

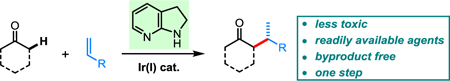
Abstract
Herein, we describe an intermolecular direct branched-selective α-alkylation of cyclic ketones with simple alkenes as the alkylation agents. Through an enamine-transition metal cooperative catalysis mode, the α-alkylation is realized in an atom- and step-economic manner with excellent branched selectivity for preparing β-branched ketones. Employment of a pair of bulky Brønsted acid and base as additives is responsible for enhanced efficiency. Promising enantioselectivity (74% ee) has been obtained. Experimental and computational mechanistic studies suggest that a pathway via alkene migratory insertion into the Ir–C bond followed by C–H reductive elimination is involved for the high branched selectivity.
Keywords: C–C bond formation, branched-selectivity, alkylation, iridium catalysis, cyclic ketones
Entry for the Table of Contents
An intermolecular direct branched-selective α-alkylation of cyclic ketones has been achieved using simple alkenes as the alkylation agents. 7-Azaindoline is employed as a bifunctional ligand to facilitate enamine formation and guide subsequent C−H activation with an iridium catalyst. This method offers a straightforward and byproduct-free means to access ketones with β–stereocenters.
Enolate alkylation is one of the fundamental organic transformations;[1] however, direct alkylation with secondary alkyl halides remains problematic due to their relatively low electrophilicity for SN2 reactions and increased instability under strong basic conditions.[2] In order to access the β-branched ketones, indirect approaches have been developed. The use of more nucleophilic (and more basic) metalloenamines, generated via imine formation followed by deprotonation,[3,4] or an aldol condensation-Michael addition sequence has been frequently utilized (Scheme 1A).[5] Although effectively, these approaches typically require multi-step operations and stoichiometric metallic reagents. In addition, the expense of alkyl halides and consequently generated halogen-containing byproducts could not be neglected. Hence, it would be highly desirable to realize a direct ketone α-alkylation[6] with suitable alkylating agents that would 1) selectively provide branched products, 2) avoid stoichiometric metals or halogens, and 3) operate in a byproduct-free fashion. Clearly, readily available olefins become one almost ideal choice of such alkylating agents.[7]
Scheme 1.
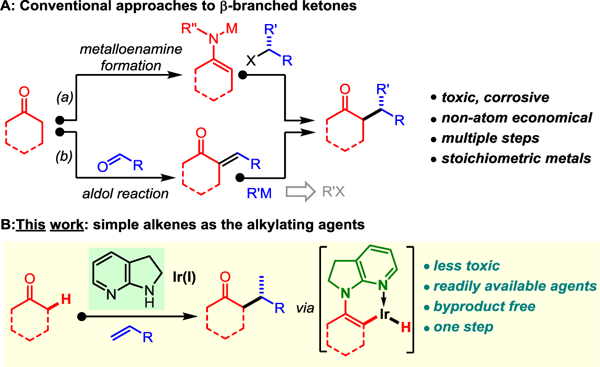
Methods for synthesizing β-branched ketones
Stimulated by the challenge of ketone alkylation with alkenes for yielding β-branched products, our laboratory has recently devised a stepwise enamide-based strategy that enables a net ketone α-alkylation with simple alkenes in high branched selectivity.[8,9] In this approach, 5-membered lactams were employed as the directing template to pre-form enamides with ketones, which further undergo Ir-catalyzed branched-selective coupling with alkenes.[10] Further hydrolysis of the enamide-alkylation products affords ketone α-alkylation products. While this is a feasible strategy, the enamide directing template nevertheless needs to be first installed and later removed in separate steps. Clearly, a direct, one-step approach for branched-selective ketone-olefin coupling without pre-installation and post-removal of directing groups (DGs) would be more attractive. Herein, we describe the initial development of a direct branched-selective α-alkylation of cyclic ketones via enamine-transition metal cooperative catalysis (Scheme 1B).[11] This method allows for a straightforward synthesis of β-branched ketones in a single-step and byproduct-free manner from simple ketones and alkenes.[12]
The challenge to realize such a direct alkylation is two-fold. First, enamines are considerably less stable towards hydrolysis than the corresponding enamides. The instability would inevitably cause a low concentration of the active enamine intermediate, which would lead to low catalytic efficiency. Second, learned from our previous Rh-catalyzed linear-selective alkylation,[13] coupling with non-ethylene olefins were inefficient with the enamine intermediates. Hence, it is not trivial to identify a catalytic system that can afford both high branched selectivity and high reactivity.
