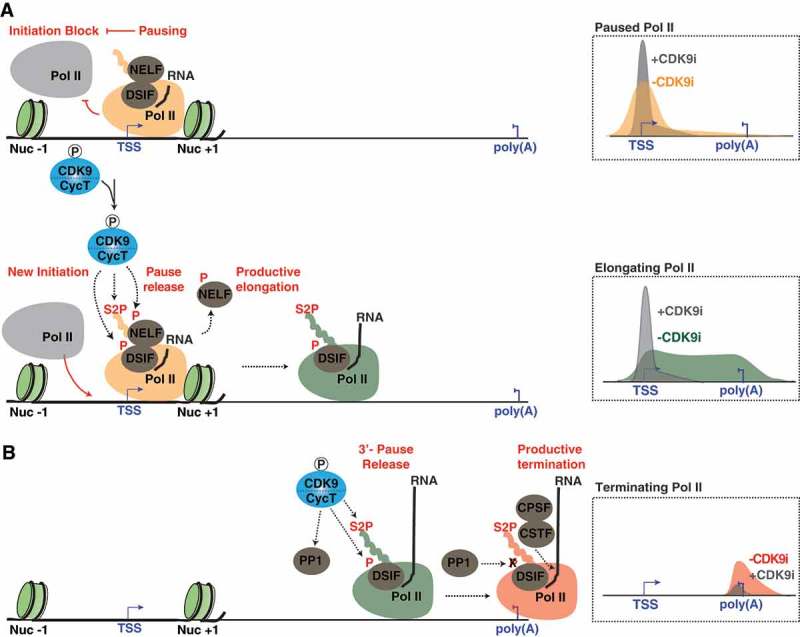
CDK9 is a central hub for proper signaling of each step in the transcription cycle. (a) CDK9 phosphorylation signals for Pol II pause release and initiation. Pausing of Pol II is facilitated by the negative elongation factors, DSIF and NELF (top) and blocks initiation of new Pol II molecules. Upon transcription stimulation, CDK9 phosphorylates the negative elongation factors and Ser2 of the Pol II CTD at promoters resulting in the removal of elongation blocks, release of Pol II into elongation, and initiation of new Pol II molecules (bottom). Inhibition of CDK9 (CDK9i) prohibits release of Pol II into elongation thereby blocking new polymerases from initiating (right). b) CDK9 is critical for proper transcription termination. Pol II pauses at poly(A) sites allowing for the CDK9-dependent phosphorylation of the Pol II CTD and recruitment of termination factors. CTD phosphorylation then provides a platform for the recruitment of termination factors at poly(A) sites and pause-release for proper termination and 3ʹ-end formation. CDK9 also phosphorylates a regulatory subunit of the PP1 phosphatase, preventing dephosphorylation of DSIF and premature termination. Once released from its paused state, Pol II transcribes through the poly(A) site resulting in the activation of PP1, dephosphorylation of DSIF, and transcription termination. CDK9 inhibitor (CDK9i) studies found that inhibition of CDK9 decreases transcript levels downstream of the poly(A) site resulting in a termination defect (right). Note that the different Pol II forms (initiating, pausing, elongating, and terminating) are color-coded for ease visualization.
CDK9 T-loop phosphorylation, and subsequent kinase activation, by CDK7 is required for the regulated release of promoter-proximal paused Pol II into the gene body [18]. The release of paused Pol II into productive elongation is facilitated by CDK9 through phosphorylation of three main substrates: 1) Ser2 residues located in the heptad repeats of Pol II C-Terminal Domain (CTD); 2) the Spt5 subunit of DSIF, transforming it into a positive elongation factor; and 3) the E- subunit of NELF, resulting in its ejection from the nascent pre-mRNA chain (Figure 2(a), bottom) [14,44,62,64–69]. Furthermore, CDK9 presence at gene promoters indirectly affects the chromatin landscape through recruitment of chromatin-remodeling and -modifying enzymes [61,70,71]. In one example, CDK9 co-operates with the histone chaperone complex FACT to relieve the NELF/DSIF-mediated transcription elongation block through the simultaneous regulation of paused Pol II and nucleosome barriers at the promoter-proximal region [61,71].
In addition to the well-established CDK9 function in transcription elongation control, a high interdependence between CDK9-mediated Pol II pause release and transcription initiation frequency has been proposed. Interestingly, two recent reports found that genes displaying a high degree of pausing tend to display a lower rate of transcriptional initiation [19,20]. The first study combined transcription initiation inhibitors (Triptolide) with the recently developed chromatin immunoprecipitation (ChIP) assay with nucleotide resolution through exonuclease, unique barcode and single ligation (ChIP-nexus) technique to identify promoters containing stably paused genes and PIC occupancy profiles in Drosophila cells [20]. Here the authors observed an anti-correlation between Pol II pausing duration and PIC occupancy, suggesting that the presence of promoter-paused Pol II inhibits subsequent rounds of transcription initiation (Figure 2(a)). Supporting this model, pharmacologic CDK9 inhibition with DRB and flavopiridol, which stabilizes paused Pol II, resulted in a decrease of transcription initiation [20]. These studies are consistent with data from the Shilatifard lab demonstrating that transcription initiation blockage with Triptolide both reduces and shifts the paused Pol II peak upstream of the TSS to the position of the PIC and/or very early transcription intermediates [72].
The second study also identified an interplay between CDK9-dependent Pol II pause release and transcription initiation in human cells [19]. Here the authors used CRISPR-Cas9 to introduce a CDK9 adenine analog sensitive mutation, allowing them to specifically and acutely inhibit endogenous CDK9 activity with the 1-NA-PP1 adenine analog. Taking a multi-omics approach to build a kinetic model, the authors found that CDK9 inhibition leads to an increase in paused Pol II duration at promoter-proximal regions and a decrease in initiation frequency, further supporting the model that CDK9 inhibition increases Pol II pausing duration thereby limiting the frequency of initiation (referred in the original work as “pause-initiation” limit) [19]. These studies illustrate an intricate mechanism for the establishment, maintenance and release of paused Pol II at promoters facilitated by CDK9 and its interplay with several positive and negative regulatory factors.
CDK9 regulation of histone modifications and RNA processing
Similar to promoters, Pol II has also been found to pause at intron-exon junctions and at the 3ʹ-ends of genes (although at much lower levels given the higher turnover of pausing at these sites) to allow for recruitment of RNA splicing and termination factors, respectively [73–76]. Furthermore, the phosphorylation pattern of the Pol II CTD acts as a platform for the recruitment of RNA processing factors at different steps of the transcription cycle [65]. Given the role of Pol II CTD phosphorylation in RNA processing and CDK9’s established role in promoting transcription through phosphorylation of the Pol II CTD at promoters, it has been proposed that CDK9 plays a role in RNA processing regulation [21,22,75].
