

ABSTRACT
induced pluripotent stem (iPS) cells can be differentiated into various cell types, including airway epithelial cells, since they have the capacity for self-renewal and pluripotency. Thus, airway epithelial cells generated from iPS cells are expected to be potent candidates for use in airway regeneration and the treatment of airway diseases such as cystic fibrosis (CF). Recently, it was reported that iPS cells can be differentiated into airway epithelial cells according to the airway developmental process. These studies demonstrate that airway epithelial cells generated from iPS cells are equivalent to their in vivo counterparts. However, it has not been evaluated in detail whether these cells exhibit physiological functions and are fully mature. Airway epithelial cells adequately control water volume on the airway surface via the function of Cl− channels. Reasonable environments on the airway surface cause ciliary movement with a constant rhythm and maintain airway clearance. Therefore, the generation of functional airway epithelial cells/tissues with Cl− channel function from iPS cells will be indispensable for cell/tissue replacement therapy, the development of a reliable airway disease model, and the treatment of airway disease. This review highlights the generation of functional airway epithelial cells from iPS cells and discusses the remaining challenges to the generation of functional airway epithelial cells for airway regeneration and the treatment of airway disease.
KEYWORDS: iPS cells, airway epithelial cells, Cl− channel, CFTR, cystic fibrosis
Introduction
There are many cases in which a central airway, such as the trachea, must be resected due to trauma, inflammation, or thyroid cancer. Although various anastomotic operation methods following airway resection have been reported [1,2], these treatments cause postoperative scarring, and granulation is sometimes observed. Autografts and allografts using costal cartilage, auricular cartilage, and skin have also been performed for airway replacement [3–5]. However, it is difficult to obtain an adequate quantity and appropriate shape of tissue for airway regeneration. Artificial material [6,7] has also been used for central airway regeneration. While this method achieves good clinical outcomes, at least two months are necessary for complete epithelial regeneration on the airway surface of the artificial trachea. The airway epithelium has important functions, such as the regulation of water volume on the airway surface via the transport function of Cl− channels to maintain the mucociliary transport system. Thus, since the regeneration of the airway epithelium must be completed as soon as possible, novel treatment methods for airway regeneration are needed.
Cystic fibrosis (CF) is an autosomal recessive, lethal genetic disease caused by mutations in the cystic fibrosis membrane conductance regulator (CFTR) gene, which encodes one of Cl− channels. Deficient and/or defective CFTR protein in airway epithelial cells results in decreased Cl− secretion and leads to a reduced water volume on the airway surface, increased viscosity, and impairment of the mucociliary transport system [8]. Therefore, lung damage and respiratory failure are caused by severe airway obstruction, bacterial infections, and chronic inflammation. Ninety percent of CF patients die due to recurrent pulmonary infections. CF disease is the most common hereditary disease in white people of northern Europe and the US, affecting approximately 1 in 2000–4000 newborns [9–11]. More than 2000 mutations in CFTR have been reported, and these mutations are divided into seven classes [12–15]. Class I mutations contribute to protein production defects and include nonsense mutations causing degradation of mRNA by nonsense-mediated decay. Class II mutations result in protein processing abnormalities leading to defects in cell surface localization. Class III mutations contribute to dysfunctional channel gating at the apical surface. Class IV mutations affect the reduction of channel conductance. Class V mutations lead to a reduced amount of CFTR protein due to abnormal RNA splicing. Class VI mutations cause protein destabilization at the apical surface due to increased protein turnover. Class VII mutations are so-called unrescuable mutations because of large deletions in the CFTR genomic sequence [15,16]. Since there is no curative therapy for CF patients in any class, symptomatic therapies involving a pharmacological approach have mainly been adopted, and effective therapies are still in the research stage. Several studies using CFTR knockout mice to test available treatments have been reported [17–19]. However, these mice do not display the CF disease-associated phenotype observed in human CF disease. Thus, a reliable CF disease model showing a phenotype similar to that of human CF disease must be constructed.
