

ABSTRACT
The Mediator-associated kinases CDK8 and CDK19 function in the context of three additional proteins: CCNC and MED12, which activate CDK8/CDK19 kinase function, and MED13, which enables their association with the Mediator complex. The Mediator kinases affect RNA polymerase II (pol II) transcription indirectly, through phosphorylation of transcription factors and by controlling Mediator structure and function. In this review, we discuss cellular roles of the Mediator kinases and mechanisms that enable their biological functions. We focus on sequence-specific, DNA-binding transcription factors and other Mediator kinase substrates, and how CDK8 or CDK19 may enable metabolic and transcriptional reprogramming through enhancers and chromatin looping. We also summarize Mediator kinase inhibitors and their therapeutic potential. Throughout, we note conserved and divergent functions between yeast and mammalian CDK8, and highlight many aspects of kinase module function that remain enigmatic, ranging from potential roles in pol II promoter-proximal pausing to liquid-liquid phase separation.
KEYWORDS: Mediator kinase, enhancer, transcription, RNA polymerase II, chromatin
Introduction
The CDK8 kinase exists in a 600 kDa complex known as the CDK8 module, which consists of four proteins (CDK8, CCNC, MED12, MED13). The CDK8 module associates with regulatory sites on a genome-wide scale [1–3], and global targeting of the CDK8 module appears to reflect its association with the 26-subunit Mediator complex[4]. CDK8 module–Mediator association is reversible [5–7] but stable, and distinct populations of “CDK8-Mediator” complexes can be biochemically purified [8,9]. The Mediator–CDK8 module interaction occurs via MED13 [10,11] and an undefined set of Mediator subunits. A paralog of CDK8, called CDK19, emerged in vertebrates and has high sequence similarity to CDK8, including near-identical cyclin binding and kinase domains. Comparatively little is known about CDK19; however, it appears to assemble into an analogous “CDK19 module” (i.e. CDK19, CCNC, MED12, MED13)[12]. In addition to CDK19, paralogs for MED12 and MED13 (MED12L and MED13L) emerged in vertebrates. Unlike CDK19, the MED12 and MED13 paralogs are more divergent in sequence, with only 59% and 53% sequence identity, respectively. The kinase module paralogs MED12L and MED13L associate in a mutually exclusive fashion with MED12 and MED13 [12], and their potential functional distinctions remain unclear.
CDK8 is considered both an oncogene [13–15] and a tumor suppressor[16], indicative of its cell-type and context-specific roles. Through mechanisms that remain incompletely understood, human CDK8 promotes cell growth via the serum response pathway [17] and also functions to maintain both tumors and embryonic stem cells in an undifferentiated state[18]. Further highlighting the basic role for CDK8 in cell proliferation and development, knockout of CDK8 in flies or mice is embryonic lethal [19,20].
In this review, we describe the functional roles of the Mediator kinases CDK8 and CDK19 and propose speculative models for how they might regulate pol II transcription in various contexts. We start with transcriptional reprogramming and enhancer-promoter looping and then transition to metabolism. Next, we discuss small molecule inhibitors, which have yielded valuable insights about CDK8 and CDK19 function. We conclude with sections devoted to the mechanism of action of Mediator kinases, in the context of the four-subunit kinase module and the 29-subunit CDK8-Mediator complex.
Mediator kinases, enhancers, and transcriptional reprogramming
Sequence-specific, DNA-binding transcription factors (TFs) are major drivers of cell physiology and cell state[21]. As examples of their biological influence, fibroblasts can be converted into myotubes upon expression of a single TF, MyoD[22], and various combinations of TFs can reprogram somatic cells to a pluripotent state [23–27]. Consistent with these themes, specific sets of TFs define each cell type and enforce expression of cell type-specific genes [28–32]. Current models of how TFs establish and maintain cell type-specific gene expression patterns involve TF binding to clustered sites at enhancer and promoter regions. This concentrated, localized TF binding recruits factors such as Mediator and cohesin to help form and stabilize enhancer-promoter loops [2,3]; these loops, in turn, promote high-level expression of cell type-specific genes [33,34], many of which include the lineage-specific TFs required to initiate the feed-forward cascade[35]. The importance of enhancer-promoter interactions (via formation of stable chromatin loops) is underscored by the fact that looped architectures change during developmental transitions [36–38] and their disruption is pathogenic [39,40]. ChIP-Seq data suggest Mediator occupies promoter and enhancer regions genome-wide, and is especially abundant at super-enhancers [41,42]. In fact, occupancy of the Mediator subunit MED1 is considered a marker for super-enhancers, along with H3K27Ac and BRD4 [43]. Super-enhancers represent clusters of enhancers that form interconnected hubs with other enhancer and promoter sequences to support high levels of gene expression[44].
The Mediator kinases CDK8 and CDK19 regulate TF function through phosphorylation [45,46]; in addition, Mediator appears to be required for expression of most, if not all, pol II transcripts in mammalian genomes, and ChIP-Seq experiments indicate that the CDK8 module co-localizes with Mediator genome-wide [2,3], including at super-enhancers [41,47,48]. Based upon these observations, a reasonable expectation is that disruption of CDK8 and/or CDK19 function would markedly impact global gene expression patterns. Contrary to these expectations, knockdown of CDK8 or CDK19 protein levels [1,17] or inhibition of CDK8 and CDK19 kinase activity [46,48,49] does not globally affect steady-state mRNA levels; rather, only subsets of genes are affected that vary with context (e.g. hypoxia) or cell type.
How can these results be reconciled? Although the mechanistic roles of Mediator kinases remain enigmatic (see below), we speculate that CDK8 and CDK19 regulate pol II transcription, in part, through enhancers and enhancer-promoter communication (Figure 1). Whereas the human genome contains an estimated fifty- to sixty-thousand enhancers[50], only a subset of these will be “active” in any given cell type[51], and this is dependent upon chromatin structure and TF binding[52]. Enhancers are cell type-specific and active enhancers reflect the lineage-specific TFs that bind enhancer sequences [38,53]. Although the process of enhancer activation remains incompletely understood, it coincides with TF binding and bidirectional transcription of enhancer RNAs (eRNAs)[54]. Accordingly, bidirectional eRNA transcription is cell type-specific[55]. Interestingly, changes in eRNA transcription appear to be a seminal event in response to signaling cascades [56–58]. This rapid, bidirectional eRNA transcriptional response, which is triggered by stimulus-specific TFs[55], can occur within minutes of a stimulus, and correlates with a re-organization of enhancer-promoter contacts and a “reprogramming” of gene expression networks [59,60].
Figure 1.