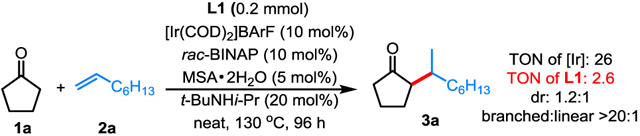
By choosing cyclopentanone 1a and 1-octene 2a as the model substrates, a variety of bifunctional directing templates, Ir precatalysts, ligands and additives were extensively examined. Ultimately, the desired alkylation product was obtained in a 40 turnover number (TON) based on iridium with complete branched selectivity (Table 1, 3a).[14] A set of control experiments revealed that 1) 7-azaindoline L1 is indispensable for this transformation.[15] The use of a series of other types of bifunctional ligands were either ineffective or resulting relative low TON; 2) an acid co-catalyst is essential for improving the efficiency while the bulky MSA (2-mesitylenesulfonic acid) was found to be superior to other sulfonic acids; 3) a catalytic amount of the bulky secondary amine, t-BuNHi-Pr, as an additive was also important to improve the efficiency, its presence was found to significantly reduce the self-aldol side reaction of cyclopentanone thereby facilitating the enamine formation process; 4) among a series of phosphine ligands tested, BINAP-family gave excellent branched selectivity with simple rac-BINAP being optimal.[16]
Table 1.
Scope of the Ir-catalyzed branched-selective α-alkylation of cyclic ketones with simple alkenes.[a]
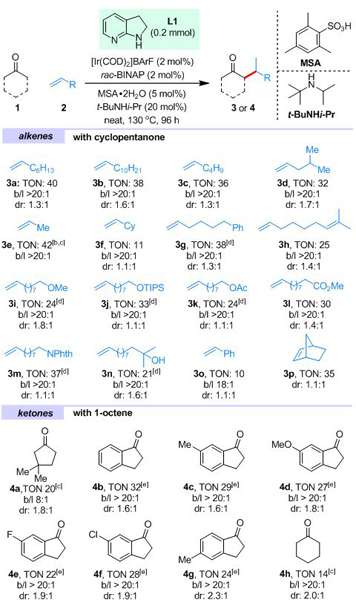 |
Unless otherwise noted, all reactions were run with 0.2 mmol of L1, 0.5 mL of 1 and 0.5 mL of alkene; loadings are based on L1; TONs were calculated based on [Ir] with isolated products; selectivities were determined by 1H NMR or GC-MS of the crude products; b/l = branched:linear ratio.
Pre-condensed propene was used.
The reaction mixture was further treated with 6 M HCl and toluene at 110 °C for 1 h.
2 mmol of alkenes were used.
2 mmol of the ketone substrate was used with 40 mol% of tert-octylamine (instead of t-BuNHi-Pr) based on L1 in 0.3 mL of toluene at 150 °C for 48 h. The reaction was further treated with 6 M HCl at 110 °C for 1 h.
