

Abstract
Fragile X syndrome (FXS), the most common form of inherited mental retardation, is a neurodevelopmental disorder caused by silencing of the FMR1 gene, which in FXS becomes inactivated during human embryonic development. We have shown recently that this process is recapitulated by in vitro neural differentiation of FX human embryonic stem cells (FX-hESCs), derived from FXS blastocysts. In the present study, we analyzed morphological and functional properties of neurons generated from FX-hESCs. Human FX neurons can fire single action potentials (APs) to depolarizing current commands, but are unable to discharge trains of APs. Their APs are of a reduced amplitudes and longer durations than controls. These are reflected in reduced inward Na+ and outward K+ currents. In addition, human FX neurons contain fewer synaptic vesicles and lack spontaneous synaptic activity. Notably, synaptic activity in these neurons can be restored by coculturing them with normal rat hippocampal neurons, demonstrating a critical role for synaptic mechanisms in FXS pathology. This is the first extensive functional analysis of human FX neurons derived in vitro from hESCs that provides a convenient tool for studying molecular mechanisms underlying the impaired neuronal functions in FXS.
SIGNIFICANCE STATEMENT Fragile X syndrome (FXS), the most common form of inherited mental retardation, is caused by silencing of the FMR1 gene. In this study, we describe for the first time the properties of neurons developed from human embryonic stem cells (hESCs) that carry the FMR1 mutation and are grown in culture for extended periods. These neurons are retarded compared with controls in several morphological and functional properties. In vitro neural differentiation of FX hESCs can thus serve as a most relevant system for the analysis of molecular mechanisms underlying the impaired neuronal functions in FXS.
Keywords: action potential, disease modeling, fragile X syndrome, human embryonic stem cells, in vitro neural differentiation, synaptic deficiencies
Introduction
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability (Penagarikano et al., 2007; Bagni and Oostra, 2013). It is a neurodevelopmental disorder that leads to abnormal neural plasticity, autism, and epilepsy. It is caused by hypermethylation-mediated silencing of the FMR1 gene and the consequent absence of its encoded protein, FMRP. Importantly, inactivation of FMR1 is developmentally regulated and, in FXS human embryos, FMR1 silencing is observed only by the end of the first trimester of pregnancy (Willemsen et al., 2002). This feature of FXS pathology is recapitulated during differentiation of FX human embryonic stem cells (FX-hESCs) (Eiges et al., 2007; Telias et al., 2013; Avitzour et al., 2014; Colak et al., 2014), but not in FMR1 knock-out animal models (Moy and Nadler; 2008; Santos et al., 2014) or in FX human induced pluripotent stem cells (FX-hiPSCs) (Urbach et al., 2010, Sheridan et al., 2011; Doers et al., 2014). Human-based FXS in vitro models have been established using neural progenitor cells established from aborted fetuses (Castren et al., 2005; Bhattacharyya et al., 2008). However, this approach uses cells from different developmental stages with a short lifespan in culture. In vitro neural differentiation (IVND) of hESCs provides a reliable model for studying human embryonic neurogenesis in health and disease (Chailangkarn et al., 2012; Wang and Doering, 2012). hESCs can be differentiated into neurons, thus recapitulating neural development and allowing for the study of the disease, not only at the neuronal end point, but also during the different stages of neurogenesis (Elkabetz and Studer, 2008; Muguruma and Sasai, 2012).
The pathology of FXS has been linked to several postsynaptic deficiencies caused by the absence of FMRP. Specifically, fmr1−/− mice show increased expression of metabotropic glutamate receptors (mGluRs) (Bear et al., 2004) and reduced expression of GABA-A receptors (Braat and Kooy, 2015). It was shown recently that fmr1−/− mice exhibit also specific presynaptic deficiencies (Deng et al., 2011; Deng et al., 2013; Wang et al., 2014; Myrick et al., 2015). However, none of these suggested mechanisms has been tested so far in a human-based model, in part because human models are scarce and generating in vitro bona fide neurons from FX-hESCs and FX-hiPSCs is challenging (Telias and Ben-Yosef, 2014). We recently reported on the generation of functional neuronal networks after IVND of FX-hESCs (Telias et al., 2013). Notably, this was the first study to produce electrophysiological data on human FXS neurons derived in vitro. We found that FXS neurons fired APs with reduced frequency and amplitude compared with controls and developed poor spontaneous synaptic activity.
In the current study, we further analyze the cellular and electrophysiological properties of human embryonic FX neurons after IVND of three different male FX-hESC lines. This is the first report in which human FX neurons are systematically analyzed at the functional level and therefore our results can shed light on the mechanism underlying the functional deficiencies of FX neurons that are probably linked to the development of the intellectual disability characterizing FXS.
Materials and Methods
hESCs and in vitro neural differentiation.
