

Abstract
TNFα is a pleiotropic cytokine which fuels tumor cell growth, invasion and metastasis in some malignancies, while in others induces cytotoxic cell death. However, the molecular mechanism by which TNFα exerts its diverse effects on breast cancer subtypes remains elusive. Using in vitro assays and mouse xenografts, we show here that TNFα contributes to the aggressive properties of triple negative breast cancer (TNBC) cell lines via upregulation of TNFAIP3(A20). In a striking contrast, TNFα induces a potent cytotoxic cell death in luminal (ER+) breast cancer cell lines which fail to upregulate A20 expression. Overexpression of A20 not only protects luminal breast cancer cell lines from TNFα induced cell death via inducing HSP70 mediated anti-apoptotic pathway but also promotes a robust EMT/CSC phenotype by activating the pStat3 mediated inflammatory signaling. Furthermore, A20 overexpression in luminal breast cancer cells induces aggressive metastatic properties in mouse xenografts via generating a permissive inflammatory microenvironment constituted by granulocytic-MDSCs. Collectively, our results reveal a mechanism by which A20 mediates pleiotropic effects of TNFα playing role in aggressive behaviors of TNBC subtype while its deficiency results in TNFα-induced apoptotic cell death in luminal breast cancer subtype.
Introduction
Despite recent advances and better diagnostics, metastatic breast cancer is still incurable and remains the leading cause of cancer related death. Unlike the luminal and HER+ breast cancer patients for whom molecularly targeted therapeutics such as the endocrine therapy and anti-HER2 agents are available respectively, patients with the basal-like triple negative breast cancer (basal/TNBC) subtype have limited treatment options and currently lack molecularly targeted therapeutics. Women with basal/TNBC subtype constitute 15–20% of breast cancer patients and are often diagnosed with aggressive/metastatic disease (1, 2). The basal/TNBCs are characterized by a distinct epithelial to mesenchymal transition (EMT) phenotype and cancer stem cell (CSC) properties (3) which we and others have shown to be driven by an inflammatory feedback loop (4–8). Consistent with these studies, we demonstrated that simultaneous knockdown of TP53 and PTEN transforms MCF10A cells resembling the molecular and functional features of triple negative breast cancer (TNBC) subtype (9). The transformed MCF10A cells (MCF10A-p53−PTEN−) and TNBCs display a rapid proteolytic degradation of SOCS3 which resulted in activation of inflammatory cytokines and induction of EMT and CSC phenotype. In line with our findings, a genome-wide siRNA screen revealed that basal/TNBC subtype is highly addicted to proteasomal degradation (10).
The TNFα induced protein 3 (TNFAIP3 also called A20), a ubiquitin-editing enzyme, is originally identified as a protein protecting cells from TNF-induced cytotoxicity(11) and thus well-known for restraining excessive inflammation via its deubiquitinase (DUB) activity (12, 13). Multiple autoimmune diseases such as lupus erythematosus are associated with polymorphisms in the A20 locus (12). In addition to its DUB domain, A20 also exhibits E3-ubiquitin ligase activity by C2-C2 zinc-finger (ZF) motifs at the N terminal (12, 14). TNFα signals through two receptors, TNFR1 and TNFR2 and activates NF-κB pathway in response to inflammation (15). A20 deficient mice demonstrate spontaneous inflammation and premature death due to multi-organ inflammation and cachexia stemming from its inability to terminate NF-κB activity (16). A20 is also required for the termination of TNF independent inflammatory signals such as Toll-like receptor (TLR) activated NF-κB activity in macrophages (13). However, some recent studies implicated a paradoxical role for A20 outside the immune system (17–20). These context-dependent diverse functions of A20 may be attributed to its dual DUB and E3-ubiquiting ligase activities (14, 21). In a striking contrast, A20 has been reported to promote liver regeneration by activating inflammatory IL6/Stat3 signaling pathway via targeting SOCS3 for proteolytic degradation (19). Consistent with these findings, elevated A20 expression, a poor prognostic factor in human cholangiocarcinoma, inversely correlated with reduced SOCS3 expression and activation of inflammatory Stat3 pathway (18). Relative A20 overexpression in glioblastoma stem cells (GSCs) compared to non-stem glioblastoma cells is shown to play a role in maintenance of self-renewing GSCs as well as protection from TNF-induced apoptosis (20). Furthermore, overexpression and prognostic utility of A20 in multiple solid tumors has also been reported (18, 20, 22). In line with these findings, a recent study demonstrated that elevated A20 levels in basal breast cancer subtypes promote the metastatic properties of this subtype by inducing an epithelial mesenchymal transition (EMT) phenotype via multi-monoubiquitylation of Snail1 (17).
Our studies here reveal that TNFα-induced A20 expression in TNBCs protects cells from cytotoxic cell death via upregulation of HSP70-mediated anti-apoptotic pathway while inducing an EMT/CSC phenotype by further activating inflammatory pStat3 pathway. Whereas in ER+ luminal cell lines, TNFα induces cytotoxic cell death due to their failure to induce A20 mediated HSP70 upregulation. We therefore provide evidence that A20 plays a critical role in regulating the pleiotropic effects of TNFα-induced responses in breast cancer subtypes.
RESULTS
A20 is highly upregulated in transformed TNBC model and patient TNBC subtype
We previously reported a transformed MCF10A model by simultaneous knockdown of p53 and PTEN (MCF10A-p53−PTEN−) which displays molecular properties of the basal/TNBC subtype (9). One of the marked characteristics of MCF10A-p53−PTEN− cells compared to parental MCF10A or single gene deleted cells was the activation of inflammatory pathways and cytokines via proteolytic degradation of SOCS3. In an effort to identify E3-ubiquitin ligase that may be responsible for SOCS3 proteolytic degradation in MCF10A-p53−PTEN− cells, we re-examined our gene expression analyses which revealed more than 500 differentially expressed genes between the parental MCF10A and MCF10A-p53−PTEN− cells. We found that TNFα-induced A20 gene, encoding E3-ubiquitin ligase is significantly upregulated in MCF10A-p53−PTEN− cells compared to parental and single gene deleted cells (Fig. 1a). Overexpression of TNFAIP3 in MCF10A-p53−PTEN− cells, compared with MCF10A, MCF10A-p53− or MCF10A-PTEN− cells is validated by qPCR and Western blot analysis (Fig.1b, c). Furthermore, A20 is significantly upregulated in human basal/TNBC subtype as shown by using TCGA data set (Fig. 1d). Elevated A20 transcripts and protein expression in basal/TNBC cell lines, MDA-MB231 and SUM159 cells compared to luminal MCF7 and ZR75.1 are confirmed by qPCR and western blotting (Fig. 1e, f).
Fig. 1. TNFAIP3/A20 is highly upregulated in transformed MCF10A-p53−PTEN−cells and patient basal/TNBC subtype.