Embryonic stem (ES) cells that are generated from the inner cell mass of blastocyst-stage embryos exhibit self-renewal and pluripotency abilities [20,21]. They can give rise to cells of all three germ layers and many different cell types under appropriate conditions, and they have been frequently suggested as a potential cell source for regenerative therapy. However, the establishment of ES cells requires the destruction of preimplantation embryos at the blastocyst stage, which is highly morally contentious. Moreover, the transplantation of ES cells for therapeutic purposes triggers host immune rejection. In 2006 and 2007, induced pluripotent stem (iPS) cells established from somatic cells by overexpression of reprogramming factors were shown to present self-renewal and pluripotency abilities similar to those of ES cells [22,23]. These cells can be induced to become various cell types with a specific function under appropriate conditions. The use of iPS cells has given rise to new possibilities for regenerative therapy based on cell/tissue transplantation as well as research on various diseases, as there have been issues of immune system rejection and ethical controversy with regard to the use of ES cells. Thus, functional airway epithelial cells derived from iPS cells are expected to be a useful cell source for airway regeneration and the treatment of airway disease (Figure 1). Several research groups have reported the generation of airway epithelial cells from iPS cells [24–35]. Here, we review recent progress focused on the generation of iPS cell-derived airway epithelial cells with physiological functions and discuss the remaining challenges to the generation of functional airway epithelial cells.
Figure 1.
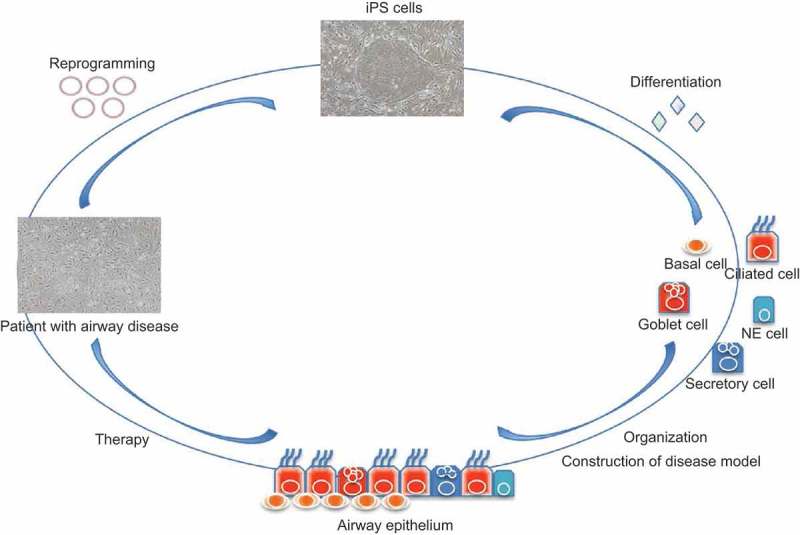
Schema of the application process for airway regeneration using iPS cell technology. iPS cells are generated from patient somatic cells by overexpression of reprogramming factors. Functional airway epithelial cells (ciliated, goblet, basal, secretory, and NE cells) are induced from iPS cells. Construction of the patterned airway epithelium and disease model is performed for airway regeneration and the treatment of airway diseases such as CF.
The various specialized cells in the airway epithelium
The upper and central airway epithelium are composed of ciliated cells, goblet cells, and basal cells. In particular, ciliated cells are the predominant cell type within the airway, accounting for over 50% of all airway epithelial cells, and these cells control the water volume on the airway surface via the transport function of Cl− channels and perform directional transport of inhaled particles via ciliary movement [36,37]. Goblet cells produce mucus to trap foreign objects [38,39], and basal cells are thought to be heterogeneous stem cell populations giving rise to ciliated cells and goblet cells [40–43]. In the distal bronchus and bronchioles, secretory cells such as Clara cells are abundant, and a small number of neuroendocrine (NE) cells are also present [44]. Secretory cells produce bronchiolar surfactant to prevent the harmful effects of the inhalation of, for example, foreign substances and carcinogens, and they serve as progenitors for both ciliated and goblet cells [45]. NE cells are thought to function as chemoreceptors and as a component of the stem cell niche, in addition to being the cells of origin in small-cell lung cancer [46,47]. However, the detailed functions of secretory cells and NE cells remain unclear, and unidentified cells may be present in the airway epithelium.