Three FX hESC lines with >200 CGG repeats in the FMR1 gene were studied: HEFX1 (male) and Lis_FX6 (male), derived at Tel-Aviv Sourasky Medical Center, and SZFX6 (male), derived in the Shaare Tzedek Medical Center. Three lines were entitled here systematically, FX1, FX2, and FX3, and were all fully characterized previously (Eiges et al., 2007; Telias et al., 2013). The hESC lines HUES-13 (male; kindly provided by the Melton Laboratory, Harvard University) and H9 (female; WiCell, University of Wisconsin) served as controls. HUES-13 was shown previously by us to include <30 CGG repeats in the FMR1 gene (Telias et al., 2013). The H9-hESC line was selected as an additional control due to its well established characterization and its wide-range use since its derivation in 1998 (Thomson et al., 1998). In addition, a very extensive study was already performed on in vitro neural differentiation of H9-hESCs (Elkabetz et al., 2008; Ziller et al., 2015), making it the “gold standard” of control hESCs in general and for neural differentiation in particular. H9 cells were analyzed and found to include <30 CGG repeats. A full list of hESC lines used in the present study, including the origin of derivation, sex, and CGG repeat size of the FMR1 gene, is described in Table 1. hESCs were cultured on feeder layers of mitomycin C (Sigma-Aldrich)-inactivated mouse embryonic fibroblasts in hES medium supplemented with bFGF (R&D Systems), as we described previously (Telias et al., 2013). Before induction of IVND, hESCs were cultured on Matrigel (BD Biosciences)-coated wells for two passages. The dual SMAD inhibition IVND protocol was applied as we described previously (Telias et al., 2014). Briefly, neural induction was achieved by gradually changing the medium from hES to N2 while adding dorsomorphin and SB431542 for 10 d; for neuronal induction, the medium was changed to N2/B27 supplemented with BDNF, GDNF, ascorbic acid, dbcAMP and DAPT for 10 additional days. At day 20, cells were dissociated using Accutase (Life Technologies) and seeded on 13 mm glass coverslips precoated with 50 μg/ml poly-d-lysine and 20 μg/ml laminin (Sigma-Aldrich) at a seeding density of ∼1.0 × 105 cells/cm2 (these conditions were selected based on a preliminary calibration study in which cells were seeded at day 20 or 35 at 3 different densities: 0.5/1/1.5 × 105 cells/cm2). From day 20, neurons were continuously grown in N2/B27 medium supplemented with 20 ng/ml BDNF, GDNF, and NT3 unless otherwise specified. Concentrations of reagents and growth factors used were as follows: 5 μm dorsomorphin (Stemgent), 10 μm SB431542 (Stemgent), 20 ng/ml BDNF (PeproTech), 20 ng/ml GDNF (PeproTech), 0.2 mm ascorbic acid (Sigma-Aldrich), 0.5 mm dbcAMP (Sigma-Aldrich), 10 μm DAPT (Tocris Bioscience), and 20 ng/ml NT3 (PeproTech). N2 medium was composed of DMEM:F12 (Life Technology) supplemented with 1% N2 (Life Technology), 1% nonessential amino acids (Biological Industries), 1% Glutamax (Life Technology), and 100 μg/ml Primocin (InvivoGen). N2/B27 medium was a 1:1 mixture of N2 and B27 media. B27 medium was composed of neurobasal (Life Technology), supplemented with 1% B27 (Life Technology), 1% nonessential amino acids (Biological Industries), 1% Glutamax (Life Technology), and 100 μg/ml Primocin (InvivoGen).
Table 1.
Full list of hESC lines used in the present study
| hESC line | Original name | Institution of derivation | Sex | CGG repeat size (FMR1 gene) | Reference |
|---|---|---|---|---|---|
| WT1 | HUES13 | Harvard University | XY | <30 | http://stemcelldistribution.harvard.edu/ (Telias et al., 2013) |
| WT2 | H9 | WiCell - University of Wisconsin | XX | <30 in both alleles | http://www.wicell.org/home/stem-cell-lines/ (Thomson et al., 1998) |
| FX1 | HEFX1 | Tel-Aviv Medical Center | XY | 200–650 | (Eiges et al., 2007; Telias et al., 2013; Avitzour et al., 2014) |
| FX2 | SZFX6 | Shaare Zedek Medical Center | XY | 200–330 | (Telias et al., 2013; Avitzour et al., 2014) |
| FX3 | Lis_FX6 | Tel-Aviv Medical Center | XY | 50–300 | (Telias et al., 2013; Avitzour et al., 2014) |
List of hESC lines studied. The number of CGG repeats was previously analyzed by us for all lines (see reference), except WT2 (H9), for which the detection of CGG repeat was performed here for the first time (by AmplideX FMR1; Asuragen).
Primary cultures from rat hippocampus.
Animal handling was done in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Weizmann Institute of Science and Israeli law. Cultures were prepared as detailed previously (Goldin et al., 2001). Briefly, rat pups were decapitated on day of birth (P0) and their brains removed and placed in a chilled (4°C), oxygenated Leibovitz L15 medium (Invitrogen) enriched with 0.6% glucose and gentamicin (Sigma-Aldrich, 20 μg/ml). Hippocampal tissue was mechanically dissociated after incubation with trypsin (0.25%) and DNAase (50 μg/ml) and passed to the plating medium consisting of 5% heat-inactivated horse serum (HS), 5% fetal calf serum, and B-27 (1 μl/1 ml) prepared in minimum essential medium (MEM) Earl salts (Invitrogen) enriched with 0.6% glucose, gentamicin (20 μg/ml), and 2 mm glutamax (enriched MEM). Cells were suspended in 1 ml of medium and plated on 12 mm round cover glasses in 24-well plates. Cells were left to grow in the incubator at 37°C, 5% CO2 for 4 d, at which time the medium was changed to 10% HS in enriched MEM plus a mixture of 5′-fluoro-2-deoxyuridine/uridine (Sigma-Aldrich, 20 μg and 50 μg/ml, respectively) to block glial proliferation. Four days later the medium was replaced with 10% HS in MEM.
Coculture of hESC-derived neurons with rat neurons.
Human neurons were stained with DiD and rat neurons with DiO (Neurotrace; Life Technologies). Neurons were incubated with 1 ml of N2/B27 medium containing 2.5% of the dye for 60 min in the dark, followed by washout using N2/B27 medium. The following day, human neurons were gently detached by mechanical pipetting, centrifuged for 5 min at 600 RPM, and reseeded on top of primary cultures of rat hippocampal neurons. Cocultures were examined by electrophysiology at least 3.5 d after preparation.
Electrophysiology.
