

Abstract
Human mutations in ZIC2 have been identified in patients with holoprosencephaly and schizophrenia. Similarly, Zic2 mutant mice exhibit holoprosencephaly in homozygosis and behavioral and morphological schizophrenic phenotypes associated with forebrain defects in heterozygosis. Despite the devastating effects of mutations in Zic2, the cellular and molecular mechanisms that provoke Zic2-deficiency phenotypes are yet unclear. Here, we report a novel role for this transcription factor in the migration of three different types of forebrain neurons: the Cajal–Retzius cells that populate the surface of the telencephalic vesicles, an amygdaloid group of cells originated in the caudal pole of the telencephalic pallium, and a cell population that travels from the prethalamic neuroepithelium to the ventral lateral geniculate nucleus. Our results also suggest that the receptor EphB1, previously identified as a Zic2 target, may mediate, at least partially, Zic2-dependent migratory events. According to these results, we propose that deficiencies in cell motility and guidance contribute to most of the forebrain pathologies associated with Zic2 mutations.
SIGNIFICANCE STATEMENT Although the phenotype of Zic2 mutant individuals was reported more than 10 years ago, until now, the main function of this transcription factor during early development has not been precisely defined. Here, we reveal a previously unknown role for Zic2 in the migration of forebrain neurons such as Cajal–Retzius cells, interneurons moving to the ventral lateral geniculate nucleus, and neocortical cells going to the amygdala. We believe that the role of this transcription factor in certain populations of migratory cells contributes to defects in cortical layering and hypocellularity in the ventral LGN and amygdala and will contribute to our understanding of the devastating phenotypes associated with Zic2 mutations in both humans and mice.
Keywords: Cajal–Retzius cells, forebrain, LOT2 amygdaloid nucleus, neuronal migration, vLGN, Zic2
Introduction
The vertebrate forebrain is subdivided into the telencephalon, which includes the future cerebral cortex and amygdala, and the diencephalon, which contains the thalamus and the prethalamus. Mutations in Zic2, a gene that encodes a member of the ZIC family of C2H2-type zinc finger proteins, cause anomalies associated with defects in forebrain development such as holoprosencephaly (Brown et al., 1998; Nagai et al., 2000; Brown et al., 2001). Zic2 mutants also exhibit defects in cortical lamination (Inoue et al., 2008) and haploinsufficient Zic2 mice and humans present schizophrenic symptoms (Hatayama et al., 2011). In advanced developmental stages, Zic2 controls the axonal navigation of retinal ganglion cells and dorsal spinal cord neurons at the midline by regulating the expression of Eph receptors (Herrera et al., 2003; García-Frigola et al., 2008; Escalante et al., 2013). However, before axon-pathfinding processes, Zic2 is expressed in other cell types such as Cajal–Retzius cells (CRCs) (Inoue et al., 2008), dorsal neural tube cells (Nagai et al., 1997; Escalante et al., 2013), or granule cell progenitors in the cerebellum (Aruga et al., 2002). Surprisingly, despite the devastating consequences of having low levels of Zic2, its precise role in early development is still poorly understood.
It has been proposed that the disorganization in cortical lamination observed in Zic2 mutant individuals could be the consequence of defects in the biology of CRCs and/or meningeal cells (Inoue et al., 2008). However, the function of Zic2 in CRCs has not been analyzed thoroughly. CRCs are an early-born neuron population with different origins, including the cortical hem and the pallial septum (Meyer et al., 2002; Takiguchi-Hayashi et al., 2004; Bielle et al., 2005; García-Moreno et al., 2007), that localize in the cortical preplate and then in the marginal zone during the development of the cortical plate (Marín-Padilla, 1998; Meyer et al., 1999). CRCs migrate tangentially to disperse over the cortical surface and it has been recently suggested that this dispersion is facilitated by random cell–cell contact repulsions mediated by Eph/ephrin signaling (Villar-Cerviño et al., 2013). Once CRCs are homogeneously dispersed in the cortex, they stimulate radial migration of late-born cortical neurons via secretion of the extracellular protein reelin, which then influences cortical layering (D'Arcangelo et al., 1995; Ogawa et al., 1995; Frotscher, 1998; Rice and Curran, 2001; Shinozaki et al., 2002; Tissir and Goffinet, 2003; Gil-Sanz et al., 2013).
Here, we report a new function for Zic2 in the migration of CRCs. Zic2 mutant embryos present anomalous morphologies and locations of CRCs, resulting in their uneven distribution along the precocious cortical surface, together with misplacement into deep cortical layers. Given the function of Zic2 in the mobility of CRCs, we aimed to investigate its role in other migrating cell types and found that this transcription factor is also essential for the proper migration of at least two additional subpopulations of forebrain neurons: neuroepithelial cells in the caudal tier of the dorsal pallium moving to the amygdala via the caudal amygdaloid stream (CAS) and prethalamic subventricular neuroepithelial cells traveling to the ventral LGN (vLGN). Our results suggest that the expression of the tyrosine kinase receptor EphB1, previously identified as a Zic2 effector in axon guidance, also depends on this transcription factor during migratory processes. In summary, at least three types of forebrain-migrating cells exhibit aberrant cell migration in Zic2 mutant mice, displaying severe difficulties in reaching their final destinations, impairing forebrain morphology, and contributing to the undesirable Zic2-associated phenotypes.
Materials and Methods
Mouse lines.
The Tg(Zic2EGFP)HT146Gsat/Mmcd line (identification number RP23-158G6) was generated by GENSAT (Gong et al., 2003) and obtained from the Mutant Mouse Regional Resource Center (http://www.mmrrc.org/strains/17260/017260.html). The hypomorphic Zic2kd mice line (Zic2tm1Jaru) was obtained from the RIKEN repository. Transgenic mice Tg(Zic2eGFP) were crossed with the heterozygous mice for Zic2 (Zic2kd/+) and the resulting F1 progeny was crossed [(Zic2+/−; Tg(Zic2eGFP) × (Zic2+/−; Tg(Zic2eGFP)] to generate [Zic2+/+; Tg(Zic2eGFP] and [Zic2kd/kd; Tg(Zic2eGFP) embryos into the same litter. These mouse lines allowed us to visualize, by eGFP fluorescence, both Zic2 cells in the control embryos and cells with no (or very low) levels of Zic2 in the Zic2 mutant embryos. All mouse lines were congenic on a C57BL/6J background and were kept in a timed pregnancy-breeding colony at the Instituto de Neurociencias. The animal protocols were approved by the Institutional Animal Care and Use Committee and met European and Spanish regulations.
BrdU injections and immunohistochemistry and in situ hybridization.
Immunostaining and in situ hybridization in mouse embryos were performed as described previously (Acloque et al., 2008). Antibodies used in this study: GFP (Aves Labs), Arx (Santa Cruz Biotechnology), Tuj1 (Covance Technology), RC2 (Developmental Studies Hybridoma Bank), Otx2 and Trb2 (Millipore), BrdU, Cofilin, and Tbr1 (Abcam), Reelin G-10 (gift from André Goffinet), Calretinin (Swant), Zic2 (home-made as described in Brown et al., 2003), COUP/TF2 (gift from Michèle Studer), and Caspase3 (Cell Signaling Technology). Probes for EphB1, ephrinB2, and ephrinB3 were described previously (Williams et al., 2003; Escalante et al., 2013).
For BrdU experiments, one single injection of BrdU (50 mg of BrdU/kg of body weight) was administrated intraperitoneally in embryonic day 10.5 (E10.5), E11.5, E12.5, or E13.5 pregnant females to label dividing cells in the embryos. Embryos were then analyzed at E14.5. Cryostat sections (20 μm) were pretreated with HCl and borate before performing BrdU and Zic2 staining. Slides were counterstained with DAPI. Fluorescence intensity (FI) obtained from immunostaining with Zic2, BrdU, or both was measured in thalamic cells using ImageJ. For quantification of BrdU cells in wild-type (WT) and mutant embryos, four/five anatomically comparable sections were analyzed per embryo. Two complementary squared areas were delineated along the migratory stream in each section as shown in Figure 8E. FI was measured in the area proximal to the ventricle (medial area) and in the area that includes the vLGN (lateral area). Corrected total area fluorescence was calculated from ImageJ measurements according to the following formula: integrated density (selected area × mean of background fluorescence).
Figure 8.

Zic2 mutant mice show lower number of eGFP thalamic prethalamic boundary (TpTB) IN in the vLGN. A, B, Coronal sections from E14.5 [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd;Tg(Zic2eGFP)] embryos through the medial diencephalon. Dashed lines delineate the MS from the third ventricle to the vLGN. White arrowheads highlight the reduction in the number of cells in the MS and vLGN of Zic2 mutant mice. Red arrowheads point to cells mislocated in the prethalamus of Zic2 mutant embryos. C, D, Close-up of the ventricular region of E14.5 [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd;Tg(Zic2eGFP)] embryos. Red arrowheads point to an area containing accumulation of cells in the ventricular wall of a Zic2 mutant embryo. E, F, Coronal sections from E14.5 control and Zic2 mutant embryos from mothers injected with BrdU at E12.5. A reduction in the number of BrdU-positive cells is observed in the vLGN area, but not in the ventricular areas of Zic2 mutant mice (arrowheads). White lines divide medial and lateral thalamic areas. G, H, Caspase3-positive cells (arrows) in coronal sections from E14.5 [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd;Tg(Zic2eGFP)] embryos. I, Graphs representing the quantification of FI from BrdU-positive cells in the MS close to the ventricular zone (medial area) or close to the vLGN (lateral area) in Zic2 mutant and control embryos. J, Graph representing the number of Caspase3-positive cells in Zic2 mutant and control embryos at E14.5. Th, Thalamus; pTh, prethalamus; TCA, thalamocortical axons.
