

Abstract
The Volume-Regulated Anion Channel (VRAC) is activated by cell swelling and plays a key role in cell volume regulation. VRAC is ubiquitously expressed in vertebrate cells and also implicated in many other physiological and cellular processes including fluid secretion, glutamate release, membrane potential regulation, cell proliferation, migration, and apoptosis. Although its biophysical properties have been well characterized, the molecular identity of VRAC remained a mystery for almost three decades. The field was transformed by recent discoveries showing that the leucine-rich repeat-containing protein 8A (LRRC8A, also named SWELL1) and its four other homologs form heteromeric VRAC channels. The composition of LRRC8 subunits determines channel properties and substrate selectivity of a large variety of different VRACs. Incorporating purified SWELL1-containing protein complexes into lipid bilayers is sufficient to reconstitute channel activities, a finding that supports the decrease in intracellular ionic strength as the mechanism of VRAC activation during cell swelling. Characterization of Swell1 knockout mice uncovers the important role of VRAC in T cell development, pancreatic β-cell glucose-stimulated insulin secretion, and adipocyte metabolic function. The ability to permeate organic osmolytes and metabolites is a major feature of VRAC. The list of VRAC substrates is expected to grow, now also including some cancer drugs and antibiotics even under non-cell swelling conditions. Therefore, a critical role of VRAC in drug resistance and cell–cell communication is emerging. This review summarizes the exciting recent progress on the structure-function relationship and physiology of VRAC and discusses key future questions to be solved.
1. INTRODUCTION
Because plasma membranes are permeable to water, decreases in extra-cellular osmolality or increases in intracellular osmolality induces a rapid net influx of water across the cell membrane, resulting in cell swelling. To achieve cell volume homeostasis, cells employ multiple ion channel and transporter mechanisms to mediate the regulatory volume decrease (RVD) through ion (mainly K+ and Cl−) efflux followed by release of osmotically obligated water (Hoffmann, Lambert, & Pedersen, 2009; Lang, 2007). A key player in RVD is the Volume-Regulated Anion Channel (VRAC), which is activated by cell swelling and mediates Cl− efflux (Hoffmann et al., 2009; Jentsch, 2016). The large VRAC current (known as ICl, swell) activated over minutes by reducing extracellular osmolality was first described in human lymphocytes and epithelial cells three decades ago (Fig. 1A) (Cahalan & Lewis, 1988; Hazama & Okada, 1988). Subsequently, it has been observed in almost every vertebrate cell type investigated (Nilius et al., 1997; Okada, 1997; Strange, Emma, & Jackson, 1996). The VRAC channel displays characteristic features, such as moderate outward rectification (larger outward currents than inward currents at positive vs. negative membrane potentials, Fig. 1B). Based on this property, VRAC is also referred as VSOR (volume-sensitive outwardly rectifying anion channel). In addition, VRAC currents inactivate at large positive membrane voltages, and have higher permeability to larger halide anions than Cl− (I− > Br− > Cl−).
Figure 1.
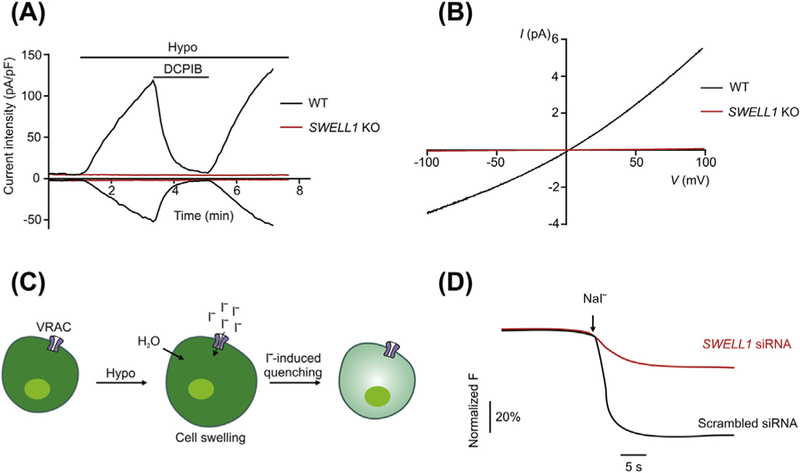
Identification of SWELL1 as an essential component of VRAC by RNAi screening. (A) Typical time course of whole-cell current densities at±100 mV induced by hypotonic solution (220 mOsm/kg) for WT and SWELL1 KO HeLa cells. DCPIB, a potent VRAC blocker, was used as indicated. (B) Representative whole-cell ICl, swell recorded by ramp protocol showing outward rectification. (C) Schematic of the I−-induced YFP quenching as the screening assay for VRAC. (D) Representative traces of I−-induced YFP quenching in HEK-YFP cells transfected with either scrambled siRNA or SWELL1 siRNA.
Although the electrophysiological characteristics of VRAC have been studied extensively, its molecular identity remained a long-standing mystery (Pedersen, Klausen, & Nilius, 2015). More than half a dozen candidates had been proposed to mediate VRAC, but none of them stood the test of time until the recent discovery of the LRRC8 (leucine-rich repeat-containing protein 8) family proteins as essential components of VRAC. In addition to cell volume regulation, VRAC has been implicated in a wide range of important physiological and cellular functions, such as regulation of membrane potential, salt and fluid secretion, glutamate release, cell proliferation and cell death (Pedersen, Okada, & Nilius, 2016). These physiological and pathological roles assigned to VRAC are mainly based on previous studies with VRAC pharmacological blockers, which often lack specificity and may also inhibit or activate other ion channels and transporters (Bowens, Dohare, Kuo, & Mongin, 2013; Fujii et al., 2015; Minieri et al., 2013). The molecular identification of SWELL1 and its associated LRRC8 homologs as essential pore-forming sub-units has transformed the VRAC research field and enabled rapid progress in elucidating the molecular mechanism and physiological role of VRAC.
2. LRRC8 HETEROMERS AS VRAC
2.1. Identification of SWELL1 (LRRC8A) by Genome-Wide RNAi Screen
There are several reasons why it was challenging to uncover the molecular identity of VRAC. First, the popular ion channel expression cloning systems, such as Xenopus oocytes and human embryonic kidney cells (HEK), express large endogenous VRAC currents (Ackerman, Wickman, & Clapham, 1994; Helix, Strobaek, Dahl, & Christophersen, 2003). This likely confounded the overexpression results of some previously proposed VRAC candidates and hampered expression cloning, a widely used approach for ion channel identification (Schroeder, Cheng, Jan, & Jan, 2008). Second, VRAC lacks a specific high-affinity channel ligand, such as toxin agonist and antagonist, which hindered direct biochemical purification of the channel protein. Lastly, unlike most cation channels, there are no sequence homologies (for example, conserved pore-lining motif) among known Cl− channel families identified to date (Duran, Thompson, Xiao, & Hartzell, 2010), including voltage-gated Cl− channel (CLCs), cAMP-activated Cl− channel (CFTR), Ca2+-activated Cl− channels (CaCC, TMEM16/Ano), and ligand-gated anion channels (GABAA and glycine receptors).
To circumvent these limitations, two research groups independently employed a loss-of-function genetics approach (Qiu et al., 2014; Voss et al., 2014) and performed a genome-wide RNA interference (RNAi) screen using HEK cells stably expressing I−-sensitive yellow fluorescent protein (YFP) (Galietta, Haggie, & Verkman, 2001). Application of hypotonic solutions and NaI triggers a rapid fluorescence quenching due to I− influx through the endogenous VRAC in HEK-YFP cells (Fig. 1C). This unbiased approach led both groups to the identification of LRRC8A, also called SWELL1 (for its activation by cell swelling), as a key regulator for hypotonicity-induced I− influx (Fig. 1D). Subsequent RNAi knockdown and CRISPR/Cas9-mediated knockout experiments revealed that SWELL1 is essential for ICl, swell (Fig. 1) and regulates the RVD in various cell types, such as HEK, HeLa and HCT11 cells (Planells-Cases et al., 2015; Qiu et al., 2014; Voss et al., 2014). These two studies highlight the power of loss-of-function genomics screen, which can be applied in the future to other widely expressed Cl− channels that are well described biophysically, yet without clear molecular identity (Duran et al., 2010). Similar genome-wide RNAi screening based on YFP quenching assay was previously conducted in Drosophila S2 cells and identified bestrophin-1 as forming the Drosophila ICl, swell channel (Stotz & Clapham, 2012). Recently, a combination of proteomics and targeted RNAi screen has also been performed to identify the prostaglandin transporter SLCO2A1 as a candidate for the maxi-anion channel (MAC) (Sabirov et al., 2017).