Functional evaluation of airway epithelial cells from iPS cells
There are several reports of the differentiation of airway epithelial cells from human/mouse iPS cells by using a stepwise developmentally guided strategy (Table 1). Green et al (2011). have succeeded in the generation of airway epithelial cells from iPS cell-derived anterior foregut endoderm using an in vivo microenvironment or niche. Mou et al (2012). have induced the differentiation of iPS cells into cells positive for Nkx2.1- and Sox2, which are indicators of airway progenitor cells, by the precisely timed addition of BMP, FGF, and Wnt. However, these reports have not included airway epithelial cell-specific functional analysis. Wong et al (2012). demonstrated the differentiation of airway epithelial cells from CF patient iPS cells, examined the maturity of the CFTR protein by immunostaining and western blot analysis, and analyzed CFTR channel activity using a halide efflux assay. Furthermore, the measurement of CFTR currents by whole-cell patch clamp experiments has also been performed by Firth et al (2014 and 2015). Additionally, Crane et al (2015). and Suzuki et al (2016). have measured the short circuit current of CFTR-dependent Cl currents by Ussing chamber analysis, and McCauley et al (2017). have reported forskolin-induced CFTR-dependent organoid swelling. On the other hand, Konishi et al (2016). examined airway epithelial cell-specific ciliary movement with a constant rhythmic rate using a high-speed camera. Our group has also investigated ciliary movement with constant rhythm using a high-speed camera and the transport function of CFTR using a halide ion sensor [35]. In all of these reports, the analyses of gene expression, proteins, and morphology indicate that the characteristics of iPS cell-derived airway epithelial cells are similar to those of their in vivo counterparts. However, there are few reports comparing the functions of iPS cell-derived airway epithelial cells with those of native airway epithelial cells. It remains unknown whether iPS cell-derived airway epithelial cells are immature or mature. A transplantation experiment using iPS cell-derived airway epithelial cells has also been reported for airway regeneration [48]. While the survival of iPS cell-derived airway epithelial cells was confirmed at the site of a defect or injury in immunodeficient mice, functional assays, such as those examining the transport function of Cl− channels to maintain the mucociliary transport system, were not performed. Hence, neither in vitro nor in vivo studies have yet included detailed evaluations of whether iPS cell-derived airway epithelial cells exhibit physiological functions such as the transport function of Cl− channels and whether these cells are fully mature.
Table 1.
Functional assessment of iPS cell-derived airway epithelial cells.
| Author (Year) | Functional assessment | Function |
|---|---|---|
| Green et al. (2011) | − | − |
| Mou et al. (2012) | − | − |
| Wong et al. (2012) | + | CFTR |
| Huang et al. (2014) | − | − |
| Firth et al. (2014 and 2015) | + | CFTR |
| Dye et al. (2015) Crane et al. (2015) Konishi et al. (2016) Suzuki et al. (2016) McCauley et al. (2017) Yoshie et al. (2019) |
− + + + + + |
− CFTR Ciliary Movement CFTR CFTR, Ciliary Movement CFTR, Ciliary Movement |
Function of Cl− channels on the airway epithelium
The airway epithelium exhibits important functions such as the regulation of mucus volume and water volume on the airway surface to maintain mucociliary transport system function. The luminal side of airway epithelium is covered with airway surface liquid (ASL), which is composed of a mucus layer that traps inhaled particles from the external environment, and a periciliary liquid layer (PCL), which is a water layer that maintains a reasonable distance between the mucus and epithelium [49,50]. The regulation of PCL volume is indispensable for the maintenance of the mucociliary transport system, and the transfer of water molecules to the luminal side mainly depends on Cl− transport via Cl− channels [51]. When Cl− is secreted to the luminal side via CFTR and/or other Cl− channels, such as TMEM16A, which is a calcium-activated Cl− channel, the electric potential on the luminal side temporarily shifts to a negative charge. The electrochemical gradient due to Cl− secretion promotes Na+ transport to the luminal side, and the resultant osmotic pressure difference between the luminal and serosal sides causes the transport of water molecules to the luminal side. Conversely, when Na+ and Cl− are absorbed into the cell via epithelial Na+ channels (ENaC) and Cl− channels, the absorption of water molecules is promoted, and the PCL volume is decreased. In particular, since CF patients exhibit dysfunction of CFTR, Cl− secretion to the luminal side is decreased compared to that