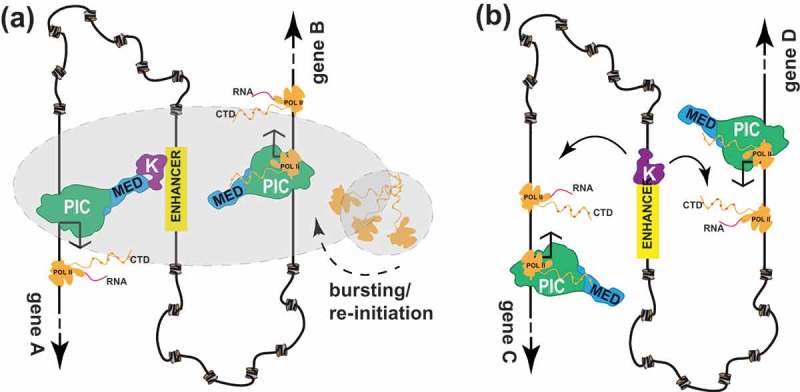
Speculative models for CDK8 or CDK19 module function at mammalian enhancers. a) CDK8 or CDK19 module (“K”) association with an enhancer (e.g. via TF binding) enables its interaction with promoters that are juxtaposed via chromatin loops. This co-localization may be facilitated by liquid-liquid phase separation (LLPS)[137], which is represented by grey shading. At gene A (left), CDK8/19 module–Mediator binding occurs after pol II clears the promoter. Whereas this interaction prevents rapid re-initiation by another pol II complex, promoter-bound CDK8-Mediator can regulate pol II pausing and/or elongation, through physical or functional interactions with the Super-Elongation Complex (SEC) or other factors [17,62]. At gene B (right), transcriptional bursting is depicted[137], in which multiple pol II complexes initiate in succession[70]. This process appears to be Mediator-dependent[148]. The CDK8/19 module does not associate with Mediator at this promoter, as CDK8 module–Mediator binding is mutually exclusive with Mediator–pol II binding [146,147]. Note that at a different point in time, the situation could be switched, with CDK8/19 module association with gene B and gene A undergoing transcriptional bursting. b) An alternative model enables the CDK8/19 module to function independently of Mediator[117]; the enhancer-bound kinase module may regulate transcription elongation via its juxtaposition with elongating pol II complexes at co-localized genes. In support of this model, enhancers have been observed to track with elongating pol II[149], and the CDK8 module appears to positively regulate transcription elongation[17].
The CDK8 module may be an essential component of this rapid enhancer response, based upon a number of observations [61]. CDK8 occupies enhancer elements genome-wide[48], and CDK8 (and/or CDK19) phosphorylates TFs that bind enhancer elements[62]. TF phosphorylation by Mediator kinases has been shown to alter TF activity [45,46]; TF activity, in turn, correlates with expression of bidirectional, enhancer-associated eRNAs [54,55,57]. The expression of eRNAs also correlates with formation of enhancer-promoter loops[63], and may function, at least in part, through direct interactions with the CDK8-Mediator complex[64]. Each of these activities (formation of enhancer-promoter loops, eRNA transcription, TF phosphorylation) can contribute to the establishment of new gene expression programs, whether during cellular differentiation or in response to signaling cascades. Given the potential role of enhancer transcription (e.g. eRNAs) in directing gene expression programs [56,65,66], combined with known roles for Mediator in enhancer-promoter communication [2,59,64], we hypothesize that CDK8 and CDK19 may regulate expression of eRNAs as part of Mediator’s overall regulatory regime.
Copy number estimates of CDK8 module subunits (except for CDK8 itself) are generally 5–10 times lower than Mediator subunits, as determined by quantitative proteomics in HeLa cells[67]. Paralogs CDK19, MED12L and MED13L are even less abundant. This suggests (but does not prove) a more specialized role for the CDK8 module. The copy numbers for MED12, MED13, and CCNC (ca. 3000 to 5000) are roughly consistent with the number of active cell type-specific enhancers and with the estimated number of pol II foci in HeLa cells[68]. Emerging evidence suggests that a single enhancer is capable of simultaneous activation of multiple genes (Figure 1) [69,70]; thus, reduced copy numbers of CDK8 module subunits (vs. Mediator, which is about 10x less abundant than pol II) [67] is consistent with this model.
The model proposed in Figure 1 is speculative and further work is needed to test and develop these concepts. An implication of the model is a role for CDK8 and/or CDK19 in transcriptional reprogramming. That is, activation of gene networks that were previously dormant. By this definition, transcriptional reprogramming occurs during developmental and cell state transitions or in response to extracellular stimuli (e.g. cytokines or hormones). In support of this model, Mediator kinase inhibition or CDK8/CDK19 knockdown typically has minimal impact on basal gene expression and generally is well-tolerated in cells under normal growth conditions [1,17,48,71]. By contrast, activation of gene sets in response to stress [1,72–74] or developmental cues [17,20] shows a dependence upon CDK8 or CDK19. This effect likely derives from their kinase function, which may support the establishment of new transcriptional programs through TF phosphorylation.
To a degree, data from yeast support the findings in mammalian cells; however, the mechanistic connections are limited by the distinct regulatory requirements between yeast and mammals. Presumably as a consequence of their smaller and more compact genomes, yeast lack enhancer elements that define mammalian gene regulation [75] and that enable regulatory interactions at extended distances in mammalian genomes [65,76,77]. Accordingly, yeast generally lack bidirectional eRNA transcription [78,79] and chromatin architectural proteins such as CTCF. Instead, yeast possess upstream activating sequences (UAS) that are only a few hundred base pairs upstream of the promoter. Despite these differences, genetic experiments have implicated yeast (S. cerevisiae) Mediator and its CDK8 ortholog in UAS-dependent regulation of transcription[80]. Furthermore, data suggest that UAS-bound kinase module transiently interacts with Mediator at promoters [81–83].
CDK8 and transcriptional memory
Related to transcriptional reprogramming is the concept of transcriptional memory, in which a transcriptional response to a stimulus is more rapid in cells that have previously been exposed to the stimulus. A study by the Brickner group, in S. cerevisiae, showed that loss of the CDK8 ortholog Srb10/Ssn3 negatively affected transcriptional memory at the INO1 locus [84]. In particular, wild-type cells showed more rapid transcriptional responses upon re-introduction of the stimulus. This “memory” persisted for 3–4 cell generations (about 6 hours). Importantly, Brickner and co-workers demonstrated similar results in human (HeLa) cells upon stimulation with IFNγ, which led the authors to conclude that CDK8 may be a conserved regulator of transcriptional memory [84].