Supporting a regulatory role of CDK9 in transcription termination, pharmacologic inhibition of CDK9 with KM05382 (KM) and DRB, leads to a decrease of nascent transcription downstream of the poly(A) site, indicative of a termination defect (Figure 2(b)) [75]. Additionally, a reduction in phosphorylated Spt5/DSIF (CDK9 substrate) and CDK9 occupancy at 3ʹ-ends of genes coincides with a decrease in transcription levels downstream of the poly(A) site upon CDK9 inhibition, suggesting that the observed decrease in transcription downstream of the poly(A) site is due to a Pol II pause release defect [75]. Furthermore, CDK9 activity also links deposition of H2B mono-ubiquitination (H2Bub1), an epigenetic mark necessary for termination, at 3ʹ-ends of genes, especially at histone genes [70]. Thus, CDK9 integrates phosphorylation events with chromatin modifications to favor co-transcriptional mRNA processing.
Additional links between CDK9 function and correct Pol II transcription termination were provided by the Fisher lab in two recent studies. First, using a chemical-genetic screen, Protein Phosphatase 1 (PP1) was identified as a CDK9 substrate [21]. Second, PP1 was found to dephosphorylate DSIF at 3ʹ-ends of genes in Drosophila, leading to transcription termination (Figure 2(b)) [22]. Interestingly, CDK9 phosphorylation inactivates PP1 activity until the appropriate time for transcription termination, allowing for sustained Spt5/DSIF phosphorylation at the 3ʹ-ends of genes and transcription through the poly(A) site (Figure 2(b)). Additionally, CDK9 is required to maintain Ser2P levels downstream of the poly(A) site, allowing for the recruitment of cleavage and polyadenylation-specific factor (CPSF) and cleavage-stimulatory factor (CSTF), which are critical for proper transcription termination (Figure 2(b)) [75]. The fact that CDK9 operates at multiple locations throughout the transcription unit suggests that it either co-transcriptionally travels with Pol II or is independently recruited to each site. Evidence that CDK9 occupies entire transcriptional units, like Pol II [44,77], and that Ser2P Pol II levels gradually increase as Pol II progresses through a gene may support the model that CDK9 travels with Pol II during transcription [44,77].
Taken together, the studies mentioned above provide compelling evidence that CDK9-dependent pause release is a critical rate-limiting step in both transcription elongation and termination and further illustrate that CDK9 is the central hub in transcription regulation that correctly signals for the execution of different steps in the transcription cycle and not only Pol II pause release as originally thought.
CDK9 regulation of enhancer transcription
CDK9 has been traditionally viewed as a transcription factor that operates exclusively at promoters; however, recent insights into the mechanisms governing enhancer regulation have since expanded our view of how and where in the genome CDK9 activity regulates gene expression. Active enhancers are regions of open chromatin defined by the presence of several features including chromatin signatures (H3K27ac and H3K4me1), the assembly of transcription factors and co-activators (p300/CBP, Mediator, BRD4, and Pol II) and symmetrical bi-directional enhancer RNA (eRNA) transcription [45,78]. Enhancers can promote gene expression both locally and distally (thousands of kilobases away) relative to promoters by delivering Pol II and other transcription factors to promoters through a chromatin looping mechanism; however, it remains unclear if enhancer-promoter looping is a universal mechanism of enhancer function regulation [79]. Importantly, mammalian genomes contain millions of cell-type specific enhancers thereby explaining their key roles in cell identity control, differentiation, and development [45,80,81].
Originally thought to be a mere staging area for the assembly of transcription factors, recent work has found that enhancers operate in a manner analogous to promoters [82,83]. For instance, Pol II mediated transcription of short (<2-kb) eRNAs has been shown to be a pre-requisite for the expression of enhancer-associated genes [84–86]. Given that enhancer associated Pol II is capable of transcribing eRNAs and that CDK9 is required for release of paused Pol II at promoters, CDK9 has been previously proposed to play a role in regulating eRNA transcription [45,87,88]. In support of this model, eRNA transcription is dependent on DSIF/NELF-mediated Pol II pausing, indicating a need for a positive elongation factor, such as CDK9, to release Pol II into active transcription [82,89,90]. Indeed, CDK9 occupies promoter-distal intergenic regions, including traditional enhancers (TE) and super-enhancers (SE) [45,87]. Whereas TE contain the classic composition of features indicated above, SE are locally grouped clusters of enhancers (defined as H3K27ac domains within 12.5-kb of each other) driving high levels of transcription of nearby cell-identity genes [91]. In support of a functional role of CDK9 at enhancers, treatment of cells with the CDK9 inhibitor flavopiridol leads to a decrease in eRNA transcription elongation [88].
Despite these great past achievements, future work delineating the precise contributions of CDK9 to promoter-enhancer communication and/or its role in eRNA-mediated promoter transcription stimulation is much needed. Given CDK9 is required for both mRNA and eRNA transcription and is also potentially required for promoter-enhancer communication, it will be imperative to determine if gene-looping is a direct consequence of CDK9 presence and/or activity at promoters and enhancers. CDK9 inhibition experiments coupled with chromatin conformation capture assays will enable one to uncouple the assembly of enhancer complexes and gene looping from eRNA synthesis. In addition, understanding whether CDK9 function at enhancers (and thus eRNA synthesis) is required for promoter activation and/or whether promoter and enhancer bound CDK9 have different complex arrangements and/or functions will be critical to hone our understanding of the transcriptional programs regulated by CDK9 in normal and disease states.
CDK9 delivery to genome regulatory regions
For CDK9 to properly activate transcription it must be delivered to a precise genomic region at the right time. In this section, we discuss the functional interactions between CDK9 and several co-factors (Figure 3(a)). Early work identified several transcription factors that could directly bind CDK9 and are necessary for CDK9 promoter occupancy [10,14]. Perhaps the first transcription factor implicated in CDK9 delivery to promoter regions was nuclear factor (NF)-κB [10]. Here the authors found that, when co-expressed, the RelA subunit of NF-κB could be immunoprecipitated along with both CDK9 and CycT1 [10]. Furthermore, the authors found that CDK9 is necessary for NF-κB recruitment to the IL-8 promoter, illustrating a requirement for CDK9 in the activation of NF-κB target genes [10].
Figure 3.