With the optimized reaction conditions in hand, the scope of alkenes was first investigated. Simple alkyl-substituted alkenes all gave α-alkylated cyclopentanones in good efficiency with complete branched selectivity (3b-d). Propene is an excellent coupling partner that introduces an isopropyl group (3e). When vinylcyclohexane was employed, the alkylation product (3f) was obtained in a low TON, probably owing to the steric hindrance of the cyclohexyl group. With diene 2h, the reaction occurred chemoselectively at the mono-substituted alkene. Alkenes bearing a phenyl group or different oxygen/nitrogen-based functional groups, e.g. OMe, OTIPS, OAc, ester, phthalic amine or even free tertiary alcohol, are all compatible in moderate to good efficiency without compromising the regioselectivity (3g, 3i-n). Styrene also worked albeit with a lower TON and a slightly reduced branched selectivity (3o). Di-substituted olefins are generally not reactive under the current conditions; however, norbornene was effectively coupled to afford the desired α-alkylation product (3p) in 35 TON.[17]The poor diastereo-selectivity is likely caused by the enamine hydrolysis that led to thermodynamic products. Nevertheless, the diastereomeric ratio (dr) of the product could be improved through forming the corresponding silyl enol ether followed by a kinetic protonation with a Lewis acid-assisted chiral Brϕnsted acid (eq 1).[18,19]
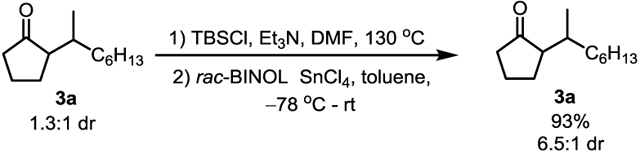 |
(1) |
The ketone scope was also explored. 3,3-Dimethyl cyclopentanone was alkylated exclusively at the less sterically hindered C5 position with a moderate branched selectivity (8:1) (4a). For this substrate, one-pot hydrolysis was required to completely release the alkylated ketone product. 1-Indanones also worked well under slightly modified reaction conditions, and the corresponding α-alkylation products (4c-g) were obtained in moderate to good efficiency. Note that alkylation at the ortho aromatic C−H bond was not observed. Cyclohexanone also gave the desired product albeit in a lower efficiency (4h). Compared to cyclic ketones, linear ketones are much less reactive probably owing to the difficulty to form corresponding enamine intermediates.[20]
 |
(2) |
When increasing the [Ir] loading from 2 mol% to 10 mol% (relative to L1), the reaction gave 2.6 turnovers of L1, indicating certain catalytic ability of the bifunctional ligand (eq 2). Encouraged by this result, we next investigated the alkylation reaction using ketones as the limiting reagent. Gratifyingly, the addition of bulky 2,6-di-t-Bu-4-methylpyridine was found to be the key for increasing the reaction efficiency. Upon further optimizations,[14] the branched product 3a was isolated in 68% yield with 10 mol% of the iridium catalyst (Table 2). With this protocol in hand, 1-hexene and several other terminal alkenes bearing different functional groups as well as norbornene were tested, and the corresponding alkylation products were obtained in 39–62% yields with excellent branched selectivities. Using 1-indanone as the limiting reagent was also successful under similar conditions. The corresponding alkylated enamine was found to be stable under the reaction conditions. After a one-pot hydrolysis, the desired alkylated indanone 4b was isolated in 43% yield. Other substituted 1-indanones were also suitable substrates.
Table 2.
Alkylation with ketones as the limiting reagent. [a]
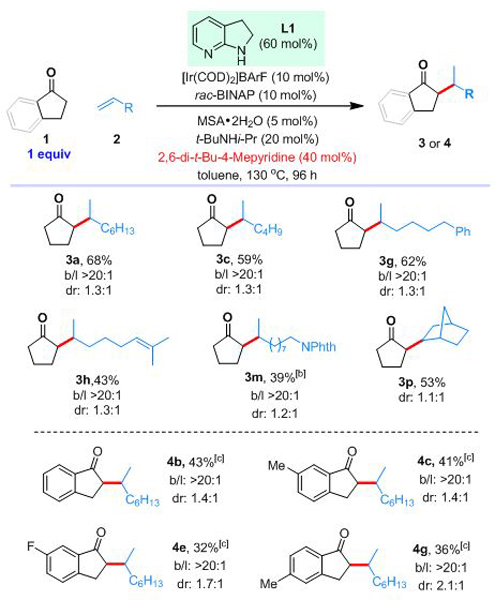 |
Unless otherwise noted, all reactions were run with 0.2 mmol of 1, 0.12 mmol of L1, 13 equiv of alkene in 0.1 mL of toluene; all yields are isolated yields; selectivities were determined by 1H NMR or GC-MS of the crude products.