Telencephalic vesicles preparation and quantification.
E12.5 [Zic2+/+; Tg(Zic2eGFP) and Zic2kd/kd; Tg(Zic2eGFP)] embryos were dissected and telencephalic vesicles were transferred to Krebs' solution. The meninges were dissected out and the telencephalic vesicles were flattened by placing four cuts around the external area (see Fig. 2A). Flattened vesicles were then fixed with 4% PFA and incubated with anti-GFP antibodies. After incubation with secondary antibodies, vesicles were mounted and photographed. For quantification, flattened telencephalic vesicles were adapted into a rectangular ROI. The ROI was longitudinally divided in 11 columns of 1040 × 60 pixels each, with the first one being most proximal to the hem. Average FI was measured with ImageJ, obtaining a numeric value for each column that was then compared one by one between WT and mutants.
Figure 2.
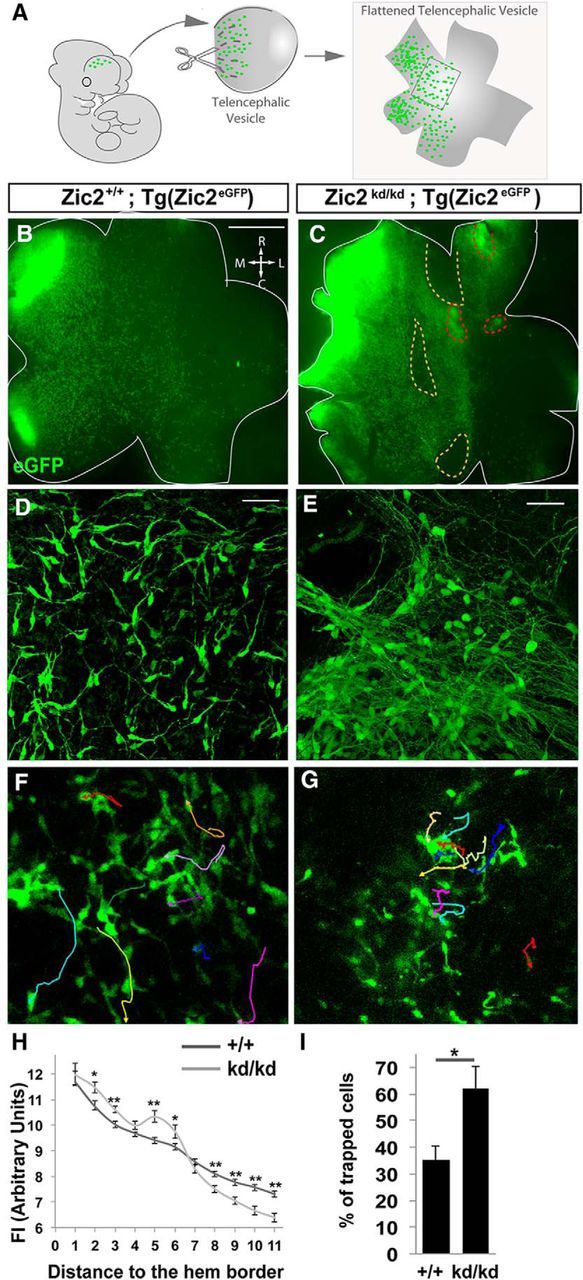
Zic2 mutants exhibit defects in the distribution of CRCs. A, Scheme summarizing the experimental procedure. Telencephalic vesicles were isolated, cut, and flattened on a dish. B, C, Images showing flattened telencephalic preparations from [Zic2kd/kd; Tg(Zic2eGFP)] and control E12.5 embryos. Note that, whereas in the control preparation, eGFP cells delineate a mediolateral gradient with homogeneously distributed CRCs, in the mutant vesicle, eGFP cells are unevenly distributed. Yellow-dashed areas in C are devoid of eGFP cells. Red-dashed areas delineate places where green cells had migrated into deeper layers. D, E, Images showing high magnification of cells from flattened telencephalic preparations. Zic2 mutant CRCs show longer processes compared with the control cells. Note that in, some areas, mutant CRCs were grouped or attached to neighbor cells at the expense of other areas devoid of cells. F, G, Time-lapse sequence of the surface of whole-mount telencephalic preparations from [Zic2kd/kd; Tg(Zic2eGFP)] and control E12.5 embryos. Cell trackings are indicated in different colors. H, Graph representing the distribution of eGFP CRCs measured by FI in flattened telencephalic vesicles of [Zic2kd/kd; Tg(Zic2eGFP)] and control E12.5 embryos. Note the higher FI in the regions close to the hem in the Zic2 mutant embryos compared with the controls and a lower FI in the regions distal to the hem. I, Graph representing the percentage of trapped cells that form aggregates and therefore do not move or move for very short distances in the time-lapse videos. R, Rostral; C, caudal; M, medial; L, lateral. Scale bars: B, C, 500 μm; D–G, 50 μm.
Culture and analysis of CRC in hem explants.
[Zic2+/+; Tg(Zic2eGFP)] and [Zic2kd/kd; Tg(Zic2eGFP)] embryos were dissected and telencephalic vesicles were included in 4% low-melting-point agarose to section them in slides of 250 μm as described previously (Anderson et al., 1997). Vibrosections were collected in Krebs' solution and the ones containing the hem were transferred to a Track-Etched (Nucleopore) membrane, incubated 1 h with MEM, and transferred to neurobasal media to be incubated for 30 min. The hem region was dissected, incubated in neurobasal for 15 more minutes and placed in Poly-l-Lysine Cell-ware 12 mm round Coverslips-BD BioCoat previously treated with 20 μg/ml laminin. Hem explants were then incubated with 0.4% neurobasal methylcellulose medium for 30–40 h. All incubations were performed at 37°C and 5% CO2. The ImageJ plug-in MTrack was used to determine the longest neurite (LN) and the shorter neurite (SN) and to quantify the complexity of CRCs. Neurites were divided into two groups: (1) first-order neurites (1ONs), those growing directly from the cell soma, and (2) second-order neurites (2ONs), those arising from first-order neurites. Cell quantification was performed in four ROIs/explant and explants from six embryos/genotype.
CRCs were quantified by manually counting the total cells/explant. A circle was delineated to delimit the explant border. A second circle located concentrically 150 μm from the first one was drawn to divide the explant in two areas: the inner part closest to the explant and the outer part distal to the explant. The cells in the culture were counted manually and the percentage of cells located in each area was referred to the total number of cells in the explant. The amount of cells attached to the explant was calculated by delineating the area covered by cells still attached to the explant and measuring FI using ImageJ. FI was then normalized to the total area of the explant.
Time-lapse imaging and cell tracking.
[Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd; Tg(Zic2eGFP)] embryos were dissected to flatten telencephalic vesicles (see Fig. 2A) or to be included in 4% low-melting-point agarose to obtain 250 μm coronal slides. Flattened telencephalic vesicles or prethalamic slides were cultured in membrane wells with MEM for 45 min and incubated in neurobasal medium. eGFP cells were recorded with a Leica laser scanning spectral confocal microscope (TCS SP2) using resonant scanning with long working distance and water-immersion 20× objectives. The ImageJ plug-in MTrack was used to track cell trajectories as described previously (Villar-Cerviño et al., 2013).
Quantitative and statistical analysis.
All quantifications were presented as the mean ± SEM. Minimal statistical significance was fixed at p < 0.05 for the results of the Student's t test and are represented in the figures as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
Zic2 is required for the proper migration of CRCs
To investigate the function of Zic2 in CRCs, we took advantage of a previously described reporter mouse line that expresses eGFP under the regulatory sequences of Zic2 [Tg(Zic2eGFP)] (Gong et al., 2003; Escalante et al., 2013). We observed eGFP cells in the surface of the telencephalic vesicles of E12.5 Tg(Zic2eGFP) embryos (Fig. 1A). These eGFP cells expressed CRC markers such as calretinin, reelin (del Río et al., 1995; Alcántara et al., 1998), COUP-TF2 (Tripodi et al., 2004), and Tbr1 (Hevner et al., 2001, 2003; Fig. 1B–E′). As expected, these eGFP cells were also positive for Zic2, confirming that Tg(Zic2eGFP) mice reproduce the endogenous expression of this transcription factor in the CRCs (Fig. 1F–G′) and demonstrating that Zic2 is expressed in these cells, as was proposed previously by in situ hybridization (Inoue et al., 2008). All the eGFP cells were reelin positive at E12.5, but eGFP was detected in only the 60.20 ± 4.14% of all the reelin-positive cells based on the observation that eGFP was detected in both the hem and the septum but not in the pallial/subpallial boundary. Zic2 was also expressed in a subpopulation of meningeal cells at E15.5 (Fig. 1G), as reported previously (Inoue et al., 2008), but eGFP was not detected in meningeal cells (Fig. 1G).
Figure 1.