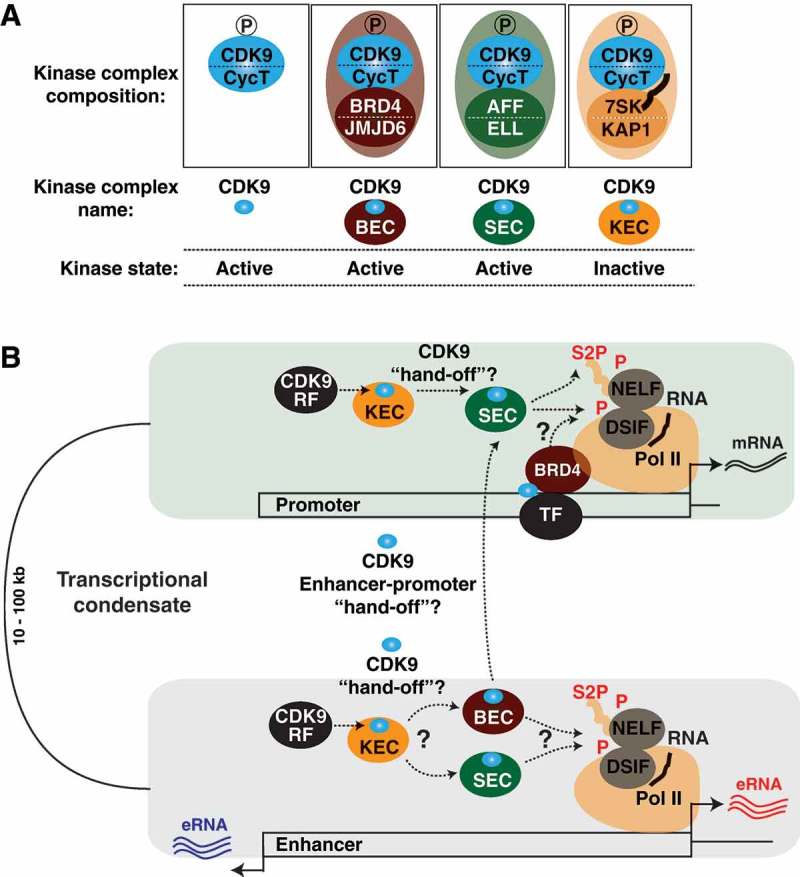
CDK9-containing elongation complexes and their functional interplay. (a) CDK9 assembles into multiple elongation complexes. Active CDK9 is bound to the SEC and BEC. Inactive CDK9, as part of the 7SK snRNP, is bound to KAP1 forming the KEC “pre-elongation” complex. (b) CDK9-containing complexes deliver CDK9 to multiple genomic loci The BEC and SEC deliver CDK9 to enhancer and super-enhancer regions. Additionally, both the SEC and KEC have been shown to deliver CDK9 to gene promoters. Also, BRD4 has been shown to recruit CDK9 to gene-specific promoters through interactions with gene-specific transcription factors. One hypothetical model of cooperation among the CDK9 delivery complexes is that positioning of inactive CDK9 by the KEC could allow for timely release and incorporation of active CDK9 into the SEC and/or BEC at enhancers and/or promoters.
Another transcription factor, c-Myc, is also known to utilize CDK9 to activate its target genes [14,92,93]. Like NF-κB, immunoprecipitation experiments also identified an interaction between c-Myc and CDK9 [14,92,93]. Additionally, CDK9 and c-Myc are find to co-occupy promoter regions of c-Myc target genes and CDK9 is sufficient to activate c-Myc gene expression [92,93]. Finally, inhibition of CDK9 leads to decreased Pol II occupancy at c-Myc target genes [14]. Besides NF-κB and c-Myc, other sequence-specific transcription factors, such as p53, among others, have been shown to interact with CDK9 to facilitate target gene activation [94,44].
Recent work delineating the mechanisms involved in delivering CDK9 to genomic loci have led to the identification of additional factors and protein complexes capable of interacting with and delivering CDK9 in both its active and inactive kinase state to specific genomic locations. One of these additional proteins implicated in CDK9 delivery to the genome was Bromodomain-containing protein 4 (BRD4). Biochemical and immunofluorescence data identified an interaction between BRD4 and the active kinase form of CDK9 in the nucleus of human cells [95,96]. Furthermore, this interaction was apparently required for Pol II-mediated transcription and CDK9 recruitment to both the HIV promoter and SE [45,95–97]. Interestingly, recent data indicate that BRD4 occupies phase-separated droplets (referred to “transcriptional condensates”) in living cells (Figure 3(b)) and disruption of these droplets results in a decrease of both BRD4 and Pol II occupancy at SE, further illustrating BRD4’s role in transcriptional regulation at enhancers [98]. However, CDK9 enhancer occupancy levels were not measured in this study. Therefore, future work is required to determine if BRD4 plays a role in CDK9 delivery to transcriptional condensates in cells or if BRD4 assembles with CDK9 after both proteins are localized to droplets.
Interestingly, the transcription factor p53 was identified as a BRD4-bound protein using an unbiased candidate screen [94]. Here the authors utilized various biochemical techniques to demonstrate a direct interaction between BRD4 and p53. Furthermore, they found BRD4 regulated genes within the p53 pathway require the BRD4-p53 interaction for activation under stress conditions [94]. This is one example of how BRD4 might cooperate with other transcription factors to position and activate CDK9 at specific genomic loci (Figure 3(b)).
The above mentioned BRD4 studies have led to the proposal that CDK9 recruitment to promoters and enhancers, as well as CDK9-mediated Pol II elongation relies on BRD4; however, recent conflicting reports demonstrate that CDK9 is recruited to the genome independently of BRD4 [87,99]. Treatment of cells with the bromodomain and extra-terminal domain (BET) inhibitor JQ1 reduced transcription elongation and BRD4 genome occupancy but had little effect on CDK9 recruitment [99]. Furthermore, acute depletion of BRD4 using a degron system reduced transcription levels independently of CDK9 recruitment, potentially indicating that BRD4 function in Pol II elongation control is unrelated to CDK9 interaction/recruitment to chromatin [87].
Given that BRD4 localizes to promoters throughout the genome, yet its depletion does not affect CDK9 recruitment at every gene, there must be additional functions for BRD4 in transcriptional regulation at promoters. Remarkably, BRD4 is considered to be an atypical kinase that binds and phosphorylates Ser2 of the Pol II CTD, suggesting BRD4 itself could act as a positive elongation factor in a context-dependent manner, consistent with the model of CDK9-independent functions in the transcription activation cycle [100]. Furthermore, BRD4 stimulates CDK9 kinase activity in vitro [101], opening the possibility that BRD4 regulates transcription activation through stimulation of CDK9; however, further studies are needed to determine if BRD4 is capable of activating CDK9 in living cells. Given the complexity of the data and the intertwined connections between BRD4 and CDK9 with the transcription elongation machinery additional investigations are required to obtain a clearer picture of the mutual and independent functions of BRD4 and CDK9 in Pol II CTD phosphorylation and elongation control.