Whereas the mechanisms remain to be established, it is plausible that the CDK8–Mediator complex may help establish long-distance enhancer-promoter loops in human cells [2,3], with potentially a simpler bridged interaction between the INO1 promoter and its UAS in S. cerevisiae. Formation of enhancer-promoter loops has been observed prior to gene activation by extracellular stimuli [52] and prior to expression of lineage-specific genes during mammalian development [85]. Transcriptional memory may also require chromatin modifications[86]. Re-activation of the INO1 gene in S. cerevisiae correlated with dimethylation of histone H3K4; in an unrelated study, Srb10/Ssn3 kinase activity was genetically linked to Set1-dependent H3K4 methylation in S. cerevisiae [87]. In human cells, CDK8 is able to phosphorylate histone H3S10, perhaps concurrently with acetylation of H3K14 [9]. Establishment of such chromatin marks may represent a mechanism by which transcriptional memory or transcriptional reprogramming could be enforced.
Mediator kinases and metabolism
Metabolites represent the biochemicals that – together with proteins and nucleic acids – comprise the entire repertoire of molecules in a cell. As such, metabolic changes are arguably as important as gene expression changes or proteome changes in controlling cell fate or disease pathogenesis [88,89], and metabolic changes are widely recognized as drivers of cancer and cell differentiation [90–94]. Cancer cells rely heavily upon glycolysis (commonly known as the Warburg effect) [88,90] whereas differentiated, non-proliferating cells divert metabolites toward oxidative phosphorylation[91].
The Mediator kinase CDK8 has diverse and conserved links to metabolism. CDK8 orthologs in Drosophila and yeast have been linked to lipid and glucose metabolism and regulation of cellular responses to nutrient depletion [95–102]. In the yeast S. cerevisiae, the Young lab completed gene expression analyses (microarray; normal vs. starved state) with a kinase-inactive mutant CDK8 ortholog, Srb10[103]. This revealed that about 3% of genes were regulated by Srb10 kinase activity, and that normal kinase function repressed their expression. Most genes affected by kinase-dead Srb10 were involved in cellular response to starvation or nutrient stress[103]. Other studies in S. cerevisiae have established Srb10 kinase-dependent regulation of DNA-binding TFs that regulate metabolic pathways [104–108], suggesting ancient origins for Mediator kinases in response to nutrient stress.
In mammalian cells, knockdown experiments have shown a role for the CDK8 protein in the induction of serum response genes[17], and chemical genetics has revealed that CDK8 kinase activity up-regulates expression of glycolytic enzymes in HCT116 cells[109]. This up-regulation of glycolysis is consistent with a role for CDK8 as an oncogene [13,14] and for stem cell maintenance[18]. In addition, CDK8 is important for cellular response to hypoxia [1] which, like serum response, induces extensive metabolic changes. Moreover, CDK8 protein levels correlate with mTOR signaling in mammals[110], and recent data from our lab [62,72] and others link Mediator kinase function to cholesterol metabolism[99].
The ability of Mediator kinases to regulate cell metabolism likely derives from phosphorylation of DNA-binding TFs, such as SREBP [99] and STAT1 [45,48]. However, other Mediator kinase substrates that do not affect transcription may contribute, such as direct modification of metabolic enzymes and/or signaling proteins (e.g. IPMK or SIRT1)[62].
In mammals, the kinase module subunit MED13 has profound effects on metabolism in vivo[111]. Using mouse models, the Olson lab has demonstrated that systemic metabolic processes are sensitive to Med13 protein levels in the heart[112]. For instance, cardiac-specific Med13 over-expression improved insulin sensitivity and conferred resistance to obesity, whereas cardiac-specific Med13 deletion had the opposite effect. Remarkably, mice with cardiac-specific Med13 over-expression showed no changes in food uptake or physical activity, but exhibited increased oxygen consumption and carbon dioxide production[112]. This suggests increased flux through the citric acid cycle and enhanced electron transport, processes that take place in mitochondria. Subsequent studies revealed these metabolic effects manifested in liver and adipose tissue; that is, increased lipid uptake, β-oxidation, and mitochondrial content was observed in these tissues when Med13 was over-expressed in the heart[113]. These results help explain the lean phenotype of these mice[112]. Parabiosis experiments implicated a secreted, cardiac-derived circulating factor but its identity remains unknown [113,114]. Mechanistically, Med13 appears to exert these effects by affecting gene expression patterns[111]. Because MED13 represents the key interface between the kinase module and Mediator [10,11], its effects on metabolism in vivo may depend in part upon targeting Mediator kinases to specific genomic loci via its interaction with the Mediator complex.
Mediator kinase inhibitors and their therapeutic potential
A number of Mediator kinase inhibitors have been discovered and developed over the past few years, and these are summarized in Table 1. Among them, the natural product cortistatin A (CA) stands apart based upon its potency and selectivity for CDK8 and CDK19[48]. Initial isolation of CA identified other kinases, such as ROCK1, as potential targets in addition to CDK8 and CDK19[115]. A limitation of the initial screen was that it measured binding to the kinase protein itself[116]; that is, CDK8 was not tested as an active kinase[115]. CDK8 lacks measurable kinase activity unless it associates with CCNC, and CCNC + MED12 increases CDK8 activity about 30-fold[117].
Table 1.
Mediator kinase inhibitors as chemical probes Data listed are for CDK8, but similar results are reported for CDK19 unless otherwise stated. All compounds are reversible inhibitors (i.e. non-covalent) and bind competitively with ATP. Each compound has varying levels of off-target effects and the extent of off-target kinase inhibition (i.e. kinases other than CDK8 or CDK19) was tested more rigorously with some compounds compared with others.
| Name | structure | IC50a | other data | REF |
|---|---|---|---|---|
| BRD6989 | 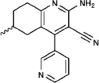 |
500nM in vitro kinase w/CDK8-CCNC |
selective for CDK8 vs. CDK19 | 49 |
| CCT251545 | 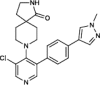 |
5nM in vitro kinase w/ CDK8-CCNC |
Kd = 3.8nM (CDK8-CCNC) IC50 = 65nM (luciferase reporter) PDB: 5BNJ |
122 |
| CCT251921 | 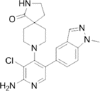 |
2.3nM Lanthascreen or reporter displacement (CDK8-CCNC) |
IC50 ~ 20 nM (luciferase reporter) PDB: 5HBJ |
153 |
| compound 2b | 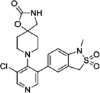 |
1.8nM Lanthascreen or reporter displacement (CDK8-CCNC) |
GI50 = 2nM+ (many lines tested) IC50 ~ 10nM (luciferase reporter) |
123 |
| compound 18 | 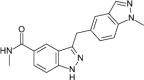 |
10nM Reporter displacement 53nM Lanthascreen (CDK8-CCNC) |
IC50 = 65nM (luciferase reporter) |
154 |
| compound 20 | 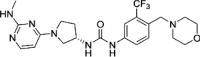 |
17.4nM Lanthascreen (CDK8-CCNC) |
IC50 = 6.5nM (luciferase reporter) PDB: 5HVY |
155 |
| compound 25 | 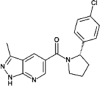 |
2.6nM Lanthascreen or reporter displacement (CDK8-CCNC) |
IC50 = 6.5nM (luciferase reporter) PDB: 5IDN |
156 |
| compound 32 | 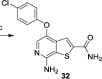 |
1.5nM assay method unclear |
GI50 = 5.4µM* (HCT116) selective for CDK8 vs. CDK19 |
71 |
| compound 51 | 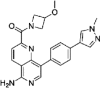 |
5.1nM Lanthascreen or reporter displacement (CDK8-CCNC) |
IC50 = 7.2nM (luciferase reporter) |
157 |
| cortistatin A | 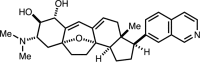 |
12nM in vitro kinase w/ CDK8 module# 100µM ATP |
Kd = 0.2nM (CDK8-CCNC) GI50 = 5nM (MOLM-14) PDB: 4CRL |
48 |
| SEL120-34A | 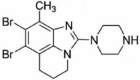 |
4.4nM in vitro kinase w/ CDK8-CCNC 10µM ATP |
Kd = 3nM (CDK8-CCNC) GI50 ~ 12nM (SKNO-1) |
158 |
| senexin A | 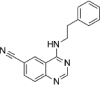 |
280nM in vitro kinase w/ CDK8-CCNC |
Kd ~ 800nM (CDK8-CCNC) |
159 |
| sorafenib | 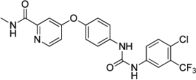 |
32.5nM Lanthascreen (CDK8-CCNC) |
Kd = 100nM (CDK8-CCNC) PDB: 3RGF |
155 160 |
aThe IC50 results determined from kinase assays will be dependent on ATP concentration, with higher [ATP] yielding higher IC50 values. [ATP] used in the assays is noted if reported.
