

Abstract
Whether interactions between synaptotagmin-1 (syt-1) and the soluble NSF attachment protein receptors (SNAREs) are required during neurotransmission is debated. We examined five SNAP-25 mutations designed to interfere with syt-1 interactions. One mutation, D51/E52/E55A, targeted negative charges within region II of the primary interface (Zhou et al., 2015); two mutations targeted region I (D166A and D166/E170A) and one mutation targeted both (D51/E52/E55/D166A). The final mutation (D186/D193A) targeted C-terminal residues not expected to interact with syt-1. An in vitro assay showed that the region I, region II, and region I+II (D51/E52/E55/D166A) mutants markedly reduced the attachment between syt-1 and t-SNARE-carrying vesicles in the absence of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. In the presence of PI(4,5)P2, vesicle attachment was unaffected by mutation. When expressed in Snap-25-null mouse autaptic neurons, region I mutations reduced the size of the readily releasable pool of vesicles, whereas the region II mutation reduced vesicular release probability. Combining both in the D51/E52/E55/D166A mutation abrogated evoked release. These data point to a division of labor between region I (vesicle priming) and region II (evoked release). Spontaneous release was disinhibited by region I mutations and found to correlate with defective complexin (Cpx) clamping in an in vitro fusion assay, pointing to an interdependent role of synaptotagmin and Cpx in release clamping. Mutation in region II (D51/E52/E55A) also unclamped release, but this effect could be overcome by synaptotagmin overexpression, arguing against an obligatory role in clamping. We conclude that three synaptic release functions of syt-1, vesicle priming, spontaneous release clamping, and evoked release triggering, depend on direct SNARE complex interaction.
SIGNIFICANCE STATEMENT The function of synaptotagmin-1 (syt-1):soluble NSF attachment protein receptor (SNARE) interactions during neurotransmission remains unclear. We mutated SNAP-25 within the recently identified region I and region II of the primary synaptotagmin:SNARE interface. Using in vitro assays and rescue experiments in autaptic neurons, we show that interactions within region II of the primary interface are necessary for synchronized calcium-triggered release, whereas region I is involved in vesicle priming. Spontaneous release was disinhibited by region I mutation and found to correlate with defective complexin (Cpx) clamping in vitro, pointing to an interdependent role of synaptotagmin and Cpx in release clamping. Therefore, vesicle priming, clamping spontaneous release, and eliciting evoked release are three different functions of syt-1 that involve different interaction modes with the SNARE complex.
Keywords: autaptic neuron, glutamatergic synapse, patch-clamp, SNAP-25, SNARE, synaptotagmin
Introduction
The machinery for synaptic release includes the soluble NSF attachment protein receptor (SNARE) complex formed from syntaxin-1, SNAP-25, and VAMP2 (Jahn and Fasshauer, 2012; Fang and Lindau, 2014; Rizo and Xu, 2015). Synaptotagmin-1 (syt-1) is the calcium sensor for synchronous neurotransmitter release (Fernández-Chacón et al., 2001). Multiple syt-1 interaction partners were identified, among them phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) (Brose et al., 1992; Davletov and Südhof, 1993; Schiavo et al., 1996), phosphatidylserine (Fukuda et al., 1996; Zhang et al., 2010), AP-2 (Zhang et al., 1994), Ca2+ channels (Leveque et al., 1992; Kim and Catterall, 1997), syntaxin-1 (Bennett et al., 1992; Chapman et al., 1995; Li et al., 1995; Kee and Scheller, 1996), SNAP-25 (Gerona et al., 2000; Zhang et al., 2002), and the SNARE complex (Söllner et al., 1993; Davis et al., 1999; Bai et al., 2004; Pang et al., 2006b; Lynch et al., 2008; Choi et al., 2010; Brewer et al., 2015). A recent crystallographic study revealed three separate syt-1:SNARE interfaces: primary, secondary, and tertiary (Zhou et al., 2015). The primary interface includes the C2B domain (the second C2 domain) of syt-1 and is subdivided into two regions dominated by polar interactions. Region I includes several charges in SNAP-25, among them D166, which interacts with Y338 in syt-1 C2B, whereas region II includes the D51/E52/E55-patch in SNAP-25 (Rickman et al., 2006), which interacts with R398 and R399 at the “bottom” (i.e., opposite to the Ca2+-binding top loops) of the C2B domain. R398 and R399 had been identified previously as being critical for release (Gaffaney et al., 2008; Xue et al., 2008a; Kedar et al., 2015). The secondary interface includes interactions between the C2B bottom domain and syntaxin-1 and the tertiary interface is formed between C2A and syntaxin-1/VAMP2. Mutagenesis of syt-1 implicated the primary and secondary interfaces in evoked release (Zhou et al., 2015). However, the syt-1:SNARE interaction has been questioned and it was suggested that syt-1 binds to PI(4,5)P2 at physiological ionic strength (Park et al., 2015).
Physiologically, the deletion of syt-1 or syt-2 eliminates synchronous release (Nonet et al., 1993; DiAntonio and Schwarz, 1994; Geppert et al., 1994; Fernández-Chacón et al., 2001; Sun et al., 2007). In most cases, syt-1 or syt-2 deletion causes a simultaneous increase in spontaneous release rates (Broadie et al., 1994; Geppert et al., 1994; Pang et al., 2006a; Kerr et al., 2008; Liu et al., 2009; Kochubey and Schneggenburger, 2011; Wierda and Sørensen, 2014). Mutagenesis studies have shown that clamping spontaneous release involves a separate syt function (Kochubey and Schneggenburger, 2011; Lee et al., 2013; Liu et al., 2014), which might include an association with the SNAREs in a different configuration than during evoked release (Bai et al., 2016). The inhibitory effects of syt-1 on release after train stimulation (Maximov and Südhof, 2005) has been attributed to the clamping of a secondary calcium sensor (Südhof, 2013), presumably syt-7 (Bacaj et al., 2013), but the origin of syt-1 clamping of unstimulated spontaneous release remains unclear. Complexin (Cpx) is another SNARE-interacting fusion clamp, which exerts species specific positive and negative effects on synaptic release (Xue et al., 2009). The negative effects are induced by the “accessory helix,” which extends along the C terminus of the SNARE complex and might inhibit exocytosis electrostatically (Trimbuch et al., 2014). Recently, a redundant function for syt-1 and syt-7 in maintaining the size of the readily releasable vesicle pool (RRP) was identified, which correlated with the ability of syt-1 to drive the formation of SNARE complexes (Bacaj et al., 2015).
Most mutagenesis studies exploring the syt:SNARE interaction have used syt-1 mutations. Because syt-1 binds to many effectors, it is important to examine the syt:SNARE interaction from the SNARE complex side. This is also necessary to understand how syt-1:SNARE-interaction is involved in clamping spontaneous release: if competition with another sensor associating to the same site underlies the increase in spontaneous release in the syt-1 knock-out, then this increase would not be expected when mutating the SNAREs to eliminate the interaction surface.
Materials and Methods
Reconstituted small unilamellar vesicle (SUV)–giant unilamellar vesicle (GUV) attachment and membrane fusion (lipid-mixing) assays.
The DNA construct (pTW34) encoding both untagged syntaxin 1A and His6-tagged SNAP-25B was described previously (Weber et al., 1998). pTW34 was used as template DNA to introduce mutations into SNAP-25 by QuikChange DNA mutagenesis (Qiagen). The v-SNARE constructs were GST-tagged mouse VAMP2 (pSK28; Kedar et al., 2015) and His6-tagged rat syt-1 lacking the lumenal domain (pLM6; Mahal et al., 2002). For Cpx, a DNA construct (pMDL80) encoding His6-tagged human Cpx II was used (Malsam et al., 2012). All constructs were expressed and purified as described previously (Weber et al., 1998; Malsam et al., 2012; Kedar et al., 2015). The concentration of purified proteins was determined using Coomassie blue-stained SDS-PAGE using bovine serum albumin as the standard protein and ImageJ software for quantification.
Unlabeled lipids were from Avanti Polar Lipids. 3H-DPPC (3H-1,2-dipalmitoyl phosphatidylcholine) was from GE Healthcare/Pharmacia Biotech. Atto448-DPPE (Atto448-1,2-dipalmitoyl phosphatidylethanolamine) and Atto550-DPPE were from Atto-Tec. The v-SNARE/syt-1 lipid mixture contained the following: 28.5 mol% POPC (1-palmitoyl-2-oleoyl-SN-glycero-3-phosphocholine), 15 mol% DOPS (1,2-dioleoyl-SN-glycero-3-phosphoserine), 25 mol% POPE (1-hexadecanoyl-2-octadecenoyl-SN-glycero-3-phosphoethanolamine), 5 mol% liver PI (L-α-phosphatidylinositol), 25 mol% cholesterol (from ovine wool), 0.5 mol% Atto488-DPPE, 0.5 mol% Atto550-DPPE and trace amounts of 3H-DPPC, 3 μmol of total lipid. Syntaxin1/SNAP-25 lipid mix: 35 mol% POPC (1-palmitoyl-2-oleoyl-SN-glycero-3-phosphocholine), 15 mol% DOPS, 20 mol% POPE, 3 mol% liver PI, 2 mol% brain PI(4,5)P2 (L-α-phosphatidylinositol-4,5-bisphosphate), 25 mol% cholesterol (from ovine wool), and 5 μmol of total lipid. Proteoliposomes and GUVs were prepared as described previously (Kedar et al., 2015) except that 1 mm EDTA was added to the reconstitution buffer. GUV sedimentation and quantitation were performed as described previously (Malsam et al., 2012). The protein:lipid ratios were as follows: VAMP2, 1:350; syt-1, 1:800; and t-SNARE, 1:1000. The vesicle–vesicle fusion assay and data analysis were performed as described previously (Malsam et al., 2012); the assay as performed here monitors lipid mixing.
Temperature-dependent dissociation of the SNARE complex in SDS.
Full-length t-SNARE complex (pTW34; 15 μm final concentration) was combined with the cytoplasmic domain of VAMP2 (pSK74, aa1–94; 50 μm final concentration) after desalting into 25 mm MOPS (3-(N-morpholino)propanesulfonic acid)-KOH, pH 7.4, 135 mm KCl,1% Octyl β-d-glucopyranoside, 1 mm Dithiothreitol, 10 mm TECEP (Tris(2-carboxyethyl)phosphine hydrochloride)-KOH, pH 7.4, 1 mm EDTA-NaOH, pH 7.4, and incubated overnight at 0°C. The incubation was continued for 1 h at 37°C to complete complex formation. 25 μl of reaction mixture (50 μg of total protein) was diluted with 225 μl of 1× Laemmli buffer (62.5 mm Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 50 mm β-mercaptoethanol, 0.1% bromphenol blue) and aliquots (15 μl) were incubated at the indicated temperature for 5 min. Quantification of syntaxin 1 was performed using ImageJ version 1.43u after separation of samples on 15% SDS-PAGE gels, followed by Coomassie brilliant blue staining. Data were normalized to the maximum value of a measurement set.
Cell culture and lentiviral constructs.