In addition to sequence-specific transcription factors and the BRD4 co-factor, CDK9 was, most recently, found to interact with several elongation complexes. Below, we propose a new nomenclature of CDK9-containing elongation complexes to start integrating the datasets generated by many different labs in different systems (Figure 3(a)).
The Jumonji C-domain-containing protein 6 (JMJD6) demethylase has recently been found in complex with BRD4 and CDK9 [102], a protein complex we refer here to as the BEC for BRD4-containing Elongation Complex (Figure 3(a)). JMJD6 is capable of demethylating both the γ-mono-methylphosphate cap of 7SK RNA and symmetric/asymmetric H4R3me2 (an epigenetic mark apparently read by the 7SK RNA) on enhancers, resulting in the eviction of CDK9 from the 7SK snRNP and ejection of 7SK RNA from chromatin ultimately facilitating gene promoter activity and Pol II pause release from distal, “anti-pause” enhancers (Figure 3(b)) [102]. While earlier work implicated BRD4 in direct recruitment of CDK9, the recent work described here suggests that BRD4’s role in transcription activation is to promote the release and activation of CDK9 at gene promoters and enhancers.
In addition to the BEC, CDK9 assembles into a second macromolecular complex, named SEC (Super Elongation Complex), for Pol II phosphorylation and elongation control (Figure 3(a)). The SEC, which is composed of CycT/CDK9, the eleven-nineteen Lys-rich leukemia (ELL) family members (ELL1, ELL2 and ELL3), either AF9 or the eleven-nineteen leukemia (ENL) family members and the AF4/FMR2 (AFF) family members (AFF4 or AFF1), delivers active CDK9 to both promoters and enhancers (Figure 3(b)) [103–105].
While the mechanism of SEC genomic recruitment has been extensively studied in the context of leukemia and HIV infection (discussed in the next section), the mechanism by which it is normally recruited to the genome under both basal and signal-regulated physiologic conditions are still murky [105,106]. Thus, it remains unclear whether the SEC is a global regulator under basal conditions and whether it fulfills a pathway-specific regulatory function during cell stimulation conditions in various physiologic contexts such as differentiation, development and cell fate responses.
Beyond the canonical SEC, at least two additional SEC-like complexes (SEC-L2/L3) have been described [107]. SEC-L2 and L3 contain AFF4 and AFF3, respectively, ENL family members and lack the ELL components found in the canonical SEC [107]. Furthermore, work in mouse ES cells proposed that the SEC interacts with master transcription factors, such as the Notch complex [108], thus exemplifying an interesting scenario for dictating target gene specificity.
In addition to its interaction with master regulators, recent work has shown that the SEC interacts with acetylated chromatin through an AFF4 – H3K27ac interaction, potentially providing timing and/or a secondary binding interface for selective and high-affinity SEC recruitment to active loci [109]. Together, the different SEC compositions and their interactions with tissue specific factors and epigenetic marks, suggests that the mechanism by which the SEC is recruited to the genome in a normal cell state is tissue- and context-dependent. Nonetheless, additional work is needed to precisely define the normal mechanism of SEC and SEC-like delivery of CDK9 to target loci for transcription activation.
While delivery of active CDK9 to the genome through the BEC and SEC is required to stimulate transcription elongation, components of the 7SK snRNP complex have also been found to occupy gene promoters and enhancers, suggesting that a reservoir of inactive or primed CDK9 is also present at these genomic loci [77,102,110]. This observation has challenged the long-standing view that the main function of 7SK is to solely inactive the kinase, arguing for a revised model in which the 7SK snRNP functions as a “vehicle” to deliver primed kinase for activation at target gene promoters. Delivery of primed CDK9 to promoters could “time” the transition from pausing to pause release and productive elongation [28,43,111]. Therefore, the 7SK snRNP could maintain CDK9 in its inactive state until it is recruited to the right location, namely chromatin domains and promoters containing CDK9 substrates, at the right time (e.g., for the rapid response to environmental induction).
If the 7SK snRNP complex is recruited to chromatin to promote CDK9 kinase function, one would expect that one or more factors could fulfill this function. The Kruppel-associated box-protein 1 (KAP1) interacts with the 7SK snRNP, forming what we have termed “KEC” for KAP1-7SK Elongation Complex (Figure 3(a)), and is necessary for 7SK snRNP delivery to promoters of cellular and viral NF-κB inducible genes [43]. Furthermore, the KEC occupies promoters of most, if not all, active genes containing paused Pol II, indicating that KEC-mediated delivery of CDK9 to promoters is a widespread phenomenon (Figure 3(b)) [43]. However, whether the KEC is required for global or pathway-specific CDK9 recruitment for Pol II elongation control in basal- and/or signal-regulated transcription still needs to be addressed. Additionally, the mechanism by which KAP1 itself interacts with target promoter regions to tether the 7SK snRNP complex to promoters remains an open paradigm. It should also be noted that a recent study discovered that subunits of 7SK localize to sn/snoRNA and promote their transcription independently of CDK9, suggesting multiple transcriptional roles for the 7SK snRNP complex [112].
At difference to the BEC, which appears to assemble only at enhancers to promote enhancer-driven promoter activation, the SEC delivers active CDK9 to both promoters and enhancers to potentially control mRNA and eRNA synthesis, respectively (Figure 3(b)) [95,96,102–105]. While the BEC and SEC deliver active CDK9 to target genomic regions, the KEC brings inactive CDK9, as part of the 7SK snRNP, to gene promoters, priming them for subsequent activation (Figure 3(b)) [43]. As such, it is possible that the KEC functions as a “pre-elongation complex” to deliver primed kinase for “on site” activation in either the free state or through a “hand-off” mechanism to promote BEC and/or SEC assembly at promoters and/or enhancers (Figure 3(b)).
The identification of multiple protein complexes and transcription factors capable of delivering CDK9 to gene promoters and enhancers in different contexts and in response to signal-regulated transcription introduces the intriguing possibility that these complexes could cooperate to regulate Pol II pause release and elongation. However, most of the work characterizing the SEC, BEC and KEC has taken place in diverse cellular systems under different stimulus conditions, thus impeding an integrated analysis to start answering these key points. In support of a co-operative model of SEC and BEC gene regulation, a recent proteomic analysis discovered that several BET proteins, including BRD4, are bound to the SEC, suggesting that the BEC and SEC may co-operate in gene expression regulation [113]. Another study proposed that the SEC and BRD4 cooperatively regulate the release of paused Pol II through the recruitment of multiple CDK9 molecules to promoters; however, the lack of gene expression data in this study makes it difficult to conclude that these complexes cooperatively activate gene transcription [114].
