

Abstract
Chronic kidney disease (CKD) is a clinical model of premature ageing characterized by progressive vascular disease, systemic inflammation, muscle wasting and frailty. The predominant early vascular ageing (EVA) process mediated by medial vascular calcification (VC) results in a marked discrepancy between chronological and biological vascular age in CKD. Though the exact underlying mechanisms of VC and EVA are not fully elucidated, accumulating evidence indicates that cellular senescence - and subsequent chronic inflammation through the senescence-associated secretary phenotype (SASP) - plays a fundamental role in its initiation and progression. In this review, we discuss the pathophysiological links between senescence and the EVA process in CKD, with focus on cellular senescence and media VC, and potential anti-ageing therapeutic strategies of senolytic drugs targeting cellular senescence and EVA in CKD.
Keywords: Chronic kidney disease, Early vascular ageing, Vascular calcification, Senescence, Senolytic drugs
Graphical Abstract
1. Introduction
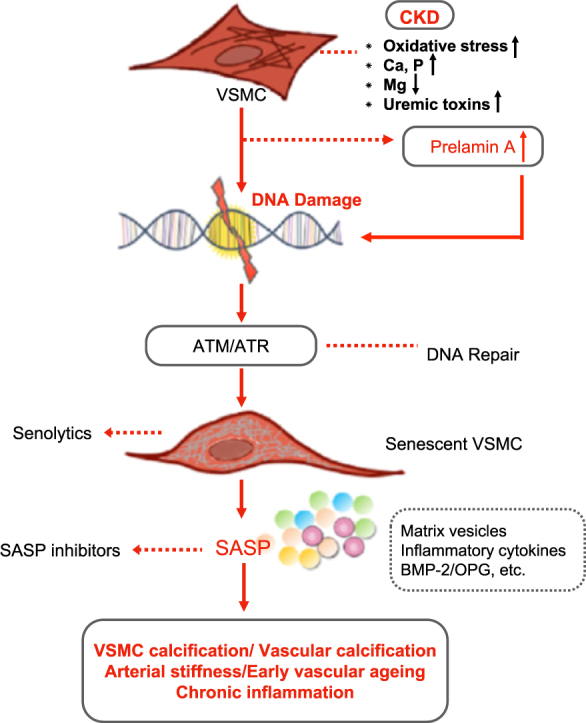
Ageing - a universal finding in humans - is the leading risk factor for deterioration of structure and function of tissues and organs in most of the chronic conditions that limit lifespan and quality of life [1]. As compared to the general population, patients with chronic kidney disease (CKD) have a much accelerated ageing process characterized by progressive vascular disease, persistent uremic inflammation, muscle wasting, osteoporosis and frailty, even before the onset of terminal renal failure necessitating renal replacement treatment by dialysis or kidney transplantation [[2], [3], [4], [5], [6]]. Early vascular ageing (EVA) is an evolving construct that has been growing around accumulating evidence showing arterial stiffness as an intermediate end-point and independent predictor of mortality due to cardiovascular disease (CVD) [7]. In CKD, the process of EVA is predominantly characterized by media vascular calcification (VC), a cell-mediated process primarily driven by alterations in vascular smooth muscle cells (VSMCs), and the extent of VC may be used as a measure of biological vascular age [8,9]. In CKD, the arterial wall appears older than its chronological age due to the impact of concomitant chronic inflammatory status, reflecting premature adaptive changes as a result of persistent increase in allostatic load, repeated cellular insults, and imbalance of pro-ageing and anti-ageing systems [5,6]. Although the exact underlying mechanisms of EVA in CKD have as yet not been fully elucidated, cellular senescence - and the subsequent senescence-associated secretary phenotype (SASP) causing chronic inflammation - appears to play a fundamental role in its initiation and progression [[10], [11], [12], [13]].
In this review, we discuss the pathophysiological links between senescence and EVA in CKD, with a focus on cellular senescence and media VC, and potential anti-ageing therapeutic strategies of senolytic drugs targeting cellular senescence and EVA in CKD.
2. Cellular Senescence
Cellular senescence is referred to as the irreversible growth arrest and apoptosis resistance occurring in response to oncogenic mutations, metabolic insults and DNA damage [[14], [15], [16]]. In fact, the concept of cellular senescence was discovered nearly 50 years ago [17,18], and since then studies have associated cellular senescence with pathological ageing mechanisms that may represent potential therapeutic targets for delaying and attenuating age-related phenotypes and organ dysfunction [[19], [20], [21], [22], [23], [24], [25]]. Similar to replication, differentiation and apoptosis, senescence is a cell event that can occur at any point of life [26]. While acute senescence in development, regeneration and oncogene-activation is regarded as beneficial [[27], [28], [29], [30]], chronic senescence - due to a wide range of intra- and extracellular insults or combinations, including DNA damage and mutations, oncogenic stimuli, reactive metabolites, proteotoxic stress and pro-inflammatory mediators - is considered as detrimental [[31], [32], [33], [34]]. These inducers activate two main pathways governed by the gatekeeper tumor suppressor proteins p53 and pRB (retinoblastoma protein), both of which are transcriptional regulators at the heart of pathways that include a plethora of upstream regulators and downstream effectors. These trigger a senescence response, and depending on tissue and cell type diversity, it takes days to weeks for senescent cells to become fully established and irreversible [11,35] (Fig. 1). Senescent cells are resistant to apoptosis and potentially cleaned by the immune system. The excessive accumulation of senescent cells in various tissues correlates well with age-related metabolic disorders and the development of premature chronic disease [35]. Of note, concomitant with cellular senescence, another common feature of ageing-related conditions is persistent low-grade sterile inflammation, “inflammaging” [36,37]. The source(s) of chronic inflammation in premature ageing processes remains to be determined, but one possible origin could be derived from senescent cells that develop a senescence-associated secretory phenotype (SASP), characterized by a secretion profile with excretion of pro-inflammatory cytokines, growth factors and soluble receptors that poison the surrounding tissues and affect their function [11,35,38].
Fig. 1.