For electrophysiology and immunocytochemistry, isolated hippocampal neurons were plated on astrocyte microislands (Bekkers and Stevens, 1991) to generate autaptic hippocampal cultures. Snap-25 heterozygote mice (Washbourne et al., 2002) were crossed to generate Snap-25-null embryos. Females carrying embryonic day 18 (E18) pups were cervically dislocated and embryos were obtained via caesarean section. Embryos of either sex were killed by decapitation and the hippocampi were dissected and collected in HBSS (Sigma-Aldrich), buffered with 7 mm HEPES, pH 7.35. The hippocampi were then incubated for 15 min in HBSS/HEPES containing 0.25% trypsin in a shaking water bath at 37°C. After washing with HBSS/HEPES, the neurons were triturated, counted, and plated in Neurobasal medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), 1.8% HEPES, 1% Glutamax (Invitrogen), 1% penicillin/streptomycin (Invitrogen), and 0.2% β-mercaptoethanol. Neurons were plated at a density of 1000/cm2 on microislands of mouse glia. To obtain glial islands, cleaned glass coverslips were first coated with 0.15% agarose. After drying and UV sterilization, custom-made rubber stamps were used to print dots with a diameter of 200–250 μm using a substrate mixture containing 0.4 mg/ml poly-d-lysine and 0.25 mg/ml rat tail collagen dissolved in 17 mm acetic acid. Mouse glial cells isolated from CD1 mice were plated at 4900/cm2. The lentiviral GFP-SNAP-25 WT construct has been described previously (Delgado-Martínez et al., 2007). GFP-SNAP-25B mutants were generated by site-directed mutagenesis using overlapping primers or by the QuikChange II XL Site-directed mutagenesis kit (Agilent Technologies) and subcloned into the pLenti vector under control of a CMV promotor. Viral particles were produced as described previously (Naldini et al., 1996). For syt-1 knock-down experiments, a lentiviral m-Cherry-tagged shRNA construct was kindly provided by the Südhof laboratory (Xu et al., 2012). Neurons were transduced at 0 d in vitro (DIV0).
Electrophysiological recordings.
Isolated hippocampal neurons were recorded between DIV10 and DIV14. Glial islands containing one single neuron were first identified by visual inspection under the microscope. Upon successful whole-cell patch-clamp of a cell, the presence of additional connected neurons on the same or neighboring island was ruled out based on the absence of postsynaptic currents containing multiple peaks upon stimulation of action potentials in the patched cells. Excitation of a neighboring, unclamped neuron results in a recurrent postsynaptic response, visible as a multicomponent postsynaptic current at the clamped cell. The intracellular pipette solution contained the following (in mm): 136 KCl, 18 HEPES, 15 creatine phosphate, 4 Na-ATP, 4.6 MgCl2, 1 EGTA, and 50 U/ml phospocreatine kinase (300 mOsm), pH 7.3. The external medium used for recordings contained the following (in mm): 140 NaCl, 2.4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 14 glucose (300 mOsm), pH 7.4. For the calcium gradient recordings in Figure 5, the medium contained the following (in mm): 127 NaCl, 2.4 KCl, 4 MgCl2, 10 HEPES, 12 glucose, and supplementary 1–12 CaCl2. For sucrose measurements, the external medium was supplemented with an additional 500 mOsm sucrose.
Figure 5.
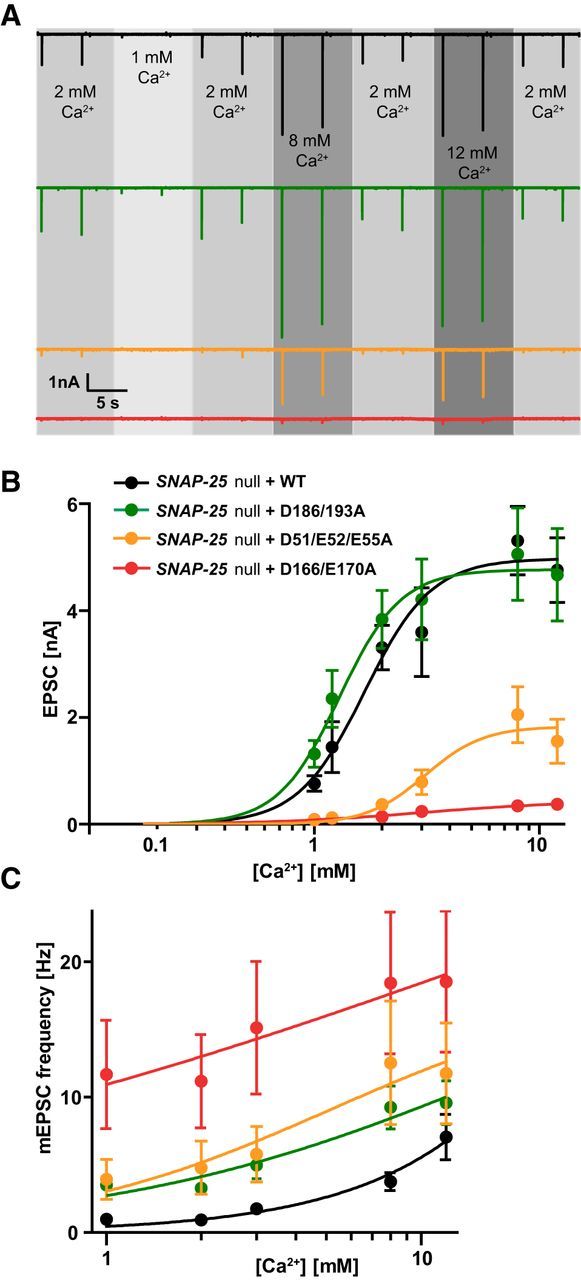
Ca2+ sensitivity of evoked and spontaneous release. A, Representative traces of 0.2 Hz train stimulation in extracellular [Ca2+] ranging between 1 and 12 mm, with constant 4 mm Mg2+. Each experimental [Ca2+] is bracketed by two single stimulations in 2 mm Ca2+ to ensure recording stability and quality. B, Linear log plot of EPSC amplitudes and Ca2+ concentrations. Lines are fits of the Hill equation. EPSCs were pooled from dataset 1 [1 mm Ca2+, 2 mm Ca2+, 8 mm Ca2+, 12 mm Ca2+. n = 18 (WT), 10 (D186/D193A), 11 (D51/E52/E55A), 13 (D166/E170A)] and dataset 2 [1 mm Ca2+, 1.2 mm Ca2+, 2 mm Ca2+, 3 mm Ca2+. n = 12 (WT), 13 (D186/D193A), 10 (D51/E52/E55A), 12 (D166/E170A)], with EPSC amplitude at 2 mm Ca2+ not significantly different between same conditions from dataset 1 and 2. Plot displays mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn's post test). C, Lin-log plot of mEPSC frequencies and Ca2+ concentrations. Plot displays mean ± SEM.
Cells were patch clamped at room temperature in whole-cell voltage-clamp mode with a holding potential of −70 mV by an EPC 10 amplifier (HEKA Elektronik), supplied with Patchmaster version version 2x53 software (HEKA Elektronik). Recorded currents were low-pass filtered at 2.9 kHz and sampled at 20 kHz. Patch pipettes had a resistance between 2 and 5 MΩ and only cells with a series resistance <10 MΩ were included in the analysis. The compensation of the series resistance was set to 70%. Action potentials and resulting EPSCs were evoked using a brief 2 ms depolarization of the soma to 0 mV. Sucrose and different calcium concentrations were applied via a custom-made barrel system digitally controlled by the trigger interface TIB 14S (HEKA Elektronik) and the SF-77B Perfusion Fast-Step (Warner Instrument). The mean EPSC amplitudes for the different calcium concentrations in each condition were fitted with the Hill function, I = (Imax × [Ca2+]c)/(KDc + [Ca2+]c), where I is the recorded EPSC amplitude, Imax the maximal current measured at the individual condition, KD the apparent calcium affinity, c the apparent cooperativity, and [Ca2+] the calcium concentration.
The identity of recorded glutamatergic neurons was determined by visual analysis of decay time kinetics in spontaneous and evoked release (Wierda and Sørensen, 2014). Spontaneous events were detected and analyzed using the Mini Analysis program (Synaptosoft). Igor Pro version 6.21 was used for the analysis of evoked recordings and sucrose stimulation responses. All autaptic cells that displayed either spontaneous or evoked release were included in the analysis.
Immunocytochemistry.
Autaptic cultures were fixed in 4% PFA at DIV10–14 for 20 min. After washing with PBS, neurons were blocked for 1 h in 0.25% Tween 20 (Sigma-Aldrich) in PBS containing 4% normal goat serum (NGS). Cells were incubated overnight at 4°C with primary antibodies α-GFP (1:2000, mouse, 132011; Synaptic Systems), α-MAP2 (1:1000, chicken, AB15452; Millipore), and α-VGLUT1 (1:4000, rabbit, 135303; Synaptic Systems) in 4% NGS/0.02% Tween 20/PBS. After washing with 0.02% Tween 20/PBS, the secondary antibodies goat α-mouse-Alexa Fluor 488 (A11029; Invitrogen), goat α-rabbit-Dylight 650 (ab96950; Abcam), and goat α-chicken-Alexa Fluor 546 (A-11010; Thermo Fisher) each at 1:1000 were applied for 2 h at room temperature. After the washing steps, the coverslips were mounted onto glass slides using FluorSave (345789; Calbiochem). Z-stack images were acquired at a Zeiss LSM 710 confocal microscope with a Plan-Apochromat 20× air objective, numerical aperture 0.8 (420650–9901-000; Zeiss) using ZEN imaging software (black edition). Maximum intensity projection was performed in ImageJ.
Synapses were detected using the automated synapse recognition software SynD (Schmitz et al., 2011). First, the dendritic arborization of an isolated neuron was manually traced by MAP2 staining. Second, the threshold for synapse detection was set to 0.6 μm2 to exclude dots representing smaller intracellular compartments or noise. Last, only dots appearing on the dendritic mask or in a maximal distance of 1 μm were included.
Western blot.
High density cortical cultures were harvested on DIV 10–13 with RIPA (Sigma-Aldrich)/proteinase inhibitor (1 mg/ml) and a plastic cell scraper and then centrifuged for 10 min at 17,000 × g (4°C). The supernatant's protein concentration was visualized with a Pierce BCA protein assay kit and read by a POLARstar Omega (BMG Labtech). A constant amount of reduced and denatured protein (supplied with LDS sample buffer, LDS sample reducing agent; Thermo Fisher) was run on a Novex Bolt 4–12% Bis-Tris Plus Gel at 120 mV along with SeeBlue Plus2 Prestained Standard (Thermo Fisher). Proteins were afterward transferred to a Hybond LFP PVDF membrane (GE Healthcare) at 16 V via an SD semidry transfer cell (Bio-Rad) and the membrane was blocked in 5% milk in TBS-Tween 20 (TBST) 0.1% for 1 h at room temperature. Incubation in primary antibodies (α-SNAP-25: rabbit, 1:1000, ab5666, Abcam; α-Syt-1: mouse, 105011; Synaptic Systems) was performed overnight with 50 rpm shaking at 4°C, followed by washing in TBST (0.1%) and a 1 h incubation in secondary antibody (goat α-rabbit-HRP: 1:10,000, P0448, Dako; goat α-mouse-HRP: 1:10,000, P0447, Dako). After washing, Pierce ECL Western blotting substrate was added and chemiluminescence was visualized with FluorChem E (Proteinsimple). Actin was detected with α-β actin-HRP (mouse, 1:25000, A3854; Sigma-Aldrich).