Finally, a systematic identification of the promoters and enhancers occupied and regulated by the three elongation complexes (SEC, BEC, and KEC) in the same cellular system is an urgent need in the field. Furthermore, given the genome-wide localization of these complexes it will be important to determine whether sequence-specific transcription factors (such as c-Myc, NF-κB, and p53) dictate sequence specific recruitment of the three elongation complexes. This knowledge will provide us with a more complete understanding of how these macromolecular complexes facilitate CDK9 delivery and activity to promote transcription under basal and/or signal-regulated conditions, and thus offer a better view of transcriptional regulation in normal and diseases states.
CDK9 malfunction in disease
Properly maintaining transcriptional homeostasis is critical for cell function. CDK9 is a global transcriptional regulator; therefore, any abnormal gene expression programs will lead to unchecked cell growth, chronic inflammation, and metabolic disorders. Indeed, malfunction of CDK9 activity can promote the development of several diseases including cardiac hypertrophy and leukemia [105,106,115,116]. Furthermore, viruses co-opt transcription elongation complexes to transcribe their genomes and induce a permissive environment for their own replication, thereby causing disease [8,25,117].
As discussed previously, SEC components are frequently fused to MLL due to chromosome rearrangements [105]. Normally, in ES cells, MLL recruits SET methyltransferases to HOX promoters, leading to their activation and thereby promoting cell differentiation (Figure 4(a)) [118]. Once differentiated, the cell then turns off expression of HOX genes (Figure 4(a)). However, in leukemia cells expressing MLL/AFF4 fusion chimeras, but not in cells expressing wild-type MLL, CDK9 (SEC) is mis-localized to promoters of developmental (HOX) genes leading to their abnormal induction and oncogenic “addiction” to the MLL fusion protein (Figure 4(b), left) [105,119]. Interestingly, HOX gene activation by MLL/AFF4 fusion proteins occurs independently of H3K4me3 deposition, indicating a mechanism of activation different from what occurs in non-cancerous stem cells [118].
Figure 4.
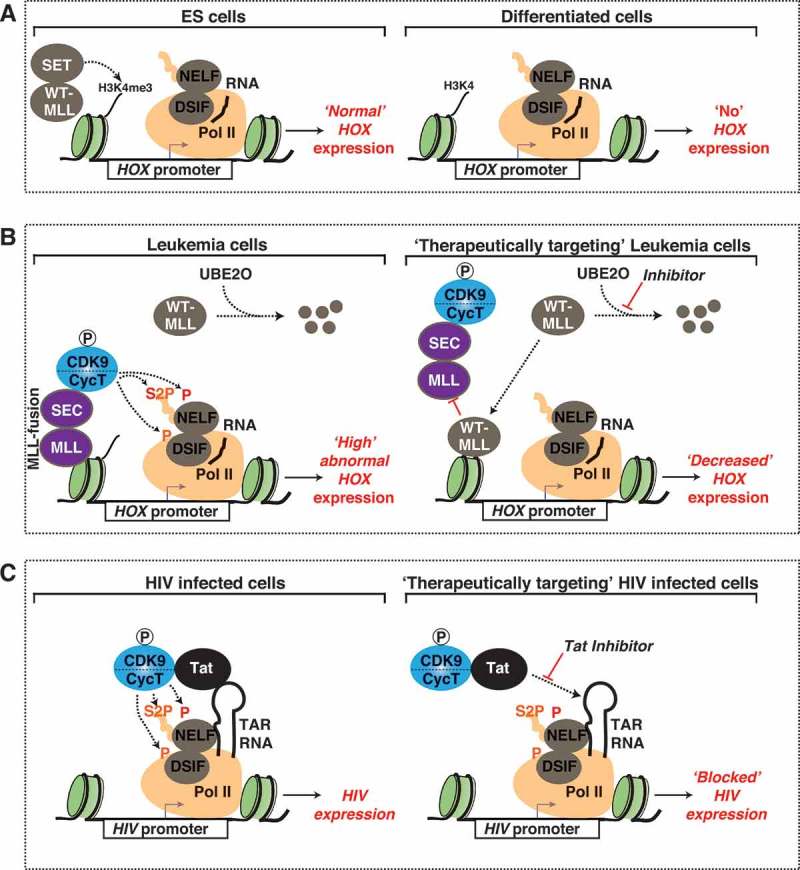
Diseases driven by malfunctioning CDK9 and therapeutic strategies (a) MLL promotes differentiation of stem cells. In ES cells, MLL recruits SET methyltransferases to promoters of HOX genes, leading to deposition of H3K4me3, gene activation, and proper differentiation. (b) MLL malfunction and therapeutic strategies in disease. In leukemia cells that express MLL/SEC fusion protein, WT-MLL is targeted by the UBE2O ubiquitin ligase for degradation, leading to an increase in SEC occupancy at HOX gene promoters and aberrant HOX gene activation. A promising new therapeutic strategy for leukemia inhibits the degradation of WT-MLL leading to increased occupancy of WT-MLL at HOX gene promoters, which in turn displaces MLL/SEC fusion protein leading to HOX downregulation. (c) HIV hijacks CDK9 for transcription of its own genome. The HIV Tat protein tethers CDK9/CycT to the HIV promoter through its interaction with the HIV TAR RNA (left). Tat inhibitors suppress HIV and are crucial for the success of the “block and lock” curative strategy of HIV infection (right).
Recent therapeutic strategies for leukemia have focused on targeting these MLL/AFF4 fusion proteins. Depletion of AFF4 leads to downregulation of HOX genes thus ameliorating disease outcome, demonstrating the necessity of MLL/AFF4 fusions for MLL disease progression [105]. Additionally, small-molecule inhibition of BET proteins, such as BRD4, in a leukemia cell culture model suppresses expression of MLL target genes and reduces occupancy of BRD4 and CDK9 (as part of the SEC) levels at their promoters thereby dampening HOX transcription [113]. However, a recent study found that resistance to BET inhibitors arises in stem cells thereby leading to decreased efficacy of BET inhibitors in a leukemia disease model [120]. Therefore, further development of small-molecules targeting the CDK9 delivery machinery may still be necessary to cope with the mechanisms of resistance.
In an effort to identify therapeutic targets of MLL, a recent study found that wild-type MLL is less stable in leukemia cells relative to MLL fusion proteins [121]. Interestingly, the UBE2O ubiquitin ligase associates with wild-type MLL, but not MLL oncogenic-chimeras, promoting its degradation in leukemia cells (Figure 4(b), left) [121]. Pharmacologic inhibition of the pathways involved in the degradation of wild-type MLL increases its stabilization and promotes displacement of fused MLL chimeras, AFF4 and AFF1, from gene promoters in leukemia cells expressing MLL fusion proteins (Figure 4(b), right) [121]. Furthermore, stabilization of wild-type MLL in a leukemia mouse model delays disease progression and increases survival [121]. Replacement of MLL fusion proteins with wild-type MLL leads to a decrease in occupancy of SEC components and a downregulation of MLL target genes, therefore reversing the oncogenic “addiction” displayed by leukemia cells. Taken together, stabilization of wild-type MLL is a promising therapeutic strategy for leukemia patients given the striking decrease in leukemia progression observed in both human cell culture and mouse models.