bOther compounds with similar activity were tested in this study.
*In this study, the GI50 was determined to be due to an off-target effect based upon studies in CDK8 and/or CDK19 knockout cell lines.
#CDK8 module = CDK8, CCNC, MED12, MED13
In vitro kinase experiments with CA and the four-subunit, 600 kDa CDK8 module (MED12, MED13, CDK8, CCNC) revealed an IC50 of about 10nM, whereas the Kd of CA for the CDK8–CCNC dimer was determined to be 0.2nM (Table 1)[48]. Crystal structure data revealed the structural basis for CA selectivity, further verified by kinome profiling assays (KiNativ and ProQinase; collectively testing about 400 distinct kinases). This selectivity for CDK8 and CDK19 was observed even when CA was evaluated at 1µM, which is 5000-times the measured Kd. Importantly, CDK8 and CDK19 were verified as the biologically relevant targets in MOLM-14 cells via site-directed mutagenesis (W105M) that enabled CDK8 or CDK19 resistance to CA inhibition[48]. Although these data cannot rule out CA binding to non-kinase targets, its potency and selectivity for CDK8 and CDK19 is unmatched compared with other chemical probes currently available (Table 1).
Another chemical probe that stands out is JH-XI-10–02, with a structural scaffold based upon the CA steroid core[118]. Whereas this core structure selectively targets JH-XI-10–02 to CDK8 and CDK19, JH-XI-10–02 also contains pomalidomide tethered via a flexible polyethylene glycol linker. As an analog of thalidomide, pomalidomide enables recruitment of Cereblon, an E3 ubiquitin ligase[119]. This, in turn, triggers ubiquitination of the targeted protein (i.e. CDK8 or CDK19) and subsequent degradation by the proteasome. This PROteolysis TArgeting Chimera (PROTAC) strategy of selectively targeting enzymes for proteolytic degradation has emerged as a promising therapeutic approach, in part because a single bi-functional molecule can repeatedly target its substrate for degradation [120]. Furthermore, with respect to the Mediator kinases, markedly different effects have been observed upon kinase inhibition vs. knockdown of CDK8 or CDK19 protein levels[62]. Thus, PROTACs should yield therapeutic advantages that are distinct from targeted Mediator kinase inhibition. PROTACs may be especially relevant for therapeutic targeting of CDK19, which has shown kinase-independent effects in the regulation of p53 response[72].
A growing number of studies directly link CDK8 and CDK8 module subunits to specific types of cancer (reviewed in [121]) and the development of selective inhibitors of CDK8 and CDK19 has helped establish Mediator kinases as therapeutic targets [48,49,150]. Studies that have used a variety of methods to disrupt CDK8 and/or CDK19 function have generally shown that kinase inhibition does not markedly affect normal cellular function [46,48,73]. Instead, Mediator kinase activity appears more critical for transcriptional “reprogramming” in response to developmental or environmental cues (see above). These characteristics may be beneficial in the clinic, as they suggest that Mediator kinase inhibitors may be well-tolerated in vivo. Available pre-clinical data in mouse models support this conclusion in some cases[48], but other challenges such as bioavailability and therapeutic index remain to be resolved[151].
Inhibition of Mediator kinases may also have therapeutic value in preventing multi-drug resistance, a near-universal obstacle in cancer medicine. The development of drug resistance requires reprogramming of signaling cascades and gene expression networks to circumvent the vulnerability exploited by the treatment[122]. The CDK8 module subunit MED12 has been implicated in multi-drug resistance in numerous cancer cell types [123,124]. In colon, lung, and other cancer cell types, its role appears to involve TGFβ signaling; specifically, under conditions of drug selection, MED12 knockdown induced TGFβR2 expression and triggered activation of MEK/ERK pathways[123]. This was observed in response to a variety of chemotherapeutics, ranging from cisplatin and 5-fluorouracil to more targeted ALK (crizotinib) and BRAF (vemurafenib) inhibitors[123]. Notably, MED12 was still required for proliferation in these resistant cell lines and near-complete depletion was broadly cytotoxic.
Because MED12 activates human CDK8 and CDK19 kinase function [117,125], the development of multi-drug resistance may be due, in part, to changes in CDK8 and/or CDK19 kinase activity. Inhibitors of other transcriptional CDKs, such as CDK7 and CDK12, have been shown to prevent the onset of multi-drug resistance[126], presumably because inhibition of CDK7 and/or CDK12 hinders the establishment of new gene expression programs. Likewise, Mediator kinase inhibitors may prevent the development of multi-drug resistance by similar means; however, this hypothesis remains to be tested.
CDK8 and CDK19 enzymatic activity vs. scaffold function
Among proteins with enzymatic activity, it is commonly observed that the cellular or physiological effects of enzyme inhibition do not match protein depletion or deletion. For transcriptional regulators, this disparity has been demonstrated across different types of enzymes, such as kinases [103,127], acetyltransferases[128], and ubiquitin ligases[129]. These results underscore the importance of the physical presence of an enzyme for structural or scaffolding purposes. In agreement, several studies have documented markedly distinct effects upon knockdown or knockout of CDK8 or CDK19 vs. targeted kinase inhibition [62,72]. Because enzymes are typically components of multi-subunit assemblies, removal by knockdown or knockout can adversely affect the stability or function of the other subunits. This has been consistently observed for the CDK8 module[130]. For example, knockdown of CDK8 can reduce MED12 protein levels in HCT116 cells [17,61]. In contrast to what has been observed in mammalian cells, genetic disruption of srb10/cdk8 kinase activity in yeast phenocopies srb10/cdk8 deletion mutants[103]. Thus, structural/scaffolding roles for Mediator kinases do not appear to be conserved in yeast.