2.2. LRRC8 Heteromers as Essential Components of VRAC
SWELL1 overexpression, although detected on cell surface, didn’t enhance but rather decreased the endogenous ICl, swell (Qiu et al., 2014; Voss et al., 2014), which suggested that SWELL1 could be one of the sub-units in a multimeric channel complex. In this scenario, SWELL1 overexpression may result in a stoichiometry that lacks VRAC activity. Indeed, co-immunoprecipitation experiments in transfected HEK cells showed extensive interactions among SWELL1 and its four homologous family members (LRRC8B, 8C, 8D, and 8E) (Lee, Freinkman, Sabatini, & Ploegh, 2014; Voss et al., 2014). Affinity purification of SWELL1 close to the endogenous expression level in HeLa cells revealed only one prominent high molecular weight protein band after native gel electrophoresis (Syeda et al., 2016). Mass spectrometry analysis detected all five LRRC8 proteins, but no other proteins, as specific components within SWELL1-containing protein complexes (Syeda et al., 2016). CRISPR/Cas9-mediated disruption of four LRRC8 homologs (8B-8E) collectively also abolished ICl, swell, indicating SWELL1 and at least one other LRRC8 subunit are required for VRAC activity (Syeda et al., 2016; Voss et al., 2014). Consistently, co-expression of SWELL1 and another LRRC8 member, but not SWELL1 alone reconstituted ICl, swell in cells lacking all five LRRC8 proteins (Voss et al., 2014). SWELL1 is important for the surface expression of VRAC, as its LRRC8 homologs remained intra-cellular when transfected alone but were partially trafficked to the plasma membrane when co-transfected with SWELL1 (Voss et al., 2014).
LRRC8 proteins form a conserved protein family in chordates (Abascal & Zardoya, 2012). They are composed of two main parts: N-terminal 4 transmembrane helices (TM) and C-terminal 17 leucine-rich repeats (LRRs) (Fig. 2). The LRRs were initially presumed to face the outside of the cells as potential ligand binding domains (Sawada et al., 2003), however, immunofluorescent staining of non-permeabilized cells indicated that the putative TM1-TM2 and TM3-TM4 loops are extracellular and the N- and C-termini are cytoplasmic (Qiu et al., 2014; Voss et al., 2014). The cytoplasmic localization of LRR domains of SWELL1 and LRRC8D was also confirmed by protease protection assay (Lee et al., 2014). Based on weak but significant sequence similarities, the TM half of LRRC8 proteins is evolutionarily related to pannexins (Abascal & Zardoya, 2012). Pannexins are the vertebrate homologs of invertebrate gap junction-forming innexins and are structurally related to vertebrate gap junction-forming connexins. Pannexins form hexameric large-pore channels that can release ATP during autocrine and paracrine signaling (Dahl, 2015). Biochemical purification, cross-linking, and single-step photobleaching experiments indicated multi-subunit assemblies of SWELL1-containing protein complexes, probably as hexamers like pannexins (Gaitan-Penas et al., 2016; Syeda et al., 2016). In addition to the similar transmembrane topology, both LRRC8 proteins and pannexins are N-glycosylated and possess two pairs of conserved cysteine residues in their extracellular loops (Fig. 2) (Abascal & Zardoya, 2012; Voss et al., 2014), which likely mediate inter-or intra-molecular disulfide bond formation between different subunits.
Figure 2.
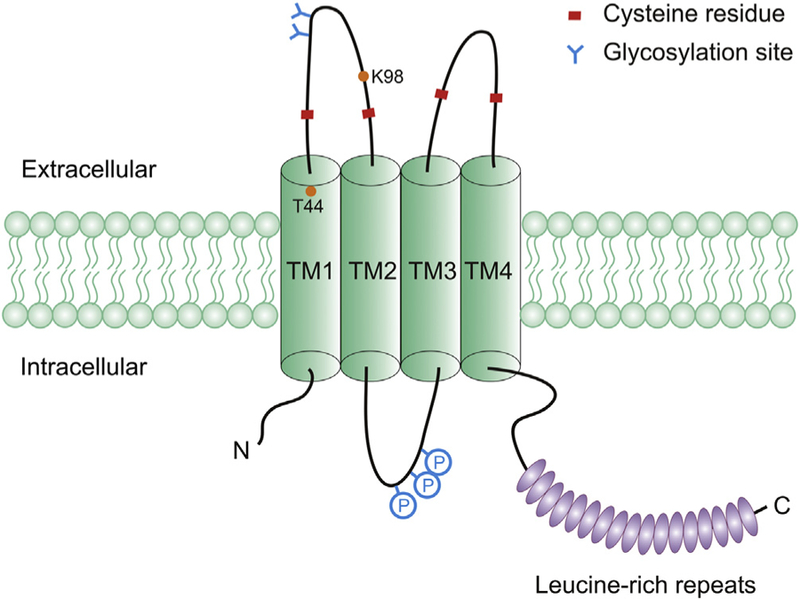
Schematic representation of SWELL1 protein. The diagram contains N-terminal 4 transmembrane helices (TM1–4), C-terminal 17 leucine-rich repeats (ovals), two pairs of conserved cysteine residues in the extracellular loops, and putative N-linked glycosylation sites and phosphorylation sites (P). Threonine (T44) located at the external boundary of TM1 and lysine (K98) within the TM1-TM2 extracellular loop were identifies as potential pore-lining residues.
The exact stoichiometry and subunit arrangement of functional LRRC8 heteromers are not known. Native gel electrophoresis indicated that the oligomeric state of SWELL1-containing complexes remains the same, despite the deletion of three or all four LRRC8 homologs (Syeda et al., 2016). At least in overexpression systems, sequential co-immunoprecipitation in HEK cells and high-resolution co-localization study in Xenopus oocytes suggested that individual VRAC channel may contain SWELL1 and more than one (in theory, up to four) LRRC8 homologs (Gaitan-Penas et al., 2016; Lutter, Ullrich, Lueck, Kempa, & Jentsch, 2017). Most cells and tissues express SWELL1 and all four LRRC8 subunits albeit at variable expression levels (The Human Protein Atlas). Given the potential flexibility in stoichiometry and subunit arrangement within the channel complexes, individual cells may express a large variety of different VRAC channels. Several ligand-gated ion channel families, such as GABAA receptors, exhibit similar flexibility in subunit composition and their arrangement. Nevertheless, this structural heterogeneity with its consequential functional diversity in channel biophysical properties (discussed in Section 2.4) makes VRAC very unusual among the large ion channel families.
2.3. LRRC8 Heteromers Form the VRAC Channel Pore
Based on the homology with pannexins, LRRC8 proteins are likely pore-forming subunits of VRAC rather than essential auxiliary channel regulators (Abascal & Zardoya, 2012). Both mutagenesis study and in vitro reconstitution of purified proteins support this conclusion. Ion selectivity of a channel is typically determined by its pore (Isacoff, Jan, & Minor, 2013). For VRAC, a slight permeability preference of I− over Cl− (PI/PCl: ~1.2–1.4) is ones of its characteristics. To perform structure–function analysis, mutants of SWELL1 and other LRRC8 proteins were expressed or co-expressed in cells lacking SWELL1, LRRC8B-8E, or all five LRRC8 proteins (Qiu et al., 2014; Voss et al., 2014). To identify SWELL1 mutants that might change this selectivity, cysteine mutations were systematically introduced in individual amino acids of all 4 putative TM domains of SWELL1. This screen led to the identification of threonine at position 44 (T44) (Fig. 2), whose mutations caused significant alterations in anion permeability ratios (Qiu et al., 2014). The T44 residue is also conserved in the other LRRC8 subunits except LRRC8B. Like SWELL1 T44C, mutations at T44 in LRRC8C-8E proteins also increased PI/PCl (Syeda et al., 2016), indicating these LRRC8 subunits are also contributing to the VRAC pore. T44 is predicted to be located at the external boundary of TM1, and intriguingly, the similar regions of connexins and pannexins were shown to line part of their channel pores (Maeda et al., 2009; Wang & Dahl, 2010).