Glioblastoma, an aggressive brain cancer for which there is no efficient treatment, also shows dependency on CDK9-containing complexes. An shRNA loss-of-function functional screen identified the BEC component, JMJD6, as a factor necessary for proliferation of patient-derived glioblastoma cells [116]. Additionally, JMJD6 occupies promoter and enhancer regions in patient-derived glioblastoma cells and JMJD6 depletion leads to decreased growth of glioblastoma cells both in culture and in mouse models. Other factors that were identified in the screen to be necessary for proliferation of glioblastoma, but not followed up, include the second BEC component (BRD4) and the SEC component AFF4 [116]. Given JMJD6’s role as a CDK9 release factor from the inactive, 7SK snRNP state, and CDK9 association with both the BEC and SEC, it is plausible that aberrant localization of CDK9 to promoters and enhancers drives the progression of glioblastoma and could thus be targeted therapeutically.
Given CDK9’s central role in transcriptional regulation, viruses have evolved to hijack the kinase to promote their replication and survival in host cells. The most well known virus to utilize CDK9 is HIV [8,25,40,54]. The HIV transcription activator Tat tethers CDK9 to the 5ʹ-end of viral nascent pre-mRNAs (Trans-activation response element: TAR RNA) at the HIV promoter through direct interaction with CycT to promote viral transcription elongation (Figure 4(c), left) [8,25,122]. Additionally, Tat has been implicated in the release and activation of CDK9 from the 7SK snRNP by both competing with HEXIM for CDK9 binding and recruiting CDK9 RFs (Figure 1(d,e)) to the 7SK snRNP complex [28,40,41].
Current anti-retroviral therapy (ART) treatments target active HIV; however, a latent reservoir of HIV in resting CD4 T cells evades treatment, preventing complete eradication of the virus. A recently proposed “block and lock” approach aims at locking HIV in a permanently silent state refractory to signal-regulated transcription as a potential curative strategy for HIV infection [123]. When used in parallel with ART, inhibition of Tat leads to a decrease in both HIV activation and rebound after removal of ART (Figure 4(b), right). The success of this strategy will depend on the efficiency and specificity of small molecules inhibiting Tat by blocking its association with the TAR RNA. Refinement of Tat inhibitors and a better understanding on the mechanisms of action and side effects on the host will be critically required.
Concluding remarks
Since its discovery as a transcription activator, CDK9 has emerged as a critical regulator of transcriptional homeostasis. Genome-wide studies have expanded our view of CDK9 as a broad transcription activator operating from both promoters and enhancers. Initially thought to only regulate the elongation step of transcription, recent work suggests that CDK9 is responsible for coupling different steps of the transcription cycle (from initiation to termination). These findings have provided fundamental principles of transcription regulation.
While much progress has been made in understanding how CDK9 regulates gene expression, more work is needed to gain a complete picture of the mechanisms governing CDK9 recruitment to genomic loci in both normal and disease states. The SEC, BEC and KEC are three elongation complexes that have been implicated in CDK9 activation and delivery to promoters and enhancers. Whether these complexes operate in a co-dependent or independent manner and/or in different contexts under basal- and signal-regulated transcription remain open questions. Comprehensive studies in the same cellular context of how these complexes regulate CDK9 function will have broad implications in our understanding of transcription activation under both normal and disease states, leading to the development of improved therapeutics to treat diseases characterized by CDK9 malfunction.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases [R01AI114362];National Institute of Allergy and Infectious Diseases [R33AI116222];National Science Foundation [2016220513];Welch Foundation [I-1872].
Acknowledgements
We are indebted with members of the D’Orso laboratory for review of the manuscript and Ashwini Challa for providing the western blot image for Figure 1. We apologize with colleagues whose work could not be cited due to space constraints. This study was supported by the National Institute of Allergy and Infectious Diseases of the NIH under award numbers R01AI114362 and R33AI116222, and Welch Foundation grant number I-1782 (to ID), and NSF Graduate Research Fellowship number 2016220513 (to CWB).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hochegger H, Takeda S, Hunt T.. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;11:910–916. PMID: 18813291. [DOI] [PubMed] [Google Scholar]
- [2].Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;3:155–166. PMID: 25693131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Soutourina J. Transcription regulation by the Mediator complex. Nat Rev Mol Cell Biol. 2018;4:262–274. PMID: 29209056. [DOI] [PubMed] [Google Scholar]
- [4].Liang K, Gao X, Gilmore JM, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;6:928–938. PMID: 25561469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bartkowiak B, Liu P, Phatnani HP, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;20:2303–2316. PMID: 20952539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marshall NF, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;5:2078–2090. PMID: 1569941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;21:12335–12338. PMID: 7759473. [DOI] [PubMed] [Google Scholar]
- [8].Mancebo HS, Lee G, Flygare J, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;20:2633–2644. PMID: 9334326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;8:2629–2634. PMID: 10733565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barboric M, Nissen RM, Kanazawa S, et al. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;2:327–337. PMID: 11545735. [DOI] [PubMed] [Google Scholar]
- [11].Lis JT, Mason P, Peng J, et al. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000;7:792–803. PMID: 10766736. [PMC free article] [PubMed] [Google Scholar]
- [12].Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 2014;e02407 DOI: 10.7554/eLife.02407 PMID: 24843027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;34:31793–31799. PMID: 11431468. [DOI] [PubMed] [Google Scholar]
- [14].Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;3:432–445. PMID: 20434984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ghamari A, van de Corput MP, Thongjuea S, et al. In vivo live imaging of RNA polymerase II transcription factories in primary cells. Genes Dev. 2013;7:767–777. PMID: 23592796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lu H, Yu D, Hansen AS, et al. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature. 2018;7709:318–323. PMID: 29849146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wada T, Takagi T, Yamaguchi Y, et al. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. Embo J. 1998;24:7395–7403. PMID: 9857195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Larochelle S, Amat R, Glover-Cutter K, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;11:1108–1115. PMID: 23064645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gressel S, Schwalb B, Decker TM, et al. CDK9-dependent RNA polymerase II pausing controls transcription initiation. Elife. 2017. DOI: 10.7554/eLife.29736 PMID: 28994650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shao W, Zeitlinger J. Paused RNA polymerase II inhibits new transcriptional initiation. Nat Genet. 2017;7:1045–1051. PMID: 28504701. [DOI] [PubMed] [Google Scholar]
- [21].Sanso M, Levin RS, Lipp JJ, et al. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 2016;1:117–131. PMID: 26728557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Parua PK, Booth GT, Sanso M, et al. A Cdk9-PP1 switch regulates the elongation-termination transition of RNA polymerase II. Nature. 2018. DOI: 10.1038/s41586-018-0214-z PMID: 29899453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu H, Herrmann CH. Differential localization and expression of the Cdk9 42k and 55k isoforms. J Cell Physiol. 2005;1:251–260. PMID: 15452830. [DOI] [PubMed] [Google Scholar]