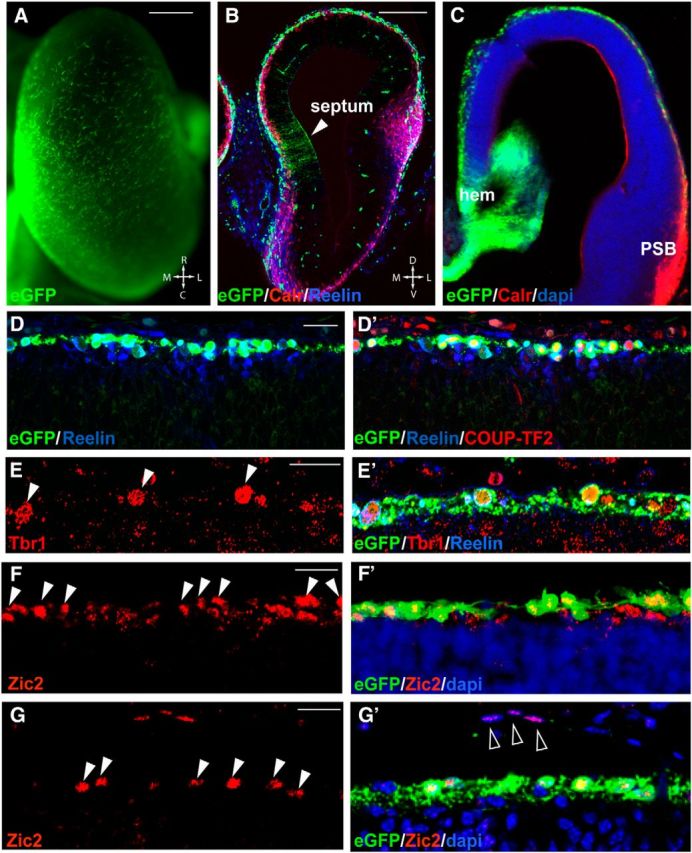
Zic2 is expressed in CRCs from hem and septum. A, Whole-mount telencephalic hemisphere of a E12.5 Tg(Zic2eGFP) embryo. B, Calretinin and reelin immunostaining in coronal sections through the rostral telencephalon of E12.5 Tg(Zic2EGFP). Note eGFP expression in the septum region. C, Calretinin immunostaining in coronal sections through the telencephalon of E12.5 Tg(Zic2EGFP) mouse embryos revealed that, whereas a large number of calretinin cells are found in the dorsolateral superficial cells, eGFP cells only localize in medial areas, close to the hem region. D–D′′, Reelin and COUP-TF2 stainings in coronal sections through the telencephalon of E12.5 Tg(Zic2EGFP) mouse embryos show that the majority of Zic2/eGFP cells express these CRC markers. E, E′, Tbr1 and reelin staining in coronal sections of E14.5 Tg(Zic2EGFP) embryos confirm that Zic2/eGFP cells still express this CRCs marker at later stages. Arrowheads point to Tbr1 positive cells that are also positive for reelin and eGFP. F–G′, Zic2 staining in coronal sections through the telencephalon of E12.5 and E15.5 Tg(Zic2EGFP) mouse embryos demonstrate that eGFP expression reproduces the endogenous expression of Zic2 in the CRCs (white arrowheads). Note that, at E15.5, Zic2 but not eGFP is expressed in some DAPI-stained meningeal cells (empty arrowheads). R, Rostral; C, caudal; M, medial; L, lateral; PSB, palium subpalium boundary. Scale bars: A–C, 200 μm; D–G′, 20 μm.
To further analyze the distribution of eGFP CRCs over the developing cortex in WT and Zic2 mutant mice, we dissected and flattened the telencephalic vesicles of control [Zic2+/+;Tg(Zic2eGFP)] and hypomorphic Zic2 [Zic2kd/kd;Tg(Zic2eGFP)] mice (Nagai et al., 2000; Escalante et al., 2013; see Materials and Methods; Fig. 2A–E). The total number of eGFP cells, measured by FI, was similar in flattened vesicle preparations from the Zic2 mutants [average FI in the WT vesicles (FIWT) = 26.94 ± 2.50 arbitrary units (a.u.); n = 6] or from the controls (FIMUT = 26.17 ± 3.10 a.u.; n = 7; p = 0.85). However, although flattened vesicles from E12.5 control embryos showed a high density of eGFP cells at the hem/septum that spread gradually to lateral areas, eGFP cells in the Zic2 mutants were unevenly distributed over the cortical surface. In fact, an accumulation of eGFP cells was detected into the areas proximal to the hem/septum in the mutants compared with the control littermates, whereas fewer eGFP cells reached the lateral regions of the vesicles in the mutants (Fig. 2B,C,H). We also observed that in the Zic2-hypomorphic embryos eGFP cells show very long processes and often form circled paths or aggregates compared with control cells (Fig. 2D,E). This phenotype suggested defects in cell migration, so we decided to analyze the behavior of eGFP CRCs in the telencephalic vesicles of Zic2 mutant and WT E12.5 embryos by time-lapse imaging. Video-tracking analysis revealed that, in contrast to the behavior of eGFP in the controls, in the Zic2 mutants, many eGFP CRCs were trapped, forming aggregates (Fig. 2F,G,I). In addition, in the mutant preparations, many cells seemed to migrate to deeper layers. To confirm whether this was the case, we sectioned the cortex of E12.5 embryos and observed that, in fact, eGFP CRCs were occasionally spreading over deeper layers of the cortical primordium (Fig. 3A–C). At later stages (E15.5), reelin-expressing eGFP CRCs were found in deep cortical layers in the Zic2 mutant mice, whereas in the controls, CRCs were located exclusively below the pial surface (Fig. 3D,E). Using markers specific for different cell types in the developing cortex (Tbr1 to label layer 6 and Tbr2 to label intermediate progenitors in the subventricular zone), we confirmed that, as soon as E12.5 and coinciding with the CRCs mislocalization phenotype, cortical lamination was severely affected in the rostral telencephalon of mutant mice (Fig. 3F–K), showing a cobblestone-like appearance, as reported previously (Inoue et al., 2008).
Figure 3.
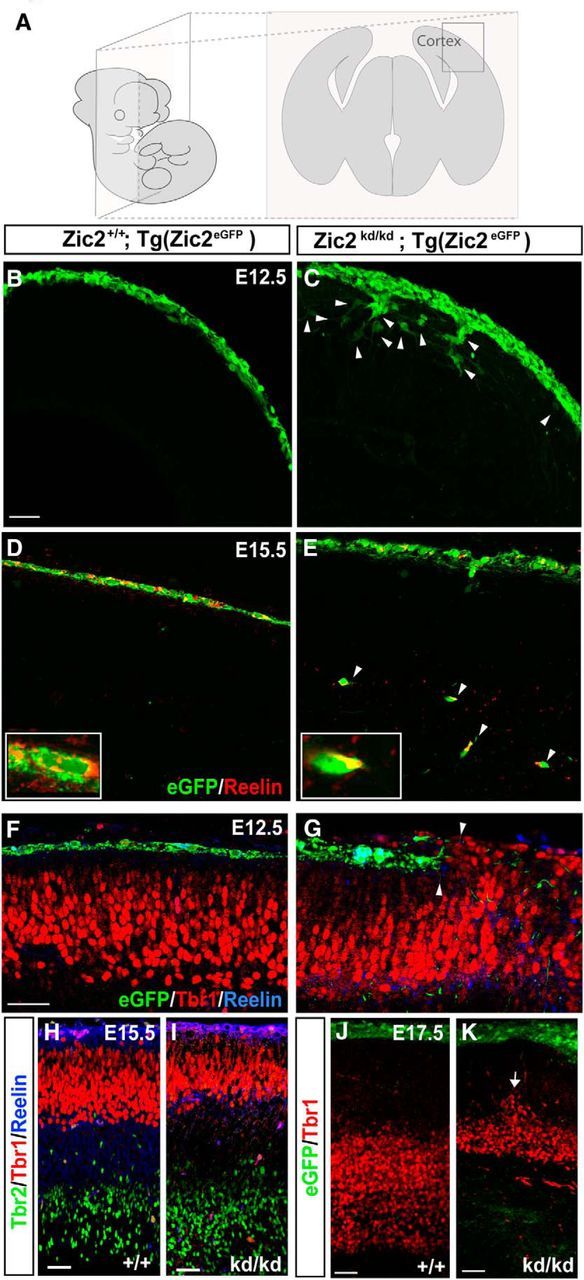
Zic2 mutants show cortical lamination defects. A, Schematic drawings representing the orientation and level of sectioning in the rest of the figure. B, C, Coronal sections of E12.5 Tg(Zic2eGFP) embryos showing that Zic2 CRCs are located in the superficial layer of the telencephalic vesicle, whereas in the Zic2 mutant embryos, eGFP cells were not restricted to the most superficial cortical layer. White arrowheads point to mislocated eGFP cells. D, E, Coronal sections of E15.5 Tg(Zic2eGFP) embryos showing that Zic2 CRCs are located in the telencephalic superficial layer. In contrast, in the Zic2 mutant embryos, many eGFP cells expressing reelin reached deep cortical laminae. White arrowheads point to mislocated eGFP/reelin cells. F–J, Inmunohistochemistry for Tbr1, Tbr2, and reelin in coronal sections of embryos at the indicated stages show that cortical layering, particularly the Tbr1 laminae, are affected from early stages of development (arrows) in the Zic2 mutant mice. Scale bars in B–K indicate 100 μm.
To better visualize the morphology of individual CRCs and to facilitate cell movement analysis in a 2D surface, we dissected the hem regions of E12.5 Zic2 mutant (n = 6) and control embryos (n = 6) and cultured them for 36 h on wells preincubated with laminin to evaluate the number of CRCs migrating from the hem explant. A total of 348.80 ± 2.38 CRCs per explant were evenly dispersed through the well surface in control cultures and 19.47 ± 2.02% of them migrated >150 μm away from the explant. In contrast, in the Zic2 mutants, despite the fact that the size of mutant and WT explants was similar, the number of CRCs moving away from the explant was consistently lower (107.67 ± 1.30) and only 10.25 ± 1.17% (p < 0.002) of them migrated >150 μm away from the explant (Fig. 4A,B,K). In addition, many more cells were attached to the Zic2 mutant explants (IFMUT: 239.93 ± 19.10) compared with the controls (IFWT: 185.44 ± 14.87 a.u.; p < 0.05; Fig. 4A′–B′,L), suggesting that Zic2kd/kd CRCs migrate shorter distances or do not even leave the explant. Moreover, many mutant cells looked longer and exhibited excessive sprouting compared with control cells (Fig. 4C–J). Measurements of the LNs and SNs in individual cells confirmed that CRCs are significantly longer when Zic2 is downregulated (LN: WT = 33.15 ± 0.75 μm, mutant = 37.55 ± 1.4 μm, p < 0.003; SN: WT = 9.73 ± 0.36 μm, mutant = 12.75 ± 0.65 μm, p < 0.001). To quantify the sprouting phenotype, we counted the 1ONs and 2ONs. Most cells were bipolar in both the Zic2 mutant and control cultures (1ON: WT = 2.00 ± 0.08 neurites/cell, mutant = 1.89 ± 0.04 neurites/cell, p = 0.302). However, the number of 2ONs or lamellipodia/filopodia arising from 1ONs was significantly higher in the mutant compared with the control CRCs (2ON: WT = 2.56 ± 0.20 neurites/cell, mutant = 3.59 ± 0.17 neurites/cell, p < 0.002; Fig. 4C–J,M).
Figure 4.
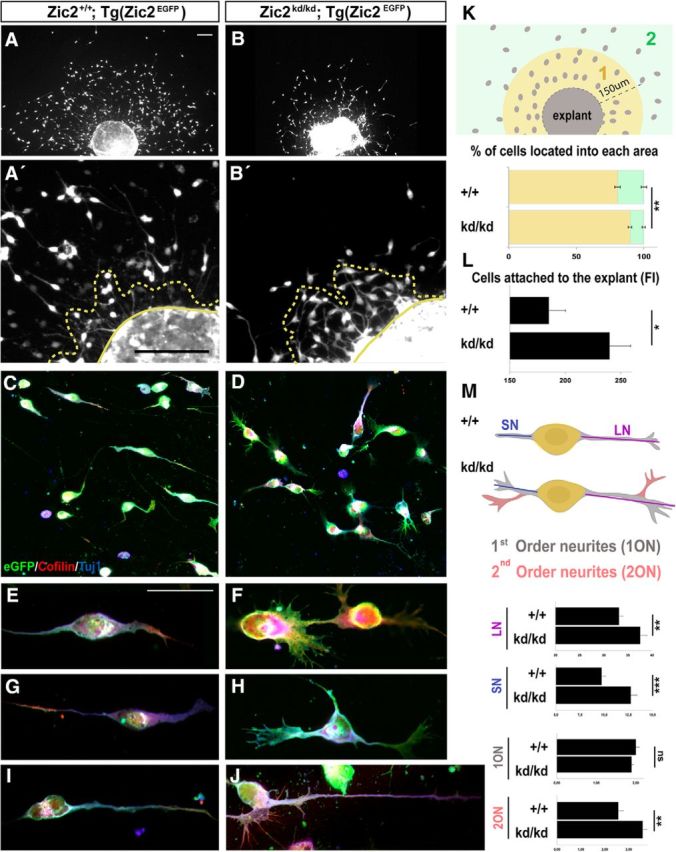
Zic2 is essential to maintaining proper morphology and migration of CRCs. A, B, Hem explants from E12.5 [Zic2+/+;Tg(Zic2eGFP)] or [Zic2kd/kd; Tg(Zic2eGFP)] embryos cultured on laminin showed a reduced number of cells migrating away from the explant. A′–B′, High magnification of representative hem explants from E12.5 embryos showing a higher number of eGFP CRCs attached to the explant in the [Zic2kd/kd; Tg(Zic2eGFP)] explant than in the controls. Yellow dashed lines include cells that have not completely detached from the explant. C, D, Immunostaining against cofilin and Tuj1 in migrating cells from the explants showing cytoskeleton differences between Zic2 mutant and control cells. E–J, Three representative examples of WT and Zic2 mutant CRCs. K, Drawing representing the analyzed areas in hem explant cultures. The graph shows the percentage of cells located within the 150 μm close to the explant (area 1, yellow) and the percentage of cells located in area 2 (green). Note that, in the Zic2 mutant explants, most cells are located in area 1 and there are very few cells located in area 2. L, Graph quantifying eGFP CRCs not completely detached from the explant measured by FI (in a.u.). M, Schematic drawing representing a control (+/+) and a Zic2 mutant (kd/kd) CRC to clarify the analysis approach. Light blue line represents the SN and the dark blue line the LN. Neurites growing directly from the cell body (yellow) were considered 1ONs (gray) and any other neurites were considered 2ONs (pink). Graphs show the quantification of 1ONs and 2ONs and the average length of the LNs and SNs in CRCs from E12.5 Zic2 mutant and control cells from hem explants. ns, Nonsignificant. Scale bars in A–J indicate 50 μm.
Together, both explant cultures and in vivo results suggested that Zic2 is required for proper morphology, migration, and dispersion of CRCs in the telencephalic vesicles during early embryonic development.
Zic2 is necessary for the migration of another population of telencephalic neurons, the caudal amygdaloid stream
While analyzing the CRCs, we observed another population of Zic2-expressing cells that appeared to delaminate from the neuroepithelium of the dorsal pallium at caudal levels and occupied a conspicuous curve-shaped stripe that resembles what has been described as the caudal amygdaloid stream (CAS). CAS cells migrate toward a cortical amygdaloid nucleus, the nucleus of the lateral olfactory tract (nLOT) (Remedios et al., 2007). The nLOT is connected to the olfactory bulb and piriform cortex and is composed of three layers (Santiago and Shammah-Lagnado, 2004). Layer 2 (nLOT2) differs from the other nLOT layers in its distinct embryonic origin in the most caudal part of the dorsal telencephalon (Fig. 5A). nLOT2 cells are generated between E11.5 and E12.5 (Remedios et al., 2007; Soma et al., 2009) and then migrate rostrally along the CAS to reach the amygdala (Remedios et al., 2007).
Figure 5.

Zic2 is expressed in CAS cells migrating from the caudal telencephalon to the amygdaloid nucleus. A, Schematic drawing representing the area of CAS cells migrating from the lateral ventricle to nLOT2. B, C, Zic2 and Tbr1 staining in sagittal sections of E15.5 [Zic2+/+;Tg(Zic2eGFP)] embryos demonstrating that eGFP expression reproduces the endogenous expression of Zic2 in the area where Tbr1 CAS cells are migrating to nLOT2. D, E, Representative sagittal sections of Zic2 E15.5 [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd; Tg(Zic2eGFP)] embryonic brains revealing a defect in the trajectories of eGFP positive cells in the Zic2 mutant mice. D′–E′, Tbr1 immunostaining in sagittal sections of Zic2 E15.5 [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd; Tg(Zic2eGFP)] embryonic brains. High-magnification images are shown in the squared areas in D and E. D′′–E′′, High magnification of the squared areas in D and E reveal aberrant morphologies of eGFP cells in the Zic2 mutant mice. Scale bars: B, C, E, 100 μm; D–E′′, 50 μm.
Similarly to CRCs, control Tg(Zic2eGFP) mice reproduce Zic2 endogenous expression in CAS cells (Fig. 5B–B′′). Sagittal sections from Tg(Zic2EGFP) embryos showed that the Zic2/eGFP cells delaminating from the telencephalic neuroephitelium were positive for Tbr1, which also labels nLOT2 cells (Medina et al., 2004) (Fig. 5C), indicating that Zic2 cells were indeed migrating neurons navigating toward the amygdala. When hypomorphic [(Zic2kd/kd;Tg(Zic2eGFP)] mutant mice were analyzed, we noticed that the CAS cell population was less abundant, occupied a wider area, and did not form the curved-shaped stream typical in control mice (Fig. 5D,E). In addition, as was the case for CRCs, many CAS cells still expressed Tbr1 but presented anomalous morphologies in the mutants compared with the controls (cf. Fig. 5D′–E′, D′′–E′′).
These observations indicate that Zic2 is necessary for the proper migration of a second population of telencephalic neurons, the cells integrating the CAS that will populate the amygdaloid nLOT2 nucleus.
Zic2 also controls cell migration in a population of diencephalic interneurons that will end at the vLGN
We wondered whether Zic2 plays a role in the migration of nontelencephalic neural types during development. We knew that Zic2 is expressed in postmitotic thalamocortical neurons (Escalante et al., 2013), but we also noticed another population of Zic2-expressing cells in the diencephalon that formed a narrow stream running from the prethalamic neuroepithelium to the vLGN (Fig. 6A). This Zic2 expression was also recapitulated by the expression of eGFP in the Tg(Zic2eGFP) mice (Fig. 6C,D,F–F′′′). At E13.5, Zic2/eGFP cells in this stream coincided with a region positive for Arx, a transcription factor that marks the prethalamus and its boundary with the thalamus (Kitamura et al., 1997; Fig. 6B). One day earlier (E12.5), Zic2 was expressed rostral to the expression of Sonic Hedgehog (data not shown) in the zona limitans intrathalamica (ZLI), a signaling center compartment located between the thalamus and the prethalamus. Altogether, these data showed that Zic2-positive cells localize in the prethalamic area adjacent to the ZLI. To further characterize this Zic2 population in the prethalamus, we performed immunostaining for Tuj1 and RC2 in E15.5 coronal sections from Tg(Zic2eGFP) embryos to distinguish neurons from radial glial cells (Fig. 6E,F). Zic2 cells were identified as neurons because they expressed Tuj1. We then used Otx2 staining to label a population of interneurons (INs) located between the thalamic and the prethalamic area (Golding et al., 2014) and observed that the Zic2 cells that form a narrow stripe in the prethalamus are indeed Otx2 positive and are therefore INs (Fig. 6G).
Figure 6.
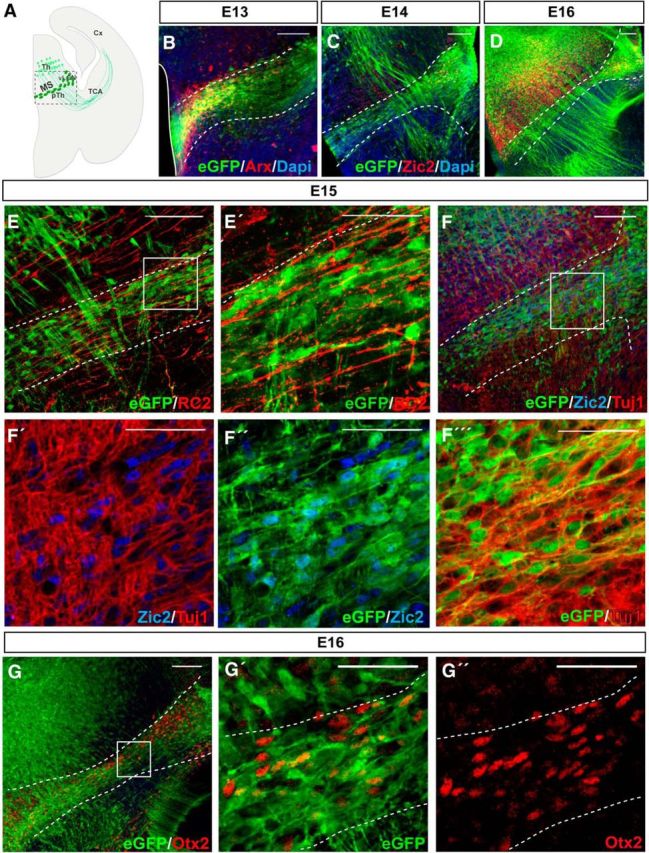
Zic2 is expressed in thalamus–prethalamus boundary INs. A, Scheme representing a coronal section from an E13.5 mouse brain. The squared area delimits the zone depicted in all the images of this figure. B, Arx immunostaining in coronal sections of E13.5 Tg(Zic2eGFP) embryos confirm the existence of a population of Zic2/eGFP cells in the thalamic-prethalamic boundary. White dashed lines delimit the thalamic prethalamic boundary (TpTB) Zic2-positive area. C, D, Zic2/eGFP cells in the thalamic–prethalamic boundary at E15.5 and E16.5. Note that Zic2 is also expressed in the thalamus and that thalamocortical axons are also positive for eGFP and visible from E14.5 in advance. E, E′, Immunostaining for RC2 in coronal sections of E15.5 Tg(Zic2eGFP) embryos demonstrate that Zic2/eGFP cells are not glial cells. F–F′′′, Zic2 and Tuj1 immunostaining in coronal sections of E15.5 Tg(Zic2eGFP) embryos demonstrate that Zic2/eGFP cells in the TpTB are neurons. Note that eGFP cells present Zic2 staining in the nucleus. G–G′′, Otx2 immunostaining in coronal sections of E15.5 Tg(Zic2eGFP) embryos demonstrate that Zic2/eGFP cells are INs. Th, thalamus; pTh, prethalamus, Cx, cortex; TCA, thalamocortical axons. Scale bars: B–G, 100 μm; E′–G′′, 50 μm.
Although previous evidence has suggested that a subpopulation of vLGN INs originate in the prethalamic subventricular zone (Kitamura et al., 1997; Vue et al., 2009; Golding et al., 2014), their movement from the neuroepithelium has never been confirmed. To confirm the existence of such cell migration, we first determined, by BrdU uptake experiments, the birthdate of most Zic2 IN at E12.5 (Fig. 7A,B). Next, we performed time-lapse imaging experiments in coronal slides taken from E13.5 Tg(Zic2eGFP) embryos, when most Zic2 INs are already differentiated. Analysis and quantification of time-lapse videos (n = 5) in coronal slices demonstrated that Zic2/eGFP cells arising in the subventricular zone form a migratory stream (MS) of cells that moves along the prethalamus to the vLGN with an average speed ratio of 7.27 ± 0.28 μm/h (Fig. 7C). Then, to investigate whether Zic2 plays a role in the migration of these prethalamic INs, we analyzed E13.5 embryos and found that the number of eGFP cells in the MS and in the vLGN of Zic2 mutants was significantly lower than in the control embryos (Fig. 8A,B). However, this phenotype was variable, ranging from just a narrower MS (6 embryos of 11 mutants) compared with the WT embryos (n = 14) to almost no cells in the MS (5 embryos of 11 mutants). In addition, we observed eGFP cells located ectopically in the prethalamus that appeared to be MS cells that escaped from the stereotyped pathway (Fig. 8A,B). A number of Zic2 mutant mice also presented cell accumulations in the subventricular area (7 of 11 mutants; Fig. 8C,D). BrdU uptake experiments showed a higher number of BrdU cells in the subventricular region of mutant embryos (FI:7.57 ± 0.38 a.u.; n = 5) than in the controls (5.91 ± 0.43 a.u.; n = 5) and fewer BrdU cells in the vLGN area of mutants (3.71 ± 0.22 a.u.) than in the WT littermates (4.74 ± 0.41 a.u.) (Fig. 8E,F,I). However, analysis of cycling cells in the subventricular zone of E13.5 embryos, by counting phosphohistone (pH3)-positive cells, did not reveal significant differences between WT and mutant embryos (WT: 0.34 ± 0.17 cells/μm2, n = 5; mutant: 0.38 ± 0.12 cells/μm2, n = 3; p = 0.62), indicating that proliferation is not grossly altered in the ventricular wall of Zic2 mutant mice. In agreement with previous reports showing similar number of apoptotic cells in zebrafish or Zic2 mutant mice compared with control animals (Inoue et al., 2008; Sanek and Grinblat, 2008), we did not find differences in the number of Caspase3-positive cells in the thalamus and the prethalamus of E12.5 Zic2 mutants (9.67 ± 1.20 cells/embryo; n = 3) compared with their WT littermates (9.33 ± 2.33cells/embryo; n = 3; p = 0.91; Fig. 8G,H,J). Therefore, the most likely explanation for both the accumulation of BrdU cells in the ventricular area and the ectopic cells located in the prethalamus would be a defect in the migration of eGFP cells to lateral areas. To assess whether this was in fact the case, we performed time-lapse videos in E13.5 embryos and tracked the trajectories of 121 eGFP cells in [Zic2kd/kd; Tg(Zic2eGFP)] mutants (n = 5) and 104 eGFP cells in [Zic2+/+; Tg(Zic2eGFP)] control mice (n = 4) (Fig. 9A–C). In the control embryos, most eGFP cells originated in the ventricle and migrated correctly, following a medial-to-lateral stereotyped pathway to the vLGN. Only 16.65 ± 2.60% of eGFP cells did not follow this stereotyped pathway in the WT embryos. In contrast, in the Zic2 mutants, a significant number of eGFP cells (70.96 ± 6.71%; p < 0.001) showed any of three types of aberrant migration: (1) they escaped from the stereotyped path, (2) they moved in the opposite direction to the normal path, or (3) they did not follow a linear pathway (Fig. 9D). In addition, those eGFP cells abandoning the regular trajectory exhibited an altered morphology, with many protrusions and longer processes than in control embryos. The migratory speed of the eGFP cells was significantly higher in Zic2 mutant embryos compared with the controls (average speed: WT, 7.27 ± 0.28 μm/h; mutant, 8.69 ± 0.47 μm/h, p < 0.02; maximum speed: WT, 24.47 ± 1.23 μm/h; mutant, 30.44 ± 1.38 μm/h, p < 0.002; Fig. 9D). These results indicate that Zic2 is required in a subpopulation of INs to migrate correctly from the prethalamic subventricular zone to the vLGN and also confirm a role for Zic2 in cell migration in these diencephalic migrating INs.
Figure 7.
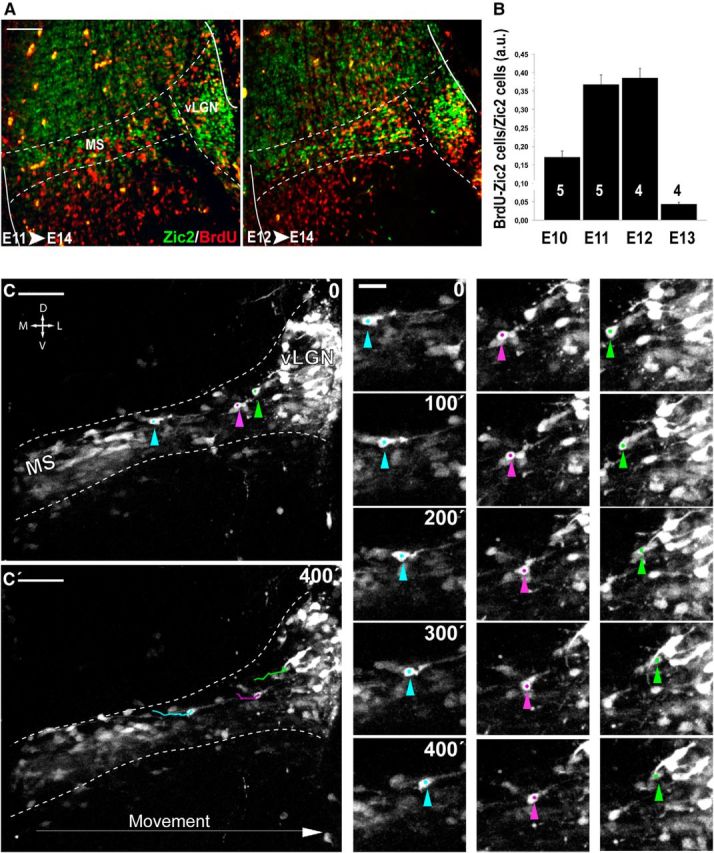
Zic2 thalamus–prethalamus INs are born between E11.5 and E13.5. A, Double staining for BrdU and Zic2 in coronal sections from E14.5 embryos with mothers that were injected with BrdU at E11.5 or E12.5. B, Quantification of the number of double-labeled Zic2/BrdU cells in E14.5 embryos from mothers injected with BrdU at E10.5, E11.5, E12.5, or E13.5. The majority of the Zic2 cells in the thalamic prethalamic boundary (TpTB) area were born between E11.5 and E12.5. Quantifications were performed in at least four sections per embryo. Numbers in the columns represent number of embryos analyzed. C, C′, Time-lapse sequence in the MS of E13.5 Tg(Zic2eGFP) embryos. At the left, color arrowheads point out the starting (top) and the ending (bottom) points of the video. At the right, the three examples of migrating eGFP cells (turquoise, violet, and green arrowheads) over time demonstrate that these cells move from lateral to medial locations. D, Dorsal; V, ventral; M, medial; L, lateral. Scale bars: A, C–C′, 100 μm; small panels, 20 μm.
Figure 9.
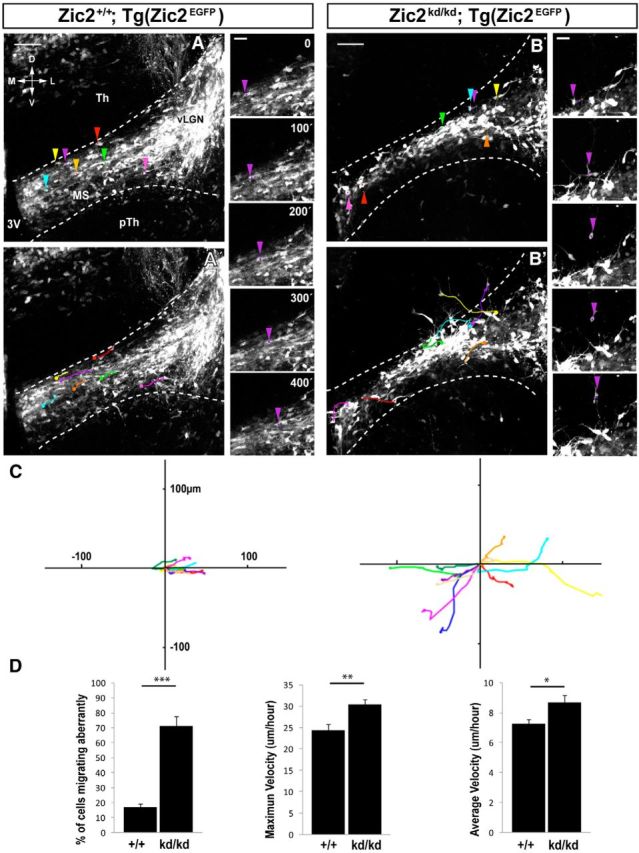
eGFP thalamic prethalamic boundary (TpTB) INs from Zic2 mutant mice show defects in migration. A–B′, Time-lapse sequence of cells in the MS of [Zic2+/+;Tg(Zic2eGFP)] and [Zic2kd/kd;Tg(Zic2eGFP)] E13.5 embryos. Color arrowheads in A and B mark the starting point of individual eGFP cells and colored lines in A′ and B′ delineate individual cell trajectories. Note that, in the Zic2 mutant mouse, many cells abandon their stereotyped mediolateral path to the vLGN. As an example, the trajectories of an eGFP-migrating cell in the control and the Zic2 mutant are shown and highlighted with a purple arrowhead. C, Tracks of 10 cells taken from three different embryos evidence aberrant trajectories in Zic2 mutant embryos (right) compared with the controls (left). Cell trajectories in the mutants delineate aberrant directions and cover longer distances. D, Quantification of the percentage of cells that migrate aberrantly and the maximum and average speed of [Zic2kd/kd;Tg(Zic2eGFP)] and control cells. D, Dorsal; V, ventral; M, medial; L, lateral; Th, thalamus; pTh, prethalamus; vLGN, ventral lateral geniculate nucleus; 3V, third ventricle. Scale bars: A–B′, 100 μm; small panels, 20 μm.
Expression of the guidance receptor EphB1 is disrupted in the migrating INs of Zic2 mutants
The tyrosine kinase receptors EphB1 and EphA4, two different receptors for ephrinB2, are respectively controlled by Zic2 in two different types of differentiated neurons, the retinal ganglion cells (García-Frigola et al., 2008) and the dorsal horn neurons in the spinal cord (Escalante et al., 2013). To investigate whether any of these two receptors are implicated in the migration of Zic2 cells, we first analyzed the diencephalic expression of EphA4 and EphB1 in the thalamic prethalamic boundary area. In situ hybridization in coronal sections showed that, whereas EphA4 is not expressed in the MS (data not shown), EphB1 mRNA was detected in E13.5 embryos in the area positive for Zic2 INs (Fig. 10A,B′′). This EphB1 mRNA expression was reduced in the Zic2kd/kd mutant (Fig. 10C,D), suggesting that, as reported previously in the visual system, Zic2 may control the expression of EphB1 in migrating Zic2 INs. We then analyzed the expression of ephrinBs in this region by in situ hybridization in coronal sections of E13.5 WT embryos. EphrinB2 was expressed near the ventricular area of the thalamus and very mildly in the prethalamus. The expression of ephrinB3 in the thalamus was similar to ephrinB2, but ephrinB3 was also expressed at basal levels in the rest of the thalamus and strongly expressed in the prethalamic area. However, the boundary between thalamus and prethalamus was devoid of ephrinB2/B3 expression. Therefore, the expression of both ephrinB2 and ephrinB3 exhibited a complementary pattern to the expression of EphB1 all along the MS pathway (Fig. 10E,F).
Figure 10.

EphB1 is expressed in thalamic prethalamic boundary (TpTB) cells and its expression is disrupted in Zic2 mutant mice. A–B′′, In situ hybridization in coronal sections through the diencephalon of E13.5 embryos showing that EphB1 mRNA is expressed in the TpTB area coinciding with the location of Zic2 cells. Arrowheads point to EphB1 expression. B–B′′, Higher-magnification of the squared areas in B. C, D, In situ hybridization in Zic2 mutant and control littermates revealing a reduction in the expression of EphB1 in embryos expressing low levels of Zic2. Arrowheads delineate the expression of EphB1 in the control and its absence in the mutant section. E, In situ hybridization in coronal sections of E13.5 embryos showing that ephrinB2 mRNA is expressed in thalamic areas proximal to the ventricular zone. Zic2 staining reveals the Zic2 MS. F, In situ hybridization in coronal sections of E13.5 embryos showing that ephrinB3 mRNA is highly expressed in thalamic and prethalamic areas proximal to the ventricular zone and at basal levels in lateral areas. Note that the stream of Zic2/eGFP-migrating cells is devoid of ephrinB3 expression. L, Lateral; M, medial; 3V, third ventricle; pTH, prethalamus; Th, thalamus. Scale bars: A–F, 100 μm; B′-B′′, 50 μm.
Discussion
We show here that the transcription factor Zic2 plays a critical role in the migration of three different types of forebrain migrating cells, the CRCs, the pallial-derived neurons that will end in the nLOT2 nucleus of the amygdala, and a diencephalic group of INs that will integrate into the vLGN of the thalamus. Low levels of Zic2 provoke similar defects in the morphology of all of these cell types, affecting migration to their final destinations. In addition, the expression of EphB1, which was implicated previously in the dispersion of CRCs (Villar-Cerviño et al., 2013), is altered in the prethalamus of Zic2 mutant mice, suggesting that Zic2 regulates EphB1 expression during cell migration, as it does during axonal navigation processes.
Zic2 in telencephalic neurons
Explant cultures obtained from the Zic2 mutants showed a lower number of migrating CRCs than the controls. However, we did not observe in vivo an obvious reduction in the number of CRCs in the telencephalic vesicles of Zic2 mutants. Therefore, the more parsimonious explanation for a lower number of migrating CRCs in the cultures was a defect in their movement and/or dispersion. In fact, cell accumulations around the explant were observed more abundantly in the Zic2 mutant than in the control explants. Because of defects in cell migration and/or dispersion, eGFP CRCs could not even leave the Zic2 mutant explant, so fewer cells were observed in the dish. This was consistent with the aberrant distribution and behavior of eGFP CRCs found in the whole-mount telencephalic vesicles in vivo, in which many CRCs are trapped, forming cell aggregates. Therefore, both the results in vivo in thelencephalic vesicles and those obtained with the explants in vitro are compatible with defects in the migration/dispersion of the CRCs in the mutant animals.
The cortical layering defects described previously in Zic2 hypomorphic mice has been attributed to a function of Zic genes in CRCs and/or meningeal cells (Inoue et al., 2008). In both the time-lapse and the in vitro experiments (Figs. 2F,G, 4), we found striking migration differences between Zic2 mutant CRCs and controls even though the meninges were dissected out in all the cases. Therefore, we favor the hypothesis that defects in the migration of CRCs in the Zic2 mutant mice are caused by the lack of Zic2 expression in the CRCs themselves rather than defects in signals coming from meningeal cells. However, because Zic2 is also expressed in the meninges, we cannot disregard the idea that the cortical layering defect observed in the Zic2 mutants is due to a putative role for this transcription factor in meningeal cells.
CRCs in the E12.5 Zic2 mutants were not localized only in the preplate, but rather were dispersed toward deeper layers of the cortical primordium. This early defect in the migration of CRCs seems to provoke mislocalization of CRCs still expressing reelin into deeper areas at later stages (Fig. 3D,E). Therefore, in principle, we tend to believe that the cortical layering phenotype in the Zic2 mutants is due to the migration defects of CRCs. However, the relationship between CRCs and cortical lamination has been challenged lately because massive reduction of CRCs in the hem-ablated mice (Yoshida et al., 2006), mice lacking Dbx1-expressing progenitors in the septum and PSB (Bielle et al., 2005), and p73-deficient mice (Meyer et al., 2002) seem to show anatomically correct neocortical layers. Therefore, given the role of Zic2 in the migration of different cell populations, another hypothesis to explain cortical layering defects in the mutants is a putative role for Zic2 in the migration of cortical progenitors at some point of their radial trajectories, an issue that will be investigated in the future.
Zic2 in diencephalic neurons
A proliferative niche in the prethalamic wall of the third ventricle has been proposed as the origin of Otx2-INs that populate the vLGN. It has been reported previously that prethalamic INs are positive for Otx2 (Golding et al., 2014), but it was never demonstrated that epithelial wall cells move adjacently to the ZLI along the prethalamic boundary to reach the vLGN. In the experiments presented here, we observe a decrease in the number of BrdU cells in the vLGN area of Zic2 mutant mice and, although we cannot discard modest alterations in the number of proliferating cells in the ventricular area of Zic2 mutant mice during early developmental stages, it is likely that the strong reduction in the number of BrdU cells in the vLGN rather results from defects in the migration of cells moving from medial to lateral prethalamic areas. Therefore, our results confirm the existence of this type of radial migration in the prethalamus and determine the speed ratio of these INs while migrating. In addition, we identify the first transcription factor involved in the migration of these prethalamic INs.
Although Zic2 controls the expression of EphA4 in dorsal spinal cord neurons to mediate axon midline avoidance (Escalante et al., 2013), in the developing retina, this transcription factor regulates the expression of another guidance receptor, EphB1 (García-Frigola et al., 2008). Here, we show that migrating prethalamic INs express EphB1 but not EphA4 and that, in the absence of Zic2, the expression of EphB1 in these cells is seriously compromised. Because the sum of ephrinB2 and ephrinB3 expression delineates a complementary pattern to EphB1 expression in the thalamus/prethalamus boundary, it is likely that EphB/ephrinB-mediated repulsion is involved in channeling Zic2 INs into the MS. In agreement with the idea that EphB1 expression plays an important role in the migration of Zic2 neurons, it seems that CAS cells also express EphB1 (see http://developingmouse.brain-map.org/experiment/siv?id=100053200&imageId=101048978&initImage=ish).
Moreover, it has been reported recently that the repulsive interactions mediated by EphB/ephrinB signaling leads to random dispersion of CRCs throughout the cortical surface (Villar-Cerviño et al., 2013), which also supports a role for Zic2 in controlling the contact cell repulsion mediated by EphB/ephrinB during the dispersion of CRCs on the telencephalic vesicles and supports the idea that Zic2 mutant CRCs fail to be repelled from each other during dispersion. Remarkably, EphB/ephrin signaling operating in the prethalamus and in CRCs may explain the apparently contradictory phenotype observed in the prethalamic Zic2 mutant IN and the defects observed in the hem explant cultures. Diencephalic GFP cells migrate into a narrow stream restricted by repulsive cues that create a repulsion-free corridor. In the mutant embryos, cells do not respond to the repulsive signals at both sides of the corridor and therefore they escape at higher speed than WT cells, which sense the repulsion and are braking all along the pathway. The consequence of decreased EphB/ephrinB signaling in CRCs is the formation of aggregates both in vivo and in vitro because these cells are unable to repel each other.
Role of Zic2 in cell migration: a putative explanation to holoprosencephaly and the other diencephalic phenotypes
In addition to the previously reported expression of Zic2 in several populations of cells at early embryonic stages (Nagai et al., 1997; Elms et al., 2003; Warr et al., 2008), we report here that this transcription factor is also expressed in CRCs, prethalamic INs, and amygdaloid CAS cells. A common feature of these three neural populations is the expression of Zic2 while navigating to their final destinations and we demonstrate here a function for this protein in cell migration. It has been suggested that mildly impaired ZIC2 functions result in psychiatric illnesses (Hatayama et al., 2011). In fact, Zic2 heterozygous mutant mice show behavioral phenotypes reminiscent of those of schizophrenia, a condition that has been related to defects in cortical cell migration during embryogenesis (Valiente and Marín, 2010). It is therefore possible that the schizophrenic features observed in Zic2 heterozygous individuals are the consequence of slight defects in the migration of CRCs and/or cortical progenitors.
A previous study showed a modest reduction in the number of BrdU-positive cells in the prethalamus of Zic2 zebrafish morphants (Sanek and Grinblat, 2008). However, further studies in different animal models, including the present work, showed evidence that alterations in cell proliferation or cell death do not fully explain the different phenotypes found in the Zic2 mutants (Elms et al., 2003; Sanek and Grinblat, 2008). Interestingly, a decreased number of neural crest cells exiting the dorsal neural tube has been reported in Zic2 mutant embryos (Elms et al., 2003), but it was not investigated whether this reduction in the number of neural crest cells in the mesenchyme is a consequence of defects in cell proliferation, cell migration, or both. Conversely, Zic2 mutant mice also exhibit important defects in cerebellar development (Aruga et al., 2002). The chemokine Cxcl12 is a strong chemoattractant to upper rombic lip cells, preventing the migration of Cxcr4-expressing premature granule cells away from the external granule layer (Zou et al., 1998). Similarly, Cxcr4, expressed in CRCs, is essential for their migration through the surface of the telencephalic vesicles that express Cxcl12 in the meningeal cells (Borrell et al., 2006). Cxcr4 expression has been shown to be strongly reduced in Zic2 mutant mice (Inoue et al., 2008). All of these observations may be reinterpreted in light of the results presented here. Both the reduction in the number of neural crest cells in mesenchymal tissues and the defects in cerebellar development observed in the Zic2 mutants could be easily explained by defects in cell migration. Zic2mRNA has been detected in prechordal plate cells even earlier, at midgastrula stages (Warr et al., 2008), but an increase in apoptosis or alterations in proliferation have not been detected in Zic2-defective embryos at these stages (Elms et al., 2003; Merzdorf, 2007; Warr et al., 2008). We speculate that Zic2 mutant prechordal plate cells may suffer defects in migration during early gastrulation and that these migratory alterations may underly the failure in telencephalic vesicle division that leads to holoprosencephaly.
Footnotes
This work was supported by the Regional Government (Grant Prometeo 2012–005 to E.H.), Spanish Ministry of Economy and Competitiveness (MINECO) (Grant BFU2010-16563 to E.H. and Grant BFU2010-17305 to A.F.), and the European Research Council (Grant ERC2011-StG201001109 to E.H.). The Instituto de Neurociencias is a Severo Ochoa Excellence Center. B.M. and N.R.R. hold Formación de Personal Investigador (FPI) fellowships from the MINECO. We thank A. Barco for critical reading of the manuscript, C. Vegar for genotyping and maintenance of the mice, G. Exposito for assistance in time-lapse imaging, the RIKEN BioResource (Japan) for providing the Zic2 mutant mice, and the Mutant Mouse Regional Resource Center Repository for providing the Tg(Zic2eGFP) mice.
References
- Acloque H, Wilkinson DG, Nieto MA. In situ hybridization analysis of chick embryos in whole-mount and tissue sections. Methods Cell Biol. 2008;87:169–185. doi: 10.1016/S0091-679X(08)00209-4. [DOI] [PubMed] [Google Scholar]
- Alcántara SA, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Aruga J, Inoue T, Hoshino J, Mikoshiba K. Zic2 controls cerebellar development in cooperation with Zic1. J Neurosci. 2002;22:218–225. doi: 10.1523/JNEUROSCI.22-01-00218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielle F, Griveau A, Narboux-Nême N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A. Multiple origins of Cajal-Retzius cells at the borders of the developing pallium. Nat Neurosci. 2005;8:1002–1012. doi: 10.1038/nn1511. [DOI] [PubMed] [Google Scholar]
- Borrell V, Marín O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat Neurosci. 2006;9:1284–1293. doi: 10.1038/nn1764. [DOI] [PubMed] [Google Scholar]
- Brown LY, Odent S, David V, Blayau M, Dubourg C, Apacik C, Delgado MA, Hall BD, Reynolds JF, Sommer A, Wieczorek D, Brown SA, Muenke M. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum Mol Genet. 2001;10:791–796. doi: 10.1093/hmg/10.8.791. [DOI] [PubMed] [Google Scholar]
- Brown LY, Kottmann AH, Brown S. Immunolocalization of zic2 expression in the developing forebrain. Gene Expr Patterns. 2003;3:361–367. doi: 10.1016/S1567-133X(03)00043-7. [DOI] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- del Río JA, Martínez A, Fonseca M, Auladell C, Soriano E. Glutamate-like immunoreactivity and fate of Cajal-Retzius cells in the murine cortex as identified with calretinin antibody. Cereb Cortex. 1995;5:13–21. doi: 10.1093/cercor/5.1.13. [DOI] [PubMed] [Google Scholar]
- Elms P, Siggers P, Napper D, Greenfield A, Arkell R. Zic2 is required for neural crest formation and hindbrain patterning during mouse development. Dev Biol. 2003;264:391–406. doi: 10.1016/j.ydbio.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Escalante A, Murillo B, Morenilla-Palao C, Klar A, Herrera E. Zic2-dependent axon midline avoidance controls the formation of major ipsilateral tracts in the CNS. Neuron. 2013;80:1392–1406. doi: 10.1016/j.neuron.2013.10.007. [DOI] [PubMed] [Google Scholar]
- Frotscher M. Cajal—Retzius cells, Reelin, and the formation of layers. Curr Opin Neurobiol. 1998;8:570–575. doi: 10.1016/s0959-4388(98)80082-2. [DOI] [PubMed] [Google Scholar]
- García-Frigola C, Carreres MI, Vegar C, Mason C, Herrera E. Zic2 promotes axonal divergence at the optic chiasm midline by EphB1-dependent and -independent mechanisms. Development. 2008;135:1833–1841. doi: 10.1242/dev.020693. [DOI] [PubMed] [Google Scholar]
- García-Moreno F, López-Mascaraque L, De Carlos JA. Origins and migratory routes of murine Cajal-Retzius cells. J Comp Neurol. 2007;500:419–432. doi: 10.1002/cne.21128. [DOI] [PubMed] [Google Scholar]
- Gil-Sanz C, Franco SJ, Martínez-Garay I, Espinosa A, Harkins-Perry S, Müller U. Cajal-Retzius cells instruct neuronal migration by coincidence signaling between secreted and contact-dependent guidance cues. Neuron. 2013;79:461–477. doi: 10.1016/j.neuron.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding B, Pouchelon G, Bellone C, Murthy S, Di Nardo AA, Govindan S, Ogawa M, Shimogori T, Lüscher C, Dayer A, Jabaudon D. Retinal input directs the recruitment of inhibitory interneurons into thalamic visual circuits. Neuron. 2014;81:1057–1069. doi: 10.1016/j.neuron.2014.01.032. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Hatayama M, Ishiguro A, Iwayama Y, Takashima N, Sakoori K, Toyota T, Nozaki Y, Odaka YS, Yamada K, Yoshikawa T, Aruga J. Zic2 hypomorphic mutant mice as a schizophrenia model and ZIC2 mutations identified in schizophrenia patients. Sci Rep. 2011;1:16. doi: 10.1038/srep00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera E, Brown L, Aruga J, Rachel RA, Dolen G, Mikoshiba K, Brown S, Mason CA. Zic2 patterns binocular vision by specifying the uncrossed retinal projection. Cell. 2003;114:545–557. doi: 10.1016/S0092-8674(03)00684-6. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Neogi T, Englund C, Daza RA, Fink A. Cajal-Retzius cells in the mouse: transcription factors, neurotransmitters, and birthdays suggest a pallial origin. Brain Res Dev Brain Res. 2003;141:39–53. doi: 10.1016/s0165-3806(02)00641-7. [DOI] [PubMed] [Google Scholar]
- Inoue T, Ogawa M, Mikoshiba K, Aruga J. Zic deficiency in the cortical marginal zone and meninges results in cortical lamination defects resembling those in type II lissencephaly. J Neurosci. 2008;28:4712–4725. doi: 10.1523/JNEUROSCI.5735-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Yanazawa M, Miyashita T, Kato K. Expression patterns of Brx1 (Rieg gene), Sonic hedgehog, Nkx2.2, Dlx1 and Arx during zona limitans intrathalamica and embryonic ventral lateral geniculate nuclear formation. 1997;67:83–96. doi: 10.1016/s0925-4773(97)00110-x. [DOI] [PubMed] [Google Scholar]
- Marín-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64–71. doi: 10.1016/s0166-2236(97)01164-8. [DOI] [PubMed] [Google Scholar]
- Medina L, Legaz I, González G, De Castro F, Rubenstein JL, Puelles L. Expression of Dbx1, Neurogenin 2, Semaphorin 5A, Cadherin 8, and Emx1 distinguish ventral and lateral pallial histogenetic divisions in the developing mouse claustroamygdaloid complex. J Comp Neurol. 2004;474:504–523. doi: 10.1002/cne.20141. [DOI] [PubMed] [Google Scholar]
- Merzdorf CS. Emerging roles for zic genes in early development. Dev Dyn. 2007;236:922–940. doi: 10.1002/dvdy.21098. [DOI] [PubMed] [Google Scholar]
- Meyer G, Goffinet AM, Fairén A. Feature article: what is a Cajal-Retzius cell? A reassessment of a classical cell type based on recent observations in the developing neocortex. Cereb Cortex. 1999;9:765–775. doi: 10.1093/cercor/9.8.765. [DOI] [PubMed] [Google Scholar]
- Meyer G, Perez-García CG, Gleeson JG. Selective expression of doublecortin and LIS1 in developing human cortex suggests unique modes of neuronal movement. Cereb Cortex. 2002;12:1225–1236. doi: 10.1093/cercor/12.12.1225. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Takada S, Günther T, Spörle R, Schughart K, Mikoshiba K. The expression of the mouse Zic1, Zic2, and Zic3 gene suggests an essential role for Zic genes in body pattern formation. Dev Biol. 1997;182:299–313. doi: 10.1006/dbio.1996.8449. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Minowa O, Sugimoto T, Ohno Y, Noda T, Mikoshiba K. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci U S A. 2000;97:1618–1623. doi: 10.1073/pnas.97.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Remedios R, Huilgol D, Saha B, Hari P, Bhatnagar L, Kowalczyk T, Hevner RF, Suda Y, Aizawa S, Ohshima T, Stoykova A, Tole S. A stream of cells migrating from the caudal telencephalon reveals a link between the amygdala and neocortex. Nat Neurosci. 2007;10:1141–1150. doi: 10.1038/nn1955. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Sanek NA, Grinblat Y. A novel role for zebrafish zic2a during forebrain development. Dev Biol. 2008;317:325–335. doi: 10.1016/j.ydbio.2008.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago AC, Shammah-Lagnado SJ. Efferent connections of the nucleus of the lateral olfactory tract in the rat. J Comp Neurol. 2004;471:314–332. doi: 10.1002/cne.20028. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Miyagi T, Yoshida M, Miyata T, Ogawa M, Aizawa S, Suda Y. Absence of Cajal-Retzius cells and subplate neurons associated with defects of tangential cell migration from ganglionic eminence in Emx1/2 double mutant cerebral cortex. Development. 2002;129:3479–3492. doi: 10.1242/dev.129.14.3479. [DOI] [PubMed] [Google Scholar]
- Soma M, Aizawa H, Ito Y, Maekawa M, Osumi N, Nakahira E, Okamoto H, Tanaka K, Yuasa S. Development of the mouse amygdala as revealed by enhanced green fluorescent protein gene transfer by means of in utero electroporation. J Comp Neurol. 2009;513:113–128. doi: 10.1002/cne.21945. [DOI] [PubMed] [Google Scholar]
- Takiguchi-Hayashi K, Sekiguchi M, Ashigaki S, Takamatsu M, Hasegawa H, Suzuki-Migishima R, Yokoyama M, Nakanishi S, Tanabe Y. Generation of reelin-positive marginal zone cells from the caudomedial wall of telencephalic vesicles. J Neurosci. 2004;24:2286–2295. doi: 10.1523/JNEUROSCI.4671-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Tripodi M, Filosa A, Armentano M, Studer M. The COUP-TF nuclear receptors regulate cell migration in the mammalian basal forebrain. Development. 2004;131:6119–6129. doi: 10.1242/dev.01530. [DOI] [PubMed] [Google Scholar]
- Valiente M, Marín O. Neuronal migration mechanisms in development and disease. Curr Opin Neurobiol. 2010;20:68–78. doi: 10.1016/j.conb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Villar-Cerviño V, Molano-Mazón M, Catchpole T, Valdeolmillos M, Henkemeyer M, Martínez LM, Borrell V, Marín O. Contact repulsion controls the dispersion and final distribution of Cajal-Retzius cells. Neuron. 2013;77:457–471. doi: 10.1016/j.neuron.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vue TY, Bluske K, Alishahi A, Yang LL, Koyano-Nakagawa N, Novitch B, Nakagawa Y. Sonic hedgehog signaling controls thalamic progenitor identity and nuclei specification in mice. J Neurosci. 2009;29:4484–4497. doi: 10.1523/JNEUROSCI.0656-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr N, Powles-Glover N, Chappell A, Robson J, Norris D, Arkell RM. Zic2-associated holoprosencephaly is caused by a transient defect in the organizer region during gastrulation. Hum Mol Genet. 2008;17:2986–2996. doi: 10.1093/hmg/ddn197. [DOI] [PubMed] [Google Scholar]
- Williams SE, Mann F, Erskine L, Sakurai T, Wei S, Rossi DJ, Gale NW, Holt CE, Mason CA, Henkemeyer M. Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron. 2003;39:919–935. doi: 10.1016/j.neuron.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor Cxcr4 in heamotopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]