Statistics.
Values shown are the mean ± SEM, with n referring to the number of recorded neurons for each condition or the number of repetitions (for the biochemical experiments). The reconstituted SUV–GUV fusion and vesicle attachment assays were analyzed with a one-way ANOVA, followed by Dunnett's post test with the WT complex as the control group. All other datasets were tested using the Kruskal–Wallis test. If a statistical difference was found within the population of these groups, then the differences between individual group medians were calculated using Dunn's method. Statistical significance was assumed when the two-tailed p < 0.05. Statistical testing was performed using GraphPad Prism version 5.01 software. In figures, the significance levels are indicated by asterisks as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Eliminating negative surface charges in SNAP-25 leads to impaired calcium-triggered membrane fusion in vitro
Here, we used five different SNAP-25 mutations to investigate the proposed interaction interface between the SNARE protein SNAP-25 and syt-1. One mutation (D51/E52/E55A; Rickman et al., 2006) targeted negative charges within region II of the primary interface (Zhou et al., 2015); two mutations targeted region I (D166A and D166/E170A; Mohrmann et al., 2013), and one mutation targeted both (D51/E52/E55/D166A). The final mutation (D186/D193A) targeted charges in the C-terminal end of the SNARE complex, which did not interact with syt-1 in recent structures (Brewer et al., 2015; Zhou et al., 2015). Our previous studies in adrenal chromaffin cells showed that D51/E52/E55A and D166/E170A interfered with calcium-triggered release (Mohrmann et al., 2013). However, because there are important differences between synaptic transmission in central neurons and chromaffin cell adrenaline release (Neher, 2006), it is important to test synaptic release. In addition, the question of how syt-1:SNARE interactions affect spontaneous release is open. Finally, it is desirable to test the functionality of these mutants in an in vitro setting to understand which parts of their physiological function are understandable in a minimal system, where confounding factors (for instance alternative calcium sensors) are absent.
We first created three mutants in SNAP-25B based on our previous work (Mohrmann et al., 2013): D51/E52/E55A, D166/E170A, and D186/D193A, as shown in Figure 1A. We used GUVs containing the t-SNAREs syntaxin-1 and SNAP-25 (WT or mutant) and 3H-labeled SUVs containing the v-SNARE VAMP2 and syt-1 to study vesicle attachment and in vitro lipid mixing indicative of vesicle fusion (Parisotto et al., 2012). For vesicle attachment, the SUVs (50 μm lipid, 143 nm VAMP2, 62 nm syt-1) were mixed with the GUVs (280 μm lipid, 280 nm syntaxin-1/SNAP-25) and incubated for 5 min on ice, followed by centrifugation at 5000 × g to reisolate the GUVs. GUV pellets were analyzed for the fraction of bound SUVs in the presence or absence of 2% PI(4,5)P2. In the presence of PI(4,5)P2, all mutants had a similar number of SUVs attached to GUVs as the WT (Fig. 1B). Including Cpx II into the mixture did not further stimulate attachment (Parisotto et al., 2012). Without PI(4,5)P2 in the GUVs, vesicle attachment in the presence of the WT SNARE complex was reduced from 71.6 ± 2.2% to 48.7 ± 0.7% (Fig. 1C). Under these circumstances, D51/E52/E55A and D166/E170A mutants showed a strong decrease in vesicle attachment; the D186/D193A mutant was less severe, but still resulted in significantly reduced vesicle attachment (Fig. 1C). Again, vesicle attachment was unaffected by the presence of Cpx II. These data are consistent with the notion that, in this in vitro setting, syt-1 drives vesicle attachment of SUVs to GUVs by binding alternatively to PI(4,5)P2 and to the SNAREs (Kim et al., 2012; Parisotto et al., 2012). It is also possible that, when both species are present, syt-1 binds simultaneously to both (Brewer et al., 2015; Zhou et al., 2015).
Figure 1.
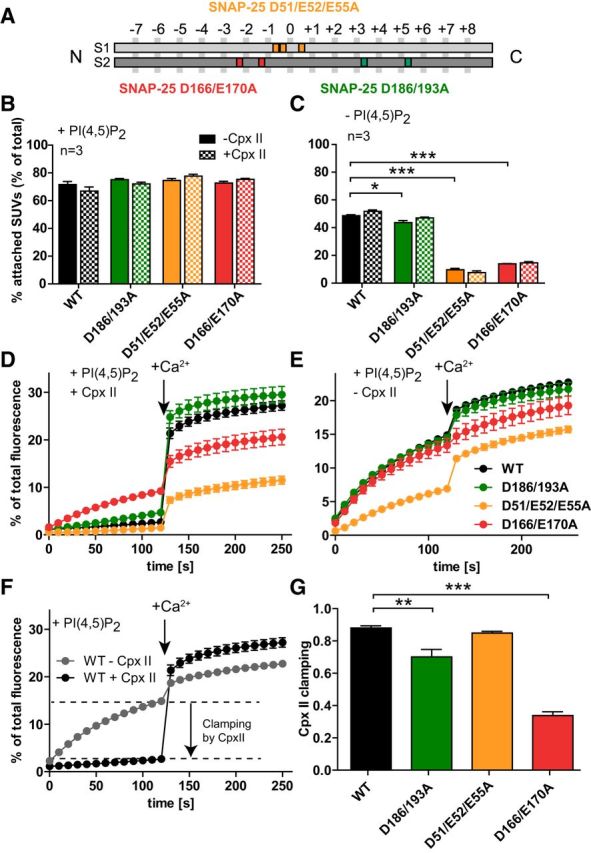
Vesicle–vesicle attachment and fusion in vitro are inhibited by the SNAP-25B mutations D166/E170A and D51/E52/E55A. A, Schematic representation of the mutants SNAP-25B D51/E52/E55A (orange), which is located around the hydrophilic zero layer of the SNARE helix 1 (S1), SNAP-25B D166A/E170A (red), and SNAP-25B D186/D193A (green), both located on the SNARE helix 2 (S2). B, C, t-SNARE-GUVs were mixed with 3H-labeled v-SNARE/syt-1-SUVs with and without Cpx II in the presence (B) or absence (C) of PI(4,5)P2. Samples were incubated for 5 min on ice to allow SUV:GUV attachment, followed by centrifugation to reisolate GUV. 3H-labeled SUVs that were attached to GUVs were quantified and normalized to total input. D, E, t-SNARE-GUVs containing SNAP-25 WT (black), SNAP-25B D186/D193A (green), SNAP-25B D51/E52/E55A (orange), or SNAP-25B D166/E170A (red) were mixed with v-SNARE-SUVs in the presence (D) or absence (E) of Cpx II. The increase of Atto488 fluorescence due to lipid mixing was monitored. After 2 min at 37°C, Ca2+ was added to a final concentration of 100 μm and the measurement continued for another 2 min. Values were normalized to the maximum fluorescence signal after detergent lysis. F, Direct comparison of lipid mixing for WT SNAP-25 in the absence (gray) and in the presence (black) of Cpx II. Cpx II clamps most of the prestimulation membrane fusion. G, Fractional clamping effect of Cpx II for the different constructs. Note that D166/E170A inhibits Cpx II clamping. All graphs display mean ± SEM (n = 3). *p < 0.05, **p < 0.01; ***p < 0.001 (one-way ANOVA, significance was calculated using Dunnett's post test).
Because vesicle attachment through a syt-1:SNARE interaction was strongly perturbed by our SNAP-25 mutations, whereas by nature the syt-1:PI(4,5)P2 interaction remained intact, we used these mutants to study specifically the role of the syt-1:SNARE-interaction. We first performed membrane fusion in an in vitro assay. 3H-labeled v-SNARE-SUVs (25 μm lipid, 71 nm VAMP2, 31 nm syt-1) contained 0.5 mol% Atto488- and 0.5%mol Atto550-labeled lipids, resulting in fluorescence resonance energy transfer (FRET) upon Atto488 excitation. The SUVs were mixed with t-SNARE-GUVs (140 μm lipid, 140 nm syntaxin-1/SNAP-25) containing either SNAP-25 WT or the SNAP-25 mutants in the presence and absence of 6 μm Cpx II to control fusion (see below). As a result of lipid mixing during vesicle–vesicle fusion, FRET efficiency is decreased and can be registered by increased donor (Atto488) fluorescence intensity. Fluorescence was measured at 37°C and, after 2 min, Ca2+ was added to a final concentration of 100 μm; the measurement continued for another 2 min. The intensity of fluorescence was normalized to the maximum fluorescent signal obtained after detergent lysis. The assays showed a severe impairment in calcium-dependent fusion for the D51/E52/E55A and the D166/E170A mutant (Fig. 1D), whereas the D186/D193A mutant displayed very similar fusion to that of the SNAP-25 WT experiments performed in parallel experiments (Fig. 1D). Strikingly, the D166/E170A and, to a lesser degree, the D186/D193A mutant also displayed increased fusion before the Ca2+ stimulus.
In Figure 1D, Cpx II was included in the lipid-mixing assay. Cpx II, together with Cpx I, is highly abundant in neurons and binds to the SNARE complex in the groove between VAMP2 and syntaxin-1 (Chen et al., 2002). Cpx reduces the frequency of spontaneous release in in vitro membrane fusion systems (Malsam et al., 2012; Lai et al., 2014); that is, it acts as a fusion clamp. As observed previously (Malsam et al., 2012), there was a high rate of fusion in the Cpx II-free environment even without the calcium trigger (Fig. 1E). Spontaneous lipid mixing was similar for the D166/E170A and D186/D193A mutants, but reduced for the D51/E52/E55A mutant. Ca2+-dependent membrane fusion was almost completely absent in the D166/E170A mutant without Cpx II (Fig. 1E).
For each construct, we compared fusion with and without Cpx II and determined the fractional reduction in lipid mixing rate by Cpx II before the Ca2+ trigger (Fig. 1F). In the WT case, Cpx II clamped 88.0 ± 1.3% of the Ca2+-independent fusion (Fig. 1F,G). This fraction was significantly reduced for the D186/D193A and even more for the D166/E170A mutant (Fig. 1G), whereas the D51/E52/E55A mutant displayed indistinguishable Cpx clamping from WT. The latter mutant had reduced calcium independent fusion both with and without Cpx (Fig. 1D,E), but the degree of Cpx clamping was intact. These data surprisingly show that some mutations in SNAP-25 affect clamping by Cpx, although Cpx does not itself interact with SNAP-25 when bound to the SNARE complex (Chen et al., 2002).