- [24].Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998;22:13855–13860. PMID: 9593731. [DOI] [PubMed] [Google Scholar]
- [25].Wei P, Garber ME, Fang SM, et al. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;4:451–462. PMID: 9491887. [DOI] [PubMed] [Google Scholar]
- [26].Taube R, Lin X, Irwin D, et al. Interaction between P-TEFb and the C-terminal domain of RNA polymerase II activates transcriptional elongation from sites upstream or downstream of target genes. Mol Cell Biol. 2002;1:321–331. PMID: 11739744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Baumli S, Lolli G, Lowe ED, et al. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. Embo J. 2008;13:1907–1918. PMID: 18566585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McNamara RP, McCann JL, Gudipaty SA, et al. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013;5:1256–1268. PMID: 24316072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garber ME, Mayall TP, Suess EM, et al. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol Cell Biol. 2000;18:6958–6969. PMID: 10958691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mbonye U, Wang B, Gokulrangan G, et al. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J Biol Chem. 2018. DOI: 10.1074/jbc.RA117.001347 PMID: 29743242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li Q, Price JP, Byers SA, et al. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem. 2005;31:28819–28826. PMID: 15965233. [DOI] [PubMed] [Google Scholar]
- [32].Barboric M, Lenasi T, Chen H, et al. 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci U S A. 2009;19:7798–7803. PMID: 19416841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nguyen VT, Kiss T, Michels AA, et al. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;6861:322–325. PMID: 11713533. [DOI] [PubMed] [Google Scholar]
- [34].Yang Z, Zhu Q, Luo K, et al. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;6861:317–322. PMID: 11713532. [DOI] [PubMed] [Google Scholar]
- [35].Brogie JE, Price DH. Reconstitution of a functional 7SK snRNP. Nucleic Acids Res. 2017;11:6864–6880. PMID: 28431135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].He N, Jahchan NS, Hong E, et al. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell. 2008;5:588–599. PMID: 18249148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jeronimo C, Forget D, Bouchard A, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;2:262–274. PMID: 17643375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Krueger BJ, Jeronimo C, Roy BB, et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008;7:2219–2229. PMID: 18281698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Michels AA, Fraldi A, Li Q, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. Embo J. 2004;13:2608–2619. PMID: 15201869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schulte A, Czudnochowski N, Barboric M, et al. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;26:24968–24977. PMID: 15855166. [DOI] [PubMed] [Google Scholar]
- [41].Faust TB, Li Y, Bacon CW, et al. The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. Elife. 2018. DOI: 10.7554/eLife.31879 PMID: 29845934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Prasanth KV, Camiolo M, Chan G, et al. Nuclear organization and dynamics of 7SK RNA in regulating gene expression. Mol Biol Cell. 2010;23:4184–4196. PMID: 20881057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McNamara RP, Reeder JE, McMillan EA, et al. KAP1 Recruitment of the 7SK snRNP complex to promoters enables transcription elongation by RNA polymerase II. Mol Cell. 2016;1:39–53. PMID: 26725010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gomes NP, Bjerke G, Llorente B, et al. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;5:601–612. PMID: 16510875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Di Micco R, Fontanals-Cirera B, Low V, et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014;1:234–247. PMID: 25263550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lin C, Garrett AS, De Kumar B, et al. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev. 2011;14:1486–1498. PMID: 21764852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen R, Liu M, Li H, et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008;10:1356–1368. PMID: 18483222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang Y, Dow EC, Liang YY, et al. Phosphatase PPM1A regulates phosphorylation of Thr-186 in the Cdk9 T-loop. J Biol Chem. 2008;48:33578–33584. PMID: 18829461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gudipaty SA, McNamara RP, Morton EL, et al. PPM1G Binds 7SK RNA and hexim1 to block P-TEFb Assembly into the 7SK snRNP and Sustain Transcription Elongation. Mol Cell Biol. 2015;22:3810–3828. PMID: 26324325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cho S, Schroeder S, Kaehlcke K, et al. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. Embo J. 2009;10:1407–1417. PMID: 19387490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Calo E, Flynn RA, Martin L, et al. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;7538:249–253. PMID: 25470060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ji X, Zhou Y, Pandit S, et al. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;4:855–868. PMID: 23663783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Budhiraja S, Famiglietti M, Bosque A, et al. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J Virol. 2013;2:1211–1220. PMID: 23152527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].O’Keeffe B, Fong Y, Chen D, et al. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem. 2000;1:279–287. PMID: 10617616. [DOI] [PubMed] [Google Scholar]
- [55].Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;10:720–731. PMID: 22986266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;3:167–177. PMID: 25693130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Muse GW, Gilchrist DA, Nechaev S, et al. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;12:1507–1511. PMID: 17994021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeitlinger J, Stark A, Kellis M, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;12:1512–1516. PMID: 17994019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gilchrist DA, Dos Santos G, Fargo DC, et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell. 2010;4:540–551. PMID: 21074046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gilchrist DA, Nechaev S, Lee C, et al. NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev. 2008;14:1921–1933. PMID: 18628398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wada T, Orphanides G, Hasegawa J, et al. FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Mol Cell. 2000;6:1067–1072. PMID: 10912001. [DOI] [PubMed] [Google Scholar]
- [62].Wada T, Takagi T, Yamaguchi Y, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;3:343–356. PMID: 9450929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wu CH, Yamaguchi Y, Benjamin LR, et al. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev. 2003;11:1402–1414. PMID: 12782658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yamaguchi Y, Takagi T, Wada T, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;1:41–51. PMID: 10199401. [DOI] [PubMed] [Google Scholar]
- [65].Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev. 2013;11:8456–8490. PMID: 23952966. [DOI] [PubMed] [Google Scholar]
- [66].Suh H, Ficarro SB, Kang UB, et al. Direct analysis of phosphorylation sites on the Rpb1 C-terminal domain of RNA Polymerase II. Mol Cell. 2016;2:297–304. PMID: 26799764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yamada T, Yamaguchi Y, Inukai N, et al. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;2:227–237. PMID: 16427012. [DOI] [PubMed] [Google Scholar]
- [68].Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3ʹ end processing. Mol Cell. 2004;1:67–76. PMID: 14731395. [DOI] [PubMed] [Google Scholar]
- [69].Cho EJ, Kobor MS, Kim ME-J, et al. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 2001;2415:3319–3329. PMID: 11751637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pirngruber J, Shchebet A, Schreiber L, et al. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3ʹ-end processing. EMBO Rep. 2009;8:894–900. PMID: 19575011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Belotserkovskaya R, Oh S, Bondarenko VA, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;5636:1090–1093. PMID: 12934006. [DOI] [PubMed] [Google Scholar]