Kinase module roles in transcription remain enigmatic
As kinases, CDK8 and CDK19 impact cellular function primarily (but not entirely) through protein phosphorylation. The large number of proteins whose phosphorylation state is affected by CDK8 and/or CDK19 implies a complex and elaborate regulatory network [62]. This complexity is compounded by the fact that Mediator kinase substrates are likely to change as a function of cell type or physiological context, and the functional consequences of Mediator kinase-dependent phosphorylation are hard to predict.
The cell type-specific and context-dependent functions of Mediator kinases offer intriguing parallels with DNA-binding TFs, which are also expressed in cell type- and context-specific ways [29]. Sequence-specific, DNA-binding TFs represent a major class of proteins that are targeted by CDK8 and CDK19 [62], and the cell-type and context-specific functions for Mediator kinases could reflect CDK8/19-dependent TF phosphorylation. In a few well-studied cases, CDK8 and/or CDK19-dependent TF phosphorylation has been shown to alter TF activity [45,46,99]. Future experiments will no doubt provide additional insights, but it is notable that even two-fold changes in the level of lineage-specifying TFs can alter cell state and induce differentiation [131,132]. Similarly, two-fold changes in TF activity could have the same effect; thus, it is plausible that TF modification by Mediator kinases underlies many of its biological functions.
Beyond DNA-binding TFs, CDK8 and/or CDK19 phosphorylate other general transcription factors, including TFIID (TAF10), the Super Elongation Complex (SEC; AFF4), NELF (NELFA), pol II (POLR2M), and Mediator itself. Chromatin remodelers and modifiers (e.g. SETD1A, CHD4, KDM3A) were also identified as high-confidence Mediator kinase substrates in HCT116 cells [62]. The functional consequences of these phosphorylation events (if any) remain to be characterized; however, several studies have implicated CDK8 in the regulation of pol II pausing and elongation [1,17,48], suggesting that phosphorylation of proteins such as AFF4, NELFA, or POLR2M may control their function. Yeast lack NELF and the pol II subunit POLR2M, and S. cerevisiae pol II does not appear to be regulated by promoter-proximal pausing [133,134]. Thus, mechanistic insights from yeast are expected to be limited in this case.
The kinase activity of CDK8 and CDK19 is regulated by CCNC and MED12 [117,125,135], and ablation of CCNC in mouse cells prevents CDK8 association with MED12 and MED13[130]. These results suggest a complex network of interactions within the CDK8 module; such interactions are likely similar with CDK19, but perhaps not identical. Although CDK19 is nearly indistinguishable in its kinase domain, about 120 residues in its C-terminus have diverged from CDK8. Low-resolution structural data [10,11,136] and in vitro mechanistic assays have shown that CDK8 module association with Mediator blocks pol II association to inhibit transcription initiation or re-initiation events, invoking a “checkpoint” model for transcription[10]. Both MED12 and MED13 appear important for this checkpoint activity. Whereas a role for MED12 remains unclear, structural and biochemical studies have established that MED13 physically links the CDK8 module to Mediator [10]. This role for MED13 is conserved going back to yeast [11].
As a “molecular switch” that governs Mediator–pol II interaction, the association of the CDK8 module with Mediator may have profound functional consequences. How CDK8 module–Mediator association and dissociation is regulated is a key question that remains incompletely resolved. MED13 contains phospho-degron motifs that trigger its ubiquitination and degradation, and this can impact relative levels of CDK8 module–Mediator association in human cells [5]. However, other means to control CDK8 module association and dissociation with Mediator on more rapid time scales (e.g. seconds to minutes) are likely to exist, but our mechanistic understanding is limited [6,7]. Reversible post-translational modifications could play a role, as well as potential tethering via enhancer-bound TFs or eRNA binding [64]. A tethering role could promote re-association with Mediator by maintaining a high local concentration of the CDK8 module near sites of active transcription. Association of the CDK8 module in phase separated condensates is conceptually similar, and is discussed further below.
Mediator kinase module and liquid-liquid phase separation
An additional means by which the CDK8 module may regulate pol II transcription is via liquid-liquid phase separation (LLPS) [137]. LLPS represents a phenomenon in which proteins and/or nucleic acids achieve higher local concentrations and form “membrane-less organelles” with properties distinct from bulk solvent[138]. Sequence characteristics of proteins that form phase-separated droplets include intrinsically disordered regions, of which Mediator [139] and the CDK8 module have in abundance (Figure 2). Although the molecular forces that contribute to LLPS in biological contexts remain incompletely understood, some basic principles are beginning to be established[140].
Figure 2.
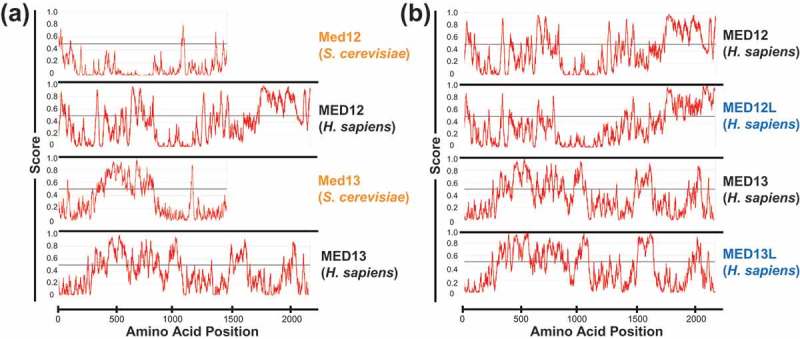
Summary of intrinsically disordered regions (IDRs) in kinase module subunits. a) Comparison of yeast (S. cerevisiae) Med12 and Med13 with human MED12 and MED13. Overall sequence identity is about 13% for yeast vs. human MED12 and MED13. b) Comparison of human MED12, MED12L and human MED13, MED13L. Not shown are CDK8, CDK19, and CCNC, which are largely structured, but each contains IDRs at their C-termini. Plots were generated with IUPred2A[152]; regions with values over 0.5 are considered disordered.
The potential for Mediator [141,142] and the CDK8 module to phase separate may facilitate functionally relevant interactions or may help target the CDK8 module (or CDK8-Mediator) to key regulatory loci. Human CDK8 module subunits – in particular MED12, MED13, and their paralogs MED12L and MED13L – possess domains that are predicted to be intrinsically disordered (Figure 2), suggesting the CDK8 module or CDK8-Mediator may have evolved to phase separate. Human MED12 and MED13 are substantially larger than their yeast counterparts, with intrinsically disordered regions that are not conserved.