Mutating the binding site for syt-1 in SNAP-25 does not alter cell survival, morphology, and synapse density
In low-density hippocampal Snap-25-null cultures most cells die after several days (Washbourne et al., 2002; Delgado-Martínez et al., 2007). Introducing the aforementioned SNAP-25 mutants fused to the C terminus of EGFP by lentiviral transduction in a Snap-25-null background led to rescue of the neuronal survival rate to the same extent as with WT EGFP-SNAP-25B (Fig. 2A). Western blotting was used to assess the expression level of all SNAP-25 mutants and it was found that their levels were similar to the WT (EGFP-SNAP-25) rescue construct (Fig. 2B). Compared with endogenous SNAP-25 expression, the virally introduced SNAP-25 was expressed in an approximately double amount (Fig. 2B). To test for normal synapse formation in autaptic hippocampal cultures, cells were fixed and stained against EGFP (a marker for SNAP-25), MAP2 (a dendritic marker), and VGLUT1 (a marker for glutamatergic vesicles) at DIV10–14 (Fig. 2C) and analyzed with SynD software (Schmitz et al., 2011), which allows automatic synapse detection. The number of synapses was determined by quantification of the presynaptic VGLUT1 puncta (red) within 1 μm distance of the dendritic mask (blue) and this number did not differ between the SNAP-25 WT rescue construct and the SNAP-25 mutants (Fig. 2D), indicating normal morphological synapse formation in all three mutants.
Figure 2.
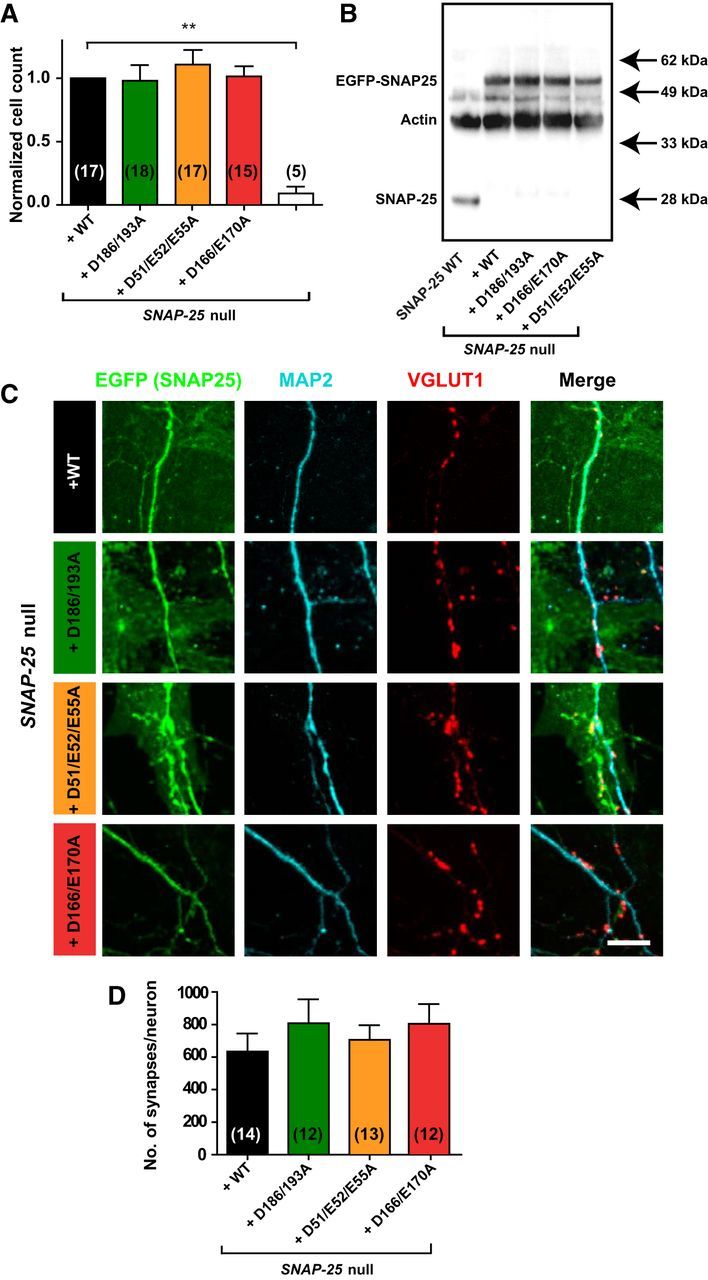
Lentiviral introduction of SNAP-25 mutants not binding syt-1 rescues cell survival and synapse formation. A, Normalized number of hippocampal neurons counted on glial islands. Between 5 and 18 coverslips were analyzed per condition. B, Western blot of high-density cultures visualizing SNAP-25 endogenous (SNAP-25 WT, gray), Snap-25-null + WT rescue, Snap-25-null + SNAP-25B D186/D193A, Snap-25-null + SNAP-25B D166/E170A, and Snap-25-null + SNAP-25B D51/E52/E55A (all as fusions with EGFP). C, Representative immunocytochemistry images of hippocampal autaptic cultures stained against GFP (green, marker for EGFP-SNAP-25), MAP2 (cyan), and VGLUT1 (red). Image acquisition by confocal microscopy was followed by maximum intensity projection and image analysis. Scale bar, 10 μm. D, Quantification of synapses within 1 μm the dendritic mask. The whole autaptic neuron was analyzed. Bar graph displays mean ± SEM. **p < 0.01 (Kruskall–Wallis test followed by Dunn's post test).
Evoked vesicle release and its kinetics depend on functional syt-1/SNAP-25 interaction
We performed whole-cell voltage-clamp recordings on Snap-25-null autaptic cultures of hippocampal cells rescued with each of the mutant constructs or the WT SNAP-25 protein to examine synaptic release (Bekkers and Stevens, 1991). Brief depolarizations from the −70 mV holding potential to 0 mV resulted in action potential generation and subsequently in a recurrent autaptic response, which was comparable between the WT rescue and the D186/D193A mutant (Fig. 3A,B). The EPSC amplitude was reduced to 14.4 ± 5.8% in the D51/E52/E55A mutant and 3.5 ± 1.5% in the D166/E170A mutant (Fig. 3A,B). In the D51/E52/E55A mutant, 12 of 59 cells did not show detectable evoked release, whereas for the D166/E170A mutant, 20 of 48 were silent (these cells were included into the statistics in Fig. 3). In contrast, 0 of 100 cells in the WT and 1 of 54 cells in the D186/D193A mutant lacked an EPSC. The kinetics were analyzed using the cumulative evoked charge (Fig. 3C,D). The evoked charge was fitted by a sum of two exponentials, representing the synchronous and the asynchronous component, respectively (Fig. 3E–G). Synchronous release represented the majority of the release for the WT and D186/D193A mutation (∼80% of the total evoked charge; Fig. 3G). In contrast, in the D51/E52/E55A and D166/E170A mutants, synchronous release was abrogated (Fig. 3E), whereas the asynchronous component (i.e., the slower exponential) was reduced in amplitude, but still substantial (Fig. 3F). Therefore, the total evoked charge of the D51/E52/E55A and D166/E170A mutants consisted to a larger degree of asynchronous release compared with the WT and D186/D193A mutant (Fig. 3G).
Figure 3.
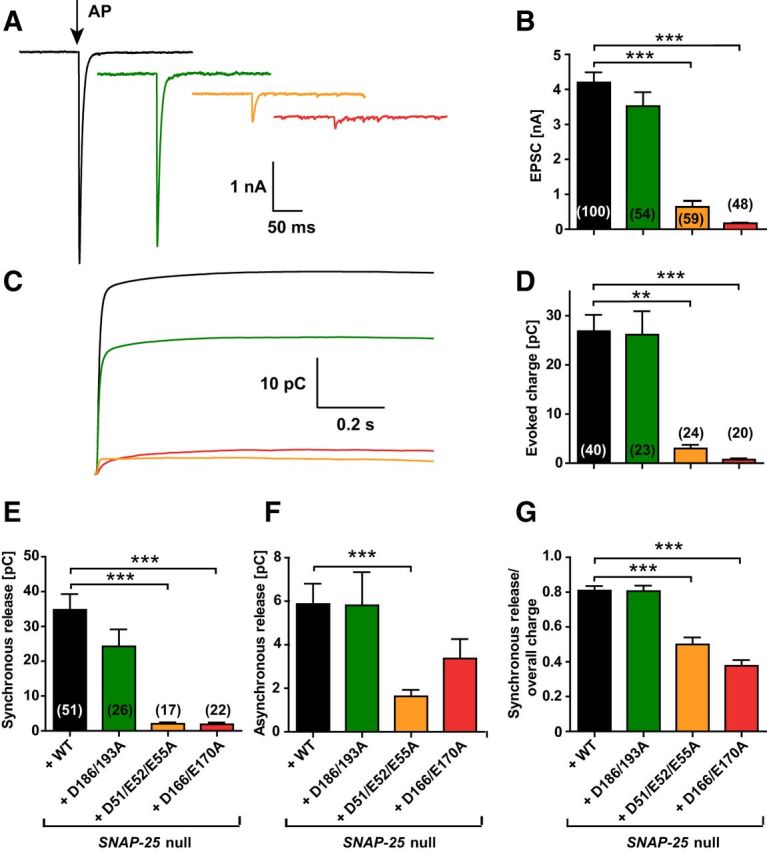
The extent and synchronicity of evoked release is affected by D166/E170A and D51/E52/E55A mutations in SNAP-25B. A, A 2 ms depolarization to 0 mV of the autaptic hippocampal neuron (indicated by arrow) leads to an EPSC (representative traces). B, Peak EPSC amplitudes were strongly reduced in the D166/E170A and D51/E52/E55A mutants. C, Cumulative charge of EPSCs. D, Total evoked charge (1 s integration) after a single depolarization. E, Synchronous release component of EPSCs. F, Asynchronous release component of EPSCs. G, Fractional contribution of the synchronous release component to overall release. Bar graphs display mean ± SEM. **p < 0.01; ***p < 0.001 (Kruskal–Wallis test followed by Dunn's post test).
Lack of syt-1/SNAP-25 interaction unclamps spontaneous vesicle release and decreases the probability of evoked release
At a constantly held voltage of −70 mV, which effectively blocks action potential generation in the autaptic configuration, all examined mutants displayed a strong unclamping effect on the frequency of spontaneous release, rising from 1.3 ± 0.2 Hz (WT, n = 62) to 8.0 ± 1.5 Hz (D186/D193A mutant, n = 41), 5.3 ± 0.9 Hz (D51/E52/E55A mutant, n = 48), or 12.5 ± 1.8 Hz (D166/E170A mutant, n = 40; Fig. 4A,B). Spontaneous release amplitudes remained constant (Fig. 4C), indicating no large postsynaptic effect of the mutants. To determine whether a change in the pool of releasable vesicles underlies the observed decrease in evoked response (Fig. 3A), we determined the RRP size with hyperosmotic sucrose applications. For this, cells were held at −70 mV while a hypertonic solution (extracellular solution including 500 mOsm sucrose) was applied locally to the neuron using a gravity-driven perfusion and a stepper motor system. Upon sucrose application, the vesicles corresponding to the RRP size are released (Rosenmund and Stevens, 1996; Schotten et al., 2015). Interestingly, the RRP charge was reduced only in the D166/E170A mutant, whereas the pool size was unaffected in the D186/D193A and D51/E52/E55A mutants (Fig. 4D,E). By dividing the sucrose pool with the mean charge of a single mEPSC from the same cell, the number of vesicles in the RRP could be calculated (Fig. 4F); this analysis resulted in the same conclusion. The release probability for a vesicle (PVR) when stimulated by a single action potential was calculated by dividing the evoked charge (Fig. 3D) by the RRP charge (Fig. 4E) of the same cell, revealing a strongly reduced probability of release in the D166/E170A and D51/E52/E55A, but no change in the D186/193A mutation (Fig. 4G).