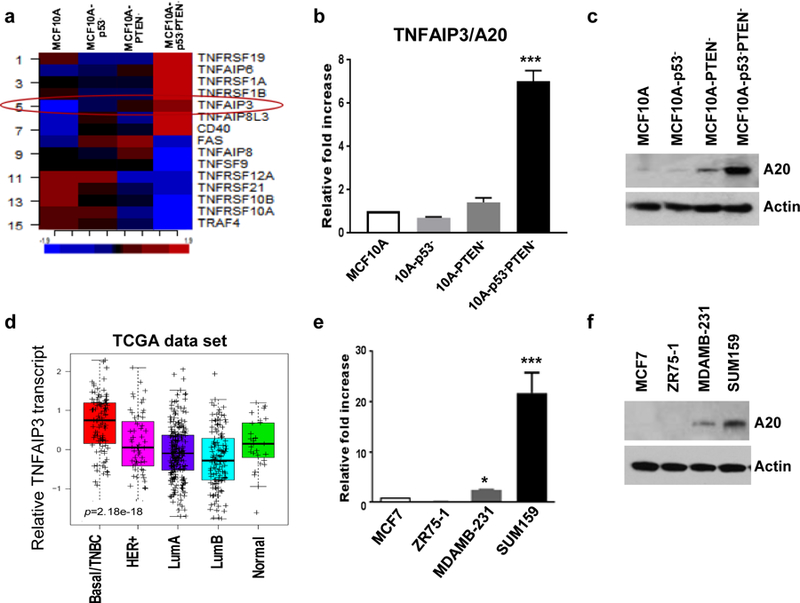
a TNFα-induced genes including TNFAIP3 (red circle) is highly expressed in MCF10A-p53−PTEN− cells. b Expression of TNFAIP3/A20 was verified by qPCR and c Western blot analyses. d TCGA data set indicates the upregulation of A20 in Basal/TNBC breast cancer subtype. e, f Higher expression of A20 in TNBC cell lines, compared with luminal subtype was confirmed by Western blot and qPCR assays. Results are presented as mean ±SD. *P<0.05, ***P<0.0001.
A20 protects luminal breast cancer cells from TNFα-induced cell death
TNFα is a pleiotropic cytokine which acts in a context dependent manner (23). However, the pleiotropic effects and mechanism of action of TNFα have not been well characterized in breast cancer subtypes. To test this, luminal and TNBC cell lines were treated with TNFα (50ng/ml for 48 hr.) and examined for their apoptotic cell death by flow cytometry analyses based on Annexin V staining. Our data show that responses of luminal and basal/TNBC subtypes to TNFα stimulation are profoundly different. While TNFα induces a significantly high apoptotic cell death (over %70) in luminal MCF7 and ZR75–1 cell lines, it shows no detectable apoptosis in basal/TNBC cell lines, MDA-MB231 and SUM159 (Fig. 2a and Supplementary Fig. 1a). We also confirmed that TNFα failed to induce cytotoxic cell death in our transformed MCF10A-p53−PTEN− cells when compared to parental MCF10A which showed more than 25% cell death (Supplementary Fig. 1b, c). In line with the preceding data, luminal breast cancer cell lines showed no detectable A20 expression or induction in response to TNFα stimulation (Fig. 2b, c). In contrast, basal/TNBC cell lines showed a baseline A20 expression which was further enhanced in response to TNFα treatment (Fig. 2b, c). TNFα induced cell death in luminal breast cancer cell lines correlated with an increased PARP1 cleavage and reduced anti-apoptotic protein BCL2, however, basal/TNBC cell lines showed a moderate increase in BCL2 level and there was no detectable PARP1 cleavage (Fig. 2c).
Fig. 2. A20 overexpression protects luminal breast cancer cells from TNFα-induced cell death.
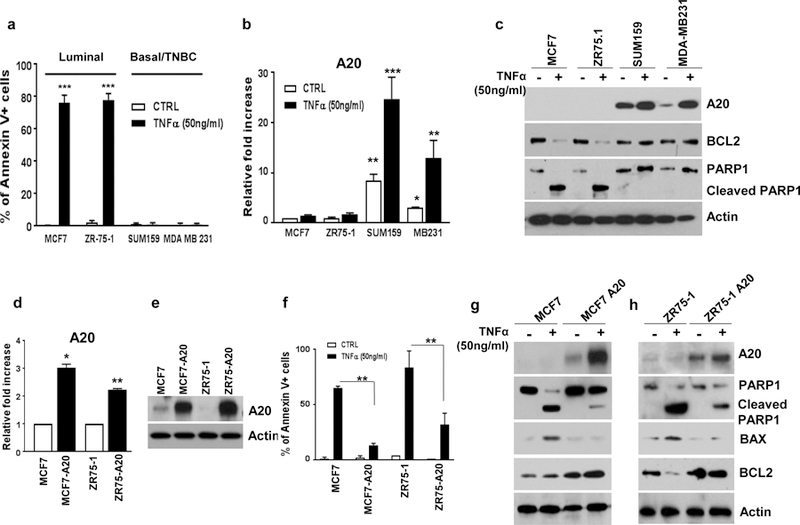
a Luminal and TNBC cell lines were treated with TNFα (50ng/ml) for 48hrs and examined for apoptotic cell death by flow cytometry analyses based on Annexin V staining. b Expression of A20 in TNFα-treated breast cancer cells was shown by qPCR assays. c TNFα-treated induced expressions of A20, BCL2 and PARP cleavage were evaluated by western blot analyses. d, e Overexpression of A20 in MCF7 and ZR75–1 cells was confirmed by qPCR and Western blotting. f Parental and A20 overexpressing MCF7 and ZR75–1 cells were treated with TNFα (50ng/ml) for 48hrs and analyzed cell death by Annexin V flow cytometry apoptotic cell staining. g, h Protein expression of A20, PARP cleavage, BAX and BCL2 was shown in parental and A20 overexpressing cells with TNFα treatment. Results are presented as mean ±SD. *P<0.05, **P<0.001, ***P<0.0001.
In order to investigate the molecular mechanisms of A20-mediated protection from TNFα induced cell death, we generated A20 overexpressing stable clones of MCF7 and ZR75–1 cell lines. Overexpression of A20 in these cell lines is confirmed by qPCR analysis and western blotting (Fig. 2d, e). We then examined whether overexpression of A20 protects cells from TNFα-induced cytotoxicity. Indeed, A20 overexpression in both luminal MCF7 and ZR75–1 cell lines protected cells from TNFα-induced cytotoxicity while parental cell lines displayed a significant apoptotic cell death under identical conditions (Fig. 2f and Supplementary Fig. 2a). This A20-mediated protection from TNFα induced cell death in A20 overexpressing MCF7 and ZR75–1 cells correlated with an increased level of A20 and anti-apoptotic BCL2 and reduced PARP1 cleavage and BAX (Fig. 2g, h).
TNFα induced post-translational upregulation of HSP70 is A20 dependent
Heat shock protein 70 (HSP70), one of the well-studied members of HSP family, is upregulated under conditions of cell stress and has been shown to protect cells from TNFα-induced cytotoxicity (24, 25). We therefore investigated the possible involvement of HSP70 in A20 mediated protection of breast cancer cells from TNFα-induced cytotoxicity. The baseline HSP70 protein levels are comparable between A20 overexpressing MCF7 and ZR75–1 cell lines and their parental counterparts (Fig. 3a, b). However, TNFα significantly upregulated the HSP70 protein levels in MCF7-A20 and ZR75–1-A20 cell lines while it was substantially downregulated in their respective parental counterparts (Fig. 3a, b). Moreover, A20 protein levels are upregulated upon TNFα stimulation in A20 overexpressing cells suggesting a post-translational protein stability (Fig. 3a, b). Consistent with the data, we further confirmed that TNFα induces the upregulations of A20 and HSP70 proteins in TNBC cell lines, MDA-MB231 and Sum159 (Fig. 3c). Altogether these data suggested that HSP70 may be involved in protection of cells from TNFα-induced cytotoxicity in an A20 dependent manner. To determine this, we stimulated cells with TNFα in the presence or absence of HSP70 inhibitor, VER155008. We found that the rate of apoptotic cell death did not significantly change when parental MCF7 and ZR75–1 cells were treated with combination of TNFα and VER155008 compared to the treatment with TNFα alone (Fig. 3d, e and Supplementary Fig. 2b, c). In contrast to parental cells, TNFα, when combined with VER155008, significantly increased the apoptotic cell death in A20 overexpressing cells which are resistant to TNFα-induced cytotoxicity (Fig. 3d, e and Supplementary Fig. 2b, c). We next determined that the combination of TNFα and VER155008 significantly downregulates the expressions of A20, HSP70 and anti-apoptotic BCL2 while increases the PARP1 cleavage in MCF7-A20 cells (Fig. 3f). Expectedly no significant changes were observed in parental MCF7 cells by inhibiting HSP70, because TNFα alone indeed downregulates the expressions of HSP70 and anti-apoptotic BCL2 and induces PARP1 cleavage (Fig. 3g). In order to further verify the role of A20-mediated HSP70 in TNFα-induced cytotoxicity, we knocked down A20 in Sum159 cell line which represents TNBC subtype. The knockdown of A20 indeed renders Sum159 cells sensitive to TNFα-induced cytotoxicity (Fig. 3h) and that significantly correlates with reduced HSP70 and BCL2 protein levels (Fig. 3k).