Although the relative contribution of LLPS to transcription regulation remains to be determined, it is notable that the pol II C-terminal domain (CTD) is a low-complexity, intrinsically disordered sequence that readily forms phase separated droplets in vitro [143] and in cells [144,145]. In the yeast S. cerevisiae, the pol II CTD consists of 26 heptapeptide repeats of the sequence YSPTSPS. In humans, the pol II CTD is twice the length (52 repeats), and its additional 26 heptapeptide repeats contain nine positively charged residues (mostly lysine) that are spaced throughout the distal 26 repeats. Phase separation is driven in part by pi-cation interactions[140], and the positive charges present in the distal half of the human CTD may promote such interactions with tyrosines spaced throughout the CTD sequence. In agreement, human pol II shows greater propensity to undergo LLPS in vitro and in cells, compared with S. cerevisiae pol II[144].
It remains to be established whether Mediator, CDK8-Mediator, or the CDK8 module can form phase separated droplets in vitro; however, a large intrinsically disordered region in MED1 shows this behavior[142], and evidence in cells suggests Mediator can phase separate [141,142]. Potentially, liquid droplets that contain the CDK8 module may disperse droplets formed with pol II or the pol II CTD. Biochemical experiments indicate that Mediator–CDK8 module association is mutually exclusive with Mediator–pol II [146,147], and it is expected that this would manifest in phase separated compartments as well. Such compartmentalization may help regulate distinct stages of transcription by physically sequestering initiation factors (e.g. TFIID, Mediator, unphosphorylated pol II) from elongation factors (e.g. P-TEFb, CDK8 module, spliceosome).
Funding Statement
This work was supported by the National Institute of General Medical Sciences [GM117370;GM008759] and the National Science Foundation [MCB-1244175].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Galbraith MD, Allen MA, Bensard CL, et al. HIF1A employs CDK8-Mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kagey M, Newman J, Bilodeau S, et al. Mediator and Cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Phillips-Cremins JE, Sauria ME, Sanyal A, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allen BL, Taatjes DJ.. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Davis MA, Larimore EA, Fissel BM, et al. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013;27:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mo X, Kowenz-Leutz E, Xu H, et al. Ras induces mediator complex exchange on C/EBPb. Mol Cell. 2004;13:241–250. [DOI] [PubMed] [Google Scholar]
- [7].Pavri R, Lewis B, Kim TK, et al. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell. 2005;18:83–96. [DOI] [PubMed] [Google Scholar]
- [8].Taatjes DJ, Naar AM, Andel F, et al. Structure, function, and activator-induced conformations of the CRSP coactivator. Science. 2002;295:1058–1062. [DOI] [PubMed] [Google Scholar]
- [9].Meyer KD, Donner AJ, Knuesel M, et al. Cooperative activity of CDK8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. Embo J. 2008;27:1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Knuesel MT, Meyer KD, Bernecky C, et al. The human CDK8 subcomplex is a molecular switch that controls Mediator co-activator function. Genes Dev. 2009;23:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tsai KL, Sato S, Tomomori-Sato C, et al. A conserved Mediator-CDK8 kinase module association regulates Mediator-RNA polymerase II interaction. Nat Struct Mol Biol. 2013;20:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Daniels DL, Ford M, Schwinn MK, et al. Mutual exclusivity of MED12/MED12L, MED13/13L, and CDK8/19 paralogs revealed within the CDK-Mediator kinase module. J Proteomics Bioinform. 2013;S2:004. [Google Scholar]
- [13].Kapoor A, Goldberg MS, Cumberland LK, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Firestein R, Bass AJ, Kim SY, et al. CDK8 is a colorectal cancer oncogene that regulates b-catenin activity. Nature. 2008;455:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Morris EJ, Ji J, Yang F, et al. E2F1 represses b-catenin transcription and is antagonized by both pRB and CDK8. Nature. 2008;455:552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McCleland ML, Soukup TM, Liu SD, et al. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J Pathol. 2015;237:508–519. [DOI] [PubMed] [Google Scholar]
- [17].Donner AJ, Ebmeier CC, Taatjes DJ, et al. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Adler AS, McCleland ML, Truong T, et al. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 2012;72:2129–2139. [DOI] [PubMed] [Google Scholar]
- [19].Loncle N, Boube M, Joulia L, et al. Distinct roles for Mediator cdk8 module subunits in Drosophila development. Embo J. 2007;26:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Westerling T, Kuuluvainen E, Makela TP. Cdk8 is essential for preimplantation mouse development. Mol Cell Biol. 2007;27:6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tapscott SJ, Davis RL, Thayer MJ, et al. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. [DOI] [PubMed] [Google Scholar]
- [23].Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. [DOI] [PubMed] [Google Scholar]
- [24].Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. [DOI] [PubMed] [Google Scholar]
- [25].Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- [26].Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- [27].Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- [28].Heinz S, Romanoski CE, Benner C, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172:650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mullen AC, Orlando DA, Newman JJ, et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011;147:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Neph S, Stergachis AB, Reynolds A, et al. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Trompouki E, Bowman TV, Lawton LN, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Deng W, Rupon JW, Krivega I, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saint-Andre V, Federation AJ, Lin CY, et al. Models of human core transcriptional regulatory circuitries. Genome Res. 2016;26:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Novo CL, Javierre BM, Cairns J, et al. Long-range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep. 2018;22:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Won H, de la Torre-Ubieta L, Stein JL, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nord AS, Blow MJ, Attanasio C, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Franke M, Ibrahim DM, Andrey G, et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016;538:265–269. [DOI] [PubMed] [Google Scholar]
- [40].Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53:859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pott S, Lieb JD. What are super-enhancers?. Nat Genet. 2015;47:8–12. [DOI] [PubMed] [Google Scholar]
- [45].Bancerek J, Poss ZC, Steinparzer I, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013;38:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nitulescu II, Meyer SC, Wen QJ, et al. Mediator kinase phosphorylation of STAT1 S727 promotes growth of neoplasms with JAK-STAT activation. EBioMedicine. 2017;26:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Aranda-Orgilles B, Saldana-Meyer R, Wang E, et al. MED12 regulates HSC-specific enhancers independently of mediator kinase activity to control hematopoiesis. Cell Stem Cell. 2016;19:784–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pelish HE, Liau BB, Nitulescu II, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526:273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Johannessen L, Sundberg TB, O’Connell DJ, et al. Small-molecule studies identify CDK8 as a regulator of IL-10 in myeloid cells. Nat Chem Biol. 2017;13:1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen H, Li C, Peng X, et al. Pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173:386–399 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Heinz S, Romanoski CE, Benner C, et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dukler N, Booth GT, Huang YF, et al. Nascent RNA sequencing reveals a dynamic global transcriptional response at genes and enhancers to the natural medicinal compound celastrol. Genome Res. 2017;27:1816–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Azofeifa JG, Allen MA, Hendrix JR, et al. Enhancer RNA profiling predicts transcription factor activity. Genome Res. 2018;28:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Arner E, Daub CO, Vitting-Seerup K, et al. Gene regulation. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347:1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Franco HL, Nagari A, Malladi VS, et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018;28:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kaikkonen MU, Spann NJ, Heinz S, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hsieh CL, Fei T, Chen Y, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A. 2014;111:7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lyu X, Rowley MJ, Corces VG. Architectural proteins and pluripotency factors cooperate to orchestrate the transcriptional response of hESCs to temperature stress. Mol Cell. 2018;71:940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kuuluvainen E, Domenech-Moreno E, Niemela EH, et al. Depletion of Mediator kinase module subunits represses superenhancer-associated genes in colon cancer cells. Mol Cell Biol. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Poss ZC, Ebmeier CC, Odell AT, et al. Identification of Mediator kinase substrates in human cells using cortistatin A and quantitative phosphoproteomics. Cell Rep. 2016;15:436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sanyal A, Lajoie BR, Jain G, et al. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lai F, Orom UA, Cesaroni M, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Plank JL, Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell. 2014;55:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Schaukowitch K, Joo JY, Liu X, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kulak NA, Pichler G, Paron I, et al. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. [DOI] [PubMed] [Google Scholar]
- [68].Iborra FJ, Pombo A, Jackson DA, et al. Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei. J Cell Sci. 1996;109(Pt 6):1427–1436. [DOI] [PubMed] [Google Scholar]
- [69].Allahyar A, Vermeulen C, Bouwman BAM, et al. Enhancer hubs and loop collisions identified from single-allele topologies. Nat Genet. 2018;50:1151–1160. [DOI] [PubMed] [Google Scholar]
- [70].Fukaya T, Lim B, Levine M. Enhancer control of transcriptional bursting. Cell. 2016;166:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Koehler MF, Bergeron P, Blackwood EM, et al. Development of a potent, specific CDK8 kinase inhibitor which phenocopies CDK8/19 knockout cells. ACS Med Chem Lett. 2016;7:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Audetat KA, Galbraith MD, Odell AT, et al. A kinase-independent role for cyclin-dependent kinase 19 in p53 response. Mol Cell Biol. 2017;37:e00626–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen M, Liang J, Ji H, et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc Natl Acad Sci U S A. 2017;114:10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Donner AJ, Szostek S, Hoover JM, et al. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol Cell. 2007;27:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014;157:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fanucchi S, Shibayama Y, Burd S, et al. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. [DOI] [PubMed] [Google Scholar]
- [77].Fullwood MJ, Liu MH, Pan YF, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rawal Y, Chereji RV, Valabhoju V, et al. Gcn4 binding in coding regions can activate internal and canonical 5ʹ promoters in yeast. Mol Cell. 2018;70:297–311 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sigova AA, Mullen AC, Molinie B, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci U S A. 2013;110:2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Reavey CT, Hickman MJ, Dobi KC, et al. Analysis of polygenic mutants suggests a role for mediator in regulating transcriptional activation distance in saccharomyces cerevisiae. Genetics. 2015;201:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Andrau J, van de Pasch L, Lijnzaad P, et al. Genome-wide location of the coactivator Mediator: binding without activation and transient cdk8 interaction on DNA. Mol Cell. 2006;22:179–192. [DOI] [PubMed] [Google Scholar]
- [82].Jeronimo C, Langelier MF, Bataille AR, et al. Tail and kinase modules differently regulate core mediator recruitment and function in vivo. Mol Cell. 2016;64:455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Petrenko N, Jin Y, Wong KH, et al. Mediator undergoes a compositional change during transcriptional activation. Mol Cell. 2016;64:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].D’Urso A, Takahashi YH, Xiong B, et al. Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memory. Elife. 2016;5:e16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dimitrova E, Kondo T, Feldmann A, et al. FBXL19 recruits CDK-Mediator to CpG islands of developmental genes priming them for activation during lineage commitment. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. [DOI] [PubMed] [Google Scholar]
- [87].Law MJ, Finger MA. The Saccharomyces cerevisiae Cdk8 Mediator Represses AQY1 Transcription by Inhibiting Set1p-Dependent Histone Methylation. G3 (Bethesda). 2017;7:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Agathocleous M, Harris WA. Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol. 2013;23:484–492. [DOI] [PubMed] [Google Scholar]
- [89].Deberardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang YH, Israelsen WJ, Lee D, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- [95].Kuchin S, Treich I, Carlson M. A regulatory shortcut between the Snf1 protein kinase and RNA polymerase II holoenzyme. Proc Natl Acad Sci U S A. 2000;97:7916–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kuchin S, Yeghiayan P, Carlson M. Cyclin-dependent protein kinase and cyclin homologs SSN3 and SSN8 contribute to transcriptional control in yeast. Proc Natl Acad Sci U S A. 1995;92:4006–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mousley CJ, Yuan P, Gaur NA, et al. A sterol-binding protein integrates endosomal lipid metabolism with TOR signaling and nitrogen sensing. Cell. 2012;148:702–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Song W, Treich I, Qian N, et al. SSN genes that affect transcriptional repression in Saccharomyces cerevisiae encode SIN4, ROX3, and SRB proteins associated with RNA polymerase II. Mol Cell Biol. 1996;16:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhao X, Feng D, Wang Q, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest. 2012;122:2417–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lindsay AK, Morales DK, Liu Z, et al. Analysis of Candida albicans mutants defective in the Cdk8 module of mediator reveal links between metabolism and biofilm formation. PLoS Genet. 2014;10:e1004567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Law MJ, Ciccaglione K. Fine-tuning of histone H3 Lys4 methylation during pseudohyphal differentiation by the CDK submodule of RNA polymerase II. Genetics. 2015;199:435–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Xie XJ, Hsu FN, Gao X, et al. CDK8-cyclin C Mediates nutritional regulation of developmental transitions through the ecdysone receptor in drosophila. PLoS Biol. 2015;13:e1002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Holstege FC, Jennings EG, Wyrick JJ, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. [DOI] [PubMed] [Google Scholar]
- [104].Chi Y, Huddleston MJ, Zhang X, et al. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 2001;15:1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hirst M, Kobor MS, Kuriakose N, et al. GAL4 is regulated by the RNA polymerase II holoenzyme-associated cyclin-dependent protein kinase SRB10/CDK8. Mol Cell. 1999;3:673–678. [DOI] [PubMed] [Google Scholar]
- [106].Nelson C, Goto S, Lund K, et al. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature. 2003;421:187–190. [DOI] [PubMed] [Google Scholar]