Electrophysiological recordings were conducted as described previously (Telias et al., 2013). In brief, neurons cultured on glass coverslips were transferred to a recording chamber in standard recording medium containing the following (in mm):10 HEPES, 4 KCl, 2 CaCl2, 1 MgCl2, 139 NaCl, and 10 d-glucose (340 mOsm, pH 7.4). Cells were patch-clamped with pipettes containing the following (in mm): 136 K-gluconate, 10 KCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 0.3 Na-GTP, 1 Mg-ATP, and 5 phosphocreatine, pH 7.2 (pipette tip resistance was 5–8 MΩ). Action potentials were evoked either by injecting depolarizing current pulses (in current-clamp mode) or by depolarizing voltage commands (in voltage-clamp mode). Membrane potential was held at −60 mV. Spontaneous synaptic currents were recorded continuously for 2 min sessions in voltage-clamp mode with a 50 μs sampling rate. Signals were amplified with a Multiclamp700B amplifier and recorded with Clampex9.2 software (Molecular Devices). Data were subjected to a 500 Hz low-pass filter and analyzed using Clampfit-9 and SigmaPlot.
FM1-43 lipophilic dye uptake and release.
Neurons seeded on laminin-coated glass coverslips in 24-well plates were incubated at 37°C for 5 min with or without the FM1-43 (5 μm) in physiological solution (PBS containing Ca+2 and Mg+2) for studying spontaneous uptake. Addition of 50 mm KCl was used to examine active uptake. After incubation, cells were washed 10 times with physiological solution and live imaged continuously on the stage of an inverted fluorescent microscope. Each coverslip was randomly imaged in >5 different fields and mean gray value was calculated for each field in spontaneous and active uptake and normalized to the values obtained in controls.
FM1-43 release.
After active uptake of FM1-43 and washout (as described above), neurons were videorecorded using an inverted fluorescent microscope for up to 5 min while being exposed to 50 mm KCl, or to physiological solution (PBS containing Ca+2 and Mg+2). Movies were analyzed offline and mean gray values were calculated as described above.
Immunofluorescence.
Immunostaining was performed as described previously (Telias et al., 2013). Briefly, cells were fixed for 15 min at room temperature (RT) using Cytofix (BD Biosciences) and washed with PBS. Primary antibodies were incubated overnight at 4°C, in a PBS solution containing 2.5% BSA and 0.1% Triton. Staining with secondary antibody was performed for 1 h at RT in the dark. Primary antibodies used were as follows: rabbit anti-human FMRP (Abcam), mouse anti-human NeuN (GeneTex), mouse anti-human TUJ1 (Millipore), rabbit anti-human phosphorylated CaMK-II (Millipore), and rabbit anti-human MAP2 (Santa Cruz Biotechnology). Primary antibodies were detected using sheep anti-mouse Cy2-conjugated or goat anti-rabbit Cy3-conjugated secondary antibodies (Jackson Laboratories). Nuclei were stained with DAPI (Sigma-Aldrich).
qRT-PCR and Western blot.
qRT-PCR and Western blot analysis were conducted as described previously (Eiges et al., 2007; Telias et al., 2013). The sequences of primers used were as follows: GAPDH Fwd 5′-agccacatcgctcagacacc-3′, Rev 5′-atacgaccaaatccgttgactc-3′; FMR1 Fwd 5′-cacctcaaagcgagcacat-3′, Rev 5′-aatagcagtgaccccaggt-3′. Primary antibodies used for Western blot analysis were rabbit anti-FMRP (Abcam, #ab17722) and mouse anti-β-Actin (Abcam, ab#8224). HRP-conjugated secondary antibodies were purchased from Jackson Laboratories. Working concentrations of primary and secondary antibodies were as recommended by the manufacturer.
Imaging and microscopy.
Bright-field, phase, and fluorescence images of cells were obtained using an Olympus IX51 inverted light microscope or a Zeiss LSM 700 confocal microscope. Morphological assessment of human in vitro neurons was performed using confocal images of MAP2-stained neurons by the LSM Image Browser (Zeiss), CellA (Olympus), and ImageJ software.
Statistical analysis.
Statistical analyses (Student's t test, ANOVA, and χ2 tests) were performed using SPSS, SigmaPlot, and GraphPad QuickCalcs (http://www.graphpad.com/quickcalcs).
Results
Differentiation of human neurons from FX-hESCs
hESCs were differentiated into neurons using the dual anti-SMAD IVND protocol (Chambers et al., 2009), which aims at in vitro generation of forebrain neurons, as we described previously (Telias et al., 2014). In this study, we report the results obtained from IVND experiments performed with FX-hESC lines compared with a control wild-type (WT)-hESC line with normal (<50 CGG repeats) FMR1 gene. The WT-hESC lines used in the present study (see also Materials and Methods and Table 1) were shown previously by us to differentiate into bona fide neurons exhibiting electrical activity indistinguishable from primary neurons in culture (Telias et al., 2013, 2014). In addition, we have already shown that inactivation of FMR1 is a developmentally regulated process that can be recapitulated by IVND of FX-hESCs and that FMR1 is completely inactivated only in mature neurons (Eiges et al., 2007; Telias et al., 2013). Consistent with this, we show here that, although some NeuN-positive cells were observed already by day 23 of differentiation, complete inactivation of FMR1 was observed only by day 60. By day 23, the very few FX neurons derived still expressed FMR1, whereas by day 48, although most cells expressed NeuN, they still expressed FMRP (Fig. 1A). However, by day 60, FMR1 expression levels were negligible in FX neurons compared with WT (FMR1-mRNA expression levels, Fig. 1B; FMRP protein levels, Fig. 1C). In addition, at day 60, WT and FX neurons were positively stained for the neuronal cytoskeletal protein TUJ1 and for the forebrain-specific enzyme CaMK-II (Fig. 1D), indicating that these are bona fide human forebrain/cortical neurons (Shcheglovitov et al., 2013). In our previous report, we performed the MAP2/GFAP ratio bioassay (Luo et al., 2010; Briggs et al., 2013) to measure the efficiency of IVND in both WT and FX lines and showed a significant reduced neuronal yield in all three FX-hESC lines (Telias et al., 2013). Despite this significant difference in total yield, spacing between neurons was similar for FX and WT cultures (Fig. 1D) because, in both, neurons were found close to the attached neurospheres, whereas glia and nonneuronal cells were found in the periphery of the coverslip (see also Figs. 4B, 5B in Telias et al., 2013). In the present study, WT and FX neurons were stained for MAP2 (Fig. 1E), and were morphologically analyzed and compared in confocal microscopy imaging (Fig. 1F). The results show that human FX neurons have significantly smaller somata and shorter and less tortuous neurites than their WT counterparts, suggesting that downregulation of FMR1 impairs the process of neuronal maturation even at this early stage. Nevertheless, neuronal arborization was not different between FX and WT, with similar average numbers of branch points and formation of secondary projections for each neurite (p = 0.33 and p = 0.065, respectively; two-tailed t test analysis). These results suggest that initial neurite outgrowth might be unaffected by the FMR1 mutation, as was previously reported on human FX neuroblasts derived from FX-hESCs (Telias et al., 2013) and contrary to what was recently found in FX-hiPSCs (Doers et al., 2014). Together, these results suggest that FMRP inactivation affects neuronal morphology but not dendritic branching.