- [72].Chen F, Gao X, Shilatifard A. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015;1:39–47. PMID: 25561494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Carrillo Oesterreich F, Bieberstein N, Neugebauer KM. Pause locally, splice globally. Trends Cell Biol. 2011;6:328–335. PMID: 21530266. [DOI] [PubMed] [Google Scholar]
- [74].Kuehner JN, Pearson EL, Moore C. Unravelling the means to an end: RNA polymerase II transcription termination. Nat Rev Mol Cell Biol. 2011;5:283–294. PMID: 21487437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Laitem C, Zaborowska J, Isa NF, et al. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol. 2015;5:396–403. PMID: 25849141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mayer A, Di Iulio J, Maleri S, et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;3:541–554. PMID: 25910208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].D’Orso I, Frankel AD. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol. 2010;7:815–821. PMID: 20562857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;8:1273–1283. PMID: 21632746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;4:272–286. PMID: 24614317. [DOI] [PubMed] [Google Scholar]
- [80].Heinz S, Romanoski CE, Benner C, et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;3:144–154. PMID: 25650801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;4:934–947. . PMID: 24119843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Henriques T, Scruggs BS, Inouye MO, et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;1:26–41. PMID: 29378787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kim TK, Shiekhattar R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell. 2015;5:948–959. PMID: 26317464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;4:207–223. PMID: 26948815. [DOI] [PubMed] [Google Scholar]
- [85].Melo CA, Drost J, Wijchers PJ, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;3:524–535. PMID: 23273978. [DOI] [PubMed] [Google Scholar]
- [86].Li W, Notani D, Ma Q, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;7455:516–520. PMID: 23728302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Winter GE, Mayer A, Buckley DL, et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell. 2017;1:5e19–18e19. PMID: 28673542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kaikkonen MU, Spann NJ, Heinz S, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;3:310–325. PMID: 23932714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Galli GG, Carrara M, Yuan WC, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell. 2015;2:328–337. PMID: 26439301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ghavi-Helm Y, Klein FA, Pakozdi T, et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;7512:96–100. PMID: 25043061. [DOI] [PubMed] [Google Scholar]
- [91].Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;2:307–319. PMID: 23582322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem. 2002;42:40156–40162. PMID: 12177005. [DOI] [PubMed] [Google Scholar]
- [93].Gargano B, Amente S, Majello B, et al. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle. 2007;16:2031–2037. PMID: 17700062. [DOI] [PubMed] [Google Scholar]
- [94].Wu SY, Lee AY, Lai HT, et al. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;5:843–857. PMID: 23317504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jang MK, Mochizuki K, Zhou M, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;4:523–534. PMID: 16109376. [DOI] [PubMed] [Google Scholar]
- [96].Yang Z, Yik JH, Chen R, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;4:535–545. PMID: 16109377. [DOI] [PubMed] [Google Scholar]
- [97].Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;2:320–334. PMID: 23582323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018. DOI: 10.1126/science.aar3958 PMID: 29930091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kanno T, Kanno Y, LeRoy G, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol. 2014;12:1047–1057. PMID: 25383670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Devaiah BN, Lewis BA, Cherman N, et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A. 2012;18:6927–6932. PMID: 22509028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Itzen F, Greifenberg AK, Bosken CA, et al. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014;12:7577–7590. PMID: 24860166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Liu W, Ma Q, Wong K, et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;7:1581–1595. PMID: 24360279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lin C, Garruss AS, Luo Z, et al. The RNA Pol II elongation factor Ell3 marks enhancers in ES cells and primes future gene activation. Cell. 2013;1–2:144–156. . PMID: 23273992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;9:543–547. PMID: 22895430. [DOI] [PubMed] [Google Scholar]
- [105].Lin C, Smith ER, Takahashi H, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;3:429–437. PMID: 20159561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;7:661–672. PMID: 21460034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Luo Z, Lin C, Guest E, et al. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;13:2608–2617. PMID: 22547686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Liu K, Shen D, Shen J, et al. The super elongation complex drives neural stem cell fate commitment. Dev Cell. 2017;6:537e6–551e6. PMID: 28350987. [DOI] [PubMed] [Google Scholar]
- [109].Zhang Z, Nikolai BC, Gates LA, et al. Crosstalk between histone modifications indicates that inhibition of arginine methyltransferase CARM1 activity reverses HIV latency. Nucleic Acids Res. 2017;16:9348–9360. PMID: 28637181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Flynn RA, Do BT, Rubin AJ, et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat Struct Mol Biol. 2016;3:231–238. PMID: 26878240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].D’Orso I. 7SKiing on chromatin: move globally, act locally. RNA Biol. 2016;6:545–553. PMID: 27128603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Egloff S, Vitali P, Tellier M, et al. The 7SK snRNP associates with the little elongation complex to promote snRNA gene expression. Embo J. 2017;7:934–948. PMID: 28254838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;7370:529–533. PMID: 21964340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lu X, Zhu X, Li Y, et al. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res. 2016;14:6853–6867. PMID: 27353326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sano M, Abdellatif M, Oh H, et al. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. 2002;11:1310–1317. PMID: 12368904. [DOI] [PubMed] [Google Scholar]
- [116].Miller TE, Liau BB, Wallace LC, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature. 2017;7663:355–359. PMID: 28678782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Alfonso-Dunn R, Turner AW, Jean Beltran PM, et al. Transcriptional elongation of hsv immediate early genes by the super elongation complex drives lytic infection and reactivation from latency. Cell Host Microbe. 2017;4:507e5–517e5. PMID: 28407486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Milne TA, Briggs SD, Brock HW, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;5:1107–1117. PMID: 12453418. [DOI] [PubMed] [Google Scholar]
- [119].Guenther MG, Lawton LN, Rozovskaia T, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;24:3403–3408. PMID: 19141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;7570:538–542. PMID: 26367796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Liang K, Volk AG, Haug JS, et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell. 2017;1–2:59e13–72e13. PMID: 28065413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Tahirov TH, Babayeva ND, Varzavand K, et al. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;7299:747–751. PMID: 20535204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kessing CF, Nixon CC, Li C, et al. In vivo suppression of hiv rebound by didehydro-cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017;3:600–611. PMID: 29045830. [DOI] [PMC free article] [PubMed] [Google Scholar]