In another set of studies of chimeras between LRRC8C and LRRC8E (discussed in the Section 2.4), a conserved lysine residue (K98) within the TM1-TM2 extracellular loop was identified (Fig. 2), which upon charge reversal decreased PI/PCl (Ullrich, Reincke, Voss, Stauber, & Jentsch, 2016). The effect on selectivity was further augmented when the same mutation was present in both SWELL1 and LRRC8C or 8E (Ullrich et al., 2016). Collectively, these mutagenesis results support the idea that SWELL1 and its LRRC8 homologs are pore-forming subunits of VRAC. It’s unclear why residues from two different regions both play a role in anion selectivity. It has to be noted that although significant, the effects of these mutations on PI/PCl are relatively small (decreased to ~1.1 or increased to ~1.6) (Qiu et al., 2014; Ullrich et al., 2016). Furthermore, no effect on cation selectivity was observed (Ullrich et al., 2016). So it’s possible that other critical pore-lining residues are yet to be discovered.
In vitro reconstitution provides direct and definitive evidence for pore-forming ability of ion channel candidates. Purified SWELL1-containing protein complexes were reconstituted into droplet interface lipid bilayers assembled from two monolayers (Syeda et al., 2016). Robust Cl− channel activities, induced by osmotic gradient and sensitive to the VRAC inhibitor DCPIB, were observed (Syeda et al., 2016). This demonstrated that purified SWELL1 and associated LRRC8 proteins are sufficient to recapitulate the key VRAC channel properties. The activation mechanisms of VRAC by cell swelling have long been debated (Hoffmann et al., 2009; Pedersen et al., 2016). Hypotonicity-induced cell swelling evoke numerous changes in cell physiology, which can be generally categorized into three main aspects, including: (1) mechanical changes in the plasma membrane or cytoskeleton; (2) decreased ionic strength or concentrations of specific ions; (3) other signaling pathways downstream of cell swelling. The reconstitution of purified SWELL1 and associated LRRC8 proteins in vitro excluded an essential role of other cellular components (such as cytoskeletons) and signaling pathways (such as kinases) in hypotonicity-induced channel activation. Increasing the droplet volume and the subsequent mechanical changes in the lipid bilayers failed to stimulate channel activities. Instead, lowering the ionic strength (from 150 mM KCl to 70 mM KCl) in the absence of an osmotic gradient was sufficient to activate SWELL1-containing protein complexes (Syeda et al., 2016). This is consistent with the seminal finding in endothelial cells that a decrease of intercellular ionic strength rather than an increase in cell volume per se is the initial trigger for activation of ICl, swell (Voets, Droogmans, Raskin, Eggermont, & Nilius, 1999). It should be noted that VRAC can also be activated without cell swelling (Fig. 3, see further discussion in Section 4.1). Their mechanisms of activation are still unclear but unlikely involve ionic strength change. Together, both mutation analysis and in vitro reconstitution demonstrated that SWELL1 and associated LRRC8 proteins are pore-forming subunits of VRAC, which is also supported by the contribution of LRRC8 subunits to other VRAC properties discussed in the following two sections.
Figure 3.
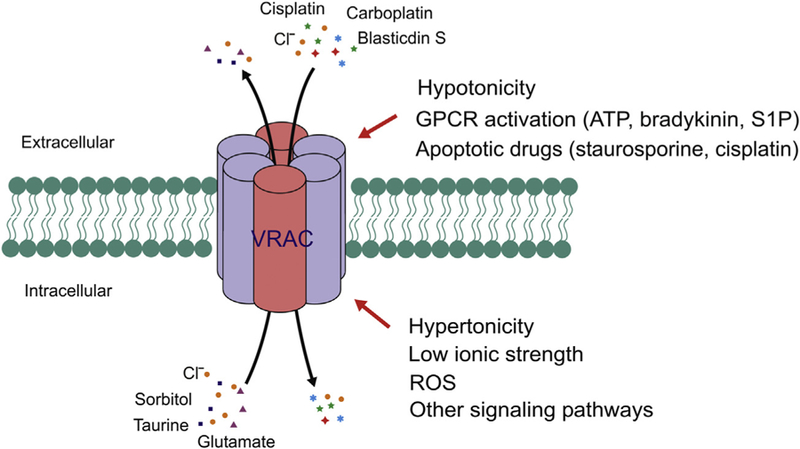
Diverse activation mechanisms and substrates suggesting an important role for VRAC in cell–cell communication. VRAC can be activated by extracellular hypertonicity, GPCR activation, and apoptosis-inducing drugs; and intracellular hypertonicity, reduced ionic strength, and reactive oxygen species (ROS). S1P, sphingosine-1-phos-phate. In addition to conducting Cl−, VRAC also permeate many organic osmolytes, metabolites, cancer drugs and antibiotics.
2.4. LRRC8 Subunit Compositions Determine Key VRAC Properties
Although ICl, swell was widely recorded, some differences of VRAC channels in various tissues and cell types were observed over the years, including: the degree of outward rectification, inactivation kinetics in positive membrane potentials, and even the single channel conductance (Nilius et al., 1997; Okada, 1997; Strange et al., 1996). This suggested potential molecular diversity of VRAC and added to the confusion to its molecular identification. Identification of LRRC8 heteromers as VRAC essential components and appreciation of their biochemical heterogeneity immediately suggested a compelling explanation for the differences in native VRAC currents (Voss et al., 2014). Indeed, co-expression of SWELL1 and LRRC8C in cells lacking all five LRRC8 proteins generated ICl, swell with slow inactivation kinetics, whereas SWELL1 and LRRC8E yielded fast inactivating currents (Voss et al., 2014). Subsequent chimeric experiments between LRRC8C and LRRC8E identified two amino acid residues in a highly conserved region of TM1-TM2 extracellular loop as the main determinant of VRAC inactivation kinetics (Fig. 2) (Ullrich et al., 2016). There is also a good correlation between the endogenous expression levels of LRRC8C and LRRC8E with differences in ICl, swell inactivation kinetics. Strikingly, lipid bilayer reconstitution of SWELL1-containing protein complexes generated a spectrum of intermediate single-channel conductances ranging from w10 to 50 pS with various open probabilities (Po) (Syeda et al., 2016). The single-channel conductance range became narrow and non-overlapping when SWELL1 with only one of LRRC8 subunits was incorporated in vitro, similar to the observation with cell-attached single-channel recordings on the corresponding LRRC8 triple KO cells (Syeda et al., 2016). Compared to LRRC8C and LRRC8E, LRRC8D-mediated VRAC displayed greater outward rectification and smaller permeability preference of I− over Cl− (Syeda et al., 2016), although the change of anion selectivity was relatively small and was not observed in another study (Voss et al., 2014). Finally, the sensitivity to oxidative stress for VRAC heterologously expressed in Xenopus oocytes is also dependent on the composition of LRRC8 subunits (Gradogna, Gavazzo, Boccaccio, & Pusch, 2017).
2.5. VRAC as the Conducting Channel for Large Organic Compounds: Amino Acids, Cancer Drugs and Antibiotics
In addition to ions (mainly K+ and Cl−), vertebrate cells, in response to cell swelling, also release various organic osmolytes and metabolites to lower the intracellular osmolality, which drives secondary water efflux and facilitates regulatory volume decrease (Hoffmann et al., 2009). These small organic osmolytes and metabolites include sorbitol, myo-inositol, choline, non-essential amino acids and their derivatives (most prominently taurine). The corresponding transport pathway was termed VSOAC (volume-sensitive organic osmolyte anion channel) ( Jackson, Morrison, & Strange, 1994). Both VRAC and VSOAC are activated by cell swelling and are inhibited by a similar panel of non-specific anion channel inhibitors. The diameter of the VRAC pore was estimated in the range of 1.2–1.4 nm (Droogmans, Maertens, Prenen, & Nilius, 1999; Ternovsky, Okada, & Sabirov, 2004), large enough to be permeable to larger (and not necessarily negatively charged) molecules, including taurine, glutamate, sugars, and even ATP. Therefore VRAC and VSOAC were thought to share the same molecular identity. However, discrepancies over the time course for their activation and sensitivity to some inhibitors led to the proposal that VSOAC and VRAC were separate entities (Lambert & Hoffmann, 1994; Stutzin et al., 1999). After the successful molecular identification of VRAC, SWELL1 was shown to be essential for VSOAC activities, that is, swelling-induced efflux of a wide range of organic osmolytes, such as taurine, D-aspartate, and myo-inositol (Hyzinski-Garcia, Rudkouskaya, & Mongin, 2014; Lutter et al., 2017; Planells-Cases et al., 2015; Qiu et al., 2014; Schober, Wilson, & Mongin, 2017; Voss et al., 2014). This strongly suggested that VRAC and VSOAC are identical channels or at least share the same essential SWELL1 subunit.
Compositions of the other LRRC8 subunits not only determine VRAC anion-conducting properties, but also significantly contribute to the selectivity of organic osmolytes. Although not absolutely essential, LRRC8D was important for swelling-induced efflux of taurine and myo-inositol in both HEK cells and primary rat astrocytes (Lutter et al., 2017; Planells-Cases et al., 2015; Schober et al., 2017). However, for swelling-induced release of D-aspartate (a non-metabolizable analogue and reporter for glutamate), LRRC8C, 8D and 8E all seem to contribute to some extent depending on the cell types under examination (Lutter et al., 2017; Schober et al., 2017). Heterozygous expression of SWELL1 and LRRC8 subunits in Xenopus oocytes also mediated fluxes of amino acids and ATP with somewhat different selectivity compared those in the mammalian cells (Gaitan-Penas et al., 2016). Additionally, two unbiased haploid cell loss-of-function genetic screens surprisingly uncovered an important function of LRRC8D in the uptake of antibiotic blasticidin and cancer drugs (cisplatin and carboplatin) under isotonic culture conditions (Lee et al., 2014; Planells-Cases et al., 2015). These discoveries significantly expanded the role of VRAC channel, especially composed of SWELL1 and LRRC8D, in transporting larger organic compounds in mammalian cells. Importantly, they also suggested alternative activation mechanisms of VRAC under physiological conditions and without cell swelling (Fig. 3).
3. PHYSIOLOGICAL ROLE OF SWELL1 AND VRAC
The discovery of LRRC8 heteromers as VRAC provides tremendous opportunities to evaluate previously proposed physiological role and to discover novel function of this important channel (Fig. 4). Swell1 knockout mice had high embryonic and postnatal lethality, growth retardation, and abnormalities in multiple tissues, including curly hair, thin skeletal muscle bundles, and vacuolated renal tubular cells (Kumar et al., 2014). In zebrafish, Swell1 knockdown caused pericardial edema and defects in trunk elongation and somatogenesis (Yamada, Wondergem, Morrison, Yin, & Strange, 2016). These severe phenotypes highlight the importance of VRAC in animal development and tissue homeostasis. Exciting progress has been made on the role of VRAC in the immune system and metabolism.
Figure 4.
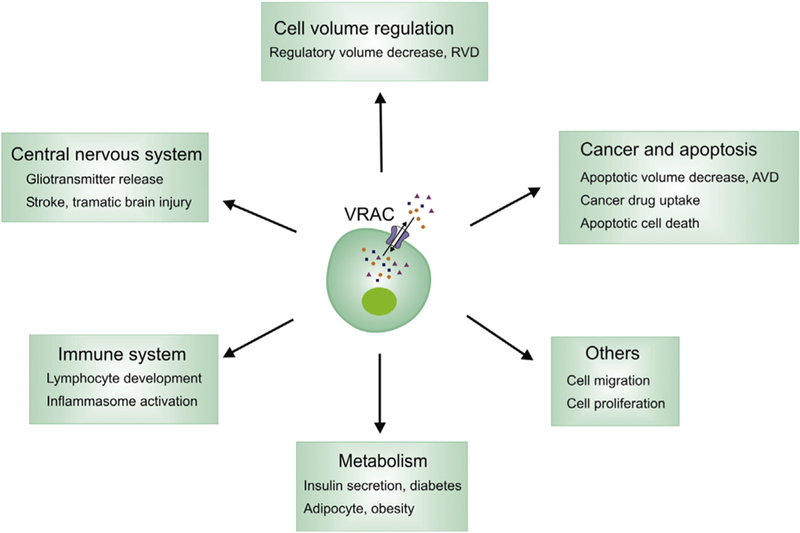
Proposed role of VRAC in cell biology, physiology, and disease.
3.1. Role of SWELL1 and VRAC in the Immune System
SWELL1 was first cloned from a female patient with congenital agammaglobulinemia and markedly reduced circulating B cells (Sawada et al., 2003). A balanced chromosome translocation was identified which leads to the deletion of the C-terminal two-and-a-half LRRs of SWELL1 (91 aa) and the addition of 35 aa derived from an intronic sequence (SWELL1Δ91/+35). Bone marrow transplantation in mice with progenitors overexpressing SWELL1Δ91/+35 resulted in a severe block in B cell development at the pro-B cell to pre-B cell transition and reduced numbers of T cells (Sawada et al., 2003). Thus a potential dominant-negative effect of this heterozygous SWELL1 mutation was proposed as the cause of agammaglobulinemia in this single patient (Sawada et al., 2003). However, reduced number of T cells was not observed in this human case. Additionally, while SWELL1Δ91/+35 was unable to form functional VRAC, no apparent dominant-negative or gain-of-function effect on ICl, swell was observed in overexpressing cells (Qiu et al., 2014). Therefore the link between the mutant SWELL1 and agammaglobulinemia is not clear at this point.
Swell1 knockout mice exhibited a modest block in B cell development but normal B cell function (Kumar et al., 2014). In contrast, the knockout mice displayed a profound cell-intrinsic block in T cell development at the double-negative stage with decreased proliferation and increased apoptosis, and reduced phosphorylated AKT levels (Kumar et al., 2014). The mechanistic interpretation of this T cell defect was based on a very unusual dual-membrane topology hypothesis that the C-terminal LRR domains of maybe a fraction of SWELL1 is extracellular and interact with a putative SWELL1 ligand, resulting in LCK–ZAP-70–GAB2–PI3K-mediated activation of AKT and T cell survival (Kumar et al., 2014). Recently, a hypomorphic spontaneous mutation in Swell1, designated ébouriffé (ebo), was reported (Platt et al., 2017). This allele was caused by a 2-bp deletion that was predicted to truncate the 15C-terminal LRRs of Swell1. ebo/ebo mice shared some similar features as Swell1 knockout mice, but the overall phenotype is much milder. In contrast to Swell1 knockout mice, ebo/ebo mice had intact T cell development and function, associated, however, with a dramatic reduction of VRAC current in T cells (Platt et al., 2017). It was therefore concluded that the critical role Swell1 plays in T cell development is independent of its VRAC activity, suggesting the involvement of potential channel-independent functions (Platt et al., 2017). However, it should also be noted that as indicated by some residual ICl, swell activity, there might be low levels of intact Swell1 protein expressed in ebo/ebo mice (though not detected by western blot). Furthermore, the different genetic backgrounds of Swell1 knockout (C57B/6) and ebo (FVB/N) mice could also account for the differential phenotypes observed. Although the underlying mechanisms still require future investigation, both the human and mouse studies strongly suggested an important role of SWELL1 in the development and function of lymphocytes. In addition to the adaptive immune system, the pharmacological studies also implicated VRAC in the activation of NLRP3 inflammasome in macrophages, which is a critical component of innate immune response and may play a role in the pathogenesis of Alzheimer’s disease (Compan et al., 2012; Daniels et al., 2016).
3.2. Role of SWELL1 and VRAC in Metabolism
Most vertebrate animals maintain a stable osmolality in the extracellular fluid (in mammals, close to 300 mOsm/kg of water). Thus extracellular hypotonicity is thought to be unlikely the physiological stimulus to VRAC in most tissues. Instead, VRAC can be activated by increased intracellular osmolality (hypertonicity) resulted from uptake and accumulation of ions or organic osmolytes (Fig. 3). For example, in insulin-secreting pancreatic β-cells, glucose uptake and metabolism, the subsequent intracellular accumulation of glucose metabolites and osmotic entry of water, can induce β-cell swelling which is sufficient to activate VRAC (Best, 2005; Best, Brown, Sener, & Malaisse, 2010; Best, Yates, Decher, Steinmeyer, & Nilius, 2004; Miley, Sheader, Brown, & Best, 1997). In the classic model of glucose-stimulated insulin secretion, glucose metabolism increases the intra-cellular ATP/ADP ratio, which leads to the closure of the ATP-sensitive K+ channels (KATP), thereby inducing membrane depolarization (Ashcroft & Rorsman, 2013). This in turn opens the voltage-gated Ca2+ channels, leading to Ca2+ entry into the cell, thus triggering insulin release by exocytosis (Ashcroft & Rorsman, 2013). However, pharmacological studies suggested that VRAC may cooperate with KATP and mediate additional glucose-sensing mechanism (Best et al., 2004). Similar to KATP closure, glucose-induced VRAC opening was proposed to assist membrane depolarization (due to Cl− efflux) and thus lead to Ca2+ influx and insulin secretion (Best et al., 2010). Consistent with this model, a recent study showed that SWELL1 is required for hypotonicity- and glucose-stimulated ICl, swell in both mouse and human β-cells (Kang et al., 2018). Very strikingly, SWELL1 depletion completely abolished hypotonicity- and glucose-stimulated b-cell membrane depolarization and intracellular calcium signaling (Kang et al., 2018). Furthermore, glucose-stimulated insulin secretion both in vitro and in vivo was eliminated upon SWELL1 deletion. Finally, β-cell-specific Swell1 knockout mice exhibited impaired glucose tolerance (Kang et al., 2018). While future work is warranted to determine the exact relationship and relative contribution of the classical KATP and VRAC in glucose-stimulated insulin secretion, this study established a critical role for SWELL1 in regulating β-cell excitability and insulin secretion.