Figure 1.

Differentiation and characterization of WT and FX neurons. A, Representative images of neurons derived from FX-hESCs at day 23 (left) and 48 (right) after IVND in the FX2 line (repeated for all lines), stained for FMRP (red), NeuN (green), and DAPI (blue). Scale bars, 50 μm. B, qRT-PCR analysis of FMR1 transcription in neurons at day 60 of IVND; WT (blue), FX (red, average of all three FX lines). Values are mean ± SEM. **p < 0.01, t test. C, Western blot analysis of FMRP translation at day 60 of IVND, as described in B shown for WT and FX1 line (performed 2–3 times in all lines). D, Representative images of neurons at day 60 of IVND shown for WT (top) and FX1 (bottom, repeated for all three FX lines) stained for TUJ1 (green) and CaMK-II (red). Scale bars, 50 μm. E, Representative images of WT (left) and FX2 (right) neurons at day 60 of IVND stained for MAP2 (red), repeated also in line FX3. F, Quantification of neuronal morphology in MAP2-stained neurons as shown in E, including: soma area and perimeter, neurite length, neurite tortuosity (AU, arbitrary units), and the number of branch points and secondary projections. E, F, Number of cells included in the analysis: WT, n = 43; FX2, n = 17; FX3, n = 22. WT is shown in blue and the average value for FX2 and FX3 is shown in red. Values are mean ± SEM. *p < 0.05; **p < 0.01, t test.
Figure 4.

Voltage-clamp recordings of net inward current in WT and FX neurons. A, Representative traces of net inward current obtained from voltage-clamp recordings of a WT neuron (left) and a FX3 neuron (right) after 90–100 d of in vitro neural differentiation. Bottom, Illustration of 8 consecutive 10 mV voltage steps from −40 to 30 mV. Holding potential was −60 mV for both cells. B, I–V curve for net inward current (left) and activation curve (right) for WT (blue) and FX (red). Values are mean ± SEM. The numbers of cells included in the analysis were as follows: WT, n = 55; FX1, n = 23; FX2, n = 21; FX3, n = 25.
Figure 5.

Voltage-clamp recordings of K+ currents in WT and FX neurons. A, Representative traces of K+ currents obtained from voltage-clamp recordings of a WT neuron (left) and a FX3 neuron (right) after 90–100 d of in vitro neural differentiation after treatment with TTX. Bottom, Illustration of 11 consecutive 10 mV voltage steps from −60 to 40 mV. Holding potential was −60 mV for both cells. B, I–V curve for IA (left) and IK (right) for WT (blue) and FX (red). Values are mean ± SEM. The numbers of cells included in the analysis were as follows: WT, n = 55; FX1, n = 23; FX2, n = 21; FX3, n = 25.
Action potential discharges in human FX neurons
Using current-clamp recording mode (holding potential Em = ∼−60 mV), we analyzed the basic properties of AP firing in FX compared with WT neurons (>60 d after IVND). Of the WT neurons, 65% (WT1, 34 of 55; WT2, 7 of 8) fired repetitive AP's. In contrast, none of the FX neurons (FX1, 0 of 23; FX2, 0 of 21; FX3, 0 of 25) derived from the three FX-hESC lines fired more than a single AP to a prolonged (300 ms) depolarizing current pulse (Fig. 2A). Nonetheless, spikes in both WT and FX were similarly suppressed by addition of the Na+ channel blocker tetrodotoxin (TTX), indicating that APs in FX result from the activation of Na+ channels, as in WT, despite their incapacity for generating trains of APs (Fig. 2A, FX+TTX). These results were reproducible in all three different FX-hESC lines studied and are consistent with our results obtained initially with the FX3 line (Telias et al., 2013), indicating that this is a recurring motif in the functional deficiencies of embryonic FX human neurons. We then analyzed the basic electrical properties of WT and FX neurons (Fig. 2B). Quantification of neuronal properties from current-clamp recordings demonstrated that input resistance and spike threshold were similar for WT and FX. In contrast, spike amplitude, duration, and after-hyperpolarization were all significantly altered in all three FX lines (Fig. 2B). Specifically, although spike amplitude was reduced in FX compared with WT, spike duration was increased. Furthermore, although WT neurons expressed after-hyperpolarization, FX neurons displayed after-depolarization. These results are similar to those found in neurons of the fmr1−/− mice, in which AP width was shown to be significantly increased (Deng et al., 2013). Together, these results suggest that, in human neurons, inactivation of FMR1 affects the ability of neurons to fire trains of APs.
Figure 2.