3 equiv of 2m was used.
The reaction was run at 150 °C and the mixture was further treated with 6 M HCl at 110 °C for 1 h.
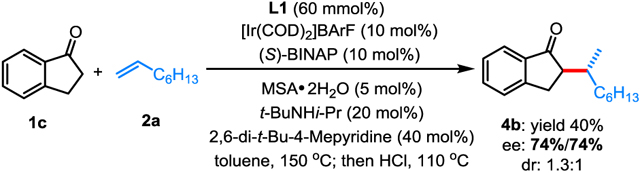
The feasibility of developing an enantioselective version of this transformation was then explored. Employing (S)-BINAP as the ligand, the coupling between 1-indanone 1c and 1-octene 2a afforded product 4b in 40% yield with 1.3:1 dr, and a promising level of enantioselectivity (74% ee) has been obtained for both diastereomers (eq 3).[10i–l]
 |
(3) |
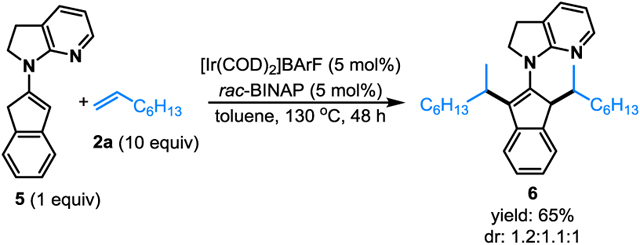
To support the proposed enamine-mediated pathway, pre-formed enamine 5 derived from 2-indanone was used as the testing substrate. Interestingly, dialkylation product 6 was efficiently formed (eq 4).[21] In addition, an Ir–H complex (7) was successfully prepared from enamine 5 with [Ir(coe)2Cl]2 and PMe3 (eq 5).[22,23] Although complex 7 did not react with 1-octene likely owing to the use of strongly coordinating PMe3 ligand, it nonetheless shows that a low-valent iridium complex can insert into enamine vinyl C–H bonds via oxidative addition.[7,9,15]
 |
(4) |
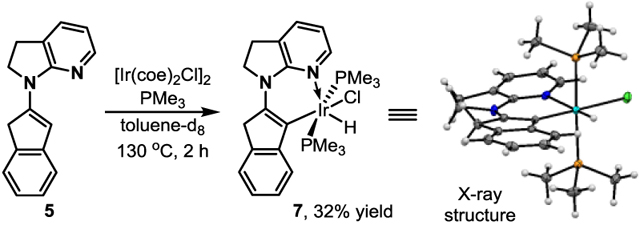 |
(5) |
To gain further mechanistic understandings, the reaction between 2,2,5,5-D4-cyclopentanone (d-1a) and 1-octene was conducted with N-deuterated 7-azaindoline (d-L1), and stopped early (Scheme 2A). The deuterium incorporation at both α and β positions of unreacted 1-octene and the deuterium loss at all the α-positions of the product ketone are consistent with a hypothesis that oxidative addition into the vinyl C–H bond of the in situ formed enamine is reversible and Ir–H migratory insertion into the alkene occurs in both 1,2 and 2,1-addition fashions and is also reversible. In the generated product d-3a, deuterium was transferred to both the terminal methyl and its adjacent methine positions. In addition, the side-by-side reactions of 1a and d-1a were conducted (Scheme 2B). A low intermolecular kinetic isotope effect (KIE) of 1.7 was observed, indicating that the oxidative addition into the enamine vinyl C–H bond is unlikely to be the rate-determining step.
Scheme 2.
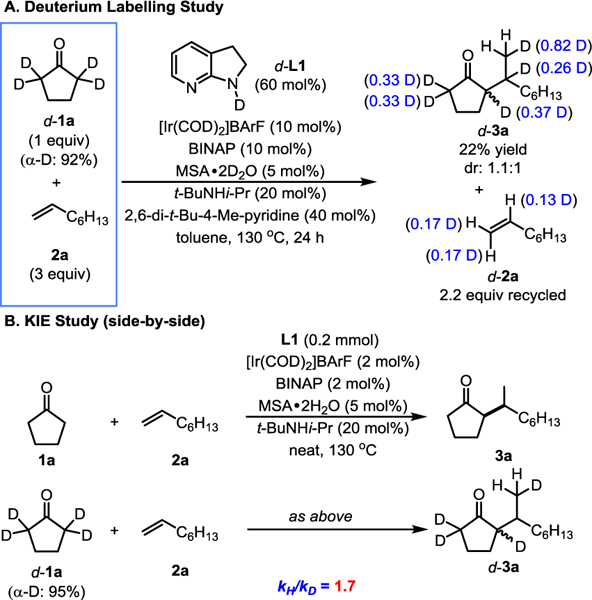
Deuterium labelling studies.
To understand the origin of the high branched selectivity, density functional theory (DFT) calculations were performed on the reaction of ketone 1a and olefin 2c (Figure 1). The computational results indicate the enamine C(sp2)–H oxidative addition to form Ir(III)-hydride 10 and the subsequent alkene migratory insertion into the Ir–H bond (via 15-TS or 15L-TS) both require relatively low kinetic barriers and are reversible, in accordance with the deuterium labelling experiments described above (Scheme 2). However, these Ir–H migratory insertion pathways cannot lead to the alkylation products because of the high barriers of the subsequent C–C reductive elimination (> 30 kcal/mol for 17-TS or 17L-TS). The most favorable pathway involves migratory insertion into the Ir–C bond followed by C–H reductive elimination (via 11-TS and 13-TS). This mechanism is consistent with Huang’s recent study of the enamide system.[9] The regioselectivity is determined in the alkene migratory insertion step, which favours C–C forming with the internal carbon of the alkene (via 11-TS). The 2,1-migratory insertion transition state 11L-TS is destabilized by steric repulsions with the BINAP ligand.[24]
Figure 1.
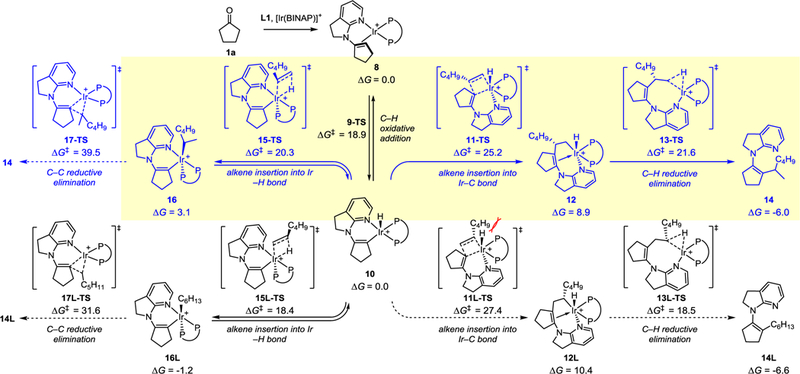
Computed reaction pathways for the formation of branched and linear alkylation products. For details, see the supporting information.
In summary, a direct branched-selective α-alkylation of cyclic ketones has been realized using simple alkenes as the alkylation agents, which offers a byproduct-free access to β-branched ketones. While the efficiency of the reaction remains to be further improved, the dual activation mode demonstrated and mechanistic understanding obtained here should have implications beyond this transformation.
Supplementary Material
Acknowledgements
Financial supports from NSF (CHE-1254935) (G.D.), NSFC (21772043) (D.X.) and NIH (R35GM128779) (P.L.) are acknowledged. D.X. thanks the International Postdoctoral Exchange Fellowship Program 2015 from the Office of China Postdoctoral Council (OCPC, document 38, 2015). Mr Ki-Young Yoon is thanked for the X-ray structure analysis. Ms. Lin Deng is thanked for checking the experiments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dong Xing, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China.