The volume of adipocyte, another metabolically active cell type, undergoes considerable expansion in the setting of obesity. Although VRAC is not known to be mechanosensitive, ICl, swell-like current was recorded by enlarging adipocytes with the application of positive pressure via the patch pipette (Zhang et al., 2017). Both this mechanically-activated “swell” current and the hypotonicity-activated ICl, swell in adipocytes were shown to be dependent on SWELL1. Interestingly, the hypotonicity-activated ICl, swell activities were augmented in hypertrophic adipocytes of obese mice and humans (Zhang et al., 2017). Functionally, it was shown that SWELL1 positively regulates adipocyte glucose uptake and lipogenesis via insulin-PI3K-AKT2 signaling through LRR domains-mediated interactions with GRB2, a negative regulator of insulin signaling (Zhang et al., 2017). It’s still unclear what the physiological stimuli for VRAC activation might be (adipocyte volume expansion or maybe increased intracellular osmolality during lipogenesis) and what the contribution, if any, of VRAC channel function itself is. Importantly, adipocyte-specific Swell1 knockout reduced adiposity and adipocyte size in obese mice while inducing systemic glucose intolerance and insulin resistance (Zhang et al., 2017). On the other hand, high-fat diet-induced obesity is associated with the upregulation of Swell1 protein expression in adipose, which may be beneficial and contribute to improved systemic glycemia by enhancing insulin sensitivity (Xie et al., 2017).
Intriguingly, SWELL1 expression in human islets is significantly decreased in hyperglycemic individuals based on a global gene expression analysis (Taneera et al., 2012), which may lead to impaired insulin secretion from β-cells. Based on the studies on adipose and liver, it would be important to determine whether VRAC activity or SWELL1 expression is also reduced in these tissues during the development of Type 2 diabetes; if so, this could contribute to insulin resistance. Thus it’s possible that enhancing VRAC activity in both insulin-secreting (β-cells) and insulin-sensitive (adipose and liver) tissues may provide a novel therapeutic strategy for Type 2 diabetes and other metabolic disorders.
3.3. Role of VRAC in Apoptosis and Cancer Drug Resistance
Several apoptosis-inducing drugs, such as staurosporine and cisplatin, can slowly activate ICl, swell-like currents under isotonic conditions (Fig. 3) (Ise et al., 2005; Maeno, Ishizaki, Kanaseki, Hazama, & Okada, 2000; Poulsen et al., 2010; Shimizu, Numata, & Okada, 2004). Like RVD, these currents may mediate the efflux of intracellular Cl– and organic osmolytes, which contributes to apoptotic volume decrease (AVD), a critical early step facilitating the progression of apoptosis. VRAC blockers were shown to protect cells from drug-induced apoptosis (Ise et al., 2005; Maeno et al., 2000; Poulsen et al., 2010). Several drug-resistant cancer cell lines also exhibited decreased cisplatin-induced apoptosis, which is associated with reduced VRAC activity, RVD, and hypotonicity-stimulated taurine release (Lee et al., 2007; Min et al., 2011; Poulsen et al., 2010; Sorensen, Thorsteinsdottir, & Lambert, 2014). Indeed, upon the identification of SWELL1 as an essential VRAC component, SWELL1 knockout or knockdown significantly diminished cisplatin- or staurosporine-induced VRAC activity, caspase-3 induction and apoptosis (Planells-Cases et al., 2015; Sorensen, Nielsen, Thorsteinsdottir, Hoffmann, & Lambert, 2016). In addition to facilitating AVD-dependent apoptosis, VRAC channels, especially those formed by SWELL1 and LRRC8D, also mediate uptake of platinum-based cancer drugs, including cisplatin and carboplatin (Planells-Cases et al., 2015). The acquirement of cisplatin resistance in cancer cell lines is associated with the downregulation of SWELL1 expression (Sorensen, Dam, Sturup, & Lambert, 2016). Further supporting a clinically relevant role of VRAC in cancer drug sensitivity, low LRRC8D expression was shown to correlate with shorter survival of ovarian cancer patients treated with platinum-based drugs (Planells-Cases et al., 2015).
4. FUTURE STUDIES OF VRAC
Since the first descriptions of ICl, swell more than three decades ago, a large body of literature has accumulated on various aspects of VRAC in diverse tissues and cell types. This includes its electrophysiological characteristics, pharmacology, activation and regulatory mechanisms, cell biological function, and potential physiological and pathological roles of VRAC, but confusion and unanswered questions persisted due to the lack of a molecular identity. The molecular identification of SWELL1 and its associated LRRC8 homologs as essential VRAC pore-forming subunits represents a breakthrough that has moved the once stagnant field forward. It provides the basis to examine its structure-function relationships and to unambiguously assign cellular and physiological functions to VRAC. It will also likely lead to discoveries of unexpected functions of this important channel family.
4.1. Structural and Molecular Basis of VRAC Permeation, Activation and Regulation
How many SWELL1 and LRRC8 subunits are present in the VRAC channel complex? How their relative positions are arranged in functional VRAC channels? How much flexibility of subunit stoichiometry (composition and position) it exists in the native channel complexes? To address these questions, a combination of single-molecule and structural studies (cryo-EM in particular) on both overexpressed and endogenous channel complexes is required. The structure information and subsequent mutagenesis studies will also provide molecular insights in many fundamental questions about the conducting and gating mechanisms of VRAC. First, what are the pore-lining permeation pathways for both Cl− and organic compounds? What is the molecular basis for anion and substrate selectivity of different LRRC8 subunit compositions? Second, lowering ionic strength has been shown to activate VRAC both in vitro constitution and in whole-cell recording. Does a decrease in ionic strength cause conformational change and open the VRAC channel pore? Are the LRR domains the potential ionic strength sensor? Third, how is VRAC activity modulated by pharmacological inhibitors (such as DCPIB and DIDS), ATP and lipids? The interaction between ATP and VRAC is particularly interesting. Intracellular ATP but not its hydrolysis is required for maintaining VRAC activity ( Jackson et al., 1994; Oike, Droogmans, & Nilius, 1994). This ATP dependence was postulated as a self-protection mechanism preventing cells from losing too much organic osmolytes during RVD. However, it’s unclear whether it’s due to direct ATP binding to VRAC. ATP can also permeate the VRAC pore, but a high concentration of extracellular ATP acts as a voltage-dependent VRAC blocker, reminiscent of an open-pore blocker ( Jackson & Strange, 1995; Nilius, Oike, Zahradnik, & Droogmans, 1994; Tsumura, Oiki, Ueda, Okuma, & Okada, 1996). Specific and potent chemical antagonists and agonists have been indispensable in elucidating the mechanism of action and probing physiological and pathological function of ion channels. Such reagents are still not available for VRAC. High-throughput YFP quenching assay and the now known molecular identity of VRAC will undoubtedly facilitate the identification of better and more useful chemical probes for VRAC channels.
In addition to cell swelling, VRAC can also be activated by other physiologically relevant stimuli, such as apoptosis-inducing drugs (Maeno et al., 2000; Shimizu et al., 2004) and G protein-coupled receptor (GPCR) activation downstream of ATP, bradykinin, or sphingosine-1-phosphate (Burow, Klapperstuck, & Markwardt, 2015; Liu, Akita, Shimizu, Sabirov, & Okada, 2009; Takano et al., 2005) (Fig. 3). Unlike the classical hypotonicity-activated ICl, swell, these VRAC currents typically take longer time to develop and are smaller in amplitude. These features indicate alternative mode of VRAC activation with potential involvement of second messenger systems. Indeed, reactive oxygen species (ROS) and various protein kinases were proposed to either activate or modulate VRAC activity (Browe & Baumgarten, 2006; Pedersen et al., 2016; Shimizu et al., 2004; Varela, Simon, Riveros, Jorgensen, & Stutzin, 2004). Future biochemical study, especially on the post-translational modification, of SWELL1 and LRRC8 subunits may provide clues on the mechanisms and signaling cascades leading to VRAC activation under non-swelling conditions.
Up until now, co-expression of SWELL1 and LRRC8 homologs in mammalian cells failed to significantly increase ICl, swell (Voss et al., 2014), although purified LRRC8 heteromers were sufficient to reconstitute VRAC channel activity in vitro (Syeda et al., 2016). It is therefore possible that there is yet another factor or maybe cellular mechanism to limit the amplitude of ICl, swell. This is also supported by a recent study that a pair of closely related cells with very similar LRRC8 gene expression nevertheless had a big difference in their VRAC activity (Okada, Islam, Tsiferova, Okada, & Sabirov, 2017). Alternatively, it’s still a possibility that overexpression of mixed LRRC8 proteins may fail to generate large amounts of correctly assembled functional heteromers due to a stricter stoichiometry than expected. Regardless, it’s important to understand the basis for this observation and examine what cellular factor or mechanism may modulate or limit the VRAC amplitude.
4.2. Role of SWELL1 and VRAC in the Brain Physiology and Disease
Robust VRAC activity is ubiquitously observed and future studies will surely uncover many interesting physiological functions in different tissues and organs (Catalan et al., 2015). Its significance in the brain is particularly intriguing (see two excellent recent reviews on this topic (Akita & Okada, 2014; Mongin, 2016)). Astrocytes undergo dramatic and sustained swelling in a number of pathological conditions, such as ischemic stroke, traumatic brain injury, hyponatremia, brain tumor, and epilepsy (Kimelberg, 2005). Due to VRAC’s unusual ability to support the permeation of organic compounds, activation of VRAC in swelling astrocytes is proposed to be a major pathway for the release of glutamate, the primary excitatory neurotransmitter (Feustel, Jin, & Kimelberg, 2004; Kimelberg, 2005; Kimelberg, Goderie, Higman, Pang, & Waniewski, 1990; Liu, Tashmukhamedov, Inoue, Okada, & Sabirov, 2006). Increase in extracellular glutamate and subsequent excessive activation of neuronal glutamate receptors, known as excitotoxicity, has emerged as an important mechanism of neuronal cell damage in brain diseases (Lai, Zhang, & Wang, 2014). In addition to its role in astrocytic glutamate release, VRAC activation in neurons may mediate Cl− influx and further contribute to cell damage by promoting neuronal swelling (Inoue & Okada, 2007), although a recent study suggested the involvement of a new type of Cl− channel (Rungta et al., 2015). Consistently, VRAC blockers inhibited the release of glutamate and dramatically reduced brain damage in rodent models of ischemic stroke (Alibrahim et al., 2013; Zhang, Zhang, Feustel, & Kimelberg, 2008). With the establishment of the essential role of SWELL1 in swelling-induced neurotransmitter release in vitro (Hyzinski-Garcia et al., 2014; Lutter et al., 2017; Schober et al., 2017), brain-specific Swell1 knockout mouse models will be useful to evaluate its importance in many neurological diseases associated with astrocyte swelling.
One of the paradigm shifts in neuroscience over the last two decades is the appreciation of gliotransmission, the active modulation of neuronal synaptic transmission by astrocytes (Gundersen, Storm-Mathisen, & Bergersen, 2015). However, the mechanism of astrocytic release of gliotransmitters (such as glutamate, D-serine, ATP) has been heavily debated (Fiacco & McCarthy, 2018; Hamilton & Attwell, 2010; Savtchouk & Volterra, 2018). Whereas astrocyte release of neurotransmitters by vesicular exocytosis is controversial, there is strong evidence for the involvement of ion channel-mediated release, such as VRAC, which can be activated by physiological stimuli, including GPCR activation (Liu et al., 2009; Takano et al., 2005). Future studies will elucidate the contribution of SWELL1 and other LRRC8 subunits to the communication between neurons and astrocytes and their physiological and behavioral significance.
4.3. Contribution of LRRC8 Subunits to VRAC Physiology and Beyond
So far, most physiological studies have been focused on the essential VRAC component SWELL1 except for the previous reports on the link between LRRC8C and adipose metabolism (Hayashi et al., 2011; Tominaga et al., 2004). Although the contribution of the other LRRC8 subunits to VRAC diversity is well established, what is the physiological relevance of the electrophysiological and substrate selectivity differences? In the future, thorough analyses on their tissue expression profiles and knockout mouse models will help answer this question. In addition to forming cell surface VRAC channels, it’s conceivable that SWELL1 and its LRRC8 homologs may also form volume-regulated channels on intracellular organelles, such as the endoplasmic reticulum (ER), and that LRRC8B-8E may even have VRAC-independent functions. Interestingly, LRRC8B, which has a minimal role in various aspects of VRAC activity, was recently shown to mediate ER Ca2+ leak in HEK cells (Ghosh, Khandelwal, Kumar, & Bera, 2017). Whether it functions directly as an ER Ca2+ channel or regulates ER Ca2+ levels by indirect mechanisms still requires further investigation. Three groups recently reported the cryo-EM structures of homomeric mouse and human SWELL1 channels, revealing the overall hexameric architecture and ion-conducting pore of VRAC (Deneka et al., 2018; Kefauver et al., 2018; Kasuya et al., 2018). These structures provide a solid foundation for further dissecting the heterogeneity and mechanisms of activation and regulation of VRAC.
ACKNOWLEDGMENTS
We thank the reviewers for their many insightful comments and suggestions. The work in the senior author’s (ZQ) laboratory is supported by a grant from the NIH (R35 GM124824).
REFERENCES
- Abascal F, & Zardoya R (2012). LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 34(7), 551–560. 10.1002/bies.201100173. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Wickman KD, & Clapham DE (1994). Hypotonicity activates a native chloride current in Xenopus oocytes. The Journal of General Physiology, 103(2), 153–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, & Okada Y (2014). Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience, 275, 211–231. 10.1016/j.neuroscience.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Alibrahim A, Zhao LY, Bae CY, Barszczyk A, Sun CL, Wang GL, & Sun HS (2013). Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacologica Sinica, 34(1), 113–118. 10.1038/aps.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, & Rorsman P (2013). K(ATP) channels and islet hormone secretion: New insights and controversies. Nature Reviews. Endocrinology, 9(11), 660–669. 10.1038/nrendo.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best L (2005). Glucose-induced electrical activity in rat pancreatic beta-cells: Dependence on intracellular chloride concentration. The Journal of Physiology, 568(Pt 1), 137–144. 10.1113/jphysiol.2005.093740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best L, Brown PD, Sener A, & Malaisse WJ (2010). Electrical activity in pancreatic islet cells: The VRAC hypothesis. Islets, 2(2), 59–64. 10.4161/isl.2.2.11171. [DOI] [PubMed] [Google Scholar]
- Best L, Yates AP, Decher N, Steinmeyer K, & Nilius B (2004). Inhibition of glucose-induced electrical activity in rat pancreatic beta-cells by DCPIB, a selective inhibitor of volume-sensitive anion currents. European Journal of Pharmacology, 489(1–2), 13–19. 10.1016/j.ejphar.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Bowens NH, Dohare P, Kuo YH, & Mongin AA (2013). DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Molecular Pharmacology, 83(1), 22–32. 10.1124/mol.112.080457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM, & Baumgarten CM (2006). EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. The Journal of General Physiology, 127(3), 237–251. 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burow P, Klapperstuck M, & Markwardt F (2015). Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflügers Archiv, 467(6), 1215–1226. 10.1007/s00424-014-1561-8. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, & Lewis RS (1988). Role of potassium and chloride channels in volume regulation by T lymphocytes. Society of General Physiologists Series, 43, 281–301. [PubMed] [Google Scholar]
- Catalan MA, Kondo Y, Pena-Munzenmayer G, Jaramillo Y, Liu F, Choi S, & Melvin JE (2015). A fluid secretion pathway unmasked by acinar-specific Tmem16A gene ablation in the adult mouse salivary gland. Proceedings of the National Academy of Sciences of the United States of America, 112(7), 2263–2268. 10.1073/pnas.1415739112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compan V, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Martinez CM, Angosto D, & Pelegrin P (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity, 37(3), 487–500. 10.1016/j.immuni.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Dahl G (2015). ATP release through pannexon channels. Philosophical Transactions of the Royal Society of London B Biological Sciences, 370(1672). 10.1098/rstb.2014.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, & Brough D (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nature Communications, 7, 12504 10.1038/ncomms12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneka D, Sawicka M, Lam AKM, Paulino C, & Dutzle R (2018). Structure of a volume-regulated anion channel of the LRRC8 family. Nature, 558, 254–259. 10.1038/s41586-018-0134-y. [DOI] [PubMed] [Google Scholar]