Figure 4.
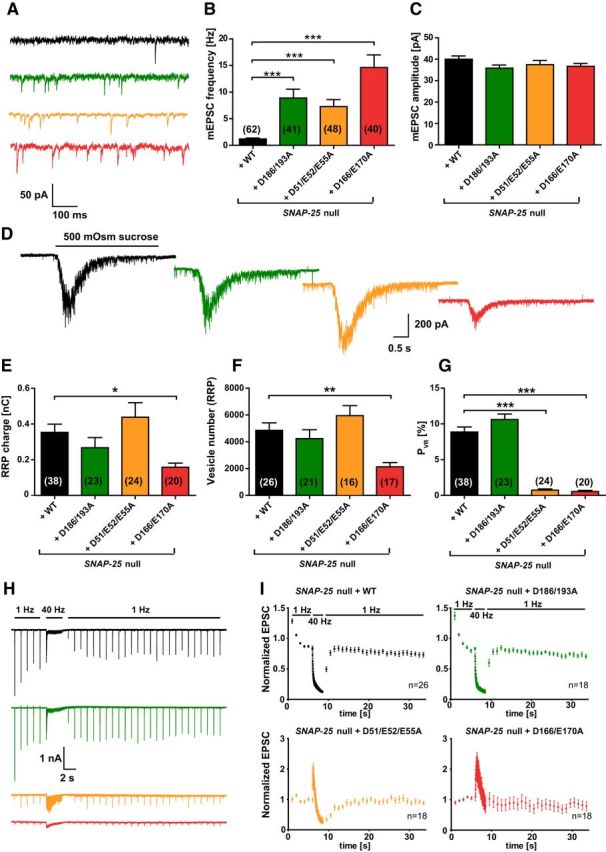
Spontaneous release is unclamped by SNAP-25B mutants not binding syt-1. A, Example traces of spontaneous vesicle release at a holding potential of −70 mV. B, Spontaneous release frequencies were increased in all three SNAP-25 mutants. C, Amplitudes of spontaneous release events. D, Representative responses upon 5 s perfusion with hyperosmotic sucrose solution (0.5 m). E, Overall RRP charge in mutant conditions, derived from the sucrose response. F, Number of vesicles in the RRP, calculated by the ratio of the RRP charge (E) and the mean charge of a spontaneous event from the same cell. G, PVR, derived from the ratio of the single evoked charge (Fig. 3D) and the RRP charge (E) of the same cell. The PVR was strongly reduced by D51/E52/E55A and D166/E170A mutations. H, Example traces of 100 depolarizations (40 Hz) preceded and followed by 1 Hz stimulations. I, Train stimulations (40 Hz) normalized to the mean amplitude of the first five EPSCs. Bar graphs display mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn's post test).
The lowered release probability in the D166/E170A and D51/E52/E55A mutant should be accompanied by facilitation of release (Dobrunz and Stevens, 1997; Zucker and Regehr, 2002). To test this, we performed high-frequency train stimulation experiments (Fig. 4H). The WT and the D186/D193A mutant displayed a moderate amplitude depression upon 1 Hz train stimulation and a further increased depression during the subsequent 40 Hz train stimulation (100 stimuli). Recovery set in immediately after returning to the 1 Hz stimulation and was fully established 2 s after the 40 Hz stimulation (Fig. 4I). In contrast, the D166/E170A and D51/E52/E55A mutants did not depress during 1 Hz train stimulations but displayed an initial facilitation of vesicle release at 40 Hz train stimulations, followed by depression (Fig. 4I). However, the facilitation never restored the amplitudes to WT values (data not shown). Bronk et al. (2007) showed previously that release does not facilitate in Snap-25-null cultures and the subpopulation of cells that do display a small EPSC depressed rapidly. This indicates that the introduction of the D166/E170A and D51/E52/E55A mutants is not equal to the Snap-25-null, consistent with the notion that the phenotype is due to a specific lack of interaction with syt-1 and is not caused by a total lack of SNAP-25 function. The same is indicated by the rescue of survival by all mutants studied (Fig. 2A) and the increased spontaneous release, which is contrasted with a decreased spontaneous release rate in the Snap-25-null (Washbourne et al., 2002; Bronk et al., 2007; Delgado-Martínez et al., 2007). This difference allows us to conclude that a SNARE complex with compromised syt-1 interaction is sufficient to support spontaneous release, whereas evoked release is specifically impaired. In summary, interfering with the binding between SNAP-25 and syt-1 through mutation of SNAP-25 led to a different result for spontaneous and evoked release: all three mutants unclamped spontaneous release, whereas only the D166/E170A and D51/E52/E55A mutants affected evoked release. For the D51/E52/E55A mutant, the reduction in evoked release is due to a reduction in PVR, whereas for the D166/E170A mutant, both the RRP and PVR were reduced.
Phenotype of the D51/E52/E55A mutant is partially rescued upon high extracellular Ca2+ concentrations
The Ca2+ influx into the presynapse during an action potential correlates directly with the EPSC size until 10 mm extracellular [Ca2+], where saturation sets in (Ikeda et al., 2008). The observed difference in EPSC amplitude between the syt-1-binding deficient SNAP-25 mutants and the WT protein could be due to a change in calcium affinity of the release machinery. To test this possibility, we investigated the effect of different extracellular calcium concentrations on the EPSC amplitude, bracketed by control measurements at 2 mm Ca2+. At 1 mm extracellular calcium, nearly all cells in the D166/E170A and D51/E52/E55A mutant condition failed to generate a postsynaptic response, whereas most WT or D186/D193A mutant cells displayed a decrease in EPSC amplitude (Fig. 5A,B; 1 mm). Increasing the [Ca2+] up to 8 mm increased the EPSC amplitude in all mutants (Fig. 5A,B; 8 mm). Remarkably, for the D51/E52/E55A mutant, evoked release was restored to 39 ± 15% of the WT amplitude at 8 mm (compared with 11 ± 4% at 2 mm extracellular calcium). At 12 mm, there was no further significant increase in EPSC size for any condition (Fig. 5A). The Hill function was fitted to the data points of each condition (Fig. 5B). The apparent calcium affinity was slightly higher for the D186/D193A mutant (KD = 1.3 ± 0.2 mm) than for the WT rescue (KD = 1.7 ± 0.2 mm); both showed a higher calcium apparent affinity than the D51/E52/E55A and D166/E170A mutant (KD = 3.1 ± 2.1 vs KD = 3.0 ± 0.6 mm, respectively). In contrast, the Hill coefficient c, reflecting the apparent cooperativity, was comparable for the WT (2.9 ± 0.8), D186/D193A mutant (3.1 ± 1.2), and D51/E52/E55A mutant (3.4 ± 1.7), whereas its value was decreased in the D166/E170A mutant (1.3 ± 0.7). In short, the calcium experiments exposed a lower apparent calcium affinity in the D51/E52/E55A. Increasing calcium concentrations resulted in a partial rescue of release for the D51/E52/E55A mutant.
Titration of spontaneous release with external calcium showed that the higher spontaneous release rate of the three mutants persisted at calcium concentrations between 1 and 12 mm (Fig. 5C). The spontaneous release rate in the WT had a higher slope, which is indicative of a higher cooperativity for calcium than the three mutants. A decreased cooperativity of spontaneous release has also been found when mutating syt-1 (Xu et al., 2009). However, because the mEPSC rate did not saturate even at 12 mm [Ca2+], we could not fit the Hill equation to these data.
Knock-down of syt-1 occludes the phenotypes of SNAP-25 mutants
If the mutations tested here work primarily by reducing the interaction between SNAP-25 and syt-1, then the effect of the mutations should be occluded by removal of syt-1. To test this, we knocked down syt-1 in Snap-25-null neurons expressing the mutants. The knock-down construct led to undetectable expression of syt-1 (Fig. 6A). In previous studies, knock-out of syt-1 led to increased spontaneous release frequency in mixed cortical cultures and slices (Kerr et al., 2008; Xu et al., 2009). In contrast, autaptic cultures of Syt-1 knock-out cells did not display an increase in spontaneous release frequency (Geppert et al., 1994; Liu et al., 2009; Wierda and Sørensen, 2014). Here, we observed that lentiviral knock-down of syt-1 in autaptic neurons increased spontaneous release frequency (syt-1 knock-down: 5.0 ± 0.9 Hz, n = 38; control: 1.3 ± 0.2 Hz, n = 62, p < 0.0001, unpaired t test). Surprisingly, it also reduced RRP size by ∼50%, which is also seen in syt-1 knock-out networks, but not in autapses (Liu et al., 2009). Therefore, syt-1 knock-down seems to better reproduce the network condition.
Figure 6.

Knock-down of syt-1 occludes the phenotype of SNAP-25 mutations. A, Western blot of neuronal high-density control (ctrl) cultures without (left) and with (right) a lentiviral syt-1 knock-down (KD) construct (B) Example traces of spontaneous vesicle release at a holding potential of −70 mV. Neurons were cultured from Snap-25-null embryos and simultaneously transduced with a lentiviral vector encoding a SNAP-25 construct and the syt-1 KD construct. C, Spontaneous release frequencies in the SNAP-25 mutants. D, Amplitudes of mEPSCs. E, Single stimulations (indicated by arrows) led to very small EPSCs (representative traces), where miniature events were visible. F, Averaged peak EPSC amplitudes. G, Example traces of EPSCs in the presence of syt-1 (same data as in Fig. 3A and rescaled as in E) for comparison. H, Representative responses upon 5 s perfusion with 500 mOsm hyperosmolar sucrose solution. I, RRP charges. Bar graphs display mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn's post test).
After knock-down of syt-1, spontaneous release frequencies were similar between constructs (Fig. 6B,C), although the D51/E52/E55A mutation had on average higher rates. The amplitude of miniature events was 38.1 ± 1.2 pA for the WT construct with syt-1 knock-down and did not differ significantly from the other conditions (Fig. 6D). We observed strongly decreased and very asynchronous EPSC amplitudes upon stimulation (Fig. 6E,F) compared with the evoked stimulation responses of autaptic hippocampal cultures in the presence of syt-1 (Fig. 6G). However, all mutants and the WT had indistinguishable EPSC amplitudes (Fig. 6F). The size of the RRP also did not display significant differences between the SNAP-25 constructs (Fig. 6H,I).