in healthy patients. Furthermore, ENaC activation by CFTR dysfunction causes Na+ absorption from the luminal side, and the resultant environment increases viscosity on the airway surface [52–54]. Thus, CFTR and other Cl− channels have an important function in maintaining the mucociliary transport system on the airway surface. We evaluated the transport function of Cl− channels in iPS cell-derived airway epithelial cells with mutant YFP (mYFP) containing two point mutations at position 148 (histidine) and 163 (valine), which shows high sensitivity to halide ions. The absorption spectrum of mYFP modified with halide ions is decreased, and its fluorescence is quenched [55–57]. Hence, following the fluorescence intensity of mYFP can indicate the transport function of Cl− channels in iPS cell-derived airway epithelial cells. The extracellular fluid of iPS cell-derived airway epithelial cells was replaced with Cl–free HBS from differentiation medium. After 1 hr, Cl–free HBS was replaced with HBS or HBS including a cocktail (10 μM Forskolin, 100 μM IBMX, and 1 mM dbcAMP) that can activate CFTR. When iPS cell-derived airway epithelial cells were continuously incubated with Cl–free HBS, there was no notable change in fluorescence intensity (Figure 2a). On the other hand, when Cl–free HBS was replaced with HBS, the fluorescence intensity was decreased in a time-dependent manner (Figure 2b), and a significant difference was confirmed between 0 min and 10 min after buffer replacement. Furthermore, when iPS cell-derived airway epithelial cells were incubated with HBS including the cocktail, the fluorescence intensity was immediately decreased (Figure 2c). A significant difference in the fluorescence intensity was shown between cells treated with Cl–free HBS, HBS, and HBS with the cocktail after 10 min of medium replacement (Figure 2b). The data presented here indicate that CFTR and other Cl− channels in iPS cell-derived airway epithelial cells transported Cl− into the cells according to the concentration gradient of Cl−. Since these results were similar to those of a previous report [35], this assay is considered to be an effective method for evaluating the transport function of Cl− channels.
Figure 2.
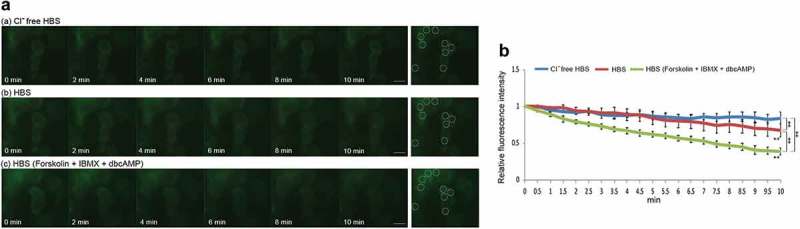
Transport function of the Cl− channels and CFTR in iPS cell-derived airway epithelial cells. (a) Time-lapse fluorescent images of mYFP-labeled iPS cell-derived airway epithelial cells in Cl–free HBS (a), normal HBS (b), and normal HBS plus 10 μM Forskolin, 100 μM IBMX and 1 mM dbcAMP (c). (b) Time-dependent relative fluorescence intensity of regions of interest (ROIs) in mYFP-labeled iPS cell-derived airway epithelial cells in various HBS conditions. The fluorescence intensity at each time point was compared with that at 0 min. **P < 0.01 vs time 0; ‡P < 0.01.
Establishment of iPS cells from CF patients
iPS cells are effective tools not only for use in regenerative medicine, such as cell/tissue replacement, but also as a disease model. CFTR knockout mice cannot mimic the human-specific CF phenotype due to species differences. Thus, the establishment and use of CF-specific human iPS cells is expected to provide a powerful tool for the treatment of CF. The most common cause of CF is the deletion of phenylalanine 508 in CFTR. The deletion of phenylalanine at residue 508 in the amino acid sequence of the protein occurs in at least 1 allele in approximately 90% of CF patients [58]. This mutation causes misfolding, misassembly, mistrafficking, and dysfunction of the CFTR protein [59–61]. For CF patients with this mutation, the dual combination of a CFTR corrector, which improves the transport of protein to the cell surface, and a CFTR potentiator, which increases in the time in which the channel is in the open state, results in an increase in PheCFTRdel protein activity and is more effective than either treatment alone [62]. Furthermore, it has been reported that treatments involving triple combinations, such as two correctors and a potentiator, result in increased CFTR function [63,64]. Recently, the establishment of iPS cells from CF patients carrying PheCFTRdel has been reported to facilitate the discovery of new mutation-targeted therapies. Wong et al (2012)., Firth et al (2015)., Crane et al (2015)., Suzuki et al (2016)., and Fleischer et al. [65]. established CF iPS cells with PheCFTRdel and confirmed defects in the normal expression pattern, plasma membrane localization, and CFTR activity. Moreover, since they indicated that functional recovery of CFTR was observed following the use of a CFTR corrector and genome editing, patient-derived iPS cell-derived airway epithelial cells have been shown to recapitulate the phenotypes of CF in vitro. However, it remains unknown whether these cells were mature airway epithelial cells with clonal properties.