Xiaotian Qi, Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Daniel Marchant, Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Peng Liu, Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Guangbin Dong, Department of Chemistry, University of Chicago, Chicago, IL 60637, USA.
References
- [1].Cain D, Carbon-Carbon Bond Formation (Ed.: Augustine RL), Marcel Dekker; New York, 1979; vol. 1, p. 85. [Google Scholar]
- [2].Smith MB, March J, March’s Advanced Organic Chemistry, 7th Ed; Wiley: New York, 2001. [Google Scholar]
- [3].Whitesell JK, Whitesell MA, Synthesis 1983, 1983, 517. [Google Scholar]
- [4].a) Stork G, Dowd SR, J. Am. Chem. Soc. 1963, 85, 2178; [Google Scholar]; b) Corey EJ, Enders D, Tetrahedron Lett. 1976, 17, 3; [Google Scholar]; c) Hatakeyama T, Ito S, Nakamura M, Nakamura E, J. Am. Chem. Soc. 2005, 127, 14192. [DOI] [PubMed] [Google Scholar]
- [5].a) Perlmutter P, Conjugate Addition Reactions in Organic Synthesis, Pergamon Press, Oxford, 1992; [Google Scholar]; b) Yamamoto Y, Formation of C-C Bonds by Addition to α,β-Unsaturated Carbonyl Compounds in Methods of Organic Chemistry (Ed. Helmchen G, Hoffmann RW, Mulzer J, Schaumann E), Thieme, Stuttgart, 1995, v E21b, p. 2041. [Google Scholar]
- [6].For a review on C−H functionalization of ketones, see: Huang Z, Lim HN, Mo F, Young MC, Dong G, Chem. Soc. Rev. 2015, 44, 7764. [DOI] [PubMed] [Google Scholar]
- [7].Dong Z, Ren Z, Thompson SJ, Xu Y, Dong G, Chem. Rev. 2017, 117, 9333. [DOI] [PubMed] [Google Scholar]
- [8].Xing D, Dong G, J. Am. Chem. Soc. 2017, 139, 13664. [DOI] [PubMed] [Google Scholar]
- [9].For a DFT calculation, see: Li X, Wu H, Lang Y, Huang G, Catal. Sci. Technol. 2018, 8, 2417. [Google Scholar]
- [10].For a recent review on branched-selective hydroarylation of alkenes, see: G. E. M. Crisenza, J. F. Bower, Chem. Lett. 2016, 45, 2. For examples of iridium-catalyzed branched-selective hydroarylation of alkenes, see: [Google Scholar]; a) Tsuchikama K, Kasagawa M, Hashimoto Y-K, Endo K, Shibata T, J. Organomet. Chem. 2008, 693, 3939; [Google Scholar]; b) Pan S, Ryu N, Shibata T, J. Am. Chem. Soc. 2012, 134, 17474; [DOI] [PubMed] [Google Scholar]; c) Sevov CS, Hartwig JF, J. Am. Chem. Soc. 2013, 135, 2116; [DOI] [PubMed] [Google Scholar]; d) Sevov CS, Hartwig JF, J. Am. Chem. Soc. 2014, 136, 10625; [DOI] [PubMed] [Google Scholar]; e) Crisenza GE, McCreanor NG, Bower JF, J. Am. Chem. Soc. 2014, 136, 10258; [DOI] [PubMed] [Google Scholar]; f) Crisenza GE, Sokolova OO, Bower JF, Angew Chem Int Ed. 2015, 54, 14866; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Shirai T, Yamamoto Y, Angew. Chem. Int. Ed. 2015, 54, 9894; [DOI] [PubMed] [Google Scholar]; h) Ebe Y, Nishimura T, J. Am. Chem. Soc. 2015, 137, 5899; [DOI] [PubMed] [Google Scholar]; i) Hatano M, Ebe Y, Nishimura T, Yorimitsu H, J. Am. Chem. Soc. 2016, 138, 4010; [DOI] [PubMed] [Google Scholar]; j) Ebe Y, Onoda M, Nishimura T, Yorimitsu H, Angew. Chem. Int. Ed. 2017, 56, 5607; [DOI] [PubMed] [Google Scholar]; k) Yamauchi D, Nishimura T, Yorimitsu H, Chem. Commun. 2017, 53, 2760; [DOI] [PubMed] [Google Scholar]; l) Grélaud S, Cooper P, Feron LJ, Bower JF, J. Am. Chem. Soc. 2018, 140, 9351. [DOI] [PubMed] [Google Scholar]
- [11].For selected reviews and recent examples on enamine-transition metal catalysis, see: [Google Scholar]; a) Afewerki S, Córdova A, Chem. Rev. 2016, 116, 13512; [DOI] [PubMed] [Google Scholar]; b) Kim D-S, Park W-J, Jun C-H, Chem. Rev. 2017, 117, 8977; [DOI] [PubMed] [Google Scholar]; c) Xu Y, Su T, Huang Z, Dong G, Angew. Chem. Int. Ed. 2016, 55, 2559; [DOI] [PubMed] [Google Scholar]; d) Liu R-R, Li B-L, Lu J, Shen C, Gao J-R, Jia Y-X, J. Am. Chem. Soc. 2016, 138, 5198; [DOI] [PubMed] [Google Scholar]; e) Yang C, Zhang K, Wu Z, Yao H, Lin A, Org. Lett. 2016, 18, 5332; [DOI] [PubMed] [Google Scholar]; f) Manzano R, Datta S, Paton RS, Dixon DJ, Angew. Chem. Int. Ed. 2017, 56, 5834. [DOI] [PubMed] [Google Scholar]
- [12].For examples of intramolecular alkylation of ketones with tethered alkenes that give branched products, see: [Google Scholar]; a) Xiao Y-P, Liu X-Y, Che C-M, Angew. Chem. Int. Ed. 2011, 50, 4937; [DOI] [PubMed] [Google Scholar]; b) Lim HN, Dong G, Angew. Chem. Int. Ed. 2015, 54, 15294. [DOI] [PubMed] [Google Scholar]
- [13].a) Mo F, Dong G, Science 2014, 345, 68; [DOI] [PubMed] [Google Scholar]; b) Mo F, Lim HN, Dong G, J. Am. Chem. Soc. 2015, 137, 15518. [DOI] [PubMed] [Google Scholar]
- [14].For detailed condition optimizations, see the Supporting Information.
- [15].For a DFT calculation on the indispensable role of 7-azaindoline, see: Dang Y, Qu S, Tao Y, Deng X, Wang Z-X, J. Am. Chem. Soc. 2015, 137, 6279. [DOI] [PubMed] [Google Scholar]
- [16].While excess 1-octene was used to ensure the high TON, only a very small portion of the alkene isomerized to internal octenes with most being intact.
- [17].The exo/endo selectivity for the norbornene-derived alkylation product 3p is unclear yet.
- [18].Nakamura S, Kaneeda M, Ishihara K, Yamamoto H, J. Am. Chem. Soc. 2000, 122, 8120. [Google Scholar]
- [19].Attempts to determine the configuration of the major diastereomer by 2D NMR or X-ray analysis of its corresponding 2,4-dinitrophenyl hydrazone product were unsuccessful.
- [20].Both 1- and 2-tetralones also failed to provide desired products.
- [21].Dialkylation products were not observed for other cyclic ketones being tested.
- [22].Using the [Ir(COD)2]BArF/BINAP system, a tiny yet clear metal-hydride peak was observed by in situ 1H NMR analysis. However, attempts to isolate and characterize this metal-hydride intermediate was unfruitful yet.
- [23].CCDC 1819814 (7) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- [24].Huang G, Liu P, ACS Cat. 2016, 6, 809. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.