Fig. 3. TNFα-induced post-translational upregulation of HSP70 is A20 dependent.
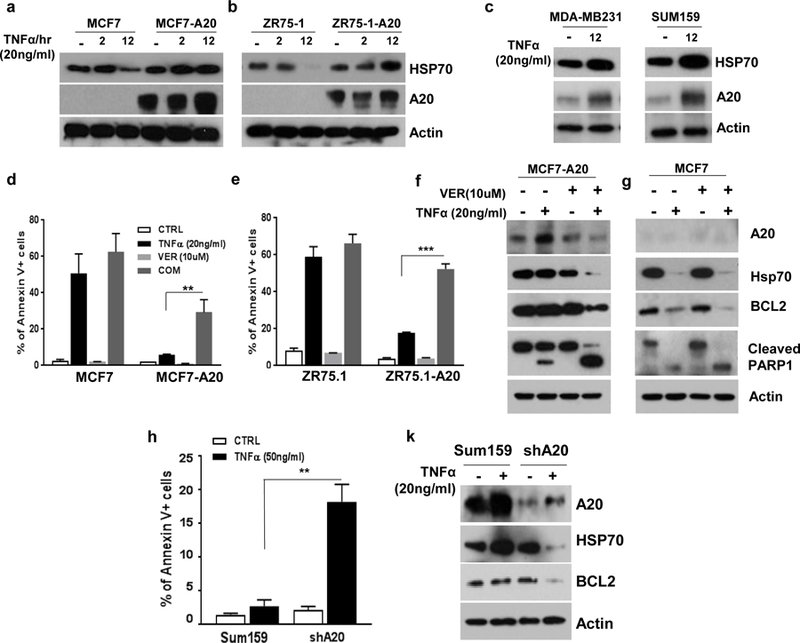
a, b Parental and A20 overexpressing MCF7 and ZR75–1 cells were treated with TNFα (20ng/ml) for 2 and 12hrs and examined the protein expression of HSP70 and A20. c TNFα increased the expression of A20 and HSP70 proteins in MDA-MB231 and Sum159 d, e Parental and A20 overexpressing MCF7 and ZR75.1 cells were treated with TNFα (20ng/ml) with or without VER155008 (10uM) for 24hrs and analyzed cell death by Annexin V flow cytometry apoptotic cell staining. f, g Protein expression of A20, HSP70, BCL2 and PARP cleavage was measured by Western blot assay. h Parental and A20-knockdown Sum159 cells were treated with TNFα (50ng/ml) for 24hrs and analyzed cell death by Annexin V flow cytometry apoptotic cell staining. k Protein expression of A20, HSP70 and BCL2 was measured by Western blot assay. Results are presented as mean ±SD. **P<0.001, ***P<0.0001.
TNFα induces HSP70 degradation in luminal breast cancer subtype
Due to the fact that A20 is a ubiquitin-editing enzyme, next we investigated whether A20 regulates TNFα-induced HSP70 proteolytic degradation. We first determined that although basal/TNBCs cell lines showed significantly higher baseline HSP70 mRNA expression compared to luminal cell lines, they failed to increase the HSP70 transcription in response to TNFα stimulation (Fig. 4a, b). In contrast, luminal cell lines, MCF7 and ZR75–1 showed a significant upregulation of HSP70 mRNA upon TNFα stimulation (Fig. 4c). Interestingly, overexpression of A20 in luminal MCF7 and ZR75–1 cell lines increased the baseline HSP70 transcript levels albeit a reduced response to TNFα in A20 expressing cells compared to parental counterparts (Fig. 4d, e). These findings warranted further analyses of post-translational regulation of HSP70 in different breast cancer subtypes upon TNFα stimulation. It has been reported that HSP70 may be targeted for proteasomal degradation by E3 ligase (26, 27). Consistent with these reports, TNFα induces the proteasomal degradation of HSP70 in luminal breast cancer cell lines, MCF7 and ZR75–1 as evidenced by the accumulation of HSP70 protein when cells were co-treated with MG132, an inhibitor of proteasome complex (Fig. 4f, g). This was further validated by the rapid HSP70 protein turnover when protein synthesis was inhibited by cycloheximide (CHX) in MCF7 and ZR75–1 cell lines upon stimulation with TNFα (Fig. 4h, i). These findings prompted us to examine the interaction between A20 and HSP70 in TNBC cell lines which show upregulation of both proteins upon TNFα stimulation as shown in figure 3c. In line with our hypothesis, A20 binds to HSP70 in Sum159 cells as shown by immunoprecipitation (IP) and this interaction is enhanced when cells were treated with TNFα in a time dependent manner (Fig. 4j). To determine poly-ubiquitination of HSP70, we performed an IP pull-down of HSP70 and immunoblotted with anti-poly-ubiquitin antibody. We indeed confirmed significantly higher levels of poly-ubiquitinated HSP70 in parental MCF7 cells upon TNFα stimulation, while this was reversed in A20 overexpressing MCF7 cells (Fig. 4k). We further corroborated these findings by overexpressing His-tag-Ubiquitin in parental MCF7 and MCF7-A20 cells. Notably, TNFα induced a significant poly-ubiquitination of HSP70 at 2 and 6-hr time points in parental MCF7 cells compared to MCF7-A20 cells (Supplementary Fig. 3). Collectively our studies provide a compelling evidence that A20 protects HSP70 from TNFα-induced proteolytic degradation in luminal breast cancer subtype.
Fig. 4. TNFα induces HSP70 degradation in luminal breast cancer subtype.
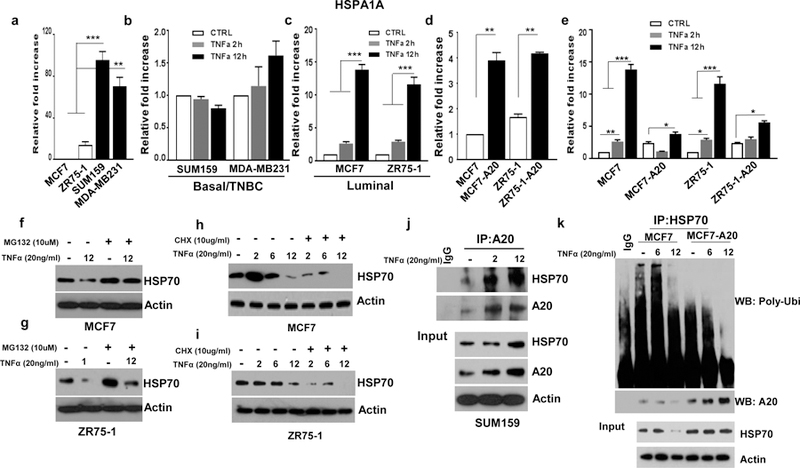
a Different mRNA expression of HSP70 in breast cancer cell lines was determined by qPCR. b, c Luminal MCF7 and ZR75–1 and basal/TNBC Sum159 and MDA-MD 231 cells were treated with TNFα (20ng/ml) for 2 and 12hrs and examined the HSP70 mRNA expression. d, e HSP70 mRNA levels were measured by qPCR in A20 overexpressing MCF7 and ZR75–1 cells with or without TNFα (20ng/ml) treatment. f, g HSP70 expression in MCF7 and ZR75–1 cell was evaluated by Western blotting following treatment with TNFα (20ng/ml) and MG132 (10uM). h, i MCF7 and ZR75–1 cells were treated with TNFα (20ng/ml) and CHX (10ug/ml) for 2, 6 and 12hrs and examined the HSP70 protein expression. j Sum159 cells were treated with TNFα (20ng/ml) for 2 and 12hrs. Cell lysates were immunoprecipitated with anti-A20 antibody and Western blotting was performed with anti-HSP70 antibody. k MCF7 and MCF7-A20 cells were treated with TNFα (20ng/ml) for 6 and 12hrs and performed immunoprecipitation (IP) to pull-down HSP70 and immunoblotted with anti-ploy-ubiquitin antibody. Results are presented as mean ±SD. *P<0.05, **P<0.001, ***P<0.0001.
TNFα induced HSP70/A20/Stat3 pathway drives an EMT/CSC phenotype
The context dependent paradoxical roles of A20-mediated activation of inflammatory IL6/pStat3 pathway via downregulation of SOCS3 in liver regeneration and cancer have been previously reported (17–20). We therefore examined the pStat1 and pStat3 pathways and expressions of cytokines in A20 overexpressing cells compared to their parental counterparts. MCF7-A20 and ZR75–1-A20 cells exhibited a higher Stat1 and Stat3 phosphorylation and downregulation of SOCS3 compared to their respective parental lines (Fig. 5a). To show whether A20 binds to SOCS3 for possible proteolytic degradation, an IP was performed from lysates of MCF7 and MCF7-A20 cells treated with MG132, a proteasome inhibitor. Expectedly there was an increased interaction between A20 and SOCS3 protein in MCF7-A20 cells upon MG132 treatment (Fig. 5b). This suggested to us that A20 targets SOCS3 for proteolytic degradation. Consistent with the activation of inflammatory pStat3 pathway, A20 moderately increased the expressions of inflammatory cytokines such as IL6, IL8, CCL5, TGFB1 and TNFA which were further increased by TNFα stimulation compared to the parental cells (Fig. 5c and Supplementary Fig. 4a).
Fig. 5. A20 overexpression induces the inflammatory Stat1/Stat3 pathway driving an EMT/CSC phenotype.
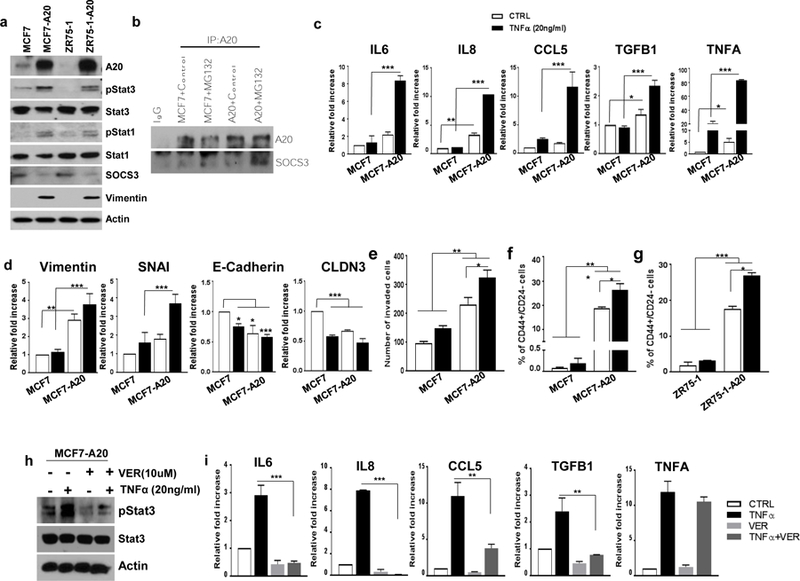
a Expression of A20, pStat1, pStat3, SOCS3 and Vimentin was shown by Western blot in parental and A20 overexpressing MCF7 and ZR75–1 cells. b A20 pulls down SOCS3 protein in MCF7-A20 cells upon MG132 treatment compared to parental MCF7 cells. c mRNA expression of IL6, IL8, CCL5, TGFB1 and TNFA was shown by qPCR in parental and A20 overexpressing MCF7 cells after treating TNFα. d Expression of EMT and epithelial markers was analyzed by qPCR in parental and A20 overexpressing MCF7 cells with TNFα stimulation. e Transwell invasion assay showed an increased invasive ability of MCF7-A20 cells. f, g Flow cytometry analysis of CSC phenotype in A20 overexpressing MCF7 and ZR75–1 cells as assessed by CD44 and CD24 staining. h Expression of pStat3 and Stat3 was shown by Western blot in MCF7-A20 cells after treating TNFα and VER155008. i mRNA expression of IL6, IL8, CCL5, TGFB1 and TNFA was shown by qPCR in MCF7-A20 cells with TNFα and VER155008 treatment. Results are presented as mean ±SD. *P<0.05, **P<0.001, ***P<0.0001.
We and others previously demonstrated that activation of inflammatory cytokines induces an EMT phenotype and expands the CSC population (4, 6, 9, 28). Therefore, we examined whether A20 overexpression in luminal breast cancer subtypes may contribute to induction of EMT/CSC phenotype. We evaluated expressions of EMT related markers in parental and A20 overexpressing cells by qPCR and western blotting analyses. Both MCF7-A20 and ZR75–1-A20 cells showed an upregulation of EMT makers, Vimentin and Snail and down regulation of epithelial markers, E-Cadherin and CLDN3 when compared to their parental counterparts (Fig. 5d and Supplementary Fig. 4b). Interestingly TNFα further enhanced the expression of EMT markers and downregulated the expression of epithelial markers in only A20 overexpressing cells (Fig. 5d and Supplementary Fig. 4b). Expectedly, A20 overexpression significantly increased the invasive potential of MCF7 cells compared to parental MCF7 and that TNFα stimulation further increased their invasive properties (Fig. 5e).
It is well established that the mesenchymal transition induces cancer stem cell (CSC) phenotype in breast cancer cells (29). To confirm the EMT induced CSC expansion in our model, we examined the CSC content in parental and A20 expressing cells by flow cytometry utilizing the CD44+CD24− phenotype. As expected, A20 expression significantly increased the CSC population which was further expanded upon TNFα stimulation in both cell lines compared to the parental cells (Fig. 5f, g).
HSP70 has been shown to activate Stat3 phosphorylation and downstream signaling pathway (30, 31). Therefore, we examined whether TNFα induced HSP70 play any role in pStat3 phosphorylation in our model. On the basis of preceding experiments, inhibition of HSP70 by VER155008 significantly suppressed TNFα-induced pStat3 phosphorylation (Fig. 5h). In addition, the latter also resulted in downregulation of inflammatory cytokines such as IL6, IL8, CCL5 and TGFb (Fig. 5i).