Future directions
Previous studies have suggested that iPS cells can be differentiated into airway epithelial cells [24–35], but it has not been clarified in detail whether these cells exhibit physiological functions such as the transport function of Cl− channels and are mature. Thus, in addition to the evaluation of iPS cell-derived airway epithelial cells by analyses of gene expression, proteins, and morphology, functional analysis is also indispensable, and the establishment of a functional assay system is essential for verification. Furthermore, since it is difficult to induce iPS cells to differentiate into mature airway epithelial cells with clonal properties, it is necessary to investigate the mechanism of terminal differentiation and search for reliable maturation markers. Hence, single-cell analysis might provide valuable information for the functional assessment of iPS cell-derived airway epithelial cells. The method for evaluation of the transport function of Cl− channels using mYFP described in this review can be analyzed at the single-cell level, so it is considered a powerful assay. The airway epithelium is mainly composed of ciliated cells, and functional analysis of these cells has been performed. On the other hand, goblet cells, secretory cells, and NE cells also exhibit important functions, such as mucus production, surfactant secretion, and chemoreceptor functions. Thus, the function of the cells differentiated from iPS cells must be evaluated. However, in the airway epithelium in vivo, the number of secretory cells and NE cells is a small compared to that of ciliated cells, and their detailed roles have not been clarified. Their functions must be further elucidated for airway regeneration to be achieved. Additionally, the construction of a patterned functional airway epithelium with polarity will be required for transplantation because all specialized cells forming the airway epithelium are regularly aligned on the basement membrane and must exhibit appropriate functions in the airway epithelium. Furthermore, after transplantation, not only must survival assessment be performed, but physiological functions such as Cl− channel function and ciliary movement must also be analyzed.
Therapeutic agents for the treatment of CF disease have been developed using primary airway epithelial cells. However, it is difficult to obtain an adequate quantity of cells and maintain them in culture for an extended period. Furthermore, genetic single nucleotide polymorphisms (SNPs) are partly responsible for the differences in drug effects among individuals. Thus, individual CF patient-derived iPS cells would be an effective tool for the discovery of new mutation-targeted therapies.
Funding Statement
This study was supported by the Japan Society for the Promotion of Science [17K16932] and the Takeda Science Foundation [SO29028].