Triggers, effector pathways and features of senescence in tissue dysfunction, ageing and chronic diseases. Inducers triggering senescence vary depending on the context, including DNA damage, reactive oxygen species (ROS), oncogenic mutations, metabolic insults, proteotoxic stress and other unknown factors. Cellular senescence is activated via p53/p21, p16/pRB dependent pathways. One typical feature of senescent cells, the senescent cell anti-apoptotic pathways (SCAPs), predisposes senescent cells to be apoptosis-resistant, resulting in their accumulation in tissues. The SCAPs are thus key targets of senolytic drugs for targeting and inducing senescent cells to undergo apoptosis. Another feature of senescent cells is the senescence-associated secretory phenotype (SASP), characterized by a secretion profile of pro-inflammatory cytokines, growth factors and soluble receptors, which could further result in both local and systemic inflammation and tissue damage effect. Activation of interleukin-1 (IL-1), tumor growth factor β (TGF-β), nuclear factor (NF)-кB (NF-кB), p38 mitogen-activated protein kinases (p38 MAPK) and inflammasome signaling are factors promoting generation of SASP.
2.1. The p53 Pathway
The p53 pathway mediating senescence is mainly triggered by telomere dysfunction, DNA damage and genotoxic stress [[39], [40], [41]]. p53 is a crucial mediator of cellular responses to DNA damage that could prevent cells from proliferating and induce permanent withdrawal from cell cycle and cellular senescence [42]. Together with a subsequent transcription of the gene encoding p21, a downstream target for p53 transactivation and inhibitor of cell cycle progression, the triggered p53 signaling causes cells to undergo senescence arrest [43]. It is well-established that the inactivation of p53, or the gene encoding p21, can delay the replicative senescence at least in diploid human fibroblasts cells [44].
2.2. The Retinoblastoma Protein (pRB) Pathway
The function of p53 is not sufficient to reverse senescence arrest in all cell types; this will depend on whether and to what extent cells express the cell cycle inhibitor p16, a tumor suppressor and a positive regulator of the tumor suppressor protein pRB that prevents excessive cell growth [45]. Though the exact role of the p16/pRB pathway in the senescence growth arrest is not yet fully understood, one possible mechanism could be the consequent development of pRB-dependent heterochromatic repression of genes encoding cyclins, many of which are activation targets of E2F transcription factors [46]. Also, the engagement of the p53 pathway could possibly interact with and induce the pRB pathway, although the effects of pRB activation by p21 differ from that of activation by p16, at least in some respects [45]. Interestingly, once p16/pRB pathway is activated, the senescence arrest cannot be reversed by inactivation of p53, silencing of p16, or inhibition of pRB [44]. Thus, the activation p16/pRB pathway is essentially irreversible and it is primarily triggered by oncogene mutations and various stress [47].
3. SASP
The SASP is a critical intrinsic characteristic of senescence programs and while the composition of excretory products of SASP varies depending on the cell type of senescent cells, as well as the mechanisms by which senescence is induced, SASP invariably contains a wide-range of secreted inflammatory cytokines, chemokines, tissue-damaging proteases, hemostatic and growth factors [33]. The concept of SASP is not unequivocal as it has both positive and negative effects on tissue and organ function depending on the context of cellular senescence. In the chronic senescence milieu, the abundant presence of SASP factors can induce and advance both local and systemic pathogenic effects by altering local tissue microenvironment, activating macrophage infiltration and provoking nearby malignant cells [[48], [49], [50], [51]]. By contrast, a time-limited SASP profile is beneficial in healing or repairing responses. For instance, in early stage of cutaneous wound healing, senescent cells could not only stimulate myofibroblast differentiation and promote wound closure, but also prevent tissue fibrosis by secreting anti-fibrotic matrix metalloproteinases [52,53]. Nevertheless, the dual role of senescence and SASP in cancer development remains to be elucidated. Prototypic SASP cytokines, such as interleukin (IL)-6 and IL-8 can augment the senescence growth arrest at least in some senescent cells as a cancer-protective defence [54]; on the other hand, malignant cancers exploit the SASP factors to stimulate growth, angiogenesis, and epithelial-to-mesenchymal transition (EMT) that further promote metastases and cancer progression [55,56].
The SASP is primarily mediated by nuclear factor (NF)-кB, p38 mitogen-activated protein kinases (p38 MAPK) and the inflammasome signaling, and is largely maintained through autocrine mechanisms by SASP factor IL-1, an upstream modulator of NF-кB signaling [57,58]. In addition, IL-1 and transforming growth factor-β (TGF-β) act together to mediate senescence and accounts for components of a positive feedback loop that regulates the SASP [59]. A recent study showed that SASP factors, such as IL-6, IL-8, osteoprotegerin (OPG), bone morphogenetic protein 2 (BMP-2), monocyte chemoattractant protein-1 and the protease inhibitors metalloproteinase-1 and -2 were highly expressed, both on mRNA and protein levels, by VSMCs from dialyzed children compared to control VSMCs [60]. Moreover, inhibition targeting ataxia-telangiectasia mutated (ATM) kinases reduced the accelerated VC observed in VSMCs from children on dialysis, which was mirrored by the reduced expression of SASP factors BMP-2, IL-6 and OPG [60].
4. SCAPs (Fig. 1)
Senescent cell anti-apoptotic pathways (SCAPs) shield senescent cells from apoptosis and may therefore be a critical key for targeting senescent cells and developing senolytic agents, including peptides [61]. The SCAPs required for senescent cells viability were identified as the Achilles' heel of senescent cells, and it has been verified in various RNA interference studies that knocking down expression of proteins involved in SCAPs could predispose senescent cells to death but not non-senescent cells [22]. Among agents with such properties, dasatinib, a synthesized dual Src/Abl family tyrosine kinase inhibitor, and quercetin, a plant pigment and a potent antioxidant bioflavonoid that could be found in various fruits and vegetables, were the first two senolytic agents discovered using this approach. Both agents were shown to induce apoptosis in senescent cells with dasatinib targeting the dependence receptors/tyrosine kinase SCAP and quercetin targeting the BCL-2/BCL-XL, PI3K/AKT, and p53/ p21/serpine SCAPs [22,62,63]. Since then, a growing number of senolytics, including natural products, synthetic small molecules, and peptides targeting the SCAPs have been identified [22,61,[64], [65], [66]]. Interestingly, since it was reported that muscle extracts from fish with negligible senescence, such as sturgeon, protect from senescence induced by oxidative stress [67], natural senolytics may be found in many parts of nature.
Notably, the SCAPs required for senescent cell resistance to apoptosis vary with cell type or cell strain specificity. For instance, navitoclax, targeting the B-cell lymphoma (Bcl-2) family of proteins, is effective as a senolytic in human umbilical vein endothelial and lung fibroblast-like cell strains but not in primary human fat cell progenitors nor primary human lung fibroblasts [24,68]. Also, since some senolytics can act synergistically in certain cell types, combinations of different senolytics can strengthen the effect as well as broaden the range of senescent cell types targeted [22,69].
5. Identification of Senescent Cells
No universal standard evidence or unequivocal criterion for identifying senescent cells has been established so far. Potential markers include an increase in cell size; high expression of p16INK4A and p21CIP1; elevated cellular senescence-associated β-galactosidase (SA-β-Gal) enzymatic activity; positive telomere-associated DNA damage foci and expression of SASP factors [70]. However, none of these markers are fully sensitive nor specific. Therefore, testing a combination of markers is highly recommended to identify the potential existence of senescent cells in vitro or in vivo, as well as to evaluate the therapeutic effect of senolytics on senescence [35].
6. Evidence for Senescence in CKD
CKD is characterized by an allostatic overload due to excessive oxidative stress and systemic inflammation, defective anti-ageing (e.g. decreased Klotho expression and fetuin A) and active ageing-promoting (e.g. hyperphosphatemia, angiotensin-2, inflammatory burden) systems, resulting in a marked discrepancy between biological and chronological age in CKD [5,6]. Indeed, patients with CKD are at high risk of developing a number of age-related burden of life style pathologies that occur in clusters [71], including EVA, muscle wasting, depression, heart failure, bone mineral disorders and frailty [11,72]. EVA, as one of the most typical premature ageing phenotypes in advanced CKD, contributes to the high risk of cardiovascular morbidity and mortality in the uremic milieu [7].
Persistent systemic inflammation is a cause as well as a consequence of premature ageing and senescence in CKD [37,[73], [74], [75]]. Telomere attrition is prevalent in dialysis patients and positively correlated with the inflammatory burden [76]. Oxidative stress, a main inducer of senescence signaling, is commonly observed in patients with advanced CKD and associated with its partner in crime – inflammation [77]. Inflammatory molecules, such as IL-6 and tumor necrosis factor (TNF), activate catabolism and block anabolic pathways via insulin-like growth factors (IGFs) and the mammalian target of rapamycin (mTOR) regulation [76]. In accordance with the role of SASP in the senescence process, systemic inflammation accounts for dysfunction of the immune regulatory system and reduced resilience to internal and external stressors, and also acts as a catalyst for premature ageing and subsequent adverse clinical outcomes in CKD [77].
The evidence of cellular senescence in CKD has been demonstrated in various animal models. In rats with adenine-induced uremia, an increased arterial mRNA expression of cyclin-dependent kinase inhibitor 2A (CDKN2A) was observed, and both p16Ink4a and Runt-related transcription factor 2 (Runx2) protein expression were detected in and around calcified areas of aortas [78]. Systemic bone marrow-derived mesenchymal stem cells from rats with CKD were also found to be prematurely senescent [79]. Additionally, in a feline model of CKD, shortened telomeres and higher SA-β-gal activity were demonstrated in proximal and distal tubules compared to young and biological-aged cats [80].
We reported that shortened telomere length correlated with low fetuin-A levels and high mortality in prevalent hemodialysis patients [76]. Moreover, in human uremic arterial tissue, we found that independent of chronological age, increased arterial expression of CDKN2A/p16INK4a and the number of SA-β-Gal positive cells associated with the extent of media VC in epigastric arteries [81]. In addition, emerging evidence also suggests that processes involving Klotho could provide a direct link between premature pathology and cellular senescence in CKD [82,83]. Klotho is highly expressed in the kidney, and Klotho-knockout mice develop progeria features with a shortened life span, extensive vascular calcification and other tissue/organ specific defects [84]. Since Klotho levels decrease with deterioration of renal function, CKD may be considered as a clinical model of Klotho depletion [84]. A recent study demonstrated that the antioxidant, anti-apoptotic and anti-senescence effects of soluble Klotho in human VSMC was mediated by an up-regulation of nuclear factor-erythroid related factor 2 (NRF2) signaling and induction of antioxidant enzymes [85]. In addition, Gao et al. [86] reported that the expression and activity of Sirtuin-1 (SIRT1) were significantly decreased in aortic endothelial cells and VSMC of KL+/− mice, suggesting that Klotho deficiency downregulates SIRT1. Accordingly, treatment with a specific SIRT1 activator eliminated Klotho deficiency-induced arterial stiffness and hypertension in this animal model. These studies indicate that the downregulated NRF2 and SIRT activity could potentially be involved in the pathogenesis of Klotho deficiency-related EVA and arterial stiffness. Further work is warranted to clarify the key pathways of Klotho as a contributor to premature vascular ageing in CKD.
Of note, apart from the data of ageing and senescence in CKD from in vitro, in vivo animal models and clinical investigations of adult patients, studying arteries of children with CKD should potentially highlight the accelerated EVA process and senescence in the context of renal dysfunction without confounding by the classical Framingham risk factors [60,[87], [88], [89]]. Indeed, data from pediatric arterial biopsies suggest that EVA with VSMC osteogenesis appears to occur already before it is possible to detect it by ‘gold standard’ clinical measurements, such as pulse wave velocity and computed tomography scans; only the children with the most highly calcified arteries had signs of increased aortic stiffness and elevated Agatston score [89]. The presence of EVA and VSMC dysfunction in children with CKD - with absence of most traditional cardiovascular risk factors - support the notion that that senescence and EVA are related to kidney dysfunction per se.
7. Senescence, Media VC and EVA
7.1. VSMC Calcification in CKD
Though VSMC calcification shares similarities with developmental osteogenesis and chondrogenesis, it is a distinct pathological and active alteration rather than a physiological process. Under normal physiological conditions, a competent defensive pathway would protect VSMCs from phenotypic differentiation and ectopic calcification. Accumulating evidence indicates that the uremic milieu (accumulation of uremic toxins and other factors inducing cellular oxidative stress) evokes the key pathways of VSMCs calcification and initiates tissue damage by inducing modifications in proteins and DNA [[90], [91], [92], [93]]. The inhibitory defence systems in CKD tends to be overwhelmed by hyperphosphatemia and hypercalcemia, which in combination with hyperparathyroidism and hypomagnesemia (and worsened by warfarin treatment), further challenge the VC inhibitory mechanism [92,94]. For instance, fetuin-A, a circulating glycoprotein produced by liver and adipose tissue, which is involved in insulin resistance and atherosclerosis, acts as a potent inhibitor of ectopic calcification through the binding of calcium and phosphate into calciprotein particles (CPPs) preventing its crystallization [95,96]. A recent study further demonstrated that decreased fetuin-A in extracellular vesicles (EVs) and CPPs from uremic serum could promote VC in vitro [97]. Consequently, stressed VSMCs lose their capacity to preserve a balanced level of VC inhibitors and are predisposed to undergo phenotypic differentiation from a contractile to synthetic VSMC phenotype. If this dysregulated signaling is not compensated, the activation of essential osteogenic/chondrogenic transcription factors such as Runx2, NF-κB, Msh homeobox 2 (MSX2), core-binding factor α-1 (CBFA1), serum- and glucocorticoid-inducible kinase 1 (SGK1) or osterix and SRY-Box 9 (SOX9) further induce the expression of osteogenic/chondrogenic proteins in VSMCs, such as osteocalcin, BMP-2 and type I collagen, in the context of high circulating levels of calcium and phosphate. Eventually this will promote uremic VSMCs to undergo osteochondrocytic differentiation and vessel ossification [47,[98], [99], [100], [101], [102], [103], [104]]. In fact, even in the absence of potent hyperphosphatemia and hypercalcemia, the phosphate and calcium load drives VC in both CKD and general population [105]. In accordance, a recent study in rats with adenine-induced CKD showed that protein-bound uremic toxins, indoxyl sulfate and p-cresyl sulfate per se could promote VC in the aorta and peripheral vessels via inflammatory and coagulation pathways [106].
7.2. DNA Damage and VSMC Calcification
The predominant EVA pathology in CKD, manifested as media VC and arterial stiffness, is to a large extent driven by VSMC osteogenic differentiation due to excessive DNA damage [9]. During normal cell proliferation, progressive telomere erosion exposes an uncapped free double-stranded chromosome end, triggering a DNA damage response (DDR) [107]. However, in the uremic milieu, the DDR pathways may become deficient as a result of an allostatic overload (i.e., oxidative stress, inflammation, glycation, carbamylation, accumulation of uremic toxins, hypercalcemia and hyperphosphatemia), which, in combination with hypomagnesemia, deficiency of fetuin-A and vitamin K, promote uremic cells to undergo senescence and become apoptosis-resistant [93,108]. VSMCs cultured by serial passaging acquire DNA damage features, such as accumulation of γH2AX and 53BP1 foci, as well as an elevated expression of p16INK4A [109,110]. In accordance, alkaline phosphatase (ALP) activity and Runx2 expression increase, implying that senescence may induce a pro-calcific phenotype and drive calcification in the vessel wall [110].
Another fact supporting the involvement of senescence in media VC is that the activation of ATM and ataxia telangiectasia and Rad3-related (ATR) kinases in DDR signaling acts as a key factor in osteogenic differentiation and calcification of VSMCs; inhibition of this pathway prevents osteogenesis of VSMCs in vitro [107]. In addition, co-culture experiments with mesenchymal progenitor cells showed that the activation of osteogenic differentiation was possibly mediated by secretory profiles of ageing VSMCs, i.e. SASP [33,110]. More importantly, several SASP factors released by senescent VSMCs, such as IL-6, BMP-2 and osteoprotegerin, modulate calcification processes [110,111]. These findings imply that the existence of senescent VSMCs may not only drive osteogenic differentiation and calcification locally, but also induce VSMCs calcification at remote sites, triggering both local and systemic stem cells to undergo osteogenic differentiation.
7.3. Nuclear Lamina and EVA
Recent studies revealed that VSMCs undergo nuclear defects and accelerate senescent processes in ageing diseases [109]. One typical example of this cellular change is observed in children with Hutchinson-Gilford progeria syndrome (HGPS), which is most commonly caused by mutations in the LMNA gene, encoding the nuclear lamina protein lamin A, leading to the accumulation of its precursor protein, prelamin A, particularly in VSMCs [112]. Similarly, accumulation of prelamin A has been detected in arteries of children on dialysis with EVA and arterial stiffening [110]. Studies have shown that the uremic toxin indoxyl sulphate could reduce cleavage of prelamin A into mature lamin A by endopeptidase FACE1 (also called Zmspte24), and thus cause the accumulation of prelamin A in VSMCs [110,113]. This in turn interferes with DNA repair by delaying the recruitment of key factors of DDR, resulting in persistent DNA damage and subsequent p53 and pRB senescence signaling [109]. Consequently, premature senescent VSMCs undergo further osteogenic gene expression and VSMC calcification and attrition. Although the role of prelamin A and lamin A in senescence and VSMC calcification needs further investigations, the accumulation of these proteins could be hypothesized as a potential mechanism of EVA in CKD.
8. Targeting Senescence in Media VC and EVA (Fig. 2)
Fig. 2.

Potential senotherapy: preventive strategies, SASPs inhibitors and senolytics. Adapted from Kirkland et al. [66].
Currently, the treatment of EVA and premature senescence in CKD is an unsolved issue. Potential intervention strategies include lowering phosphate burden, activating SIRT and transcription factor NRF2, increasing Klotho expression, inhibiting mTOR and reducing DNA damage, as well as avoidance of life-style related stressors by dietary phosphate and caloric restriction and physical exercise [114]. Additionally, given the role of gut-microbiota-derived uremic toxins in VC, gut microbiota modulation also represents a promising approach to alleviate EVA [106,115]. However, it could be speculated that the therapeutic goals of treating CKD-related EVA could be achieved more effectively by targeting fundamental mechanisms of ageing, such as processes leading to cellular senescence. Based on the insights of cellular senescence, SASP and SCAPs, potential strategies for alleviating the ageing effects of senescent cells include: 1) inhibition of pathways leading to senescence-associated growth arrest; 2) improving the clearance of senescent cells, and 3) interference of SASP signaling factors [35]. Of note, the first treatment strategy could be a double-edged sword and possibly problematic. On the one hand, the inhibition of p16INK4a, pRB, p53 or p21 tumor suppressor pathways could diminish the defensive effect of senescent cells against cancer and increase the risk of cancer development [116]. On the other hand, strategies that prevent senescent cell accumulation by reducing metabolic damage could limit ageing-related tissue dysfunctions. For instance, caloric restriction, which delays cellular senescence, and slows down VC and ageing, was shown to be an effective approach to extend the lifespan in animals [114,117].
The second solution - improving the clearance of senescent cells - may be more attractive, since it may not only alleviate tissue/organ inflammation and dysfunction, but also potentially may help to avoid cancer risk. The recent development of senolytic agents interfering with SCAPs pathways to induce apoptosis of senescent cells, is a promising approach [118]. Several senolytics, including dasatinib, quercetin, navitoclax and peptides targeting BCL-2 and p53-related SCAPs, have been shown to effectively eliminate senescent cells in mice, and decrease senescent markers (e.g. SA-β-Gal activity, p16INK4A, p21CIP1, and telomere-associated foci) [61,66]. Vasculature effects of senolytics achieved so far in the animal research field are: improved cardiac ejection fraction in old mice, and alleviated VC and increased vascular reactivity in hypercholesterolemic, high fat-fed apoE−/− mice [22,119]. Among the above-mentioned senolytics, the combination of dasatinib and quercetin turned out to be the most effective intervention in improving physical function and reducing VC in the aorta of aged and hypercholesterolemic mice [120]. Most notably, this combination therapy extended the lifespan of naturally aged and senescent cell transplanted young mice by 36% [120]. Also, the first-in-human pilot clinical trial of senolytics in idiopathic pulmonary fibrosis (IPF) further indicated that short-term, intermittent administration of dasatinib and quercetin may alleviate the physical dysfunction of IPF. These findings suggest that time may be ripe for randomized, controlled trials evaluating the effects of dasatinib, quercetin and other senolytics targeting cellular senescence-associated clinical conditions [121].
The third alternative approach of targeting senescence, i.e., interfering with SASP signaling to limit the development of SASP, could be achieved by inhibiting pro-inflammatory pathways, such as NF-кB, IL-6 and p38 MAPK signaling [57,58]. Targeting SASP is a logical approach since IL-6 sits at the “central hub” of factors involved in residual inflammatory risk, and has been proven to be effective as treatment with IL-1β mAb (canakinumab) reduced the risk of major adverse cardiovascular events with 30% in the CKD subgroup of the CANTOS study [122]. However, while SASP inhibitors would reduce the systemic burden of senescent cell-associated inflammation without interfering with anti-oncogenic pathways, this intervention may also act through other drug-specific mechanisms that potentially could impair immune cell clearance capacity, resulting in a risk of excessive accumulation of senescent cells [123]. Thus, we need to improve our understanding of how such unwarranted implications of SASP interventions, i.e., senescent-cell accumulation, could be avoided.
9. Conclusion
Several lines of new evidence have emerged recently, indicating that cellular senescence plays an important role in CKD-associated EVA. While more future work is warranted to identify and understand the specific key factors and ageing pathways involved in EVA and cellular senescence in CKD, interventions targeting the fundamental underlying disorders of cellular senescence and the development of SASP - rather than addressing specific risk factors, such as hyperphosphatemia and uremic toxins one at a time – appear already today as feasible strategies that could open new and effective therapeutic opportunities to prevent uremic EVA. The results of a recent clinical trial of senolytics in idiopathic pulmonary fibrosis indicate that interventions targeting senescent cells in premature ageing diseases is a transformative and promising strategy.
Disclosures
BL is affiliated with Baxter Healthcare. PS is on the scientific advisory board of REATA.
Declarations of Competing Interest
None of the other authors declare any conflict of interest.
Acknowledgements
The study benefited from generous support from Strategic Research Program in Diabetes at Karolinska Institutet (Swedish Research Council grant No 2009-1068), European Union‘s Horizon 2020 research and innovation Program under the Marie Skłodowska-Curie grant agreement No 722609; International Network for Training on Risks of Vascular Intimal Calcification and roads to Regression of Cardiovascular Disease (INTRICARE; www.intricare.eu). Baxter Novum is the result of a grant from Baxter Healthcare to Karolinska Institutet.
References
- 1.Westendorp R.G., Kirkwood T.B. Human longevity at the cost of reproductive success. Nature. 1998;396:743–746. doi: 10.1038/25519. [DOI] [PubMed] [Google Scholar]
- 2.Kooman J.P., Broers N.J.H., Usvyat L., Thijssen S., Van Der Sande F.M., Cornelis T. Out of control: accelerated aging in uremia. Nephrol Dial Transplant. 2013;28:48–54. doi: 10.1093/ndt/gfs451. [DOI] [PubMed] [Google Scholar]
- 3.Kooman J.P., Shiels P.G., Stenvinkel P. Premature aging in chronic kidney disease and chronic obstructive pulmonary disease: similarities and differences. Curr Opin Clin Nutr Metab Care. 2015;18:528–534. doi: 10.1097/MCO.0000000000000218. [DOI] [PubMed] [Google Scholar]
- 4.Girndt M., Seibert E. Premature cardiovascular disease in chronic renal failure (CRF): a model for an advanced ageing process. Exp Gerontol. 2010;45:797–800. doi: 10.1016/j.exger.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Kooman J.P., Kotanko P., Schols A.M.W.J., Shiels P.G., Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol. 2014;10:732–742. doi: 10.1038/nrneph.2014.185. [DOI] [PubMed] [Google Scholar]
- 6.Stenvinkel P., Larsson T.E. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis. 2013;62:339–351. doi: 10.1053/j.ajkd.2012.11.051. [DOI] [PubMed] [Google Scholar]
- 7.Cunha P.G., Boutouyrie P., Nilsson P.M., Laurent S. Early vascular ageing (EVA): definitions and clinical applicability. Curr Hypertens Rev. 2017;13:1–8. doi: 10.2174/1573402113666170413094319. [DOI] [PubMed] [Google Scholar]
- 8.Shanahan C.M. Mechanisms of vascular calcification in CKD - evidence for premature ageing? Nat Rev Nephrol. 2013;9:661–670. doi: 10.1038/nrneph.2013.176. [DOI] [PubMed] [Google Scholar]
- 9.Shroff R., Long D.A., Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol. 2013;24:179–189. doi: 10.1681/ASN.2011121191. [DOI] [PubMed] [Google Scholar]
- 10.Valentijn F.A., Falke L.L., Nguyen T.Q., Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018;12:69–82. doi: 10.1007/s12079-017-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturmlechner I., Durik M., Sieben C.J., Baker D.J., van Deursen J.M. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. 2017;13:77–89. doi: 10.1038/nrneph.2016.183. [DOI] [PubMed] [Google Scholar]
- 12.McGlynn L.M., Stevenson K., Lamb K., Zino S., Brown M., Prina A. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging Cell. 2009;8:45–51. doi: 10.1111/j.1474-9726.2008.00447.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Y., Armstrong J.L., Tchkonia T., Kirkland J.L. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care. 2014;17:324–328. doi: 10.1097/MCO.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 14.Braig M., Lee S., Loddenkemper C., Rudolph C., Peters A.H.F.M., Schlegelberger B. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 15.Chen J.-H., Stoeber K., Kingsbury S., Ozanne S.E., Williams G.H., Hales C.N. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J Biol Chem. 2004;279:49439–49446. doi: 10.1074/jbc.M409153200. [DOI] [PubMed] [Google Scholar]
- 16.Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 17.Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 18.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 19.Xia L., Huang Q., Chen X., Yin Z., Zhang Y., Lun L. Youthful systemic milieu alleviates renal ischemia-reperfusion injury in elderly mice. Kidney Int. 2018;94:268–279. doi: 10.1016/j.kint.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 20.Plotkin M. Young blood for old kidneys? More questions than answers so far. Kidney Int. 2018;94:235–236. doi: 10.1016/j.kint.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 21.Keung L., Perwad F. Vitamin D and kidney disease. Bone Rep. 2018;9:93–100. doi: 10.1016/j.bonr.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y., Tchkonia T., Pirtskhalava T., Gower A.C., Ding H., Giorgadze N. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGowan S.J., Gregg S.Q., Zhao J., Tchkonia T., Li X., Fuhrmann-Stroissnigg H. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00314-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y., Tchkonia T., Fuhrmann-Stroissnigg H., Dai H.M., Ling Y.Y., Stout M.B. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stout M.B., Mezera V., Jensen M.D., White T.A., Ogrodnik M., Ding H. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci. 2015;112:E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Deursen J.M. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrano M., Hannon G.J., Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 28.Demaria M., Ohtani N., Youssef S.A., Rodier F., Toussaint W., Mitchell J.R. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S.-H., Rowe J., Fujii H., Jones R., Schmierer B., Kong B.-W. Upregulation of chicken p15INK4b at senescence and in the developing brain. J Cell Sci. 2006;119:2435–2443. doi: 10.1242/jcs.02989. [DOI] [PubMed] [Google Scholar]
- 30.Storer M., Mas A., Robert-Moreno A., Pecoraro M., Ortells M.C., Di Giacomo V. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z., Peng J., Wang C.-J., Li D., Li T.-T., Hu C.-P. Accelerated senescence of endothelial progenitor cells in hypertension is related to the reduction of calcitonin gene-related peptide. J Hypertens. 2010;28:931–939. doi: 10.1097/HJH.0b013e3283399326. [DOI] [PubMed] [Google Scholar]
- 32.Childs B.G., Baker D.J., Wijshake T., Conover C.A., Campisi J., van Deursen J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York, NY) 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodier F., Coppé J.-P., Patil C.K., Hoeijmakers W.A.M., Muñoz D.P., Raza S.R. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melk A., Schmidt B.M.W., Vongwiwatana A., Rayner D.C., Halloran P.F. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant. 2005;5:1375–1382. doi: 10.1111/j.1600-6143.2005.00846.x. [DOI] [PubMed] [Google Scholar]
- 35.Tchkonia T., Zhu Y., van Deursen J., Campisi J., Kirkland J.L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123 doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrucci L., Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kooman J.P., Dekker M.J., Usvyat L.A., Kotanko P., van der Sande F.M., Schalkwijk C.G. Inflammation and premature aging in advanced chronic kidney disease. Am J Physiol Renal Physiol. 2017;313:F938–F950. doi: 10.1152/ajprenal.00256.2017. [DOI] [PubMed] [Google Scholar]
- 38.Mann J., Gackowska A., Hardy T., Anderson R., Taschuk M., Marques F.D.M. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3 doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Porath I., Weinberg R.A. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins C.J., Sedivy J.M. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell. 2003;2:145–150. doi: 10.1046/j.1474-9728.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 41.Wright W.E., Shay J.W. Historical claims and current interpretations of replicative aging. Nat Biotechnol. 2002;20:682–688. doi: 10.1038/nbt0702-682. [DOI] [PubMed] [Google Scholar]
- 42.Wahl G.M., Carr A.M. The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol. 2001;3:E277–E286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 43.Brown J.P., Wei W., Sedivy J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science (New York, NY) 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 44.Beauséjour C.M., Krtolica A., Galimi F., Narita M., Lowe S.W., Yaswen P. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherr C.J., McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 46.Narita M.M., Nũnez S., Heard E., Narita M.M., Lin A.W., Hearn S.A. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 47.Lowe S.W., Sherr C.J. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 48.Kipling D., Davis T., Ostler E.L., Faragher R.G.A. What can progeroid syndromes tell us about human aging? Science (New York, NY) 2004;305:1426–1431. doi: 10.1126/science.1102587. [DOI] [PubMed] [Google Scholar]
- 49.Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2011;21:107–112. doi: 10.1016/j.gde.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minamino T., Orimo M., Shimizu I., Kunieda T., Yokoyama M., Ito T. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 51.Rodier F., Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jun J.-I., Lau L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jun J.-I., Lau L.F. Cellular senescence controls fibrosis in wound healing. Aging. 2010;2:627–631. doi: 10.18632/aging.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young A.P., Schlisio S., Minamishima Y.A., Zhang Q., Li L., Grisanzio C. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10:361–369. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- 55.Sparmann A., Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 56.Tamm I., Kikuchi T., Cardinale I., Krueger J.G. Cell-adhesion-disrupting action of interleukin 6 in human ductal breast carcinoma cells. Proc Natl Acad Sci U S A. 1994;91:3329–3333. doi: 10.1073/pnas.91.8.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chien Y., Scuoppo C., Wang X., Fang X., Balgley B., Bolden J.E. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwasa H., Han J., Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–144. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 59.Hubackova S., Krejcikova K., Bartek J., Hodny Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging. 2012;4:932–951. doi: 10.18632/aging.100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanchis P., Ho C.Y., Liu Y., Beltran L.E., Ahmad S., Jacob A.P. Arterial ‘inflammaging’ drives vascular calcification in children on dialysis. Kidney Int. 2019;95:958–972. doi: 10.1016/j.kint.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirkland J.L., Tchkonia T., Zhu Y., Niedernhofer L.J., Robbins P.D. The clinical potential of Senolytic drugs. J Am Geriatr Soc. 2017;65:2297–2301. doi: 10.1111/jgs.14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anand David A., Arulmoli R., Parasuraman S. Overviews of biological importance of quercetin: a bioactive flavonoid. Pharmacognosy Reviews. 2016;10:84. doi: 10.4103/0973-7847.194044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lombardo L.J., Lee F.Y., Chen P., Norris D., Barrish J.C., Behnia K. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 64.Xu M., Bradley E.W., Weivoda M.M., Hwang S.M., Pirtskhalava T., Decklever T. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med Sci. 2017;72:780–785. doi: 10.1093/gerona/glw154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gradin P., Hollberg K., Cassady A.I., Lång P., Andersson G., Lang P. Transgenic overexpression of tartrate-resistant acid phosphatase is associated with induction of osteoblast gene expression and increased cortical bone mineral content and density. Cells Tissues Organs. 2012;196:68–81. doi: 10.1159/000330806. [DOI] [PubMed] [Google Scholar]
- 66.Kirkland J.L., Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–28. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mikhailova M.V., Belyaeva N.F., Kozlova N.I., Zolotarev K.V., Mikhailov A.N., Berman A.E. Protective action of fish muscle extracts against cellular senescence induced by oxidative stress. Biomeditsinskaya Khim. 2017;63:351–355. doi: 10.18097/PBMC20176304351. [DOI] [PubMed] [Google Scholar]
- 68.Schafer M.J., White T.A., Iijima K., Haak A.J., Ligresti G., Atkinson E.J. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kovacovicova K., Skolnaja M., Heinmaa M., Mistrik M., Pata P., Pata I. Senolytic cocktail Dasatinib+quercetin (D+Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front Oncol. 2018;8:459. doi: 10.3389/fonc.2018.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Childs B.G., Durik M., Baker D.J., van Deursen J.M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stenvinkel P., Meyer C.J., Block G.A., Chertow G., Shiels P.G. Understanding the role of the cytoprotective transcription factor NRF2 - Lessons from evolution, the animal kingdom and rare progeroid syndromes. Nephrol Dial Transpl. 2019 doi: 10.1093/ndt/gfz120. [In Press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Covic A., Vervloet M., Massy Z.A., Torres P.U., Goldsmith D., Brandenburg V. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol. 2018;6:319–331. doi: 10.1016/S2213-8587(17)30310-8. [DOI] [PubMed] [Google Scholar]
- 73.Chung H.Y., Cesari M., Anton S., Marzetti E., Giovannini S., Seo A.Y. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franceschi C., Capri M., Monti D., Giunta S., Olivieri F., Sevini F. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 75.Vasto S., Candore G., Balistreri C.R., Caruso M., Colonna-Romano G., Grimaldi M.P. Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007;128:83–91. doi: 10.1016/j.mad.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 76.Carrero J.J., Stenvinkel P., Fellström B., Qureshi A.R., Lamb K., Heimbürger O. Telomere attrition is associated with inflammation, low fetuin-a levels and high mortality in prevalent haemodialysis patients. J Intern Med. 2008;263:302–312. doi: 10.1111/j.1365-2796.2007.01890.x. [DOI] [PubMed] [Google Scholar]
- 77.Carrero J.J., Stenvinkel P. Persistent inflammation as a catalyst for other risk factors in chronic kidney disease: a hypothesis proposal. Clin J Am Soc Nephrol. 2009;4(Suppl. 1):S49–S55. doi: 10.2215/CJN.02720409. [DOI] [PubMed] [Google Scholar]
- 78.Yamada S., Tatsumoto N., Tokumoto M., Noguchi H., Ooboshi H., Kitazono T. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcif Tissue Int. 2015;96:347–358. doi: 10.1007/s00223-014-9929-5. [DOI] [PubMed] [Google Scholar]
- 79.Klinkhammer B.M., Kramann R., Mallau M., Makowska A., van Roeyen C.R., Rong S. Mesenchymal stem cells from rats with chronic kidney disease exhibit premature senescence and loss of regenerative potential. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quimby J.M., Maranon D.G., Battaglia C.L.R., McLeland S.M., Brock W.T., Bailey S.M. Feline chronic kidney disease is associated with shortened telomeres and increased cellular senescence. Am J Physiol Renal Physiol. 2013;305:F295–F303. doi: 10.1152/ajprenal.00527.2012. [DOI] [PubMed] [Google Scholar]
- 81.Stenvinkel P., Luttropp K., McGuinness D., Witasp A., Qureshi A.R., Wernerson A. CDKN2A/p16INK4a expression is associated with vascular progeria in chronic kidney disease. Aging. 2017;9:494–507. doi: 10.18632/aging.101173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.John G.B., Cheng C.-Y., Kuro-o M. Role of klotho in aging, phosphate metabolism, and CKD. Am J Kidney Dis. 2011;58:127–134. doi: 10.1053/j.ajkd.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drüeke T.B., Massy Z.A. Circulating klotho levels: clinical relevance and relationship with tissue klotho expression. Kidney Int. 2013;83:13–15. doi: 10.1038/ki.2012.370. [DOI] [PubMed] [Google Scholar]
- 84.Kurosu H., Yamamoto M., Clark J.D., Pastor J.V., Nandi A., Gurnani P. Suppression of aging in mice by the hormone Klotho. Science (New York, NY) 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maltese G., Psefteli P.-M., Rizzo B., Srivastava S., Gnudi L., Mann G.E. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J Cell Mol Med. 2017;21:621–627. doi: 10.1111/jcmm.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao D., Zuo Z., Tian J., Ali Q., Lin Y., Lei H. Activation of SIRT1 attenuates klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension (Dallas, Tex : 1979) 2016;68:1191–1199. doi: 10.1161/HYPERTENSIONAHA.116.07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Makulska I., Szczepańska M., Drożdż D., Polak-Jonkisz D., Zwolińska D. Advances in Clinical and Experimental Medicine. Official Organ Wroclaw Medical University; 2018. The importance of fetuin-A in vascular calcification in children with chronic kidney disease. [DOI] [PubMed] [Google Scholar]
- 88.Al-Biltagi M., ElHafez M.A.A., El Amrousy D.M., El-Gamasy M., El-Serogy H. Evaluation of the coronary circulation and calcification in children on regular hemodialysis. Pediatr Nephrol (Berlin, Germany) 2017;32:1941–1951. doi: 10.1007/s00467-017-3678-4. [DOI] [PubMed] [Google Scholar]
- 89.Shroff R.C., McNair R., Figg N., Skepper J.N., Schurgers L., Gupta A. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 90.Yamada S., Taniguchi M., Tokumoto M., Toyonaga J., Fujisaki K., Suehiro T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J Bone Mineral Res. 2012;27:474–485. doi: 10.1002/jbmr.539. [DOI] [PubMed] [Google Scholar]
- 91.McCabe K.M., Booth S.L., Fu X., Shobeiri N., Pang J.J., Adams M.A. Dietary vitamin K and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease. Kidney Int. 2013;83:835–844. doi: 10.1038/ki.2012.477. [DOI] [PubMed] [Google Scholar]
- 92.Shanahan C.M., Crouthamel M.H., Kapustin A., Giachelli C.M. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711. doi: 10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaziri N.D. Oxidative stress in uremia: nature, mechanisms, and potential consequences. Semin Nephrol. 2004;24:469–473. doi: 10.1016/j.semnephrol.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 94.Goettsch C., Weis D., Kramann R., Hellmann B., Floege J., Schurgers L. Magnesium but not nicotinamide prevents vascular calcification in experimental uraemia. Nephrol Dial Transplant. 2019:1–9. doi: 10.1093/ndt/gfy410. [DOI] [PubMed] [Google Scholar]
- 95.Holt S.G., Smith E.R. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol Dial Transplant. 2016;31:1583–1587. doi: 10.1093/ndt/gfw048. [DOI] [PubMed] [Google Scholar]
- 96.Eleftheriadou I., Grigoropoulou P., Kokkinos A., Mourouzis I., Perrea D., Katsilambros N. Association of plasma fetuin-a levels with peripheral arterial disease and lower extremity arterial calcification in subjects with type 2 diabetes mellitus. J Diabetes Complications. 2017;31:599–604. doi: 10.1016/j.jdiacomp.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 97.Viegas C.S.B., Santos L., Macedo A.L., Matos A.A., Silva A.P., Neves P.L. Chronic kidney disease circulating Calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (Gla-rich protein) Arterioscler Thromb Vasc Biol. 2018;38:575–587. doi: 10.1161/ATVBAHA.117.310578. [DOI] [PubMed] [Google Scholar]
- 98.Cai T., Sun D., Duan Y., Wen P., Dai C., Yang J. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345:206–217. doi: 10.1016/j.yexcr.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 99.Lin M.-E., Chen T., Leaf E.M., Speer M.Y., Giachelli C.M. Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. Am J Pathol. 2015;185:1958–1969. doi: 10.1016/j.ajpath.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun Y., Byon C.H., Yuan K., Chen J., Mao X., Heath J.M. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Voelkl J., Lang F., Eckardt K.-U., Amann K., Kuro-O M., Pasch A. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. 2019;76:2077–2091. doi: 10.1007/s00018-019-03054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Voelkl J., Tuffaha R., Luong T.T.D.D., Zickler D., Masyout J., Feger M. Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-kappaB. J Am Soc Nephrol. 2018;29:1636–1648. doi: 10.1681/ASN.2017050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu Z., Ji G., Shen J., Wang X., Zhou J., Li L. SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation. Biochem Biophys Res Commun. 2012;422:285–290. doi: 10.1016/j.bbrc.2012.04.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Voelkl J., Luong T.T.D.T., Tuffaha R., Musculus K., Auer T., Lian X. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J Clin Investig. 2018;128:3024–3040. doi: 10.1172/JCI96477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giachelli C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009;75:890–897. doi: 10.1038/ki.2008.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Opdebeeck B., Maudsley S., Azmi A., De Maré A., De Leger W., Meijers B. Indoxyl sulfate and p-Cresyl sulfate promote vascular calcification and associate with glucose intolerance. J Am Soc Nephrol. 2019 doi: 10.1681/ASN.2018060609. ASN.2018060609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 108.Iyemere V.P., Proudfoot D., Weissberg P.L., Shanahan C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. 2006;260:192–210. doi: 10.1111/j.1365-2796.2006.01692.x. [DOI] [PubMed] [Google Scholar]
- 109.Ragnauth C.D., Warren D.T., Liu Y., McNair R., Tajsic T., Figg N. Prelamin a acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200–2210. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y., Drozdov I., Shroff R., Beltran L.E., Shanahan C.M. Prelamin a accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013;112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543. [DOI] [PubMed] [Google Scholar]
- 111.Song Y., Shen H., Schenten D., Shan P., Lee P.J., Goldstein D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32:103–109. doi: 10.1161/ATVBAHA.111.236349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McClintock D., Gordon L.B., Djabali K. Hutchinson-Gilford progeria mutant Lamin a primarily targets human vascular cells as detected by an anti-Lamin a G608G antibody. Proc Natl Acad Sci U S A. 2006;103:2154–2159. doi: 10.1073/pnas.0511133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muteliefu G., Shimizu H., Enomoto A., Nishijima F., Takahashi M., Niwa T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin a through oxidative stress. Am J Physiol Cell Physiol. 2012;303:C126–C134. doi: 10.1152/ajpcell.00329.2011. [DOI] [PubMed] [Google Scholar]
- 114.Stenvinkel P., Painer J., Kuro-O M., Lanaspa M., Arnold W., Ruf T. Novel treatment strategies for chronic kidney disease: insights from the animal kingdom. Nat Rev Nephrol. 2018;14:265–284. doi: 10.1038/nrneph.2017.169. [DOI] [PubMed] [Google Scholar]
- 115.Jazani NH, Savoj J, Lustgarten M, Lau WL, Vaziri ND. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic Kidney Disease. Diseases (Basel, Switzerland) 2019;7. doi: 10.3390/diseases7010021. [DOI] [PMC free article] [PubMed]
- 116.Krimpenfort P., Quon K.C., Mooi W.J., Loonstra A., Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 117.Wang C., Maddick M., Miwa S., Jurk D., Czapiewski R., Saretzki G. Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging. 2010;2:555–566. doi: 10.18632/aging.100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tchkonia T., Kirkland J.L. Therapeutic approaches to aging-reply. JAMA. 2019;321:901–902. doi: 10.1001/jama.2018.20554. [DOI] [PubMed] [Google Scholar]
- 119.Roos C.M., Zhang B., Palmer A.K., Ogrodnik M.B., Pirtskhalava T., Thalji N.M. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973–977. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Inman C.L., Hubbard G.B., Kirkland J.L., Cubro H., Xu M., Robbins P.D. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24:1246–1256. doi: 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Justice J.N., Nambiar A.M., Tchkonia T., LeBrasseur N.K., Pascual R., Hashmi S.K. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–563. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ridker P.M., MacFadyen J.G., Glynn R.J., Koenig W., Libby P., Everett B.M. Inhibition of interleukin-1β by Canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol. 2018;71:2405–2414. doi: 10.1016/j.jacc.2018.03.490. [DOI] [PubMed] [Google Scholar]
- 123.Tasdemir N., Banito A., Roe J.-S., Alonso-Curbelo D., Camiolo M., Tschaharganeh D.F. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6:612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]