A20 promotes aggressive metastatic properties in mouse xenografts
Our in vitro findings suggested that A20 overexpression may promote aggressive properties of luminal breast cancer cells in mouse xenografts. To test their tumorigenic and metastatic potential, luciferase (Luc) expressing parental MCF7-Luc and stable clones of MCF7-A20-Luc cells were orthotopically implanted into the mammary fat pads of NOD/SCID mice. Tumor growth was monitored weekly in live animals by bioluminescence imaging (BLI). MCF7-A20-Luc xenografts exhibited a higher luciferase signal intensity compared to the parental MCF7-Luc xenografts (Fig. 6a, b). An increased tumor size in MCF7-A20 tumor-bearing mice was also consistent with higher Luc signal intensity (Fig. 6c, d). Most importantly, all MCF7-A20 tumor-bearing mice exhibited a widespread metastasis in liver and lungs as demonstrated by ex-vivo BLI of tissues (Fig. 6e–h). Molecular analyses confirmed overexpression of A20 and upregulation of Vimentin in MCF7-A20 tumors compared to the parental MCF7 tumors (Fig. 6i). Elevated expressions of A20, EMT markers and cytokines were further confirmed by qPCR analyses (Fig. 6j, k). We also determined that SOCS3 expression is suppressed in MCF7-A20 tumors compared to parental MCF7 tumors as assessed by Immunohistochemistry staining (Supplementary Fig. 5a). Hematoxylin and eosin (H&E) staining of formalin fixed paraffin embedded (FFPE) tissue sections exhibited metastatic lesions and immune infiltrates in lungs from MCF7-A20 tumor-bearing mice while no lesions were observed in lungs from MCF7 tumor-bearing mice (Supplementary Fig. 5b). We recently reported that infiltration of granulocytic myeloid derived suppressor cells (gMDSC) in lungs promotes pulmonary metastasis (32). We therefore examined whether pulmonary infiltrates in MCF-A20 tumor-bearing mice are indeed gMDSCs. Flow cytometry analyses of dissociated lungs showed a higher pulmonary gMDSC infiltration in MCF7-A20 tumor-bearing mice compared to parental controls (Fig. 6l, m). We further confirmed and extended our findings using ZR75–1, another luminal breast cancer cell line. We surgically implanted Luc expressing parental ZR75–1 and stable ZR75–1-A20 cells into the mammary fat pads of NOD/SCID mice, monitored the tumor growth weekly by BLI and sacrificed the animals 10 weeks post-implantation. In accordance with the MCF7-A20 data, ZR75–1-A20 cells compared to the parental ZR75–1 generated larger primary tumors as well as spontaneous pulmonary metastasis which may be due to increased gMDSC infiltrations in the lungs (Supplementary Fig. 6a-f).
Fig. 6. A20 overexpression promotes aggressive metastatic properties in mouse xenografts.
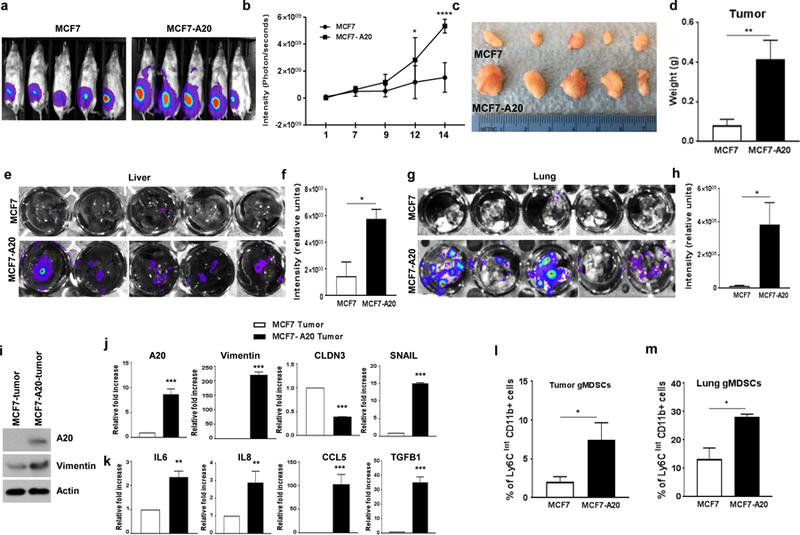
a, b MCF7-Luc and MCF7-A20-Luc cells were orthotopically implanted into the mammary fat pads of NOD/SCID mice (5 mice in each group). Enhanced primary tumor growth in MCF7-A20 mice was shown by bioluminescence imaging (BLI). c, d Primary tumors from MCF7 and MCF7-A20 tumor-bearing mice and their weights which are presented in bar graphs are shown. e-h Enhanced metastasis in MCF7-A20 tumor-bearing mice were shown by ex-vivo imaging of livers and lungs. i-k Expression of A20, vimentin, EMT markers and cytokines in primary tumors was analyzed by Western blot and qPCR. I, m Primary tumor and lung infiltrating Myeloid-derived suppressor cells were detected by flow cytometry analyses. Results are presented as mean ±SD. *P<0.05, **P<0.001, ***P<0.0001.
To provide further direct evidence for the role of A20 in aggressive behaviors of breast cancers, we downregulated A20 in Sum159 cell line which represents TNBC subtype with a higher A20 expression. We generated stable clone by knocking down A20 via lentiviral short hairpin RNA (Sum159-shA20 shown in fig. 3k) which resulted in significant suppression of EMT markers, Snail, N-cad and Vimentin, while increased the expressions of epithelial markers, EpCam and CLDN3 compared to parental SUM159 cells (Fig. 7a). The latter also correlated with reduced invasive properties of Sum159-shA20 compared to its parental counterpart (Fig. 7b, c). In addition, Sum159-shA20 cells not only showed reduced expressions of inflammatory cytokines, IL6, IL8, CCL5, TGFB1 and TNFA but also displayed decreased sensitivity to TNFα stimulation (Fig. 7d). Sum159 cells are well characterized TNBC subtype with a very high CSC compartment (over 90%) which may be attributed to the activation of multiple developmental pathways (6, 9, 33). Down regulation of A20 in Sum159 cells was able to modestly reduce the CSC population (Fig. 7e). On the basis of our in vitro data, we next examined the tumorigenic and metastatic potential of Sum159-shA20 cells. Orthotopically implanted Sum159-shA20 cells showed reduced primary tumor growth and pulmonary metastasis compared to the parental Sum159 cells (Fig.7f–i). Furthermore, the failure of Sum159-shA20 tumors to show pulmonary metastasis may be due to failure to induce gMDSC infiltration in primary tumors and lungs (Fig. 7j, k) Together these studies with A20 knockdown in TNBC subtype further validated our data acquired using overexpression of A20 in luminal cells.
Fig. 7. A20 downregulation decreases EMT/CSC phenotypes in basal/TNBC Sum159 cells.
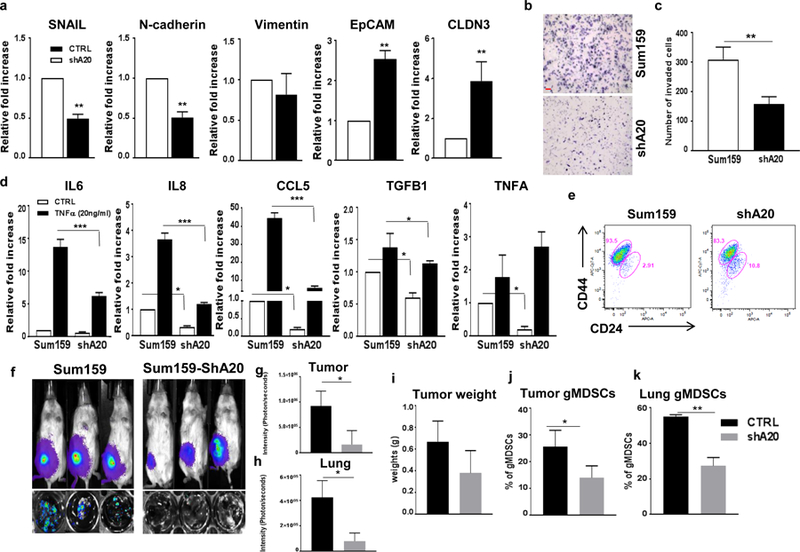
a Expression of EMT genes and pro-inflammatory cytokines was analyzed by qPCR in parental and Sum159-shA20 cells with TNFα stimulation. b, c Transwell invasion assay showed a decreased invasive ability of Sum159-shA20 cells. Scale bar, 50um. d A decreased expression of inflammatory cytokines, IL6, IL8, CCL5, TGFb1, TNFA in A20 downregulated Sum159 cells were shown by qPCR analyses. e Flow cytometry analysis of CSC phenotype in A20-depleted Sum159 cells as assessed by CD44 and CD24 staining. f-h Sum159-Luc and Sum159-ShA20-Luc cells were orthotopically implanted into the mammary fat pads of NOD/SCID mice (3 mice in each group). Suppressed primary tumor growth and lung metastasis in Sum159-ShA20 mice were shown by bioluminescence imaging (BLI). i Primary tumor weights are presented in bar graphs. j,k Primary tumor and lung infiltrating-myeloid-derived suppressor cells were analyzed by flow cytometry. Results are presented as mean ±SD. *P<0.05, **P<0.001, ***P<0.0001.
In figure 8, the summary of our data is illustrated to show that TNFα induces an apoptotic signal via proteolytic degradation of HSP70 due to the lack of A20 in luminal breast cancer cell lines. In contrast, A20 expressing basal/TNBC cells are not only protected from TNFα induced apoptotic cell death but also acquire more aggressive phenotype by activating HSP70 mediated pStat3 inflammatory pathway.
Fig. 8. Schematic illustration of diverse TNFα induced effects in luminal and basal/TNBC subtype.
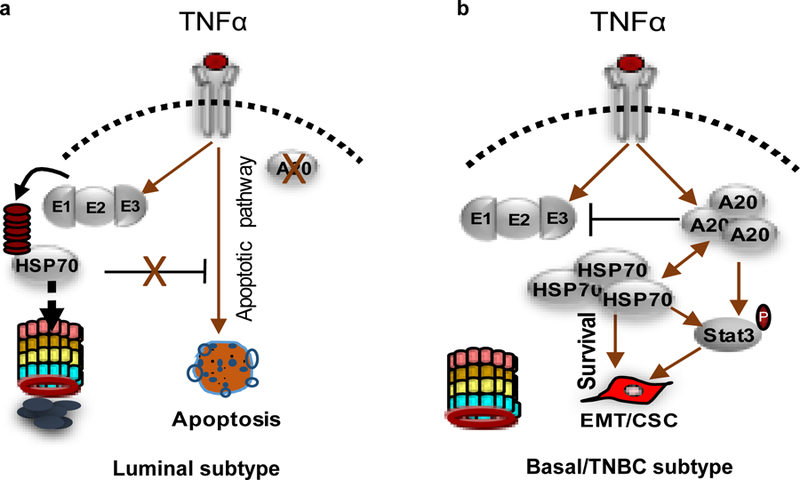
a TNFα induces an apoptotic signal via proteolytic degradation of HSP70 due to the lack of A20 in luminal breast cancer cell lines. b TNFα induces aggressive phenotype in basal/TNBC cells which are protected from apoptotic cell death via A20 mediated HSP70 upregulation.
Discussion
We previously demonstrated that simultaneous knockdown of TP53 and PTEN transforms MCF10A cells displaying the molecular features of triple negative breast cancer (TNBC) subtype such as the induction of EMT/CSC phenotype (9). The transformed MCF10A (MCF10A-p53−PTEN−) tumors in mouse xenografts exhibited aggressive properties which we attributed to the activation of inflammatory cytokines via proteolytic degradation of SOCS3 (9). The microarray analyses revealed several hundred genes that are differentially regulated in transformed MCF10A-p53−PTEN− cells compared to the parental MCF10A, single p53 or PTEN deleted clones. Among the highly upregulated genes, we identified TNFAIP3 (A20), a TNFα induced gene which encodes a ubiquitin-editing enzyme (12). The A20 gene was a particular interest because it was reported that it suppresses SOCS3 expression leading to activation of inflammatory pStat3 pathway (19). We therefore demonstrated an inverse correlation between A20 and SOCS3 protein expressions in breast cancer cell lines representing luminal and basal/TNBC subtype. Moreover, overexpression of A20 in luminal cell lines suppressed the levels of SOCS3 protein and showed enhanced binding upon treatment with MG132, a proteasome inhibitor. Given the fact that basal/TNBCs characterized with activated inflammatory signature (6–8), A20 overexpression in this subtype may play critical role in their aggressive phenotype. Consistent with the notion, we showed that A20 transcript and protein levels are highly upregulated in MCF10A-p53−PTEN− cells and TNBC subtype compared to other subtypes as shown by using the TCGA data set. Furthermore, A20 has been recently shown to be overexpressed in basal-like breast cancers and play a critical role in aggressive metastatic properties of this subtype (17).
TNFα, a pleiotropic cytokine with an important role in inflammation and host defense, exerts diverse physiological functions depending on the cellular context (12, 13, 34, 35). It induces an acute inflammatory responses via activating the inflammatory NF-κB pathway which is suppressed by A20 to protect cells from cytotoxicity (11). In contrast to its role in immune system, A20 has also been reported to play paradoxical roles in different tissues (17–20). Supporting its paradoxical role, we demonstrated that A20 regulates the pleiotropic functions of TNFα in breast cancer subtypes. TNFα induces cytotoxic cell death in luminal breast cancer cell lines which lacks A20 expression, whereas it furthers the aggressive properties of basal/TNBC cell lines expressing elevated levels of A20. Furthermore, basal/TNBC cell lines further upregulate the A20 protein expression in response to TNFα stimulation by which they are protected from cytotoxicity. These findings were further validated by overexpression of A20 in luminal breast cancer cell lines in which A20 not only protects them from TNFα-induced cytotoxic cell death but also induces aggressive properties in response to TNFα as shown by in vitro assays and mouse xenografts.
Although emerging data in cancer supports the notion that A20 may mediate TNFα-induced aggressive properties in some cancer types (17, 18, 20, 22), its central role in immune system is to protect immune cells from TNFα-induced cytotoxicity (11, 12). nduced r A20 red the potential role of rts mmatory cyttionization of MDSCs in preclinical and clinical settings. In addition, . The mechanism by which A20 protects immune cells from TNF- or TLR-induced apoptosis is by removing ubiquitin moieties from NF-kB or TRAF molecules (11, 13, 36). However, other tissues such as lung, liver, kidney and intestine may be protected from TNF- induced apoptosis by alternative mechanisms. It has been reported that HSP70 is upregulated in most tissues under conditions of cell stress and shown to protect cells from TNFα-induced cytotoxicity (37, 38). A compelling evidence supporting the latter was provided by utilizing the HSP70 deficient mice which were no longer protected from the lethal effects of TNF-induced cytotoxicity (39). We therefore hypothesized that TNFα-induced A20 may drive the inducible-HSP70 expression which may protect basal/TNBCs from cytotoxicity. We show that both A20 and HSP70 protein levels were significantly upregulated in basal/TNBCs upon TNFα stimulation while their respective transcripts were not changed. In contrast however, luminal cells lacked A20 expression and failed to upregulate baseline HSP70 protein expression in response TNFα stimulation albeit more than 10-fold increase in its transcript levels. These data were further corroborated by first using the A20 overexpressing luminal cell lines which became resistant to TNFα-induced apoptosis and exhibited enhanced HSP70 protein expression. Moreover, inhibition of HSP70 when combined with TNFα in A20 overexpressing MCF7 cells resulted in apoptotic cell death. It was also confirmed by A20 knockdown in Sum159 cells which failed to induce HSP70 protein expression and were sensitized to TNFα-induced apoptosis. Furthermore, we determined that TNFα-induced upregulation of HSP70 is regulated via post-translational modification as there was no change at the level of HSP70 transcript. Collectively our data suggest that TNFα induced A20 expression binds to HSP70 and protects it from E3-ubiquiting ligase mediated proteasomal degradation. In turn, HSP70 may be involved in stability of A20 protein.
Although the HSF1 driven transcriptional activation of HSP70 is well documented in response to stress (40–42), its post-translational modification has not been well understood in response to TNFα. As previously reported (26, 27), we demonstrated in a series of experiments that TNFα induces HSP70 proteolytic degradation in luminal breast cancer cell lines. However, A20 overexpression in luminal cell lines markedly reduced this process as evidenced by both a reduced poly-ubiquitinated HSP70 level as well as an enhanced binding of A20 to HSP70 in response to TNFα. Furthermore, our data also suggest that A20 may be a client protein for HSP70 as evidenced by reduced levels of A20 protein when cells were stimulated with TNFα in the presence of HSP70 inhibitor. Our data is consistent with the role of HSP70 to promote the proteolytic removal of damaged or improperly folded proteins (25, 43).
Accumulating evidences suggest a paradoxical role for A20 in multiple solid tumors as well as in liver regeneration (17–20, 22). Despite the central role of suppressing inflammation in immune system, A20 has been shown to activate the inflammatory IL6/pStat3 pathway during liver regeneration via suppressing SOCS3 expression (19). Furthermore, Lee at al., reported that A20 induces aggressive properties of basal-like breast cancer subtype by mono-ubiquitinating the Snail1 (17). However, these studies negated the fact that A20 is a TNF-induced gene and failed to investigate changes in breast cancer subtypes in response to TNFα. We showed here that A20 expression was further upregulated in basal/TNBCs in response to TNFα promoting their aggressive properties as evidenced by marked increase in EMT/CSC phenotype. We and others provided evidence that inflammatory cytokines play a major role in induction of EMT/CSC phenotype (6–8, 29). Consistent with these findings, A20 overexpressing cells further upregulated the expressions of inflammatory cytokines such as IL6, IL8, CCL5 and TGFb in response to TNFα stimulation. Upregulation of these inflammatory cytokines resulted in upregulation of EMT markers as well as enhancing EMT/CSC phenotype. Interestingly A20 knockdown in basal/TNBC cell line reversed the expression of inflammatory cytokines, EMT markers as well as reduced the EMT/CSC phenotype. Utilizing functional studies in mouse xenografts, we show that A20 overexpressing luminal breast cancer cell lines generated significantly larger tumors and spontaneous metastasis to liver and lung compared to their parental counterparts which grew smaller tumors and failed to show spontaneous metastasis.
In summary, our studies demonstrated how TNFα exerts its diverse effects on breast cancer subtypes. It furthers the aggressive properties of basal/TNBC subtype via A20 upregulation, whereas induces a cytotoxic cell death in luminal subtype which lacks the A20 expression. The mechanism by which A20 protects basal/TNBCs from cytotoxicity is by binding and enhancing HSP70 protein stability in response to TNFα. This results in induction of anti-apoptotic genes to overcome apoptotic cell death. In addition, A20 also promotes aggressive EMT/CSC phenotype by inducing the inflammatory Stat1 and Stat3 pathways via both downregulating SOCS3 protein expression and upregulating HSP70 expression. Collectively our studies provide a compelling evidence that A20 mediated HSP70 may be an important molecular target for women with basal/TNBC subtype. Given the early clinical developments of HSP70 inhibitors, our studies provide a strong rationale for their therapeutic utility.
Materials and methods
Cell Lines and Reagents
MCF7, ZR75–1 and MDA-MB-231 cell lines were purchased from American Type Culture Collection (ATCC), Sum159 cell line is kindly provided by Dr. Steve Either while he was in University of Michigan. All cell lines were authenticated by STR analyses and mycoplasma testing performed by qPCR analyses. MCF7 and ZR75–1 cells were maintained in RPMI supplemented with 10% fetal bovine serum, and antibiotic/antimycotic 10,000 units/ml. MDA-MB-231 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% serum and antibiotic/antimycotic. Sum159 cells were maintained in Ham’s F12 medium supplemented (with 5% fetal bovine serum, 5 mg/ml insulin, 1 mg/ml hydrocortisone and antibiotic/antimycotic 10,000 U/ml). Recombinant human TNFα was purchased from Gemini Bioproducts. VER 155008, HSP70 inhibitor, was obtained from Sigma.
Generation of A20 overexpression or knockdown cells
Lentiviral constructs were obtained from Applied Biological Materials and stable cell lines were obtained from manufacturer’s protocol. Briefly, MCF7, ZR75–1 and Sum159 cells were transfected with 2 μg of plasmid (pLenti-GIII-CMV-GFP-2A-Puro or piLenti-siRNA-GFP) with Transfection Reagent (Roche). Transduced cells were treated with puromycin and emerging colonies were selected and maintained in culture with puromycin.
Annexin-V staining and Flow cytometry
To detect apoptosis, cells were harvested in Annexin-V binding buffer (Biolgend) or PBS with 2% FBS, stained with Annexin V-APC and 7-AAD and analyzed by a FACScanto flow cytometer (BD Biosciences). For CSC analysis, the cells were labelled with anti- CD44-APC-Cy7, anti-CD24-APC antibodies. All antibodies were purchased from Biolegend. Experiments were done in triplicate and results are representative of two independent experiments.
Western Blotting and Immunoprecipitation
Cells were lysed in RIPA buffer (Sigma). 50μg of each protein with Laemmli sample buffer were boiled for 5 minutes, and subjected to SDS-PAGE. The proteins were transferred onto PVDF membrane (Bio Rad Laboratories) using semi dry Trans-Blot (Bio Rad Laboratories). Blots were first incubated in TBS blocking buffer containing either 2% milk or 2% BSA (for phospho-specific antibodies) for 1 hour at room temperature and then with the respective primary antibodies diluted in TBST (containing 0.1% Tween20 and 2% BSA) overnight at 4℃. Subsequently, blots were washed and incubated with appropriate secondary antibodies (Santa Cruz) in TBST and detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo).
For immunoprecipitation, cells were washed with cold PBS and lysed in lysis buffer (25mM Tris pH7.4, 120mM NaCl, 1mM EDTA, 1% NP-40, 5% glycerol and protein inhibitor cocktail). Cell lysates were incubated with appropriate antibodies at 4 C for 12 hrs and with Protein A/G Magnetic Beads (Pierce) at room temperature for 1h. Immuno-complexes were washed three-times with washing buffer, eluted by 2X sample buffer and boiled for 5 minutes. Immunoblot analysis was subsequently performed. TNFAIP3/A20 antibody was purchased from Thermo Fisher (Cat#MA5–16104). The antibodies to BCL2 (Cat#VMA00017) from Bio-Rad, Cleaved PARP1 (Cat#5625), BAX (Cat#5023), pStat3 (Cat#9145) and pStat1 (Cat#9167) from Cell Signaling Technology and SOCS3 (Cat#ab16030) were purchased from Abcam. HSP70 antibody (Cat#ADI-SPA-810) was from Enzo Life Sciences.
RNA extraction and real-time RT-PCR
Total RNA was extracted using RNeasy Mini kit (Qiagen) and 500 ng of RNA was used to make cDNA using iScript cDNA synthesis kit (Bio Rad). cDNA was analyzed in triplicate using real-time quantitative reverse transcription–PCR(qRT-PCR) assays (Bio-Rad). The information of the PCR primers and fluorogenic probes used are available on the Applied Biosystems website (GAPDH: Hs00266705, IL6: Hs00985641, IL8: Hs00174103, CCL5: Hs00174575, EpCAM: Hs00158980, Vimentin: Hs00185584, E-cadherin: Hs00170423, N-cadherin: Hs00983056, Claudin3: Hs00265816, Snail: Hs00195591, TGFb1:Hs00998133 and TNF:Hs01113624) KiCqStart SYBR Green predesigned primers (Sigma) were used for the following genes: TNFAIP3/A20(F: 5`-AGGCCAATCATTGTCATTTC-3`, R: 5`-AGAACAATGGGGTATCTGTAG-3`), HSP70(F: 5`-AATTTCCTGTGTTTGCAATG-3`, R: 5`-AAAATGGCCTGAGTTAAGTG-3`). The relative fold change was measured by 2-ΔΔCt formula compared to the control cells. Means and differences of the means with 95% confidence intervals were obtained using GraphPad Prism (GraphPad Software Inc.). Two-tailed student t test was used for unpaired analysis comparing average expression between conditions. P values < 0.05 were considered statistically significant. Experiments were done in triplicate and results are representative of two independent experiments.
Transcriptome expression analysis
Total RNA was extracted with RNeasy Mini kit (Qiagen) according to the manufacturere’s instructions and reverse transcribed using the IVT Express kit (Affymetrix) and hybridized on Human Genome HG-U219 Strip Arrays (Affymetrix) comprised of more than 530,000 probes covering more than 36,000 transcripts and variants, which represent more than 20,000 genes mapped through UniGene or via RefSeq annotation.
Invasion Assays
Tumor cells (5×104) were seeded into the top chamber of transwell inserts coated with Matrigel (BD). The inserts were placed in a 24-well plate containing culture media of 5% FBS RPMI. Invaded cells were counted after 16–18 hrs. Experiments were done in triplicates.
Mouse Xenografts and Tissue Analyses
All mice procedures were conducted in accordance with the Laboratory Animal Services at the Augusta University. 5 weeks old NOD/SCID female mice were purchased from Charles River at NCI. Inclusion criteria was 3 weeks and older female NOD/SCID mice due to requirement of fat pad injection in adult mammary gland. No randomization and blinding were required for our studies. Parental and A20 overexpressing MCF7 and ZR75–1 cells (100,000 cells/fat pad) expressing the luciferase gene were implanted into the fat pads of 5-week –old NOD/SCID mice. These mice were imaged utilizing the Caliper IVIS imaging systems. Primary tumors were fixed in pH7.4 buffered formalin and embedded in paraffin for histological or immunohistochemical examination. Lungs and livers were taken and processed for tissue histology to examine the metastatic lesions. Remaining fresh tumor tissues were dissociated and used for western blotting, PCR and FACS analyses.
Statistical analyses and power calculations
Results are presented as mean ± SEM, illustrated as error bars. Statistical analyses were unpaired t-test using GraphPad Prism software (GraphPad Software Inc.). A P value less than 0.05 was considered statistically significant.
Longitudinal primary tumor growth and metastasis will be modeled using repeated measure mixed models, with animal level intercept (initial tumor growth) and slope over time between the control and test groups. For the experiment’s statistical power, the differences of tumor growth between the control and test groups by 8 weeks are expected to be quite large based on our previous experiments. The difference in tumor growth as expressed in radiance (photons/sec/cm2/sr) is 4-fold and therefore, 3–5 animals per group will be yield statistical difference by 8–12 weeks.
Supplementary Material
Acknowledgements
We gratefully acknowledge the generous help from Flow Cytometry, Genomics Core facilities and Labaratory of Animal Services. This work was supported by startup funds to HK by Georgia Cancer Center. Additional research fundings to HK provided by American Cancer Society Institutional fund, Forbes Institute research fund and Augusta University Research Inc. Bridge funding. Max Wicha is supported by R35 CA197585
Footnotes
Conflict of Interest
Authors declare no conflict of interest
REFERENCES
- 1.Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR, Powe DG, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res 2009;15(7):2302–10. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007;13(15 Pt 1):4429–34. [DOI] [PubMed] [Google Scholar]
- 3.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010;12(5):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jia D, Li L, Andrew S, Allan D, Li X, Lee J, et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis 2017;8(7):e2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res 2013;73(11):3470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012;47(4):570–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest 2011;121(7):2723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin Cancer Res 2011;17(19):6125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 2014. [DOI] [PMC free article] [PubMed]
- 10.Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013;24(2):182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opipari AW Jr., Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem 1992;267(18):12424–7. [PubMed] [Google Scholar]
- 12.Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 2009;30(8):383–91. [DOI] [PubMed] [Google Scholar]
- 13.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004;5(10):1052–60. [DOI] [PubMed] [Google Scholar]
- 14.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004;430(7000):694–9. [DOI] [PubMed] [Google Scholar]
- 15.Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol 2014;35(1):22–31. [DOI] [PubMed] [Google Scholar]
- 16.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000;289(5488):2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JH, Jung SM, Yang KM, Bae E, Ahn SG, Park JS, et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. Nat Cell Biol 2017;19(10):1260–73. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Wan M, Zhou Q, Wang H, Wang Z, Zhong X, et al. The Prognostic Role of SOCS3 and A20 in Human Cholangiocarcinoma. PLoS One 2015;10(10):e0141165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Silva CG, Studer P, Skroch M, Mahiou J, Minussi DC, Peterson CR, et al. A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals. Hepatology 2013;57(5):2014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hjelmeland AB, Wu Q, Wickman S, Eyler C, Heddleston J, Shi Q, et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol 2010;8(2):e1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J 2008;409(1):77–85. [DOI] [PubMed] [Google Scholar]
- 22.Hadisaputri YE, Miyazaki T, Yokobori T, Sohda M, Sakai M, Ozawa D, et al. TNFAIP3 overexpression is an independent factor for poor survival in esophageal squamous cell carcinoma. Int J Oncol 2017;50(3):1002–10. [DOI] [PubMed] [Google Scholar]
- 23.Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med 1994;45:491–503. [DOI] [PubMed] [Google Scholar]
- 24.Gabai VL, Yaglom JA, Wang Y, Meng L, Shao H, Kim G, et al. Anticancer Effects of Targeting Hsp70 in Tumor Stromal Cells. Cancer Res 2016;76(20):5926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 2010;11(8):579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kundrat L, Regan L. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J Mol Biol 2010;395(3):587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006;440(7083):551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong H, Davis A, Ouzounova M, Carrasco RA, Chen C, Breen S, et al. A Novel IL6 Antibody Sensitizes Multiple Tumor Types to Chemotherapy Including Trastuzumab-Resistant Tumors. Cancer Res 2016;76(2):480–90. [DOI] [PubMed] [Google Scholar]
- 29.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133(4):704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diao J, Yang X, Song X, Chen S, He Y, Wang Q, et al. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med Oncol 2015;32(2):453. [DOI] [PubMed] [Google Scholar]
- 31.Bocchini CE, Kasembeli MM, Roh SH, Tweardy DJ. Contribution of chaperones to STAT pathway signaling. Jak-Stat 2014;3(3):e970459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 2017;8:14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol 2009;7(6):e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balkwill F Tumour necrosis factor and cancer. Nat Rev Cancer 2009;9(5):361–71. [DOI] [PubMed] [Google Scholar]
- 35.Balkwill F Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev 2002;13(2):135–41. [DOI] [PubMed] [Google Scholar]
- 36.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem 1992;267(25):17971–6. [PubMed] [Google Scholar]
- 37.Meng X, Harken AH. The interaction between Hsp70 and TNF-alpha expression: a novel mechanism for protection of the myocardium against post-injury depression. Shock 2002;17(5):345–53. [DOI] [PubMed] [Google Scholar]
- 38.Jaattela M, Wissing D, Bauer PA, Li GC. Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J 1992;11(10):3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K, et al. HSP70 protects against TNF-induced lethal inflammatory shock. Immunity 2002;16(5):685–95. [DOI] [PubMed] [Google Scholar]
- 40.Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT. Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell 2016;62(1):63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westwood JT, Clos J, Wu C. Stress-induced oligomerization and chromosomal relocalization of heat-shock factor. Nature 1991;353(6347):822–7. [DOI] [PubMed] [Google Scholar]
- 42.Perisic O, Xiao H, Lis JT. Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell 1989;59(5):797–806. [DOI] [PubMed] [Google Scholar]
- 43.Reeg S, Jung T, Castro JP, Davies KJA, Henze A, Grune T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med 2016;99:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.