We conclude that the knock-down of syt-1 in autaptic neurons occluded all differences between the SNAP-25 mutants when they were compared side by side (Fig. 6). Conversely, comparing the data with and without syt-1 makes it clear that syt-1 still has consequences in the presence of the D51/E52/E55A mutation, in which there is markedly more synchronized release in the presence of syt-1 than in its absence (cf. Figs. 3B, 6F). Therefore, this mutation does not entirely abolish the function of syt-1, which is expected due to the large primary interaction surface (Zhou et al., 2015). Strikingly, the D166/E170A mutation, which is located within the “region I” of the primary interaction surface (Zhou et al., 2015), appears to eliminate any effect of syt-1 on evoked release or on the RRP size (cf. Figs. 3B, 6F, 4E, 6I). The only effect of removing syt-1 in the presence of the D166/E170A mutant was a reduction in mEPSC frequency (cf. Figs. 4B, 6C; p = 0.0039, two-tailed t test). In contrast, for the D51/E52/E55A mutant, there was a tendency to a further increased mEPSC frequency upon syt-1 removal (cf. Figs. 4B, 6C; p = 0.069, two-tailed t test). Therefore, the phenotype of the D166/E170A mutant is distinct from that of the D51/E52/E55A mutant and overall more severe.
Overexpression of syt-1 clamps spontaneous release, but not evoked release, in the D51/E52/E55A mutant
In complementary experiments, we tested whether increasing the amount of syt-1 would be able to rescue the phenotypes observed in the SNAP-25 mutants. We produced a lentiviral syt-1 overexpression (OE) construct, which rescued the evoked response in six of six Syt-1 knock-out cells measured, indicating correct expression of syt-1 (Fig. 7A). The amount of expressed syt-1 was approximately three times as high upon OE compared with WT cells without the construct (Fig. 7B). In Snap-25-null neurons rescued by WT EGFP-SNAP-25, additional OE of syt-1 did not change the frequency of mEPSCs significantly [WT: 1.3 ± 0.2 Hz (Fig. 4B) vs WT+syt-1 OE: 1.2 ± 0.7 Hz (Fig. 7C), two-tailed t test, p = 0.9] or their amplitude (data not shown). In contrast, with the D51/E52/E55A mutant, the mEPSC frequency was reduced to a similar frequency as the WT by syt-1 OE (WT+syt-1 OE: 1.2 ± 0.7 Hz vs D51/E52/E55A +syt-1 OE: 1.9 ± 0.7 Hz). The decrease of frequency in the D51/E52/E55A mutant was significant when directly comparing the mutant with and without syt-1 overexpression (two-tailed t test, p = 0.0282). In contrast, for the D186/D193A and D166/E170A mutants, mEPSC frequencies remained significantly elevated even after syt-1 OE (Fig. 7C,D). No rescue of the evoked EPSC amplitude was found after syt-1 OE for any of the mutations (cf. Figs. 7E,F, 3A,B). The application of sucrose resulted in a similar RRP charge and release probability (Fig. 7G–I) as without syt-1 OE (cf. Fig. 4).
Figure 7.

Overexpression of syt-1 clamps spontaneous release frequency in the D51/E52/E55A mutant. A, EPSC amplitudes without (blue) and with (black) lentiviral syt-1 OE construct in autaptic hippocampal neurons isolated from syt-1-null mice. B, Western blot of control (ctrl) neurons without (left) and with (right) syt-1 overexpression. C, Example traces of spontaneous vesicle release at a holding potential of −70 mV. Neurons were cultured from Snap-25-null embryos and simultaneously transduced with a lentiviral vector encoding a SNAP-25 construct and the syt-1 OE construct. D, Spontaneous release frequencies in the SNAP-25 mutants with syt-1 OE. E, Single stimulation (indicated by arrow) led to a postsynaptic current (representative traces). F, Peak EPSC amplitudes. G, Representative responses upon 5 s perfusion with 500 mOsm sucrose. H, RRP charges. I, PVR calculated as the ratio of the single evoked charge and the RRP charge of the same cell. Bar graphs display mean ± SEM. *p < 0.05, ***p < 0.001 (Kruskal–Wallis test followed by Dunn's post test).
Overall, syt-1 OE clamped spontaneous release frequency of the D51/E52/E55A mutant to WT values, whereas it did not affect evoked release. This indicates that the site around D51/E52/E55 is not obligatorily coupled to limiting spontaneous release, presumably because other charges can take over this function.
Position D166 is necessary for Cpx clamping of spontaneous fusion and vesicle priming
In the above experiments, we used the D166/E170A mutant based on our previous work showing that this mutant eliminates binding to syt-1 (Mohrmann et al., 2013). However, the crystal structure published in the meantime identified only D166 as part of the region I of the primary interface (Zhou et al., 2015). We therefore tested whether mutating D166 alone might suffice to change fusion in vitro and ex vivo. At the same time, we created the D51/E52/E55/D166A quadruple mutant, which compromises both region I and II of the primary interface (Zhou et al., 2015; Fig. 8A).
Figure 8.
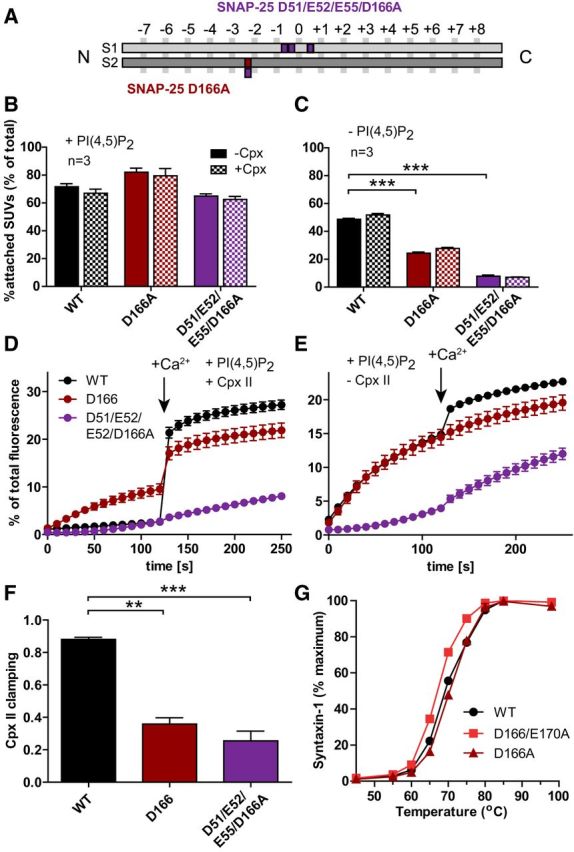
Vesicle–vesicle attachment is impaired and spontaneous fusion in vitro disinhibited by the D166A mutation. A, Schematic representation of the mutants: SNAP-25B D166A (dark red) and D51/E52/E55/D166A (violet). B, C, Attachment of SUVs to GUVs with and without Cpx II in the presence (B) or absence (C) of PI(4,5)P2. SUVs that were bound to GUVs were quantified and normalized to total input (see legend to Fig. 1). D, E, t-SNARE-GUVs containing SNAP-25 WT (black), D166A (dark red), or D51/E52/E55/D166A (violet) were mixed with v-SNARE-SUVs in the presence (D) or absence (E) of Cpx II. Lipid mixing during vesicle fusion was monitored as the increase of Atto488 fluorescence as explained in the legend to Figure 1. Values were normalized to the maximum fluorescence signal after detergent lysis. F, Fractional clamping effect of Cpx II. Note that the D166A mutation removes Cpx clamping either alone or in combination with the E170A (Fig. 1) or the D51/E52/E55A mutation. G, Unfolding of SNARE complexes formed with WT (black), D166A (dark red), or D166/E170A (red) SNAP-25B as measured by the appearance of uncomplexed syntaxin-1 at higher temperatures. Most error bars are too small to be plotted (n = 3). The D166/E170A mutation slightly destabilizes the complex, whereas D166A does not. Note that the data for WT SNAP-25B in B–F are the same as in Figure 1 because all experiments shown in Figures 1 and 8 were performed using the same vesicle preparations. All graphs display mean ± SEM (n = 3). *p < 0.05, **p < 0.01; ***p < 0.001 (one-way ANOVA, significance was calculated using Dunnett's post test).
Determination of vesicle association in vitro showed that both the D166A and the D51/E52/E55/D166A reduced vesicle association significantly in the absence of PI(4,5)P2 (Fig. 8C), but not in its presence (Fig. 8B). As before, the inclusion of Cpx II did not change vesicle attachment under any condition (Fig. 8B,C). Fusion (lipid mixing) in the presence of Cpx II, but in the absence of Ca2+, was strongly disinhibited by the D166A mutation (Fig. 8D). The D51/E52/E55/D166A displayed strongly reduced fusion both in the presence and absence of Cpx II (Fig. 8D,E). Both mutations resulted in a strongly reduced clamping by Cpx II (Fig. 8F), just as with the D166/E170A mutant (Fig. 1G). Therefore, the D166A mutant in any of those combinations (or alone) interferes with Cpx II clamping. However, D166 is located at the opposite side of Cpx on the Cpx-bound SNARE complex and does not interact with Cpx (Chen et al., 2002). Finally, we evaluated the stability of the ternary (cis-) SNARE complex and found a small but clear destabilization of the complex by the D166A/E170A mutation, whereas D166A did not compromise SNARE complex stability (Fig. 8G). This difference is likely caused by an interaction (H-bond) of the E170 side chain with the zero-layer Q-174 in SNAP-25 (Sutton et al., 1998).
In autaptic neurons expressing the D166A mutant, spontaneous release was strongly unclamped, reaching mean frequencies >50 Hz (Fig. 9A,B). Due to the high rates, we used unsupervised detection of mEPSC frequencies in this set of experiments, which leads to different absolute numbers than the supervised detection used before, but should not compromise the detection of differences. The D51/E52/E55/D166A did not further increase mEPSC frequencies (Fig. 9B). mEPSC amplitudes were unchanged by the mutations (Fig. 9C). Upon stimulation to fire an action potential, the D166A supported only small EPSCs (Fig. 9D,E), which were slightly less synchronous than in WT transduced cells recorded in parallel (Fig. 9F–I). Strikingly, the D51/E52/E55/D166A did not respond to stimulation with a detectable EPSC (Fig. 9D). Hypertonic stimulation showed that both mutations had strongly reduced RRPs (Fig. 9J,K), but the D166A displayed normal PVR (Fig. 9L). This was also reflected in the depression during a 20 Hz stimulation train, which was indistinguishable between WT and D166A neurons (Fig. 9M).
Figure 9.
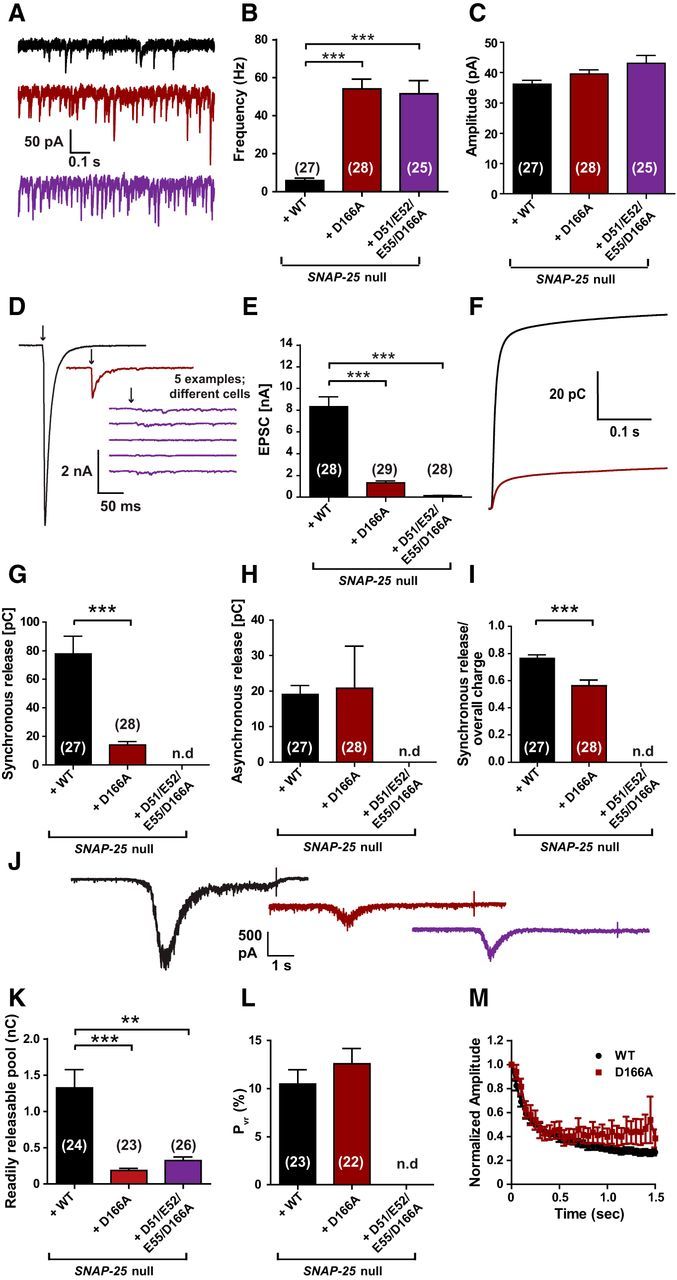
The D166A mutant unclamps spontaneous release and inhibits vesicle priming. A, Example traces of spontaneous vesicle release at a holding potential of −70 mV. B, Spontaneous release frequencies were strongly increased by SNAP-25B D166A or SNAP-25B D51/E52/E55/D166A. C, Amplitudes of spontaneous events were not significantly changed. D, Representative traces of single stimulations (indicated by arrows). Five examples are shown for the D51/E52/E55/D166A mutant, which did not have detectable evoked release. E, Peak EPSC amplitudes were reduced in the D166A mutant and absent in the D51/E52/E55/D166A mutant. Note that, in this set of experiments, EPSC amplitudes were higher than in previous experiments (Fig. 3), but the release probability was similar (L and cf. Fig. 4G). F, Cumulative charge of EPSCs used for kinetic analysis. The D51/E52/E55/D166A mutant could not be analyzed. G, H, D166A mutation reduced the synchronous release, but did not affect asynchronous release. I, Therefore, the ratio of synchronous release was decreased. J, Representative traces of hypertonic shock (500 mOsm sucrose) to measure the readily releasable pool. K, RRPs were decreased in both mutants. L, Release probability was not affected by the D166A mutation. M, Train of stimulations at 20 Hz gave similar short-term plasticity for WT and the D166A mutant. Bar and line graphs display mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test, followed by Dunn's post test for B, C, E, and K; unpaired t test for G–I and L). n.d., Not determined.
Together with the results above, these data show that D166A, either alone or in combination with E170A or D51/E52/E55A, results in unclamping of spontaneous release and a reduction in RRP. The reduced PVR found in the D166/E170A might be related to the destabilization of the SNARE complex induced by the E170A mutation, not to the lack of syt-1 interaction.
Discussion
Here, we examined five SNAP-25 mutations designed to interfere with syt-1 interactions. Our in vitro vesicle attachment assay showed that the D51/E52/E55A, D166/E170A, D166A, and D51/E52/E55/D166A mutants markedly reduced the attachment between syt-1- and t-SNARE-carrying vesicles in the absence of PI(4,5)P, whereas D186/D193A had only a small but significant effect. In the presence of PI(4,5)P2, vesicle attachment was unaffected by SNAP-25 mutation. Therefore, syt-1 can attach vesicles via interactions with PI(4,5)P2 or the SNARE complex (Kim et al., 2012; Parisotto et al., 2012; Kedar et al., 2015). Our physiological data showing profound effects of D51/E52/E55A, D166/E170A, D166A, and D51/E52/E55/D166A on evoked release, and no effect of D186/193A, are complementary to those obtained previously by mutation of syt-1 (Brewer et al., 2015; Zhou et al., 2015) and overall support the physiological relevance of syt-1:SNARE interactions (Pang et al., 2006b).
Evoked release requires SNAP-25 negative charges within the region II of the primary interface
The SNAP-25 D51/E52/E55 interaction interface (Rickman et al., 2006) was found previously to mediate syt-1-mediated vesicle attachment in vitro (Kim et al., 2012) and vesicle docking and fast vesicle fusion in adrenal chromaffin cells (Mohrmann et al., 2013). Here, we found that D51/E52/E55A reduced vesicular release probability profoundly without affecting vesicle priming. It also unclamped spontaneous release, causing a higher mEPSC frequency, but, strikingly, this effect was eliminated by overexpressing syt-1. In the presence of the D166A mutation, the D51/E52/E55A did not further increase spontaneous release. Overall, the region II of the primary interface is not essential for clamping release, but it might facilitate a collision-limited interaction of syt-1 with the SNARE complex.
Overexpression of syt-1 did not recover evoked release in the D51/E52/E55A mutant, consistent with the notion that this step is not collision limited, but rather involves a structural change in a preformed complex. In the presence of the D166A mutation, the D51/E52/E55A completely eliminated triggered release, attesting to the essential nature of these charges (Fig. 9). This aligns well with similar findings for the interacting syt-1 residues R398 and R399 (Gaffaney et al., 2008; Xue et al., 2008a; Kedar et al., 2015; Zhou et al., 2015).
Fast synchronous release driven by syt-1 was not entirely abolished upon mutation of the D51/E52/E55-site and calcium titration succeeded in partially restoring the evoked release amplitude, revealing that the calcium dependence of release is shifted, whereas the cooperativity is intact (Fig. 5). The shift in the calcium dependence of release has also been noted when mutating the interaction site within syt-1 (Borden et al., 2005; Li et al., 2006; Brewer et al., 2015). PI(4,5)P2 binding to syt-1 increases its calcium affinity in vitro (van den Bogaart et al., 2012) and it appears likely that binding to negative charges on the SNARE complex could do the same. It is an intriguing possibility that a major function of primary interface syt:SNARE-binding is to bring the Ca2+ affinity of syt-1 within a physiological feasible range upon the detection of a properly formed SNARE complex.
D166A within region I of the primary interface is required for vesicle priming and for clamping of spontaneous release
All three mutants including the D166A mutation, D166A, D166/E170A, and D51/E52/E55/D166A, were characterized by a reduced RRP and strongly increased spontaneous release rates. The lower PVR in the D166/E170A mutant might have been caused by the destabilization of the SNARE complex by the E170A mutation (Fig. 8G) because it was not found for the D166A mutant (Fig. 9).
The effect of D166A on vesicle priming was not expected as a consequence of reduced syt-1 interaction because previous studies did not identify changes in RRP size in syt-1 KO autaptic cultures (Xue et al., 2008a; Liu et al., 2009). However, it was found recently that syt-1 and syt-7 together are necessary for maintaining the RRP in mass cultures of hippocampal neurons and that syt-1 is involved in RRP formation by promoting the formation or stabilization of SNARE complexes (Bacaj et al., 2015). Another study showed that KO of syt-1 alone can decrease the RRP size in small neuronal networks (Liu et al., 2009) and we showed here that a knock-down construct of syt-1 profoundly reduced RRP size in autaptic SNAP-25 KO neurons simultaneously rescued with SNAP-25 (cf. Figs. 6, 4). Therefore, the RRP-promoting effect of syt-1 might become manifest under some experimental conditions, but not others, possibly dependent on the expression of other synaptotagmins.
The profound reduction in RRP by the D166A mutation without a change in PVR and the elimination of evoked release in the D51/E52/E55/D166A point toward a separation of function between region I (D166) and region II (D51/E52/E55) of the primary interface in vesicle priming and fusion, respectively. We cannot rule out that the D166A mutation could have a priming effect independent of syt-1, but several observations support a specific role for D166 in syt-1 binding. First, D166 was identified as interacting with syt-1 Y338 in the crystal structure (Zhou et al., 2015). Second, the D166A mutation perturbed syt-1-dependent vesicle attachment in vitro (Figs. 1, 8). Third, the D166/E170A mutation was shown to reduce syt-1 C2AB-domain pull-down (Mohrmann et al., 2013). Fourth, the SNARE complex remains stable in the presence of the D166A mutation (Fig. 8G). Finally, knock-down of syt-1 occludes the effect of D166/E170A on priming (Fig. 6I). Therefore, we propose that D166 is part of a SNARE interface that is necessary for syt-1-dependent vesicle priming, presumably by promoting SNARE complex formation or stabilization (Bacaj et al., 2015). The location of D166 to the N-terminal half of the SNARE complex (outside of layer 2) makes it a good candidate to mediate vesicle priming, which depends on N-terminal SNARE assembly (Sørensen et al., 2006; Weber et al., 2010).
A puzzling finding is that the D166A mutation, either alone or together with E170A or D51/E52/E55A, led to strong unclamping of spontaneous release (Figs. 4, 9). In D166/E170A, this unclamping was occluded by syt-1 knock-down (Fig. 6), indicating that it is a consequence of syt-1 interaction. Spontaneous release rates were not higher in the quadruple D51/E52/E55/D166A mutation than in the D166A single mutation (Fig. 9B), clearly pointing toward D166 as being causative. The unclamping of spontaneous release was also seen in our minimal in vitro fusion assay (Figs. 1D, 8D) and, indeed, the unclamping effect in the cell correlated with a lack of Cpx clamping in vitro (Figs. 1G, 8F). This correlation extends to the D186/193A mutation, which supported intact evoked release, but also unclamped spontaneous release (Fig. 4B) and reduced Cpx clamping in vitro (Fig. 1G).