- [107].Raithatha S, Su TC, Lourenco P, et al. Cdk8 regulates stability of the transcription factor Phd1 to control pseudohyphal differentiation of Saccharomyces cerevisiae. Mol Cell Biol. 2012;32:664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Vincent O, Kuchin S, Hong SP, et al. Interaction of the srb10 kinase with sip4, a transcriptional activator of gluconeogenic genes in saccharomyces cerevisiae. Mol Cell Biol. 2001;21:5790–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Galbraith MD, Andrysik Z, Pandey A, et al. CDK8 Kinase Activity Promotes Glycolysis. Cell Rep. 2017;21:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Feng D, Youn DY, Zhao X, et al. mTORC1 down-regulates cyclin-dependent kinase 8 (CDK8) and cyclin C (CycC). PLoS One. 2015;10:e0126240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Amoasii L, Holland W, Sanchez-Ortiz E, et al. A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism. Genes Dev. 2016;30:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Grueter CE, van Rooij E, Johnson BA, et al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149:671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Baskin KK, Grueter CE, Kusminski CM, et al. MED13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol Med. 2014;6:1610–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lee JH, Bassel-Duby R, Olson EN. Heart- and muscle-derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proc Natl Acad Sci U S A. 2014;111:9491–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cee VJ, Chen DYK, Lee MR, et al. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem Int Ed. 2009;48:8952–8957. [DOI] [PubMed] [Google Scholar]
- [116].Fabian MA, Biggs WH 3rd, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. [DOI] [PubMed] [Google Scholar]
- [117].Knuesel MT, Meyer KD, Donner AJ, et al. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of Mediator. Mol Cell Biol. 2009;29:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hatcher JM, Wang ES, Johannessen L, et al. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett. 2018;9:540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. [DOI] [PubMed] [Google Scholar]
- [120].Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Clark AD, Oldenbroek M, Boyer TG. Mediator kinase module and human tumorigenesis. Crit Rev Biochem Mol Biol. 2015;50:393–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Huang S, Holzel M, Knijnenburg T, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012;151:937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Luo XL, Deng CC, Su XD, et al. Loss of MED12 induces tumor dormancy in human epithelial ovarian cancer via downregulation of EGFR. Cancer Res. 2018;78:3532–3543. [DOI] [PubMed] [Google Scholar]
- [125].Park MJ, Shen H, Spaeth JM, et al. Oncogenic exon 2 mutations in Mediator subunit MED12 disrupt allosteric activation of cyclin C-CDK8/19. J Biol Chem. 2018;293:4870–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Rusan M, Li K, Li Y, et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 2018;8:59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Kanin EI, Kipp RT, Kung C, et al. Chemical inhibition of the TFIIH-associated kinase cdk7/kin28 does not impair global mRNA synthesis. Proc Natl Acad Sci U S A. 2007;104:5812–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bu P, Evrard YA, Lozano G, et al. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol. 2007;27:3405–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Illingworth RS, Moffat M, Mann AR, et al. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev. 2015;29:1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Li N, Fassl A, Chick J, et al. Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol. 2014;16:1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Karwacki-Neisius V, Goke J, Osorno R, et al. Reduced Oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by Oct4 and Nanog. Cell Stem Cell. 2013;12:531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. [DOI] [PubMed] [Google Scholar]
- [133].Mayer A, Lidschreiber M, Siebert M, et al. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. [DOI] [PubMed] [Google Scholar]
- [134].Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Turunen M, Spaeth JM, Keskitalo S, et al. Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell Rep. 2014;7:654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Elmlund H, Baraznenok V, Lindahl M, et al. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc Natl Acad Sci U S A. 2006;103:15788–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hnisz D, Shrinivas K, Young RA, et al. A phase separation model for transcriptional control. Cell. 2017;169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Bergeron-Sandoval LP, Safaee N, Michnick SW. Mechanisms and Consequences of Macromolecular Phase Separation. Cell. 2016;165:1067–1079. [DOI] [PubMed] [Google Scholar]
- [139].Toth-Petroczy A, Oldfield CJ, Simon I, et al. Malleable machines in transcription regulation: the mediator complex. PLoS Comput Biol. 2008;4:e1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wang J, Choi JM, Holehouse AS, et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018;174:688–699 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Cho WK, Spille JH, Hecht M, et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:eear3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kwon I, Kato M, Xiang S, et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Boehning M, Dugast-Darzacq C, Rankovic M, et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat Struct Mol Biol. 2018;25:833–840. [DOI] [PubMed] [Google Scholar]
- [145].Chong S, Dugast-Darzacq C, Liu Z, et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361:eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ebmeier CC, Taatjes DJ. Activator-Mediator binding regulates Mediator-cofactor interactions. Proc Natl Acad Sci U S A. 2010;107:11283–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Naar AM, Taatjes DJ, Zhai W, et al. Human CRSP interacts with RNA polymerase II CTD and adopts a specific CTD-bound conformation. Genes Dev. 2002;16:1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Tantale K, Mueller F, Kozulic-Pirher A, et al. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun. 2016;7:12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Lee K, Hsiung CC, Huang P, et al. Dynamic enhancer-gene body contacts during transcription elongation. Genes Dev. 2015;29:1992–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Dale T, Clarke PA, Esdar C, et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol. 2015;11:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Clarke PA, Ortiz-Ruiz MJ, TePoele R, et al. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife. 2016;5:e20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Dosztanyi Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018;27:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Mallinger A, Schiemann K, Rink C, et al. Discovery of potent, selective, and orally bioavailable small-molecule modulators of the Mediator complex-associated kinases CDK8 and CDK19. J Med Chem. 2016;59:1078–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Schiemann K, Mallinger A, Wienke D, et al. Discovery of potent and selective CDK8 inhibitors from an HSP90 pharmacophore. Bioorg Med Chem Lett. 2016;26:1443–1451. [DOI] [PubMed] [Google Scholar]
- [155].Bergeron P, Koehler MF, Blackwood EM, et al. Design and development of a series of potent and selective type II inhibitors of CDK8. ACS Med Chem Lett. 2016;7:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Czodrowski P, Mallinger A, Wienke D, et al. Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J Med Chem. 2016;59:9337–9349. [DOI] [PubMed] [Google Scholar]
- [157].Mallinger A, Schiemann K, Rink C, et al. 2,8-Disubstituted-1,6-Naphthyridines and 4,6-Disubstituted-Isoquinolines with potent, selective affinity for CDK8/19. ACS Med Chem Lett. 2016;7:573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Rzymski T, Mikula M, Zylkiewicz E, et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget. 2017;8:33779–33795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Porter DC, Farmaki E, Altilia S, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A. 2012;109:13799–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Schneider EV, Bottcher J, Blaesse M, et al. The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J Mol Biol. 2011;412:251–266. [DOI] [PubMed] [Google Scholar]