- Droogmans G, Maertens C, Prenen J, & Nilius B (1999). Sulphonic acid derivatives as probes of pore properties of volume-regulated anion channels in endothelial cells. British Journal of Pharmacology, 128(1), 35–40. 10.1038/sj.bjp.0702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, & Hartzell HC (2010). Chloride channels: Often enigmatic, rarely predictable. Annual Review of Physiology, 72, 95–121. 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y, & Kimelberg HK (2004). Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke, 35(5), 1164–1168. 10.1161/01.STR.0000124127.57946.a1. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, & McCarthy KD (2018). Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 38(1), 3–13. 10.1523/JNEUROSCI.0016-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Takahashi Y, Takeshima H, Saitoh C, Shimizu T, Takeguchi N, & Sakai H (2015). Inhibition of gastric H+,K+-ATPase by 4-(2-butyl-6,7-dichloro-2-cyclopenty-lindan-1-on-5-yl)oxybutyric acid (DCPIB), an inhibitor of volume-regulated anion channel. European Journal of Pharmacology, 765, 34–41. 10.1016/j.ejphar.2015.08.011. [DOI] [PubMed] [Google Scholar]
- Gaitan-Penas H, Gradogna A, Laparra-Cuervo L, Solsona C, Fernandez-Duenas V, Barrallo-Gimeno A, & Estevez R (2016). Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophysical Journal, 111(7), 1429–1443. 10.1016/j.bpj.2016.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galietta LJ, Haggie PM, & Verkman AS (2001). Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Letters, 499(3), 220–224. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Khandelwal N, Kumar A, & Bera AK (2017). Leucine-rich repeat-containing 8B protein is associated with the endoplasmic reticulum Ca(2+) leak in HEK293 cells. Journal of Cell Science, 130(22), 3818–3828. 10.1242/jcs.203646. [DOI] [PubMed] [Google Scholar]
- Gradogna A, Gavazzo P, Boccaccio A, & Pusch M (2017). Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. The Journal of Physiology, 595(21), 6719–6733. 10.1113/JP274795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Storm-Mathisen J, & Bergersen LH (2015). Neuroglial transmission. Physiological Reviews, 95(3), 695–726. 10.1152/physrev.00024.2014. [DOI] [PubMed] [Google Scholar]
- Hamilton NB, & Attwell D (2010). Do astrocytes really exocytose neurotransmitters? Nature Reviews. Neuroscience, 11(4), 227–238. 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Nozaki Y, Nishizuka M, Ikawa M, Osada S, & Imagawa M (2011). Factor for adipocyte differentiation 158 gene disruption prevents the body weight gain and insulin resistance induced by a high-fat diet. Biological and Pharmaceutical Bulletin, 34(8), 1257–1263. [DOI] [PubMed] [Google Scholar]
- Hazama A, & Okada Y (1988). Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. The Journal of Physiology, 402, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helix N, Strobaek D, Dahl BH, & Christophersen P (2003). Inhibition of the endogenous volume-regulated anion channel (VRAC) in HEK293 cells by acidic di-aryl-ureas. The Journal of Membrane Biology, 196(2), 83–94. 10.1007/s00232-003-0627-x. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, & Pedersen SF (2009). Physiology of cell volume regulation in vertebrates. Physiological Reviews, 89(1), 193–277. 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Hyzinski-Garcia MC, Rudkouskaya A, & Mongin AA (2014). LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. The Journal of Physiology, 592(22), 4855–4862. 10.1113/jphysiol.2014.278887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, & Okada Y (2007). Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 27(6), 1445–1455. 10.1523/JNEUROSCI.4694-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacoff EY, Jan LY, & Minor DL Jr. (2013). Conduits of life’s spark: A perspective on ion channel research since the birth of neuron. Neuron, 80(3), 658–674. 10.1016/j.neuron.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise T, Shimizu T, Lee EL, Inoue H, Kohno K, & Okada Y (2005). Roles of volume-sensitive Cl− channel in cisplatin-induced apoptosis in human epidermoid cancer cells. The Journal of Membrane Biology, 205(3), 139–145. 10.1007/s00232-005-0779-y. [DOI] [PubMed] [Google Scholar]
- Jackson PS, Morrison R, & Strange K (1994). The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. American Journal of Physiology, 267(5 Pt 1), C1203–C1209. 10.1152/ajpcell.1994.267.5.C1203. [DOI] [PubMed] [Google Scholar]
- Jackson PS, & Strange K (1995). Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. The Journal of General Physiology, 105(5), 661–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ (2016). VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nature Reviews. Molecular Cell Biology, 17(5), 293–307. 10.1038/nrm.2016.29. [DOI] [PubMed] [Google Scholar]
- Kang C, Xie L, Gunasekar SK, Mishra A, Zhang Y, Pai S, … Sah R (2018). SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nature Communications, 9(1), 367 10.1038/s41467-017-02664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, Yanagisawa H, Dohmae N, Hattori M, Ichijo H, Yan Z, Kikkawa M, Shirouzu M, Ishitani R, & Nureki O (2018). Cryo-EM structure of the volume-regulated anion channel LRRC8. bioRxiv. 10.1101/331207. [DOI] [PubMed]
- Kefauver JM, Saotome K, Dubin AE, Pallesen J, Cottrell CA, Hong G, Cahalan SM, Qiu Z, Crowley CS, Whitwam T, Lee W-H, Ward AB, & Patapoutian A (2018). Structure of the Human Volume Regulated Anion Channel. bioRxiv. 10.1101/323584. [DOI] [PMC free article] [PubMed]
- Kimelberg HK (2005). Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia, 50(4), 389–397. 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, & Waniewski RA (1990). Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 10(5), 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L, Chou J, Yee CS, Borzutzky A, Vollmann EH, von Andrian UH, & Geha RS (2014). Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. The Journal of Experimental Medicine, 211(5), 929–942. 10.1084/jem.20131379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Zhang S, & Wang YT (2014). Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Progress in Neurobiology, 115, 157–188. 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Lambert IH, & Hoffmann EK (1994). Cell swelling activates separate taurine and chloride channels in Ehrlich mouse ascites tumor cells. The Journal of Membrane Biology, 142(3), 289–298. [DOI] [PubMed] [Google Scholar]
- Lang F (2007). Mechanisms and significance of cell volume regulation. Journal of the American College of Nutrition, 26(5 Suppl), 613S–623S. [DOI] [PubMed] [Google Scholar]
- Lee CC, Freinkman E, Sabatini DM, & Ploegh HL (2014). The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. Journal of Biological Chemistry, 289(24), 17124–17131. 10.1074/jbc.M114.571257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EL, Shimizu T, Ise T, Numata T, Kohno K, & Okada Y (2007). Impaired activity of volume-sensitive Cl− channel is involved in cisplatin resistance of cancer cells. Journal of Cellular Physiology, 211(2), 513e521. 10.1002/jcp.20961. [DOI] [PubMed] [Google Scholar]
- Liu HT, Akita T, Shimizu T, Sabirov RZ, & Okada Y (2009). Bradykinin-induced astrocyte-neuron signalling: Glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. The Journal of Physiology, 587(Pt 10), 2197–2209. 10.1113/jphysiol.2008.165084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, & Sabirov RZ (2006). Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia, 54(5), 343–357. 10.1002/glia.20400. [DOI] [PubMed] [Google Scholar]
- Lutter D, Ullrich F, Lueck JC, Kempa S, & Jentsch TJ (2017). Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. Journal of Cell Science, 130(6), 1122–1133. 10.1242/jcs.196253. [DOI] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, & Tsukihara T (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature, 458(7238), 597–602. 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Maeno E, Ishizaki Y, Kanaseki T, Hazama A, & Okada Y (2000). Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 97(17), 9487–9492. 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miley HE, Sheader EA, Brown PD, & Best L (1997). Glucose-induced swelling in rat pancreatic beta-cells. The Journal of Physiology, 504(Pt 1), 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minieri L, Pivonkova H, Caprini M, Harantova L, Anderova M, & Ferroni S (2013). The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. British Journal of Pharmacology, 168(5), 1240–1254. 10.1111/bph.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min XJ, Li H, Hou SC, He W, Liu J, Hu B, & Wang J (2011). Dysfunction of volume-sensitive chloride channels contributes to cisplatin resistance in human lung adenocarcinoma cells. Experimental Biology and Medicine (Maywood), 236(4), 483–491. 10.1258/ebm.2011.010297. [DOI] [PubMed] [Google Scholar]
- Mongin AA (2016). Volume-regulated anion channel – a frenemy within the brain. Pflügers Archiv, 468(3), 421–441. 10.1007/s00424-015-1765-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, & Droogmans G (1997). Properties of volume-regulated anion channels in mammalian cells. Progress in Biophysics and Molecular Biology, 68(1), 69–119. [DOI] [PubMed] [Google Scholar]
- Nilius B, Oike M, Zahradnik I, & Droogmans G (1994). Activation of a Cl− current by hypotonic volume increase in human endothelial cells. The Journal of General Physiology, 103(5), 787–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike M, Droogmans G, & Nilius B (1994). The volume-activated chloride current in human endothelial cells depends on intracellular ATP. Pflügers Archiv, 427(1–2), 184–186. [DOI] [PubMed] [Google Scholar]
- Okada Y (1997). Volume expansion-sensing outward-rectifier Cl— channel: Fresh start to the molecular identity and volume sensor. American Journal of Physiology, 273(3 Pt 1), C755–C789. 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Okada T, Islam MR, Tsiferova NA, Okada Y, & Sabirov RZ (2017). Specific and essential but not sufficient roles of LRRC8A in the activity of volume-sensitive outwardly rectifying anion channel (VSOR). Channels, 11(2), 109–120. 10.1080/19336950.2016.1247133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Klausen TK, & Nilius B (2015). The identification of a volume-regulated anion channel: An amazing Odyssey. Acta Physiologica, 213(4), 868–881. 10.1111/apha.12450. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Okada Y, & Nilius B (2016). Biophysics and physiology of the volume-regulated anion channel (VRAC)/Volume-Sensitive outwardly rectifying anion channel (VSOR). Pflügers Archiv, 468(3), 371–383. 10.1007/s00424-015-1781-6. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, … Jentsch TJ (2015). Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. The EMBO Journal, 34(24), 2993–3008. 10.15252/embj.201592409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt CD, Chou J, Houlihan P, Badran YR, Kumar L, Bainter W, … Geha RS (2017). Leucine-rich repeat containing 8A (LRRC8A)-dependent volume-regulated anion channel activity is dispensable for T-cell development and function. The Journal of Allergy and Clinical Immunology, 140(6), 1651–1659. 10.1016/j.jaci.2016.12.974.e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen KA, Andersen EC, Hansen CF, Klausen TK, Hougaard C, Lambert IH, & Hoffmann EK (2010). Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: Role of chloride channels. American Journal of Physiology. Cell Physiology, 298(1), C14–C25. 10.1152/ajpcell.00654.2008. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, & Patapoutian A (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell, 157(2), 447–458. 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, & MacVicar BA (2015). The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell, 161(3), 610–621. 10.1016/j.cell.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Sabirov RZ, Merzlyak PG, Okada T, Islam MR, Uramoto H, Mori T, & Okada Y (2017). The organic anion transporter SLCO2A1 constitutes the core component of the maxi-Cl channel. The EMBO Journal, 36(22), 3309–3324. 10.15252/embj.201796685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchouk I, & Volterra A (2018). Gliotransmission: Beyond black-and-white. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 38(1), 14–25. 10.1523/JNEUROSCI.0017-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada A, Takihara Y, Kim JY, Matsuda-Hashii Y, Tokimasa S, Fujisaki H, … Hara J (2003). A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. Journal of Clinical Investigation, 112(11), 1707–1713. 10.1172/JCI18937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober AL, Wilson CS, & Mongin AA (2017). Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. The Journal of Physiology, 595(22), 6939–6951. 10.1113/JP275053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, & Jan LY (2008). Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell, 134(6), 1019–1029. 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Numata T, & Okada Y (2004). A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(–) channel. Proceedings of the National Academy of Sciences of the United States of America, 101(17), 6770–6773. 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen BH, Dam CS, Sturup S, & Lambert IH (2016). Dual role of LRRC8A-containing transporters on cisplatin resistance in human ovarian cancer cells. Journal of Inorganic Biochemistry, 160, 287–295. 10.1016/j.jinorgbio.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Sorensen BH, Nielsen D, Thorsteinsdottir UA, Hoffmann EK, & Lambert IH (2016). Downregulation of LRRC8A protects human ovarian and alveolar carcinoma cells against Cisplatin-induced expression of p53, MDM2, p21Waf1/Cip1, and Caspase-9/−3 activation. American Journal of Physiology. Cell Physiology, 310(11), C857–C873. 10.1152/ajpcell.00256.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen BH, Thorsteinsdottir UA, & Lambert IH (2014). Acquired cisplatin resistance in human ovarian A2780 cancer cells correlates with shift in taurine homeostasis and ability to volume regulate. American Journal of Physiology. Cell Physiology, 307(12), C1071–C1080. 10.1152/ajpcell.00274.2014. [DOI] [PubMed] [Google Scholar]
- Stotz SC, & Clapham DE (2012). Anion-sensitive fluorophore identifies the Drosophila swell-activated chloride channel in a genome-wide RNA interference screen. PLoS One, 7(10), e46865 10.1371/journal.pone.0046865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange K, Emma F, & Jackson PS (1996). Cellular and molecular physiology of volume-sensitive anion channels. American Journal of Physiology, 270(3 Pt 1), C711–C730. 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Stutzin A, Torres R, Oporto M, Pacheco P, Eguiguren AL, Cid LP, & Sepulveda FV (1999). Separate taurine and chloride efflux pathways activated during regulatory volume decrease. American Journal of Physiology, 277(3 Pt 1), C392–C402. [DOI] [PubMed] [Google Scholar]
- Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, … Patapoutian A (2016). LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell, 164(3), 499–511. 10.1016/j.cell.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, & Nedergaard M (2005). Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proceedings of the National Academy of Sciences of the United States of America, 102(45), 16466–16471. 10.1073/pnas.0506382102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneera J, Lang S, Sharma A, Fadista J, Zhou Y, Ahlqvist E, & Groop L (2012). A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metabolism, 16(1), 122–134. 10.1016/j.cmet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Ternovsky VI, Okada Y, & Sabirov RZ (2004). Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Letters, 576(3), 433–436. 10.1016/j.febslet.2004.09.051. [DOI] [PubMed] [Google Scholar]
- Tominaga K, Kondo C, Kagata T, Hishida T, Nishizuka M, & Imagawa M (2004). The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. Journal of Biological Chemistry, 279(33), 34840–34848. 10.1074/jbc.M312927200. [DOI] [PubMed] [Google Scholar]
- Tsumura T, Oiki S, Ueda S, Okuma M, & Okada Y (1996). Sensitivity of volume-sensitive Cl− conductance in human epithelial cells to extracellular nucleotides. American Journal of Physiology, 271(6 Pt 1), C1872–C1878. 10.1152/ajpcell.1996.271.6.C1872. [DOI] [PubMed] [Google Scholar]
- Ullrich F, Reincke SM, Voss FK, Stauber T, & Jentsch TJ (2016). Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. Journal of Biological Chemistry, 291(33), 17040–17048. 10.1074/jbc.M116.739342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela D, Simon F, Riveros A, Jorgensen F, & Stutzin A (2004). NAD(P)H oxidase-derived H(2)O(2) signals chloride channel activation in cell volume regulation and cell proliferation. Journal of Biological Chemistry, 279(14), 13301–13304. 10.1074/jbc.C400020200. [DOI] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Raskin G, Eggermont J, & Nilius B (1999). Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proceedings of the National Academy of Sciences of the United States of America, 96(9), 5298–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, & Jentsch TJ (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science, 344(6184), 634–638. 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- Wang J, & Dahl G (2010). SCAM analysis of Panx1 suggests a peculiar pore structure. The Journal of General Physiology, 136(5), 515–527. 10.1085/jgp.201010440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Zhang Y, Gunasekar SK, Mishra A, Cao L, & Sah R (2017). Induction of adipose and hepatic SWELL1 expression is required for maintaining systemic insulin-sensitivity in obesity. Channels, 11(6), 673–677. 10.1080/19336950.2017.1373225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Wondergem R, Morrison R, Yin VP, & Strange K (2016). Leucine-rich repeat containing protein LRRC8A is essential for swelling-activated Cl− currents and embryonic development in zebrafish. Physiological Reports, 4(19). 10.14814/phy2.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, & Sah R (2017). SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nature Cell Biology, 19(5), 504–517. 10.1038/ncb3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, & Kimelberg HK (2008). DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Experimental Neurology, 210(2), 514–520. 10.1016/j.expneurol.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]