In our in vitro system, syt-1 drives vesicle attachment upstream of SNARE complex formation (Parisotto et al., 2012), whereas Cpx acts as a downstream clamp, which builds up a pool of vesicles for syt-1-dependent fusion (Malsam et al., 2012). When taken together with our observation that D166 is necessary for normal vesicle priming, it is tempting to suggest that syt-1-dependent formation of SNARE complexes (Bacaj et al., 2015) might be required for the downstream association of the Cpx clamp. Syt-1 might arrest the SNARE complex in a partially assembled state (Chicka et al., 2008) for long enough that Cpx can bind and stabilize clamping. In the absence of syt-1, SNARE complexes might finalize their formation spontaneously, leading to fusion before Cpx has time to interact. If this model is correct, then Cpx deletion should have no effect on spontaneous release in the absence of syt-1, which was indeed observed in Drosophila (Jorquera et al., 2012). Evaluating this model for mammalian synapses will be more complicated because the stimulatory functions of Cpx dominate (Xue et al., 2008b; Xue et al., 2009). Furthermore, from double/triple knock-out experiments in both systems, it is clear that Cpx exerts positive effects on evoked release in the absence of syt-1 (Xue et al., 2010; Jorquera et al., 2012). More investigations will be required to reveal the interdependence between Cpx function and syt-1-dependent SNARE complex formation.
In any case, our results argue against the notion that increased spontaneous release rates upon deletion of syt-1 originate from interaction with an alternative sensor/trigger (see also Pang et al., 2011). First, our findings in the cell mirror findings in a minimal in vitro system in which no alternative sensors/triggers are present. Second, we investigated five mutants neutralizing surface charges on the SNARE complex, which would not be expected to increase association with another sensor, although the possibility remains open that, by inhibiting interaction with syt-1, they might facilitate the interaction indirectly with a sensor binding to a different site.
Conclusion: the triple life of Syt-1 depends on SNARE interactions
The triple life of syt-1 in suppressing spontaneous release, stimulating vesicle priming, and eliciting evoked release (Bacaj et al., 2015) was reproduced here by knock-down of syt-1 in autaptic neurons (Fig. 6). Moreover, our examination of five mutants of negative surface charges of the SNARE complex indicates that syt:SNARE interactions are required for all three aspects of syt-1 function. Early attachment of syt-1 to SNARE complexes as they form might set the stage for later interactions with Cpx to prevent spontaneous release and shape evoked release.
Footnotes
This work was supported by the DFG (Grant SFB/TRR83 to T.H.S.), the Danish Council for Independent Research, Medical Sciences (J.B.S.), the European Union Seventh Framework Program (Grant Agreement HEALTH-F2-2009-242167 “SynSys” project to J.B.S.), and the Lundbeck Foundation (J.B.S.). We thank Anne Marie Nordvig Pedersen for excellent technical assistance, Freddy Trinh for assisting in setting up calcium titration experiments, and Prof. Dr. Thomas Südhof (Stanford University) for providing the lentiviral m-Cherry-tagged syt-1 shRNA construct.
The authors declare no competing financial interests.
References
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron. 2013;80:947–959. doi: 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Burré J, Malenka RC, Liu X, Südhof TC. Synaptotagmin-1 and -7 are redundantly essential for maintaining the capacity of the readily-releasable pool of synaptic vesicles. PLoS Biol. 2015;13:e1002267. doi: 10.1371/journal.pbio.1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER. Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun. 2016;7:10971. doi: 10.1038/ncomms10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron. 2004;41:929–942. doi: 10.1016/S0896-6273(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- Borden CR, Stevens CF, Sullivan JM, Zhu Y. Synaptotagmin mutants Y311N and K326/327A alter the calcium dependence of neurotransmission. Mol Cell Neurosci. 2005;29:462–470. doi: 10.1016/j.mcn.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, Liu J, Zhou A, Zhou P, Barlow N, Xu J, Seven AB, Prinslow EA, Voleti R, Haussinger D, Bonvin AM, Tomchick DR, Vendruscolo M, Graham B, Südhof TC, Rizo J. Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat Struct Mol Biol. 2015;22:555–564. doi: 10.1038/nsmb.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10727–10731. doi: 10.1073/pnas.91.22.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk P, Deák F, Wilson MC, Liu X, Südhof TC, Kavalali ET. Differential effects of SNAP-25 deletion on Ca2+-dependent and Ca2+-independent neurotransmission. J Neurophysiol. 2007;98:794–806. doi: 10.1152/jn.00226.2007. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between Synaptotagmin and Syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, Südhof TC, Rizo J. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/S0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nat Struct Mol Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2- and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- Davletov BA, Südhof TC. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- Delgado-Martínez I, Nehring RB, Sørensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Schwarz TL. The effect on synaptic physiology of synaptotagmin mutations in Drosophila. Neuron. 1994;12:909–920. doi: 10.1016/0896-6273(94)90342-5. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/S0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Fang Q, Lindau M. How could SNARE proteins open a fusion pore? Physiology. 2014;29:278–285. doi: 10.1152/physiol.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Kojima T, Mikoshiba K. Phospholipid composition dependence of Ca2+-dependent phospholipid binding to the C2A domain of synaptotagmin IV. J Biol Chem. 1996;271:8430–8434. doi: 10.1074/jbc.271.14.8430. [DOI] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008;283:31763–31775. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Gerona RR, Larsen EC, Kowalchyk JA, Martin TF. The C Terminus of SNAP25 Is Essential for Ca2+-dependent Binding of Synaptotagmin to SNARE Complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Yanagawa Y, Bekkers JM. Distinctive quantal properties of neurotransmission at excitatory and inhibitory autapses revealed using variance-mean analysis. J Neurosci. 2008;28:13563–13573. doi: 10.1523/JNEUROSCI.3350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci. 2012;32:18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar GH, Munch AS, van Weering JR, Malsam J, Scheutzow A, de Wit H, Houy S, Tawfik B, Söllner TH, Sørensen JB, Verhage M. A post-docking role of Synaptotagmin 1-C2B domain bottom residues R398/399 in mouse chromaffin cells. J Neurosci. 2015;35:14172–14182. doi: 10.1523/JNEUROSCI.1911-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, Scheller RH. Localization of synaptotagmin-binding domains on syntaxin. J Neurosci. 1996;16:1975–1981. doi: 10.1523/JNEUROSCI.16-06-01975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Reisinger E, Jonas P. Differential dependence of phasic transmitter release on synaptotagmin 1 at GABAergic and glutamatergic hippocampal synapses. Proc Natl Acad Sci U S A. 2008;105:15581–15586. doi: 10.1073/pnas.0800621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Catterall WA. Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc Natl Acad Sci U S A. 1997;94:14782–14786. doi: 10.1073/pnas.94.26.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Choi BK, Choi MG, Kim SA, Lai Y, Shin YK, Lee NK. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R. Synaptotagmin increases the dynamic range of synapses by driving Ca(2)+-evoked release and by clamping a near-linear remaining Ca(2)+ sensor. Neuron. 2011;69:736–748. doi: 10.1016/j.neuron.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner RA, Padolina MS, Brunger AT. Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. Elife. 2014;3:e03756. doi: 10.7554/eLife.03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Guan Z, Akbergenova Y, Littleton JT. Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci. 2013;33:187–200. doi: 10.1523/JNEUROSCI.3214-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveque C, Hoshino T, David P, Shoji-Kasai Y, Leys K, Omori A, Lang B, el Far O, Sato K, Martin-Moutot N. The synaptic vesicle protein synaptotagmin associates with calcium channels and is a putative Lambert-Eaton myasthenic syndrome antigen. Proc Natl Acad Sci U S A. 1992;89:3625–3629. doi: 10.1073/pnas.89.8.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Südhof TC. Ca2+-dependent and -independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- Li L, Shin OH, Rhee JS, Araç D, Rah JC, Rizo J, Südhof T, Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci. 2009;29:7395–7403. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Bai H, Xue R, Takahashi H, Edwardson JM, Chapman ER. Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat Neurosci. 2014;17:670–677. doi: 10.1038/nn.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Sequeira SM, Gureasko JM, Söllner TH. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Söllner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Connell E, Pinheiro PS, Leese C, Bruns D, Davletov B, Verhage M, Sørensen JB. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci. 2013;33:14417–14430. doi: 10.1523/JNEUROSCI.1236-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Neher E. A comparison between exocytic control mechanisms in adrenal chromaffin cells and a glutamatergic synapse. Pflugers Arch. 2006;453:261–268. doi: 10.1007/s00424-006-0143-9. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-V. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006a;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Shin OH, Meyer AC, Rosenmund C, Südhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006b;26:12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Malsam J, Scheutzow A, Krause JM, Söllner TH. SNAREpin assembly by Munc18–1 requires previous vesicle docking by synaptotagmin 1. J Biol Chem. 2012;287:31041–31049. doi: 10.1074/jbc.M112.386805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Seo JB, Fraind A, Pérez-Lara A, Yavuz H, Han K, Jung SR, Kattan I, Walla PJ, Choi M, Cafiso DS, Koh DS, Jahn R. Synaptotagmin-1 binds to PIP2-containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol. 2015;22:815–823. doi: 10.1038/nsmb.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Jiménez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B. Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell. 2006;17:283–294. doi: 10.1091/mbc.E05-07-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Xu J. The synaptic vesicle release machinery. Annu Rev Biophys. 2015;44:339–367. doi: 10.1146/annurev-biophys-060414-034057. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Gu QM, Prestwich GD, Söllner TH, Rothman JE. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci U S A. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp W. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods. 2011;195:185–193. doi: 10.1016/j.jneumeth.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Schotten S, Meijer M, Walter AM, Huson V, Mamer L, Kalogreades L, ter Veer M, Ruiter M, Brose N, Rosenmund C, Sørensen JB, Verhage M, Cornelisse LN. Additive effects on the energy barrier for synaptic vesicle fusion cause supralinear effects on the vesicle fusion rate. Elife. 2015;4:e05531. doi: 10.7554/eLife.05531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Trimbuch T, Xu J, Flaherty D, Tomchick DR, Rizo J, Rosenmund C. Re-examining how complexin inhibits neurotransmitter release. Elife. 2014;3:e02391. doi: 10.7554/eLife.02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- Weber JP, Reim K, Sørensen JB. Opposing functions of two sub-domains of the SNARE complex in neurotransmission. EMBO J. 2010;29:2477–2490. doi: 10.1038/emboj.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/S0092-8674(00)81404-X. [DOI] [PubMed] [Google Scholar]
- Wierda KD, Sørensen JB. Innervation by a GABAergic neuron depresses spontaneous release in glutamatergic neurons and unveils the clamping phenotype of synaptotagmin-1. J Neurosci. 2014;34:2100–2110. doi: 10.1523/JNEUROSCI.3934-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Südhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012;73:990–1001. doi: 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008a;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008b;105:7875–7880. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat Struct Mol Biol. 2010;17:568–575. doi: 10.1038/nsmb.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Davletov BA, Südhof TC, Anderson RG. Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 1994;78:751–760. doi: 10.1016/S0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TF. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/S0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hui E, Chapman ER, Jackson MB. Regulation of exocytosis and fusion pores by synaptotagmin-effector interactions. Mol Biol Cell. 2010;21:2821–2831. doi: 10.1091/mbc.E10-04-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature. 2015;525:62–67. doi: 10.1038/nature14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]