Acknowledgments
We thank Prof. Shigeru Murono (Department of Otolaryngology, School of Medicine, Fukushima Medical University, Fukushima, Japan) for letting us use his equipment. This study was supported by Grant-in-Aid for Young Scientists (B) 17K16932 and the Takeda Science Foundation (SO29028).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Grillo HC. Surgical treatment of postintubation tracheal injuries. J Thorac Cardiovasc Surg. 1979;78(6):860–875. [PubMed] [Google Scholar]
- [2].Grillo HC. Primary reconstruction of airway after resection of subglottic laryngeal and upper tracheal stenosis. Ann Thorac Surg. 1982;33(1):3–18. [DOI] [PubMed] [Google Scholar]
- [3].Xu L, Zhang S, Li J, et al. Human tracheal allotransplant with greater omentum for revascularization. Exp Clin Transplant. 2014;12(5):448–453. [PubMed] [Google Scholar]
- [4].Martinod E, Seguin A, Radu DM, et al. Airway transplantation: a challenge for regenerative medicine. Eur J Med Res. 2013;18(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wurtz A, Porte H, Conti M, et al. Tracheal replacement with aortic allografts. N Engl J Med. 2006;355(18):1938–1940. [DOI] [PubMed] [Google Scholar]
- [6].Omori K, Nakamura T, Kanemaru S, et al. Regenerative medicine of the trachea: the first human case. Ann Otol Rhinol Laryngol. 2005;114(6):429–433. [DOI] [PubMed] [Google Scholar]
- [7].Omori K, Tada Y, Suzuki T, et al. Clinical application of in situ tissue engineering using a scaffolding technique for reconstruction of the larynx and trachea. Ann Otol RhinÄi0ol Laryngol. 2008;117(9):673–678. [DOI] [PubMed] [Google Scholar]
- [8].Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170. [DOI] [PubMed] [Google Scholar]
- [9].Cystic fibrosis foundation patient registry: annual data report to the center directors , 2014https://www.cff.org/2014_CFF_Annual_Data_Report_to_the_Center_Directors.pdf/. cited 2016 March11
- [10].Palomaki GE, FitzSimmons SC, Haddow JE. Clinical sensitivity of prenatal screening for cystic fibrosis via CFTR carrier testing in a United States panethnic population. Genet Med. 2004;6(5):405–414. [DOI] [PubMed] [Google Scholar]
- [11].Burgel PR, Bellis G, Olesen HV, et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J. 2015;46(1):133–141. [DOI] [PubMed] [Google Scholar]
- [12].McKone EF, Emerson SS, Edwards KL, et al. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet. 2003;361(9370):1671–1676. [DOI] [PubMed] [Google Scholar]
- [13].Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest. 2002;121(1):55–63. [DOI] [PubMed] [Google Scholar]
- [14].Konstan MW, Flume PA, Kappler M, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: the EAGER trial. J Cyst Fibros. 2011;10(1):54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–674. [DOI] [PubMed] [Google Scholar]
- [16].Dörk T, Macek M, Mekus F, et al. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum Genet. 2000;106(3):259–268. [DOI] [PubMed] [Google Scholar]
- [17].Snouwaert JN, Brigman KK, Latour AM, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257(5073):1083–1088. [DOI] [PubMed] [Google Scholar]
- [18].Clarke LL, Grubb BR, Gabriel SE, et al. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science. 1992;257(5073):1125–1128. [DOI] [PubMed] [Google Scholar]
- [19].Guilbault C, Saeed Z, Downey GP, et al. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol. 2007;36(1):1–7. [DOI] [PubMed] [Google Scholar]
- [20].Evans MJ, Kaufman MH. Establishment in culture of pluripo- tential cells from mouse embryos. Nature. 1981;292(5819):154–156. [DOI] [PubMed] [Google Scholar]
- [21].Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. [DOI] [PubMed] [Google Scholar]
- [22].Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. [DOI] [PubMed] [Google Scholar]
- [23].Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. [DOI] [PubMed] [Google Scholar]
- [24].Green M, Chen A, Nostro MC, et al. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat Biotechnol. 2011;29(3):267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mou H, Zhao R, Sherwood R, et al. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell. 2012;10(4):385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wong AP, Bear CE, Chin S, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol. 2012;30(9):876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang SX, Islam MN, O’Neill J, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32(1):84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Firth AL, Dargitz CT, Qualls SJ, et al. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2014;111(17):E1723–E1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Firth AL, Menon T, Parker GS, et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015;12(9):1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dye BR, Hill DR, Ferguson MA, et al. In vitro generation of human pluripotent stem cell derived lung organoids. eLife. 2015;4:e05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Crane AM, Kramer P, Bui JH, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015;4(4):569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Konishi S, Gotoh S, Tateishi K, et al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 2016;6(1):18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suzuki S, Sargent GR, Illek B, et al. TALENs facilitate single-step seamless SDF correction of F508del CFTR in airway epithelial submucosal gland cell-derived CF-iPSCs. Mol Ther Nucleic Acids. 2016;5(1):e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McCauley KB, Hawkins F, Serra M, et al. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Stem Cell Rep. 2017;20(6):844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yoshie S, Nakamura R, Kobayashi D, et al. Functional characterization of various channel‐expressing central airway epithelial cells from mouse induced pluripotent stem cells. J Cell Physiol. 2019. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [36].Harkema JR, Mariassy A, St. George J, et al. Epithelial cells of the conducting airways: aspecies comparison In: Farmer SG, Hay DWP, editors. The airway epithelium: physiology, pathophysiology and pharmacology. New York: Marcel-Dekker; 1991. p. 3–39. [Google Scholar]
- [37].Randell SH, Boucher RC. Effective mucus clearance is essential for respiratory health. Am J Respir Cell Mol Biol. 2006;35(1):20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jeffery PK. Morphologic features of airway surface epithelial cells and glands. Am Rev Respir Dis. 1983;128(2 Pt 2):S14–20. [DOI] [PubMed] [Google Scholar]
- [39].Jeffery PK. Morphology of the airway wall in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1991;143(5 Pt 1): 1152–1158. (discussion 1161). [DOI] [PubMed] [Google Scholar]
- [40].Hong KU, Reynolds SD, Watkins S, et al. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol. 2004;164(2):577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rock JR, Onaitis MW, Rawlins EL, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schoch KG, Lori A, Burns KA, et al. A subset of mouse tracheal epithelial basal cells generates large colonies in vitro. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L631–L642. [DOI] [PubMed] [Google Scholar]
- [43].Ghosh M, Brechbuhl HM, Smith RW, et al. Context-dependent differentiation of multipotential keratin 14-expressing tracheal basal cells. Am J Respir Cell Mol Biol. 2011;45(2):403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Morimoto M, Nishinakamura R, Saga Y, et al. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development. 2012;139(23):4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hong KU, Reynolds SD, Giangreco A, et al. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am J Respir Cell Mol Biol. 2001;24(6):671–681. [DOI] [PubMed] [Google Scholar]
- [46].Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015;29(14):1447–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Linnoila RI. Functional facets of the pulmonary neuroendocrine system. Lab Invest. 2006;86(5):425–444. [DOI] [PubMed] [Google Scholar]
- [48].Miller AJ, Hill DR, Nagy MS, et al. In Vitro induction and In Vivo engraftment of lung bud tip progenitor cells derived from human pluripotent stem cells. Stem Cell Rep. 2018;10(1):101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest. 2002;109(5):571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Grubb BR, Pickles RJ, Ye H, et al. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and human. Nature. 1994;371(6500):802–806. [DOI] [PubMed] [Google Scholar]
- [51].Chmiel JF, Davis PB. State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection?. Respir Res. 2003;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Knowles MR, Gatzy JT, Boucher RC. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981;305(25):1489–1495. [DOI] [PubMed] [Google Scholar]
- [53].Boucher RC, Stutts MJ, Knowles MR, et al. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest. 1986;78(5):1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mall M, Bleich M, Greger R, et al. The amiloride-inhibitable Na+ conductance is reduced by CFTR in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Galietta LJ, Haggie PM, Verkman AS. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499(3):220–224. [DOI] [PubMed] [Google Scholar]
- [56].Wachter RM, Yarbrough D, Kallio K, et al. Crystallographic and energetic analysis of binding of selected anions to the yellow variants of green fluorescent protein. J Mol Biol. 2000;301(1):157–171. [DOI] [PubMed] [Google Scholar]
- [57].Jayaraman S, Haggie P, Wachter RM, et al. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J Biol Chem. 2000;275(9):6047–6050. [DOI] [PubMed] [Google Scholar]
- [58].JL B, Macek M, JP F, et al. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. [DOI] [PubMed] [Google Scholar]
- [59].Du K, Sharma M, Lukacs GL. The delta F508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12(1):17–25. [DOI] [PubMed] [Google Scholar]
- [60].Sharma M, Pampinella F, Nemes C, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164(6):923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lukacs GL, Chang XB, Bear C, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268(29):21592–21598. [PubMed] [Google Scholar]
- [62].McColley SA, Konstan MW, Ramsey BW, et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J Cyst Fibros. 2019;18(1):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Davies JC, Moskowitz SM, Brown C, et al. VX-659-Tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Keating D, Marigowda G, Burr L, et al. VX-445-Tezacaftor-ivacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fleischer A, Lorenzo MI, Palomino E, et al. Generation of two induced pluripotent stem cell (iPSC) lines from p.F508del cystic fibrosis patients. Stem Cell Res. 2018;29:1–5. [DOI] [PubMed] [Google Scholar]