Intrinsic properties of APs in WT and FX neurons. A, Representative traces for current-clamp recordings of individual WT (blue, left) and FX1 (red, middle) neurons after 85–90 d in vitro. Voltage deflections shown in response to a current injection of ∼100 pA (bottom, left, and middle). The effect of TTX is shown for the single spike in the FX1 neuron (red, middle). Right, Spike incidence shown as percentage of cells displaying single or multiple spikes and spike frequency shown as the number of spikes per depolarization step. Values are mean ± SEM. **p < 0.01, ANOVA. B, Left, Top, Representative expanded traces of the first ∼30 ms after current injection in WT (blue) and FX2 (red). Quantification of AP parameters from current-clamp recordings in neurons at 60–120 d in vitro: input resistance, spike threshold, spike amplitude, spike duration, and after potential. Values are mean ± SEM. *p < 0.05, **p < 0.01, ANOVA. For A and B, the numbers of cells included in the analysis were as follows: WT1, n = 55; WT2, n = 10; FX1 = 23; FX2 = 21; FX3 = 25.
Analysis of Na+ and K+ currents
The possibility that the lack of repetitive firing is due to a partial inactivation of the Na+ current at resting potential was tested by holding membrane at hyperpolarizing (−80 mV) and depolarizing (−40 mV) potentials (Fig. 3). Although the responses of the WT cells to different holding potentials was as expected (inactivation at depolarizing potentials, fewer and larger APs at hyperpolarizing potentials), no difference in the number of APs was seen in FX neurons, indicating that a slow removal of inactivation is not the main cause for the lack of multiple APs in response to a prolonged depolarization in the FX neurons (n = 4–6 cells in each line). We then analyzed Na+ (Fig. 4A) and K+ (Fig. 5A) currents in voltage-clamp recording mode. Na+ I–V and activation curve of FX neurons demonstrated significantly reduced amplitudes and right-shifted activation curve compared with WT (Fig. 4B). Indeed, although the WT peak net inward current was 1134.40 ± 142.41 pA recorded at −20 mV, in FX neurons, the peak net inward current was 717.34 ± 135.33 pA recorded at −10 mV (WT n = 55, FX n = 69, p = 0.0371, two-tailed t test). Similarly, K+ currents analyzed in the TTX-insensitive region of traces obtained in voltage-clamp mode indicate reduced amplitudes of the transient and sustained components of K+ currents in FX neurons compared with WT, but this was not statistically significant (Fig. 5B). Although the IA peak in WT was 946.57 ± 73.59 pA, in FX neurons, it was 756.74 ± 81.98 pA (WT n = 55, FX n = 69, p = 0.0953, two-tailed t test). Further, although the peak of the IK current in WT was 811.38 ± 45.98 pA, in FX neurons, it was 670.16 ± 53.93 pA (WT n = 55, FX n = 69, p = 0.0555, two-tailed t test). In addition, a comparison between WT and FX corresponding I–V curves revealed no abnormal voltage dependency for IA and IK in FX neurons. Together, the results on Na+ and K+ currents on WT and FX neurons suggest that the deficiencies observed in AP firing patterns of FX neurons are linked to an intrinsic reduction in the ability of FX cells to activate Na+ and K+ currents.
Figure 3.

Voltage-dependency of APs in WT and FX neurons. A, B, Representative traces of consecutive current-clamp recordings in individual neurons from WT (A) and FX (B) after 95 and 93 d in vitro, correspondingly. Holding potential was −80 mV (left), −60 mV (middle), and −40 mV (right). Tests were repeated in all three lines. The numbers of cells included in the analysis were as follows: WT, n = 13; FX1, n = 4; FX2, n = 5; FX3, n = 5.
Synaptic content and neurotransmitter release in human FX neurons
We have shown previously that, whereas WT neurons display spontaneous EPSCs (sEPSCs), FX neurons are synaptically silent (Telias et al., 2013). Therefore, in the present study, we analyzed whether FX neurons have reduced content of synaptic vesicles and/or reduced capacity to release neurotransmitters. Using the membrane-impermeable dye FM1-43 and time-lapse live imaging (Fig. 6A), we measured the synaptic content and its release in FX versus WT neurons. Our results show that both spontaneous and active uptake of FM1-43 were significantly reduced in FX neurons (Fig. 6B). Likewise, depolarization-dependent release of FM1-43 using high (50 mm) K+ was reduced and delayed in FX neurons (Fig. 6C). In summary, the synaptically silent FX neurons showed fewer releasable synaptic vesicles and reduced release probability, suggesting a role for FMRP in presynaptic terminals. As mentioned before, we do not consider that a difference in neuronal yield would be responsible for differences in presynaptic or postsynaptic activity in FX neurons compared with controls. Although FX cultures rendered fewer neurons, these were always found in packed clusters surrounding the attached neurospheres, similar to what was found in WT cultures. Therefore, we suggest that silencing of FMR1 and lack of FMRP is probably responsible for the abnormal synaptogenesis of FX neurons.
Figure 6.

Synaptic vesicle quantification in WT and FX neurons. A, Representative images of FM1-43 uptake experiments. Phase images (left) and green fluorescent images (right) from experiments performed in WT neurons. Scale bars, 50 μm. Top, Control (no FM1-43 added). Middle, Spontaneous uptake of 5 μm FM1-43. Bottom, Active uptake of 5 μm FM1-43. B, Quantification of FM1-43 uptake, as described in A by measurement of mean gray value (shown as percentage of staining). C, Quantification of FM1-43 release by measurement of mean gray value (shown as percentage of staining) obtained from time-lapse video recordings. B, C, WT is shown in blue; average of all three FX lines is shown in red. Values are mean ± SEM. *p < 0.05, **p < 0.01, t test. The numbers of cells included in the analysis were as follows: WT, n = 12; FX1, n = 4; FX2, n = 4; FX3, n = 4.
Coculture of human neurons with rat neurons
Hypotheses regarding functional defects in fmr1−/− mouse neurons highlight specific postsynaptic deficiencies in glutamatergic and GABAergic transmission (Bagni and Oostra, 2013). However, recent studies show also a clear presynaptic deficit in the same animal model (Deng et al., 2013; Wang et al., 2014). We tested possible presynaptic effects by coculturing human FX neurons with rat hippocampal neurons (Fig. 7). Electrical recording of spontaneous synaptic activity was performed in voltage-clamp mode (Em = −60 mV) 3–7 d after coculture. Control experiments revealed that prestaining of human and rat neurons with Neurotrace did not cause any alteration in the growth, survival, or electrophysiological properties of cells and no diffusion of the dye into other cells was observed during coculture (data not shown). The results show that coculture with rat neurons increased the spontaneous synaptic activity proportionally more in FX neurons than in WT neurons (Fig. 7A–D). Although coculture increased the number of WT human neurons displaying sEPSCs compared with human WT neurons alone from 26% to 45% (WT control n = 5/19; WT coculture n = 10/22; p < 0.001, χ2 test), coculture of FX neurons increased the number of cells displaying sEPSCs more dramatically from 8% to 47% (FX control n = 2/25; FX coculture n = 9/19; p < 0.001, χ2 test). This increase in the number of cells displaying sEPSCs following coculture was concomitant with an increase in the frequency and the amplitude of sEPSCs of FX neurons to similar levels as the WT (Fig. 7C,D). In contrast, cocultures did not affect rise time and decay (τ) in either WT or FX cultures. However, this lack of change in rise time and decay could be attributed to the low number of cells with EPSCs and low frequency of EPSCs in control cultures, especially in FX lines. In addition, no spontaneous activity was detected when membrane potential was held at −20 mV or +60 mV with or without coculture for both WT and FX cells (data not shown). sEPSCs recorded in WT and FX rat cocultures (WT, n = 4; FX, n = 10) were blocked by DNQX (Fig. 7E), but were unaffected by bicuculline (data not shown), indicating that sEPSCs in cocultures were mediated by AMPA receptors. Finally, we hypothesized that the increase observed in synaptic activity in WT and FX neurons as a result of coculture with rat hippocampal neurons could be attributed to an accelerated maturation of human neurons differentiated from hESCs in the presence of rat neurons and glia, which could provide improved tissue support for their maturation by secreting neurotrophic growth factors that are not included in the in vitro differentiation protocol. However, in all coculture recordings (WT, n = 22; FX = 19), we analyzed the pattern of AP firing in WT and FX human neurons in current-clamp mode and found that they were unchanged from those obtained in cells that were not exposed to rat coculture (Fig. 2A,C), with FX neurons still firing single APs, unlike WT neurons (Fig. 7C, inserts). Together, these results show that reduced spontaneous synaptic activity in human embryonic FX neurons derived from FX-hESCs can be rescued when these cells receive synaptic input from normal mature neurons (i.e., rat hippocampal neurons). This suggests that FMRP affects synaptic mechanisms and its absence causes synaptic silence in human FX neurons.
Figure 7.
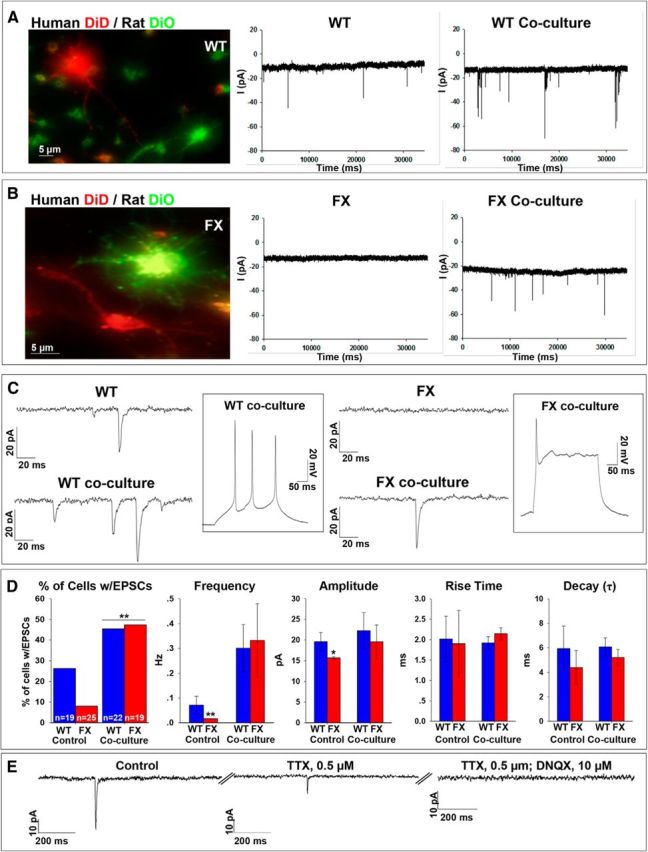
Spontaneous synaptic activity in WT and FX neurons after coculture with hippocampal rat neurons. A, B, Left, Representative images for human-rat coculture. WT (A) and FX2 (B) human neurons were stained with DiD (red) and rat hippocampal neurons were stained with DiO (green). Middle, Representative traces of spontaneous currents recorded in human WT and FX2 neurons (A and B, respectively). Right, Representative traces of spontaneous activity in human WT and FX neurons (A and B, respectively) in coculture with rat hippocampal neurons. Holding potential was −60 mV. C, Expanded traces of spontaneous currents shown in A and B, above. Inserts show representative traces from current-clamp recordings of voltage deflections evoked by the injection of ∼100 pA in WT and FX neurons in coculture. D, Quantification of synaptic parameters, including: percentage of cells displaying EPSCs showing the total number of cells analyzed for WT and FX at the bottom of each bar, frequency of EPSCs, amplitude, rise time, and decay. WT is shown in blue; average of all three FX lines is shown in red. Values are percentage of cells or mean ± SEM. *p < 0.05, **p < 0.01, χ2 test and t test. The numbers of cells included in the analysis were as follows: control cultures, WT, n = 19; FX1, n = 7; FX2, n = 10; FX3, n = 8; total FX control cultures, n = 25; human–rat cocultures, WT, n = 22; FX1, n = 5; FX2, n = 7; FX3, n = 7; total FX cocultures, n = 19. E, Representative traces obtained from voltage-clamp recording of spontaneous activity in an individual FX1 neuron in coculture with rat hippocampal neurons showing the effect of treatment with 0.5 μm TTX and 10 μm DNQX. The numbers of cells included in the analysis were as follows: WT, n = 4; FX1, n = 4; FX2, n = 3; FX3, n = 3.
Discussion
In this study, we analyze the morphological and electrophysiological properties of human FXS neurons lacking FMRP. We found that FX neurons show structural deficiencies, are unable to fire repetitive APs, display less developed inward and outward currents, and lack spontaneous synaptic activity. Altogether, these findings indicate that they are less mature than their WT counterparts. Strikingly, synaptic activity in these neurons can be restored by coculturing them with normal rat hippocampal neurons.
In FXS, the absence of FMRP causes several different cognitive impairments. However, the role of FMRP in neuronal functionality is not yet fully understood. A few theories have been developed over the last 20 years on the molecular and electrophysiological mechanisms of FXS-associated neuronal deficiencies. The most accepted is the mGluR theory, which holds that an increase in mGluRs in the lack of FMRP, causes hyperexcitation (Bear et al., 2004; Penagarikano et al., 2007). Another is the GABA theory, which states that FXS brains have reduced levels of GABA-A receptor subunits and therefore FXS brains are hypo-inhibited (Braat and Kooy, 2015; Gatto et al., 2014). However, all current theories on FXS etiology have been developed mainly from studies conducted on the fmr1−/− mouse (Wang et al., 2012; Oddi et al., 2013) and have never been validated in a human-based model. Because FMRP is present at early stages of neurogenesis in human fetuses with FXS, but not in the fmr1−/− mouse, it is important to examine the nature of the functional deficiencies of FX neurons in a human developmental model. Human in vitro models for FXS include postmortem adult neurons (Irwin et al., 2001), adult neural progenitors (Schwartz et al., 2005), and fetal neural progenitor cells (Castren et al., 2005; Bhattacharyya et al., 2008). These cells show only mild differences in their morphology and gene expression from normal human controls (Bhattacharyya et al., 2008), but significant differences from their fmr1−/− mice counterparts (Castren et al., 2005). More recently, several studies have established FX-hiPSCs and showed molecular and phenotypic abnormalities related to altered gene expression during early neurogenesis (Urbach et al., 2010; Sheridan et al., 2011; Doers et al., 2014; Halevy et al., 2015). However, FX-hiPSCs reprogrammed from FXS patients' fibroblasts do not express FMR1/FMRP even at early stages of development, resembling the fmr1−/− mouse model and unlike the FX human fetus. Most importantly, so far, there is no published report on the electrophysiological characterization of neurons differentiated from FX-human pluripotent stem cells. For these reasons, the aim of the present study was to analyze neuronal functional deficits associated with FXS for the first time in human FX neurons derived from hESCs carrying the naturally occurring full CGG expansion at the FMR1 locus.
A critical feature of neuronal connectivity is the arborization and extension of neurites, especially dendrites. Specifically, the presence, size, shape, and position of dendrites are critical to create and maintain synaptic connections. Several morphological abnormalities have been described in neurons isolated from fmr1−/− mice (Comery et al., 1997; Irwin et al., 2000; Thomas et al., 2008). However, only one study so far has reported similar results in human postmortem FXS brains (Irwin et al., 2001). Recently, a study using neurons differentiated from FX-hiPSCs showed defects in initial neurite growth (Doers et al., 2014). In contrast, we showed in our previous study that initial neurite growth in FX-hESCs undergoing IVND is not significantly altered (Telias et al., 2013). In the present study, we show that only later during neurogenesis, upon full inactivation of FMR1 and absence of FMRP, can a reduction in cell size and neurite length and tortuosity be observed. However, neurite arborization itself was not affected. This discrepancy could be attributed to the timing of FMR1 inactivation in FX-hESCs (Willemsen et al., 2002; Eiges et al., 2007; Telias et al., 2013), as opposed to the constitutive lack of FMR1 expression in FX-hiPSCs (Urbach et al., 2010; Sheridan et al., 2011; Doers et al., 2014). It is therefore possible to conclude that, together, human and mouse models of FXS show clearly that the absence of FMRP affects many crucial morphological features of neurites, which could have an important functional impact on neuronal plasticity. However, because the timing of FMRP absence is different in FX-hESCs, as opposed to fmr1−/− mice and FX-hiPSCs, the observed phenotypes vary accordingly.
So far, most studies on the role of FMRP in neuronal functionality have focused on the localized activity of FMRP as a mRNA-binding protein regulating local translation in the dendrites, showing the importance of FMRP in the “fine tuning” of synaptic activity (Bear et al., 2004; Willemsen et al., 2004; Lee et al., 2011; Swanger and Bassell, 2013). However, recent studies show new neuronal functions for FMRP, in affecting the firing of APs and neuronal excitability. One of these studies showed that neurons in the somatosensory cortex of fmr1−/− mice have significantly higher firing rates than its WT counterparts, concluding that cortical networks in FXS are hyperexcitable (Goncalves et al., 2013). Other studies have shown that a lack of FMRP in CA3 pyramidal neurons of fmr1−/− mice results in an increased AP duration, neuronal hyperexcitability, and increased neurotransmitter release (Deng et al., 2013) and that FMRP regulates neurotransmission at excitatory hippocampal synapses in a presynaptic manner (Wang et al., 2014). Furthermore, those studies on fmr1−/− mice demonstrated that this presynaptic function of FMRP takes place through its direct interaction with BK channels in a translation-independent manner (i.e., protein–protein instead of protein–mRNA). Elucidating whether FMRP plays a role in the balance between excitation and inhibition may be critical for the understanding of the etiology of mental retardation.
In the present study, we show that neurons differentiated in vitro from FX-hESCs demonstrate impaired intrinsic properties. They are unable to fire repetitive APs while displaying a significant decrease in AP amplitudes and a significant increase in AP duration while maintaining a similar spike threshold. Moreover, FX neurons displayed reduced inward and outward currents compared with WT counterparts. Based on our previous study in which we systematically dissected the timing of electrical maturation in neurons differentiated from WT-hESCs (Telias et al., 2014), we conclude that FX neurons are electrically immature compared with WT, likely as a result of loss of FMRP. In addition, their synaptic properties are also impaired. We have shown that the synaptic silence characteristic to human FX neurons in vitro is accompanied with significantly reduced synaptic content and release. However, coculture of these synaptically inactive human FX neurons with normal rat hippocampal neurons induced synaptic reactivation, but no change in the abnormal intrinsic properties was observed. It is possible that the presence of rat hippocampal neurons and supporting glia cells affected FX neurons by trophic effects (i.e., secretion of growth factors) or through other unknown mechanisms, triggering a process of cellular maturation that was preferentially localized to synaptic terminals. This partial rescue of the FXS phenotype in human neurons derived from FX-hESCs highlights differences and similarities between the mouse and the human model and sheds new light on the complexities of FXS pathophysiology.
Together, our results highlight the value of the FX-hESC model system, which needs to be further expanded and investigated. Its application to the understanding of the molecular pathophysiology of FXS can be pivotal in scientific advancement and therapeutic strategies, unveiling previously unknown aspects of FXS pathology.
Footnotes
This work was supported by the National Network of Excellency (NNE) in Neuroscience from TEVA Pharmaceuticals Ltd and by a grant from the Israel Ministry of Health. We thank Eran Parnasa for morphological analysis of neurons and Yoav Mayshar and Efrat Biton for technical assistance.
The authors declare no competing financial interests.
References
- Avitzour M, Mor-Shaked H, Yanovsky-Dagan S, Aharoni S, Altarescu G, Renbaum P, Eldar-Geva T, Schonberger O, Levy-Lahad E, Epsztejn-Litman S, Eiges R. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports. 2014;3:699–706. doi: 10.1016/j.stemcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Oostra BA. Fragile X syndrome: From protein function to therapy. Am J Med Genet A. 2013;161A(11):2809–2821. doi: 10.1002/ajmg.a.36241. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, McMillan E, Wallace K, Tubon TC, Jr, Capowski EE, Svendsen CN. Normal Neurogenesis but Abnormal Gene Expression in Human Fragile X Cortical Progenitor Cells. Stem Cells Dev. 2008;17:107–117. doi: 10.1089/scd.2007.0073. [DOI] [PubMed] [Google Scholar]
- Braat S, Kooy RF. Insights into GABAergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology. 2015;88:48–54. doi: 10.1016/j.neuropharm.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL, Nayler SP, Kao LP, Morrow CA, Thakar NY, Soo SY, Peura T, Grimmond S, Wolvetang EJ. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells. 2013;31:467–478. doi: 10.1002/stem.1297. [DOI] [PubMed] [Google Scholar]
- Castrén M, Tervonen T, Kärkkäinen V, Heinonen S, Castrén E, Larsson K, Bakker CE, Oostra BA, Akerman K. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:17834–17839. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailangkarn T, Acab A, Muotri AR. Modeling neurodevelopmental disorders using human neurons. Curr Opin Neurobiol. 2012;22:785–790. doi: 10.1016/j.conb.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343:1002–1005. doi: 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Sojka D, Klyachko VA. Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci. 2011;31:10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;77:696–711. doi: 10.1016/j.neuron.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23:1777–1787. doi: 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Studer L. Human ESC-derived neural rosettes and neural stem cell progression. Cold Spring Harb Symp Quant Biol. 2008;73:377–387. doi: 10.1101/sqb.2008.73.052. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Pereira D, Broadie K. GABAergic circuit dysfunction in the Drosophila Fragile X syndrome model. Neurobiol Dis. 2014;65:142–159. doi: 10.1016/j.nbd.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin M, Segal M, Avignone E. Functional plasticity triggers formation and pruning of dendritic spines in cultured hippocampal networks. J Neurosci. 2001;21:186–193. doi: 10.1523/JNEUROSCI.21-01-00186.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, Portera-Cailliau C. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci. 2013;16:903–909. doi: 10.1038/nn.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy T, Czech C, Benvenisty N. Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports. 2015;4:37–46. doi: 10.1016/j.stemcr.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–167. doi: 10.1002/1096-8628(20010115)98:2<161::AID-AJMG1025>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011;72:630–642. doi: 10.1016/j.neuron.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile X mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ. Advances in behavioral genetics: mouse models of autism. Mol Psychiatry. 2008;13:4–26. doi: 10.1038/sj.mp.4002082. [DOI] [PubMed] [Google Scholar]
- Muguruma K, Sasai Y. In vitro recapitulation of neural development using embryonic stem cells: from neurogenesis to histogenesis. Dev Growth Differ. 2012;54:349–357. doi: 10.1111/j.1440-169X.2012.01329.x. [DOI] [PubMed] [Google Scholar]
- Myrick LK, Deng PY, Hashimoto H, Oh YM, Cho Y, Poidevin MJ, Suhl JA, Visootsak J, Cavalli V, Jin P, Cheng X, Warren ST, Klyachko VA. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci U S A. 2015;112(4):949–956. doi: 10.1073/pnas.1423094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi D, Crusio WE, D'Amato FR, Pietropaolo S. Monogenic mouse models of social dysfunction: implications for autism. Behav Brain Res. 2013;251:75–84. doi: 10.1016/j.bbr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile X syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- Santos AR, Kanellopoulos AK, