

Abstract
Background
In animals, H3K4me2 and H3K4me3 are enriched at the transcription start site (TSS) and function as epigenetic marks that regulate gene transcription, but their functions in plants have not been fully characterized.
Results
We used chromatin immunoprecipitation sequencing to analyze the rice genome-wide changes to H3K4me1/H3K4me2/H3K4me3 following the loss of an H3K4-specific methyltransferase, SDG701. The knockdown of SDG701 resulted in a global decrease in H3K4me2/H3K4me3 levels throughout the rice genome. An RNA-sequencing analysis revealed that many genes related to diverse developmental processes were misregulated in the SDG701 knockdown mutant. In rice, H3K4me3 and H3K36me3 are positively correlated with gene transcription; however, surprisingly, the H3K4me2 level was negatively associated with gene transcription levels. Furthermore, the H3K4me3 level at the TSS region decreased significantly in the genes that exhibited down-regulated expression in the SDG701 knockdown mutant. In contrast, the genes with up-regulated expression in the mutant were associated with a considerable decrease in H3K4me2 levels over the gene body region.
Conclusion
A comparison of the genome-wide distributions of H3K4me2 in eukaryotes indicated that the H3K4me2 level is not correlated with the gene transcription level in yeast, but is positively and negatively correlated with gene expression in animals and plants, respectively. Our results uncovered H3K4me2 as a novel repressive mark in plants.
Electronic supplementary material
The online version of this article (10.1186/s13072-019-0285-6) contains supplementary material, which is available to authorized users.
Keywords: H3K4me2, ChIP-seq, RNA-seq, Gene expression, Epigenetic mark, Rice
Background
In eukaryotes, histone lysine (K) methylation is important for regulating gene expression. The K-residues at the N-terminus of histone H3 or H4 can be mono-, di-, or tri-methylated, and the methylation of a specific K-residue as well as the number of methyl groups added can mark different chromatin states [1–3]. The methylations of H3K4 and H3K36 are generally associated with gene transcriptional activation, whereas the methylations of H3K9 and H3K27 are associated with the repression of gene expression [4–7].
As an evolutionarily conserved epigenetic mark, H3K4 methylations are broadly distributed in euchromatin and are associated with actively transcribed genes in animals and plants [2, 8–10]. Studies involving chromatin immunoprecipitation (ChIP)-chip and ChIP-sequencing (ChIP-seq) technologies have provided insights into the distributions and functions of H3K4me1/H3K4me2/H3K4me3 in various species. In yeast (Saccharomyces cerevisiae), H3K4me1 and H3K4me2 appear at about 600 base pairs (bp) downstream of the transcription start site (TSS), whereas H3K4me3 is primarily enriched at 200 bp downstream of the nucleosome-free region [11]. In animals, H3K4me1/H3K4me2/H3K4me3 is enriched on both sides of the TSS, with a similar distribution pattern across the genome [12–14]. Additionally, H3K4me1 is globally associated with enhancers [15] and H3K4me2/H3K4me3 usually positively regulates gene expression in animals [8–10]. Similarly, in Arabidopsis and rice (Oryza sativa), H3K4me3 is enriched close to the TSS region and is positively correlated with gene expression levels [4, 16–18]. However, there has been some inconsistency in the reported H3K4me2 distribution pattern, with some studies indicating that the distribution pattern is similar to that of H3K4me3 [4, 16], whereas other studies have suggested that H3K4me2 is broadly distributed across the gene body region, in contrast to H3K4me3 [17, 18]. Moreover, the relationship between H3K4me2 and gene expression levels in plants remains unclear [4, 16–18].
The methylation of H3K4 is catalyzed by SET domain-containing methyltransferases. In yeast, Set1 is the sole H3K4-specific methyltransferase [19, 20] and functions as part of the COMPASS (complex proteins associated with Set1) complex together with the other subunits, Swd1, Swd2, Swd3, Bre2, Sdc1, Spp1, and Shg1 [21, 22]. Drosophila melanogaster contains three Set1 homologs [dSet1, Trithorax (Trx), and Trx-related protein], all of which can associate with the other subunit proteins to form COMPASS-like complexes, which are responsible for the methylation of H3K4 [23]. Mammals have six Set1 homologs [SET1A, SET1B, MLL1, MLL2, MLL3, and MLL4] [8], which also usually exist in COMPASS-like complexes for methylating H3K4 [24–26]. In Arabidopsis, ATX1–5 function as H3K4 methyltransferases. Additionally, ATX1 is involved in root, leaf, and floral organ development [27–30]. Previous studies revealed that ATX1 and ATX2 are functionally diverse, with ATX2 exhibiting H3K4me2 rather than H3K4me3 activity [31, 32]. Moreover, ATX3–5 has redundant functions affecting vegetative and reproductive developmental processes and the genome-wide distribution of H3K4me2 and H3K4me3 [33]. In rice, SDG723/OsTrx1 exhibits histone H3 methyltransferase activity in vitro and helps to regulate flowering time [34]. The OsTrx1 protein is specifically recruited to the target gene Ehd1 to promote rice flowering by interacting with a transcription factor, SDG723/OsTrx1/OsSET33 Interaction Protein 1 (SIP1) [35]. SDG701 is likely a major H3K4-specific methyltransferase in rice, thereby contributing to the regulation of multiple processes during rice plant growth and development, such as flowering, gametophyte development, and grain production [36].
In this study, we functionally characterized the H3K4 methylation marks (H3K4me1, H3K4me2, and H3K4me3) in rice by analyzing the SDG701 knockdown mutant in RNA-seq and ChIP-seq assays, for the SDG701 knockout mutant is lethal [36]. The SDG701 deficiency decreased the global H3K4me2/H3K4me3 levels in planta and resulted in the misregulation of many genes involved in multiple developmental processes. Interestingly, unlike the H3K4me1/H3K4me3 and H3K36me3 marks, which were positively correlated with gene transcription, H3K4me2 was negatively correlated with gene expression. Moreover, H3K4me2 levels were considerably decreased in the genes that exhibited up-regulated expression in the SDG701 knockdown mutant, further supporting a novel function of H3K4me2 to repress gene expression in rice.
Results
SDG701 is required for the proper expression of many genes involved in diverse developmental processes
We previously reported SDG701 functions as a major H3K4-specific methyltransferase that is important for rice plant development [36]. To further clarify the function of SDG701, we conducted an RNA-sequencing (RNA-seq) analysis of the SDG701 knockdown mutant 35Sp::SDG701iR1 (hereafter referred to as 701Ri-1-5) in the O. sativa spp. japonica cv. Nipponbare (NIP) background. After sequencing two biological replicates of RNA-seq libraries, more than 20 million mapped reads were obtained for a subsequent evaluation of genes that were differentially expressed between the 701Ri-1-5 mutant and the wild-type NIP (Additional file 1: Table S1). As expected, the transcription level of SDG701 (LOC_Os08g08210) decreased to half in the 701Ri-1-5 mutant (FPKM701Ri-1-5/FPKMNIP = 0.50, adjusted p value = 5.72e−4), while that of LOC_Os08g08200, a gene adjacent to SDG701, was unaffected (Fig. 1a). We previously demonstrated that SDG701 influences the expression of the flowering genes Hd3a, Ehd1, and RFT1 [36]. Thus, the RNA-seq data were analyzed to determine the expression levels of these genes. Consistent with our previous results [36], the Hd3a, Ehd1, and RFT1 expression levels were significantly lower in the 701Ri-1-5 mutant than in the wild-type NIP (Additional file 1: Fig. S1).
Fig. 1.

Transcriptome analysis of the SDG701 knockdown mutant. a IGV screen shots presenting the normalized read densities of LOC_Os08g08210 (SDG701) and LOC_Os08g08200 (gene neighboring SDG701) based on an RNA-sequencing analysis of two biological replicates of the wild-type NIP and 701Ri-1-5 mutant plants. b MA plot presenting gene transcription changes following the knockdown of SDG701. Green and red points represent the genes exhibiting down- and up-regulated expression (> 1.5-fold change), respectively, whereas the gray points represent the genes with unchanged expression, in the 701Ri-1-5 mutant compared with the wild-type NIP. The adjusted p value (p.adj) was calculated with the Benjamini–Hochberg correction. c Functional enrichment analysis of genes with significantly down- or up-regulated expression levels. The size of each point represents the number of genes, and p values are indicated with various colors
An analysis of the RNA-seq data revealed 1572 and 1508 genes whose transcription levels were up- and down-regulated by more than 1.5-fold in the 701Ri-1-5 mutant, respectively (Fig. 1b, Additional file 2: Table S2). A gene ontology (GO) enrichment analysis indicated that the misregulated genes in the 701Ri-1-5 mutant were enriched with multiple biological process GO terms, including metabolic process, biosynthetic process, response to biotic or abiotic stimuli, organ development, and signal transduction (Fig. 1c, Additional file 3: Table S3). Additionally, some of the misregulated genes in the 701Ri-1-5 mutant were related to flower development, the starch biosynthetic process, and pollen development (Additional file 3: Table S3), which is consistent with the fact that SDG701 affects gametophyte development and grain production [36]. These results imply that SDG701 is required for the proper expression of many genes and is crucial for multiple developmental processes in rice.
H3K4me2 enrichment is negatively correlated with transcription levels in rice
In rice, SDG701 is an H3K4-methyltransferase that is especially important for the tri-methylation of H3K4, and the resulting H3K4me3 serves as an active epigenetic mark for gene transcription in eukaryotes. We were interested in why many genes exhibited increased expression in the SDG701 knockdown mutant. We first performed a ChIP-seq analysis to assess whether the knockdown of SDG701 affected H3K4 methylations. Approximately 4–20 million unique reads were obtained for H3K4me1, H3K4me2, H3K4me3, and H3K36me3 in the 701Ri-1-5 mutant and wild-type NIP (Additional file 1: Table S4 and Fig. 2a). In NIP, 284 (0.5% of 55,986 annotated rice genes), 10,681 (19.1%), and 25,259 (45.1%) genes enriched with H3K4me1, H3K4me2, and H3K4me3, respectively, were identified (Additional file 1: Fig. S2A). Another 15,728 genes enriched with H3K36me3 were also detected. The peaks for enriched H3K4me2, H3K4me3, and H3K36me3 overlapped previously published ChIP-seq data (Additional file 1: Fig. S2B–D) [37–39]. As shown in Fig. 2, H3K4me1 was distributed over the gene body region at a much lower level than H3K4me2 and H3K4me3, H3K4me2 covered the entire gene body region with a weak peak close to the TSS, whereas H3K4me3 showed a sharp peak at the TSS in the wild-type NIP. As a control, H3K36me3 was mainly distributed at the 5′ end of the gene body, which was consistent with our published data [40].
Fig. 2.
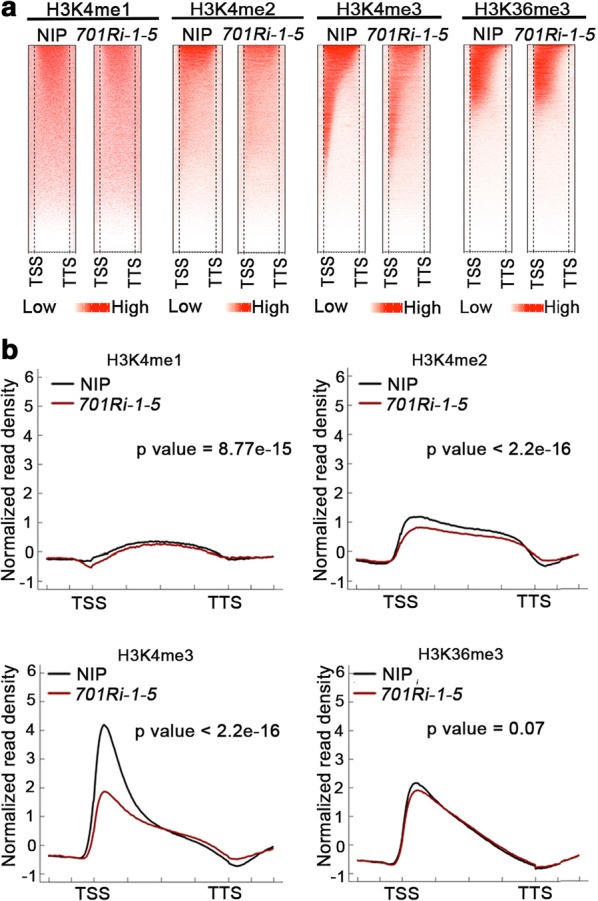
Knockdown of SDG701 results in a global decrease in H3K4me2/H3K4me3 levels in rice. a Heatmaps presenting H3K4me1/H3K4me2/H3K4me3 and H3K36me3 levels surrounding genes in the 701Ri-1-5 mutant and wild-type NIP. Each line in a heatmap represents a gene. The plots were generated from 1 kb upstream of the transcription start site (TSS) to 1 kb downstream of the transcription termination site (TTS). b Average density plots presenting the genome-wide (on 55,986 annotated rice genes) distributions of H3K4me1/H3K4me2/H3K4me3 and H3K36me3 in the 701Ri-1-5 (red) mutant and wild-type NIP (black). The p values were calculated by the Kolmogorov–Smirnov test
In the SDG701 knockdown mutant, the H3K4me1 level decreased slightly, especially close to the TSS region, while the H3K4me2 and H3K4me3 levels decreased obviously (Fig. 2b). The ChIP-seq data further confirmed that SDG701 is responsible for methylating H3K4, especially the di- and tri-methylation of H3K4 in rice (Fig. 2). We observed that the H3K36me3 level decreased very slightly close to the TSS in the 701Ri-1-5 mutant (Fig. 2b). We wondered whether this decrease was directly due to the knockdown of SDG701. Because the H3K4me1-enriched genes accounted for 0.5% of all annotated rice genes, we only selected H3K4me2-, H3K4me3-, and H3K36me3-enriched genes for further analyses. These genes were grouped into the following seven clusters based on their surrounding epigenetic marks (Additional file 1: Fig. S3A): (a) enriched with only H3K4me2, (b) enriched with only H3K4me3, (c) enriched with only H3K36me3, (d) enriched with H3K4me2 and H3K4me3, (e) enriched with H3K4me2 and H3K36me3, (f) enriched with H3K4me3 and H3K36me3, and (g) enriched with H3K4me2, H3K4me3, and H3K36me3. Heatmaps and average density plots were generated for these genes in NIP and the 701Ri-1-5 mutant (Additional file 1: Fig. S3B–E). The H3K4me2 and H3K4me3 signals were significantly decreased for the genes in clusters a and b, respectively; however, the enrichment of H3K36me3 in the genes of cluster c was unaffected, indicating that SDG701 did not directly contribute to the decrease in the H3K36me3 level in the 701Ri-1-5 mutant. Regarding the genes in cluster f (enriched with H3K4me3/H3K36me3) and cluster g (enriched with H3K4me2/H3K4me3/H3K36me3), the H3K36me3 signal decreased slightly close to the TSS, implying that the decrease in the H3K36me3 level in the 701Ri-1-5 mutant was probably related to a decrease in the H3K4me3 level. These results suggested that the knockdown of SDG701 in rice leads to a genome-wide decrease in H3K4 methylations, especially the di- and tri-methylations.
We subsequently explored the correlation between H3K4 methylations and transcript levels by analyzing the RNA-seq and ChIP-seq data for the wild-type NIP. About 90% of the H3K4me3/H3K36me3-enriched genes (cluster f) were expressed (FPKM > 1; Fig. 3a). Unexpectedly, over 80% of the H3K4me2-enriched genes (cluster a) were not expressed (0 < FPKM < 1; Fig. 3a). We also observed that if gene groups were marked with H3K4me2, the fraction of expressed genes decreased. For example, approximately 60% of the H3K4me3-enriched genes were expressed, whereas only about 30% of the H3K4me2/H3K4me3-enriched genes were expressed (Fig. 3a). Similarly, more than 60% of the H3K36me3-enriched genes were expressed, while less than 30% of the H3K4me2/H3K36me3-enriched genes were expressed (Fig. 3a). Next, we analyzed the expression levels of the seven gene clusters (Fig. 3b). The mean expression level of the H3K4me3/H3K36me3-enriched genes was the highest, and that of the H3K4me2-enriched genes was the lowest (Fig. 3b, Additional file 1: Table S5). Moreover, a comparison of the seven clusters revealed that the average expression levels were obviously lower for the genes marked with H3K4me2 than for the genes that lacked H3K4me2 (Fig. 3b, Additional file 1: Table S5), suggesting that H3K4me2 is probably related to gene repression.
Fig. 3.

H3K4me2 level is negatively correlated with the gene transcription level in rice. a Genes marked by H3K4me2 only, H3K4me3 only, H3K36me3 only, H3K4me2 and H3K4me3, H3K4me2 and H3K36me3, or H3K4me2, H3K4me3, and H3K36me3 exhibited varying transcription levels in the wild-type NIP. Genes categorized by transcription levels (FPKM values) are distinguished by different colors. b Box plots presenting the transcription levels of the genes with a differentially marked H3 in (a) (p < 0.001 according to the Wilcoxon test). c Average density plots presenting the H3K4me1/H3K4me2/H3K4me3 and H3K36me3 distribution patterns along the differentially expressed genes in the wild-type NIP. The plots were generated from 1 kb upstream of the TSS to 1 kb downstream of the TTS. The annotated rice genes were grouped based on transcription levels. Non: not expressed, 0 < FPKM < 1 (black); Low: low expression level, 1 < FPKM < 2 (green); Middle: moderate expression level, 2 < FPKM < 10 (gray); and High: high expression level, FPKM > 10 (red). d Scatter plots presenting the relationship between H3K4me1/H3K4me2/H3K4me3 and H3K36me3 levels and gene transcription levels in the wild-type NIP. The H3K4me1/H3K4me2/H3K4me3 and H3K36me3 (from the TSS to the TTS) levels were calculated as follows: ChIP-seq normalized read density−input normalized read density for expressed genes (FPKM > 1). Spearman’s rank correlation coefficient indicates the correlation between the methylation level and the gene expression level. The p value was determined based on Spearman’s rank correlation test
To further validate our results, we classified all annotated rice genes into the following four groups based on the FPKM values: Non: not expressed, 0 < FPKM < 1; Low: low expression level, 1 < FPKM < 2; Middle: moderate expression level, 2 < FPKM < 10; High: high expression level, FPKM > 10. Additionally, average density plots for H3K4me1/H3K4me2/H3K4me3 and H3K36me3 were generated for these four gene groups (Fig. 3c). A positive correlation was observed between the transcript levels and the H3K4me1, H3K4me3, and H3K36me3 levels (Fig. 3c, d). In contrast, the H3K4me2 level was negatively correlated with the transcript level, just similar as the relationship between H3K27me3 and gene transcription (Additional file 1: Fig. S4). Specifically, H3K4me3 (ρ = 0.85) and H3K36me3 (ρ = 0.96) levels were positively correlated with gene transcription levels. A weaker positive correlation was detected between H3K4me1 levels (ρ = 0.73) and gene transcription levels. Consistent with the results for H3K4me2 in NIP (ρ = -0.88), we detected a negative correlation between gene transcription levels and H3K4me2 enrichment in another rice subspecies, O. sativa ssp. japonica cv. Dongjin (ρ = − 0.89; Additional file 1: Fig. S5). Our results proved that unlike H3K4me3 and H3K36me3 modifications, H3K4me2 is negatively correlated with transcript level in rice.
H3K4me2 enrichment significantly decreases within the chromatin of the up-regulated genes in SDG701 knockdown mutant
To further test the correlation between H3K4 methylations and gene transcription, we analyzed the H3K4me1/H3K4me2/H3K4me3 and H3K36me3 signals for the misregulated genes in the 701Ri-1-5 mutant. Additionally, average density plots were generated to visualize the H3K4me1/H3K4me2/H3K4me3 and H3K36me3 distribution patterns on genes that exhibited significantly up-regulated (1572 genes), down-regulated (1508 genes), or unchanged expression levels in NIP and the 701Ri-1-5 mutant (Figs. 1b and 4). There were no obvious differences between NIP and the 701Ri-1-5 mutant regarding the H3K4me1 and H3K36me3 signals on the up- and down-regulated genes (Fig. 4a, d), indicating that the gene transcription changes in the 701Ri-1-5 mutant were not due to changes in H3K4me1 or H3K36me3 enrichment. The H3K4me2 level over the whole gene body regions of the 1572 up-regulated genes was significantly lower in the 701Ri-1-5 mutant than in NIP, while the corresponding level of the down-regulated genes was only slightly lower close to the TSS in the 701Ri-1-5 mutant than in NIP (Fig. 4b). We randomly selected eight genes, whose H3K4me2 levels significantly decreased and transcript levels increased, respectively, in 701Ri-1-5 compared to in NIP for independent validation. The RT-qPCR and ChIP-qPCR results further supported that the decreased H3K4me2 correlates with gene activation (Additional file 1: Fig. S6). Regarding the H3K4me3 level, a significant decrease was observed for both up- and down-regulated genes (Fig. 4c). Moreover, the H3K4me3 signal for the down-regulated genes was mainly a sharp peak close to the TSS, while that for the up-regulated genes was a smaller peak around the TSS and extended to the entire gene body region. Our data indicated that the genes exhibiting up-regulated expression in the 701Ri-1-5 mutant had a significantly decreased H3K4me2 level, providing further evidence that H3K4me2 represses gene transcription in rice.
Fig. 4.

Genes exhibiting up-regulated expression in the 701Ri-1-5 mutant have decreased H3K4me2 levels. a–d Average density plots presenting the H3K4me1/H3K4me2/H3K4me3 and H3K36me3 distribution patterns along genes exhibiting significantly up-regulated expression (up: 1572 genes with expression levels > 1.5-fold higher in the 701Ri-1-5 mutant than in the wild-type NIP), down-regulated expression (down: 1508 genes with expression levels > 1.5-fold lower in the 701Ri-1-5 mutant than in the wild-type NIP), and no significant changes (Invariant) in the NIP (solid line) and 701Ri-1-5 mutant (dashed line) samples. The plots were generated from 1 kb upstream of the TSS to 1 kb downstream of the TTS
Correlation between H3K4me2/H3K4me3 deposition and gene transcription
To clarify the relationship between H3K4me2/H3K4me3 enrichment and gene transcription levels regulated by SDG701, we screened for genes with H3K4me2 or H3K4me3 levels that were significantly lower in the 701Ri-1-5 mutant (> 1.5-fold) than in NIP. We detected 1885 peaks and 14,647 peaks, corresponding to 1968 of 10,681 H3K4me2-enriched (19%) and 14,473 of 25,259 H3K4me3-enriched (57.3%) genes, respectively, with significantly lower H3K4me2 or H3K4me3 levels in the 701Ri-1-5 mutant than in NIP (Additional file 1: Fig. S7 and Additional file 4: Table S6). We subsequently calculated the read density change for each peak and clustered the peaks into the following five sets with a K-means clustering algorithm (Fig. 5a): Peak Set 1 containing 1353 peaks with a slightly increased H3K4me2 level and a significantly decreased H3K4me3 level, Peak Set 2 containing 3653 peaks with an unchanged H3K4me2 level and a significantly decreased H3K4me3 level, Peak Set 3 containing 6146 peaks with a slightly decreased H3K4me2 level and a significantly decreased H3K4me3 level, Peak Set 4 containing 4737 peaks with significantly decreased H3K4me2 and H3K4me3 levels, and Peak Set 5 containing 643 peaks with a significantly decreased H3K4me2 level and an unchanged H3K4me3 level. In the wild-type NIP, the H3K4me2 levels were higher for the genes in Peak Sets 4 and 5 than for the genes in Peak Sets 1–3, and the H3K4me3 levels were higher for the genes in Peak Sets 1–4 than for the genes in Peak Set 5 (Fig. 5b). Next, we conducted an enrichment analysis of the genes in Peak Sets 1–5 and the genes that exhibited up- or down-regulated expression in the 701Ri-1-5 mutant. The corresponding genes of Peak Set 1 and Peak Set 2 were more significantly enriched in the genes with down-regulated expression than those with up-regulated expression in 701Ri-1-5 mutant (Fig. 5c). In these two peak sets, only H3K4me3 levels were significantly decreased in the 701Ri-1-5 mutant (Fig. 5a), suggesting that the decrease in the H3K4me3 level is primarily responsible for down-regulating gene expression in rice. Meanwhile, the genes of Peak Set 5 were more significantly enriched in the genes with up-regulated expression in 701Ri-1-5 mutant (Fig. 5c), indicating that the decrease in the H3K4me2 level is mainly related to gene activation in rice. Thus, H3K4me3 and H3K4me2 are apparently positively and negatively associated with rice gene transcription, respectively. Regarding the genes of Peak Sets 3 and 4, we observed that for the genes with up-regulated expression, H3K4me2 and H3K4me3 were mainly distributed over the whole gene body, whereas for the genes with down-regulated expression, H3K4me2 and H3K4me3 produced a peak close to the TSS in NIP, especially the genes of Peak Set 4 (Additional file 1: Fig. S8). These results suggested that the relative positions of H3K4me2 and H3K4me3 along genes probably also contribute to the regulation of gene expression.
Fig. 5.

Decreases in H3K4me2 and H3K4me3 levels result in up- and down-regulated gene expression, respectively. a Heatmap for five sets of peaks based on the difference in the H3K4me2/H3K4me3 levels between the 701Ri-1-5 mutant and the wild-type NIP. Significant changes to H3K4me2 and H3K4me3 peaks were based on an FDR value < 1e−4 and > 1.5-fold change (701Ri-1-5/NIP ≤ 1.5). Green, black, and red represent the peaks with decreased, unchanged, and increased H3K4me2 and H3K4me3 levels in the 701Ri-1-5 mutant relative to the levels in the wild-type NIP, respectively. b Visualization of the average normalized read intensity profile close to the H3K4me2/H3K4me3 peak center (marked as 0) for each peak set in wild-type NIP. c Enrichment analysis of the target genes of Peak Set 1–5 among the genes exhibiting up- or down-regulated expression in the 701Ri-1-5 mutant. ***p < 1e−4 and *p < 0.05 according to Fishers’ exact test
Evolutionary diversity of H3K4me2 distributions and functions in various species
The negative correlation between H3K4me2 enrichment and gene transcription levels compelled us to analyze the H3K4me2 distribution in various species. Heatmaps (Fig. 6a) and average density plots (Fig. 6b) of H3K4me2 were generated for genes from 3 kb upstream of the TSS to 3 kb downstream of the transcription termination site (TTS). We determined that H3K4me2 was mainly enriched from 500 bp upstream of the TSS to 500 bp downstream of the TTS in S. cerevisiae, but was not obviously correlated with gene transcription (Fig. 6a–c), which is consistent with published data [41]. In Caenorhabditis elegans, H3K4me2 was primarily enriched around the TSS and was positively correlated with gene expression (Fig. 6a–c). In D. melanogaster, the H3K4me2 levels on each side of the TSS were also positively correlated with gene expression (Fig. 6a–c). In a human colon cancer cell line (HCT116 cells) and mouse embryonic stem cells, H3K4me2 was sharply enriched around the TSS and was positively correlated with gene expression (Fig. 6a–c). Similar to rice, the H3K4me2 in Arabidopsis was mainly enriched in gene body regions and was negatively related to gene expression (Fig. 6a–c). Unlike H3K4me2, H3K4me3 exhibited a conserved positive correlation with gene transcription in various species (Additional file 1: Fig. S9). Thus, the H3K4me2 distribution patterns are diverse among various species. Moreover, in contrast to in animals, H3K4me2 exhibits a unique distribution pattern and functions as a repressive epigenetic mark in plants.
Fig. 6.

H3K4me2 distribution patterns and functions vary in yeast, animals, and plants. a Heatmaps presenting the genome-wide H3K4me2 distributions in various species. The plots were generated from 3 kb upstream of the TSS to 3 kb downstream of the TTS. From left to right: C. elegans, D. melanogaster, mouse embryonic stem cells (mESCs), human colon cancer cell line (HCT116 cells), S. cerevisiae, Arabidopsis, and rice. b Average density plots presenting the H3K4me2 distributions along differentially expressed genes in various species. The plots were generated from 3 kb upstream of the TSS to 3 kb downstream of the TTS. (Non: 0 < FPKM < 1; Low: 1 < FPKM < 2; Middle: 2 < FPKM < 10; and High: FPKM > 10). c Scatter plots presenting the relationship between the H3K4me2 level and the gene transcription level. The H3K4me2 levels were calculated as follows: ChIP-seq normalized read density—input normalized read density for expressed genes (FPKM > 1). Spearman’s rank correlation coefficient indicates the correlation between the methylation level and the gene expression level. The p value was determined based on Spearman’s rank correlation test
Discussion
In this study, we observed that the genome-wide levels of H3K4 modifications, especially H3K4me2 and H3K4me3, were significantly lower in the SDG701 knockdown mutant than in the wild-type NIP. Our RNA-seq analysis revealed that the knockdown of SDG701 results in more genes with up-regulated expression than genes with down-regulated expression, which is inconsistent with the fact that SDG701 functions as an H3K4-specific methyltransferase that catalyzes the epigenetic modifications that activate gene expression. Surprisingly, our combined analysis of ChIP-seq and RNA-seq data indicated that in rice, H3K4me2 enrichment is negatively correlated with gene transcript level, which differs from the positive association between H3K4me3 and H3K36me3 levels and gene expression levels. In the SDG701 knockdown mutant, the down-regulated genes were associated with a decreased H3K4me3 level close to the TSS region, while the up-regulated genes were associated with a decreased H3K4me2 level over the gene body region. A further analysis of published H3K4me2/me3 ChIP-seq and RNA-seq raw data for various species indicated that unlike in animals, H3K4me2 in rice and Arabidopsis specifically distributes over the gene body region and functions as a repressive epigenetic mark.
Although H3K4me2 serves as a repressive mark on chromatin in plants, it was unclear whether it is more common in euchromatin or heterochromatin. We observed that more than 90% of the genes marked with H3K4me2 are not transposable element (non-TE) genes (Additional file 1: Fig. S10A), suggesting that H3K4me2 occurs mainly on euchromatin in plants. We also analyzed the relationship between H3K4me2 and other repressive epigenetic marks in rice, including DNA methylations (CG, CHG, and CHH), H3K9me1/H3K9me2/H3K9me3, and H3K27me3 [42, 43]. We observed that the genes marked with H3K4me2 overlapped less with those marked with DNA methylations or with H3K9me1/H3K9me2/H3K9me3, but more with the genes marked with H3K27me3 (Additional file 1: Fig. S10B–F). Thus, H3K4me2 probably functions together with H3K27me3 to inhibit the expression of euchromatin genes in plants. Regarding the mechanism underlying the gene repression function of H3K4me2, one possibility is that H3K4me2 helps to recruit a repressive reader protein. Consistent with this possibility, the rice DNA-binding domain (CHD) protein CHR729 reportedly can bind to both H3K4me2 and H3K27me3 in vitro through its PHD finger domain [44]. Homologs of CHR729 (CHD3 subfamily) in D. melanogaster and mammals are the key subunits of NuRD complexes, which exhibit nucleosome remodeling and deacetylase activities and repress transcription [45, 46]. Future studies will need to reveal the molecular mechanisms regulating H3K4me2 functions that repress gene expression in plants.
In the SDG701 knockdown mutant, 597 genes exhibited up-regulated expression probably due to the decrease in the H3K4me2 level. More than 30% of the 597 genes are involved in plant responses to various factors, including stress, abiotic and biotic stimuli, endogenous stimuli, cold conditions, and wounding (Additional file 5: Table S7). Additionally, SDG701-mediated H3K4me2 appears to have a substantial role in plant adaptive responses to changing environmental conditions. Specifically, SDG701 can establish two epigenetic marks with distinct functions, H3K4me3 and H3K4me2, which activate and repress transcription, respectively. The data presented herein suggest there exists a new layer of complexity to histone methylation functions regarding the remodeling of chromatin. The enhanced flexibility of the chromatin remodeling function to regulate gene expression may be advantageous for plants because of the high plasticity of their growth and development and the fact they must be able to adapt to adverse environmental conditions because they are sessile organisms.
Conclusion
We characterized the novel function of H3K4me2 in rice: different from the positive correlation between H3K4me3 level and gene expression level, H3K4me2 level is negatively correlated with gene transcription level. A single enzyme SDG701 can establish two epigenetic marks with distinct functions, suggesting the complexity of histone methylations to enhance the flexibility of the chromatin and the plasticity of the environmental adaption in plants.
Materials and methods
Plant materials and growth conditions
The SDG701 knockdown mutant 35Sp::SDG701iR1 (701Ri-1-5) in the O. sativa spp. japonica cv. Nipponbare background used in this study has been previously described [36]. For the RNA-seq and ChIP-seq analyses, rice seedlings were grown in growth chambers under short-day photoperiod conditions [10-h light (30 °C):14-h dark (28 °C)].
Library construction and sequencing
To be able to compare the RNA-seq and ChIP-seq data, the same set of rice samples were used to extract the total RNA used to prepare the RNA-seq and ChIP-seq libraries. Total RNA was extracted from 30-day-old shoots with the TRIzol reagent (Invitrogen). A strand-specific RNA-seq library was constructed as previously described [47]. Additionally, ChIP assays were completed as previously described [48], with the following antibodies: anti-monomethyl-H3K4 (Millipore), anti-dimethyl-H3K4 (Millipore), anti-trimethyl-H3K4 (Millipore), and anti-trimethyl-H3K36 (Abcam, Shanghai, China). The DNA resulting from a ChIP experiment and the input control were used to construct a ChIP-seq library as previously described [6]. The sequencing of the ChIP-seq and RNA-seq libraries generated 51-bp single-end reads and 101-bp paired-end reads, respectively. Details regarding the ChIP-seq and RNA-seq mapping are summarized in Additional file 1: Tables S1 and S4, respectively.
RNA-seq analysis
More than 30-million high-quality raw reads were obtained from the sequencing of the RNA-seq library and were analyzed as previously described [49]. Briefly, the TopHat2 (version 2.0.13) [50] and Cufflinks (version 2.2.1) [51] programs were used to identify genes that were differentially expressed between the 701Ri-1-5 mutant and NIP. Significantly differentially expressed genes were defined based on the combined thresholds of > 1.5-fold change in expression level and an adjusted p < 0.05. A volcano plot was subsequently generated with the ggplot2 package [52] of the R software. To visualize the RNA-seq data, the mapping files from TopHat (version 2.0.13) were converted to BEDGRAPH format files with BEDTools (version 2.17.0) [53]. The BEDGRAPH format files were analyzed with the IGV (version 2.3.88) genome browser [54].
ChIP-seq analysis
The raw data were mapped to the reference Nipponbare (japonica) rice genome (MSU7; http://rice.plantbiology.msu.edu) with the Bowtie (version 0.7.15) program [55], and SAMtools was used to extract the unique and non-redundant mapped reads [56]. The non-redundant mapped reads were subsequently analyzed.
To examine the global distributions of H3K4me1/H3K4me2/H3K4me3 and H3K36me3, each gene (from the TSS to the TTS) longer than 500 bp was divided into 300 bins, and 1000 bp regions upstream of the TSS and downstream of the TTS for each gene were divided into 50 bins. The read number for each bin was calculated with BEDTools (version 2.17.0) [53], and the read coverage of each sample was normalized to 10 million reads and calibrated against the input control. All aggregated plots and heatmaps were generated with the R software. BED format files were produced from mapped files with BEDTools (version 2.17.0) [53]. Additionally, SICER.sh from the SICER (version 1.1) software [57] was used to identify histone methylation enrichment regions (peaks) by comparing the ChIP library with the input library (parameters: W = 200, G = 200, and FDR < 1e−2 for H3K4me1 and H3K4me2; W = 200, G = 600, and FDR < 1e−2 for H3K4me3 and H3K36me3). Peaks were considered significant if they satisfied the following thresholds: FDR < 1e−3 and IP DNA/input DNA ≥ 2. Moreover, SICER-df.sh was used to compare the differences in H3K4me2/H3K4me3 between the 701iR1-5 mutant and NIP. The following thresholds were used to determine whether peak changes were significant: FDR < 1e−4 and > 1.5-fold change (wild type/mutant ≥ 1.5). The ChIPpeakAnno package [58] from Bioconductor (http://www.bioconductor.org) was used for annotating peaks. Genes (including the 1000 bp region upstream of the TSS) that overlapped with the peaks of various histone modifications were considered as modification-enriched genes.
Gene ontology and plant ontology analysis
For the GO enrichment analysis, we analyzed the genes that exhibited down- or up-regulated expression in the 701iR1-5 mutant with the CARMO online tool, which is a comprehensive annotation platform for the functional exploration of rice multi-omics data (http://bioinfo.sibs.ac.cn/carmo/Gene_Annotation.php) [59]. The significantly enriched biological processes among the differentially expressed genes (p < 0.05) were analyzed.
Analysis of published data
We downloaded DNA methylation, H3K9me1/H3K9me2/H3K9me3, H3K27me3, H3K4me2/H3K4me3 ChIP-seq, and related gene transcription data for various species from the Sequence Read Archive (SRA) database (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra/). The SRA files were converted to FASTQ files with the fastq-dump package in the SRAToolkit (version 2.9.0) (http://www.ncbi.nlm.nih.gov/Traces/sra/?view=toolkit_doc). Distribution plots and heatmaps were generated with deepTools (version 2.0) [60]. The gene expression matrices of the other species, except for S. cerevisiae, were downloaded from the GEO database. The FPKM value of each S. cerevisiae gene was calculated with the DESeq2 package [61]. Additionally, raw DNA methylation data were re-analyzed with BatMeth package [62]. The accession numbers of the sequences analyzed in this study are as follows: GSE95356 (H3K4me2/me3) and GSE73406 (RNA) for S. cerevisiae [11]; GSE49733 (H3K4me2), GSE49739 (H3K4me3), and GSE100723 (RNA) for C. elegans (http://www.modencode.org); GSE98967 (H3K4me2/me3) and GSE98966 (RNA) for D. melanogaster testis cells (http://www.modencode.org); GSE80049 (H3K4me2/3) [10] and GSE98063 (RNA) [63] for Mus musculus embryonic stem cells; GSE51176 (H3K4me2/3) [12] and GSE54167 (RNA) [64] for the human colon cancer cell line (HCT116 cells); GSE73972 (H3K4me2/3 and RNA) for Arabidopsis [33]; and GSE81436 (H3K9me2) [43], GSE79033 (H3K9me1//me3 and H3K27me3) [42] and GSE19602 (DNA methylations) for rice [37].
RNA extraction and RT-qPCR analysis
According to the manufacturer’s instructions (Invitrogen), RNA was isolated with TRI Reagent using the same method described in RNA-seq. Reverse transcription was performed with Improm-II reverse transcriptase (Promega), and quantitative polymerase chain reaction (qPCR) was performed using specific primers listed in Additional file 1: Table S8, and Ubiquitin5 (Ubq5) was used as an internal reference gene for normalization [36].
ChIP-qPCR analysis
Chromatin immunoprecipitation (ChIP) assays were performed with the same method described in the library construction. qPCR was used to test the enrichment of DNA immunoprecipitated in the ChIP experiments. Gene-specific primers are listed in Additional file 1: Table S8.
Additional files
Additional file 1. Figures S1–S10 and Tables S1, S4, S5 and S8.
Additional file 2: Table S2. Genes up- and down-regulated by more than 1.5-fold in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 3: Table S3. Biological functions enriched in the up- and down-regulated genes in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 4: Table S6. Peaks and genes with decreased levels of H3K4me2 or H3K4me3 to more than 1.5-fold in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 5: Table S7. Biological functions enriched in 597 up-regulated genes with decreased H3K4me2 levels in 701Ri-1-5.
Acknowledgements
The research was conducted within the context of the International Associated Laboratory on Plant Epigenome Research. We thank Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac) for editing the English text of a draft of this manuscript.
Abbreviations
- TSS
transcription start site
- TTS
transcription termination site
- ChIP-seq
chromatin immunoprecipitation sequencing
- RNA-seq
RNA sequencing
- MLL
mixed lineage leukemia
- COMPASS
complex proteins associated with Set1
- Ehd1
EARLY HEADING DATE 1
- Hd3a
HEADING DATE 3a
- RFT1
RICE FLOWERING LOCUS T 1
- SIP1
SDG723/OsTrx1/OsSET33 Interaction Protein 1
- NIP
O. sativa spp. japonica cv. Nipponbare
- DJ
O. sativa ssp. japonica cv. Dongjin
- FPKM
fragments per kilobase of exon per million reads mapped
Authors' contributions
AD conceived and designed the research. KL and YL performed the experiments. YL, LY, JZ, and YZ analyzed the data. YL, YY, W-HS, JZ, YZ, and AD wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grants NSFC31570315, 31771420, 31800207, and 31571319) and the National Basic Research Program of China (2012CB910500).
Availability of data and materials
The ChIP-seq and RNA-seq data generated in this study have been deposited in NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) and are accessible through accession number PRJNA522350.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yijing Zhang, Email: zhangyijing@sibs.ac.cn.
Aiwu Dong, Email: aiwudong@fudan.edu.cn.
References
- 1.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 2.Liu CY, Lu FL, Cui X, Cao XF. Histone methylation in higher plants. Annu Rev Plant Biol. 2010;61(61):395–420. doi: 10.1146/annurev.arplant.043008.091939. [DOI] [PubMed] [Google Scholar]
- 3.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 4.Li XY, Wang XF, He K, Ma YQ, Su N, He H, Stolc V, Tongprasit W, Jin WW, Jiang JM, et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell. 2008;20:259–276. doi: 10.1105/tpc.107.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barski A, Cuddapah S, Cui KR, Roh TY, Schones DE, Wang ZB, Wei G, Chepelev I, Zhao KJ. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Zhang XY, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007;5:1026–1035. doi: 10.1371/journal.pbio.0050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81(81):65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang PF, Lin CQ, Smith ER, Guo H, Sanderson BW, Wu M, Gogol M, Alexander T, Seidel C, Wiedemann LM, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Local A, Huang H, Albuquerque CP, Singh N, Lee AY, Wang W, Wang CC, Hsia JE, Shiau AK, Ge K, et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat Genet. 2018;50:73. doi: 10.1038/s41588-017-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares LM, He PC, Chun YJ, Suh H, Kim T, Buratowski S. Determinants of histone H3K4 methylation patterns. Mol Cell. 2017;68:773. doi: 10.1016/j.molcel.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu DQ, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011;21:227–236. doi: 10.1101/gr.115519.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rickels R, Hu DQ, Collings CK, Woodfin AR, Piunti A, Mohan M, Herz HM, Kvon E, Shilatifard A. An evolutionary conserved epigenetic mark of polycomb response elements implemented by Trx/MLL/COMPASS. Mol Cell. 2016;63:318–328. doi: 10.1016/j.molcel.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heintzman ND, Stuart RK, Hon G, Fu YT, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu CX, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 16.Zhang XY, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009;10:62. doi: 10.1186/gb-2009-10-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du Z, Li H, Wei Q, Zhao X, Wang CC, Zhu QL, Yi X, Xu WY, Liu XS, Jin WW, Su Z. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L., Japonica. Mol Plant. 2013;6:1463–1472. doi: 10.1093/mp/sst018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dijk K, Ding Y, Malkaram S, Riethoven JJM, Liu R, Yang JY, Laczko P, Chen H, Xia YN, Ladunga I, et al. Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 2010;10:238. doi: 10.1186/1471-2229-10-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 20.Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SYR, Winston F, Allis CD. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA. 2001;98:12902–12907. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roguev A, Schaft D, Shevchenko A, Pijnappel WWMP, Wilm M, Aasland R, Stewart AF. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohan M, Herz HM, Smith ER, Zhang Y, Jackson J, Washburn MP, Florens L, Eissenberg JC, Shilatifard A. The COMPASS family of H3K4 methylases in drosophila. Mol Cell Biol. 2011;31:4310–4318. doi: 10.1128/MCB.06092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian set1 histone H3-Lys(4) methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 25.Lee JH, Tate CM, You JS, Skalnik DG. Identification and characterization of the human Set1B histone H3-Lys(4) methyltransferase complex. J Biol Chem. 2007;282:13419–13428. doi: 10.1074/jbc.M609809200. [DOI] [PubMed] [Google Scholar]
- 26.Wu M, Wang PF, Lee JS, Martin-Brown S, Florens L, Washburn M, Shilatifard A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez-Venegas R, Avramova Z. Methylation patterns of histone H3 Lys 4, Lys 9 and Lys 27 in transcriptionally active and inactive Arabidopsis genes and in atx1 mutants. Nucl Acids Res. 2005;33:5199–5207. doi: 10.1093/nar/gki830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumbusch LO, Thorstensen T, Krauss V, Fischer A, Naumann K, Assalkhou R, Schulz I, Reuter G, Aalen RB. The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucl Acids Res. 2001;29:4319–4333. doi: 10.1093/nar/29.21.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fromm M, Avramova Z. ATX1/AtCOMPASS and the H3K4me3 marks: how do they activate Arabidopsis genes? Curr Opin Plant Biol. 2014;21:75–82. doi: 10.1016/j.pbi.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Napsucialy-Mendivil S, Alvarez-Venegas R, Shishkova S, Dubrovsky JG. ARABIDOPSIS HOMOLOG of TRITHORAX1 (ATX1) is required for cell production, patterning, and morphogenesis in root development. J Exp Bot. 2014;65:6373–6384. doi: 10.1093/jxb/eru355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleh A, Alvarez-Venegas R, Yilmaz M, Le O, Hou GC, Sadder M, Al-Abdallat A, Xia YN, Lu GQ, Ladunga I, Avramova Z. The highly similar Arabidopsis homologs of trithorax ATX1 and ATX2 encode proteins with divergent biochemical functions. Plant Cell. 2008;20:568–579. doi: 10.1105/tpc.107.056614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pien S, Fleury D, Mylne JS, Crevillen P, Inze D, Avramova Z, Dean C, Grossniklaus U. ARABIDOPSIS TRITHORAX1 dynamically regulates FLOWERING LOCUS C activation via histone 3 lysine 4 trimethylation. Plant Cell. 2008;20:580–588. doi: 10.1105/tpc.108.058172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen LQ, Luo JH, Cui ZH, Xue M, Wang L, Zhang XY, Pawlowski WP, He Y. ATX3, ATX4, and ATX5 encode putative H3K4 methyltransferases and are critical for plant development(1[CC-BY]) Plant Physiol. 2017;174:1795–1806. doi: 10.1104/pp.16.01944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SC, Lee S, Kim SR, Lee YS, Liu CY, Cao XF, An G. Trithorax group protein Oryza sativa Trithorax1 controls flowering time in rice via interaction with early heading date3. Plant Physiol. 2014;164:1326–1337. doi: 10.1104/pp.113.228049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang PF, Wang SL, Zheng H, Li H, Zhang F, Su YH, Xu ZT, Lin HY, Qian Q, Ding Y. SIP1 participates in regulation of flowering time in rice by recruiting OsTrx1 to Ehd1. New Phytol. 2018;219:422–435. doi: 10.1111/nph.15122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu KP, Yu Y, Dong AW, Shen WH. SET DOMAIN GROUP701 encodes a H3K4-methytransferase and regulates multiple key processes of rice plant development. New Phytol. 2017;215:609–623. doi: 10.1111/nph.14596. [DOI] [PubMed] [Google Scholar]
- 37.He GM, Zhu XP, Elling AA, Chen LB, Wang XF, Guo L, Liang MZ, He H, Zhang HY, Chen FF, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Wu Y, Schnable JC, Zeng Z, Freeling M, Crawford GE, Jiang J. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012;22:151–162. doi: 10.1101/gr.131342.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu B, Wei G, Shi J, Jin J, Shen T, Ni T, Shen WH, Yu Y, Dong A. SET DOMAIN GROUP 708, a histone H3 lysine 36-specific methyltransferase, controls flowering time in rice (Oryza sativa) New Phytol. 2016;210:577–588. doi: 10.1111/nph.13768. [DOI] [PubMed] [Google Scholar]
- 40.Wei G, Liu KP, Shen T, Shi JL, Liu B, Han M, Peng ML, Fu HH, Song YF, Zhu J, et al. Position-specific intron retention is mediated by the histone methyltransferase SDG725. BMC Biol. 2018;16:44. doi: 10.1186/s12915-018-0513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramakrishnan S, Pokhrel S, Palani S, Pflueger C, Parnell TJ, Cairns BR, Bhaskara S, Chandrasekharan MB. Counteracting H3K4 methylation modulators Set1 and Jhd2 co-regulate chromatin dynamics and gene transcription. Nat Commun. 2016;7:119. doi: 10.1038/ncomms11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang Y, Wang XM, Wang L, Pan XC, Xiao J, Wang XE, Wu YF, Zhang WL. Functional characterization of open chromatin in bidirectional promoters of rice. Sci Rep. 2016;6:320. doi: 10.1038/srep32088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan F, Zhou C, Zhou QW, Zhou SL, Yang WJ, Zhao Y, Li GL, Zhou DX. Analysis of chromatin regulators reveals specific features of rice DNA methylation pathways. Plant Physiol. 2016;171:2041–2054. doi: 10.1104/pp.16.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu YF, Liu DN, Zhong XC, Zhang CJ, Zhang QF, Zhou DX. CHD3 protein recognizes and regulates methylated histone H3 lysines 4 and 27 over a subset of targets in the rice genome. Proc Natl Acad Sci USA. 2012;109:5773–5778. doi: 10.1073/pnas.1203148109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowen NJ, Fujita N, Kajita M, Wade PA. Mi-2/NuRD: multiple complexes for many purposes. Biochim Et Biophys Acta Gene Struct Expr. 2004;1677:52–57. doi: 10.1016/j.bbaexp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Hoffmeister H, Fuchs A, Erdel F, Pinz S, Grobner-Ferreira R, Bruckmann A, Deutzmann R, Schwartz U, Maldonado R, Huber C, et al. CHD3 and CHD4 form distinct NuRD complexes with different yet overlapping functionality. Nucl Acids Res. 2017;45:10534–10554. doi: 10.1093/nar/gkx711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borodina T, Adjaye J, Sultan M. A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 2011;500(500):79–98. doi: 10.1016/B978-0-12-385118-5.00005-0. [DOI] [PubMed] [Google Scholar]
- 48.Sui PF, Jin J, Ye S, Mu C, Gao J, Feng HY, Shen WH, Yu Y, Dong AW. H3K36 methylation is critical for brassinosteroid-regulated plant growth and development in rice. Plant J. 2012;70:340–347. doi: 10.1111/j.1365-313X.2011.04873.x. [DOI] [PubMed] [Google Scholar]
- 49.Li C, Liu YH, Shen WH, Yu Y, Dong AW. Chromatin-remodeling factor OsINO80 is involved in regulation of gibberellin biosynthesis and is crucial for rice plant growth and development. J Integr Plant Biol. 2018;60:144–159. doi: 10.1111/jipb.12603. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ginestet C. ggplot2: elegant graphics for data analysis. J R Stat Soc Ser a Stat Soc. 2011;174:245. doi: 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
- 53.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:212. doi: 10.1186/gb-2009-10-3-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zang CZ, Schones DE, Zeng C, Cui KR, Zhao KJ, Peng WQ. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang JW, Qi MF, Liu J, Zhang YJ. CARMO: a comprehensive annotation platform for functional exploration of rice multi-omics data. Plant J. 2015;83:359–374. doi: 10.1111/tpj.12894. [DOI] [PubMed] [Google Scholar]
- 60.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucl Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim JQ, Tennakoon C, Li GL, Wong E, Ruan YJ, Wei CL, Sung WK. BatMeth: improved mapper for bisulfite sequencing reads on DNA methylation. Genome Biol. 2012;13:82. doi: 10.1186/gb-2012-13-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, Wysocka J. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell. 2017;66:568. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Theng SS, Wang W, Mah WC, Chan C, Zhuo JL, Gao Y, Qin HN, Lim L, Chong SS, Song JX, Lee CG. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc Natl Acad Sci USA. 2014;111:E5282–E5291. doi: 10.1073/pnas.1403383111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Figures S1–S10 and Tables S1, S4, S5 and S8.
Additional file 2: Table S2. Genes up- and down-regulated by more than 1.5-fold in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 3: Table S3. Biological functions enriched in the up- and down-regulated genes in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 4: Table S6. Peaks and genes with decreased levels of H3K4me2 or H3K4me3 to more than 1.5-fold in 701Ri-1-5 mutant compared to in the wild-type NIP.
Additional file 5: Table S7. Biological functions enriched in 597 up-regulated genes with decreased H3K4me2 levels in 701Ri-1-5.
Data Availability Statement
The ChIP-seq and RNA-seq data generated in this study have been deposited in NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) and are accessible through accession number PRJNA522350.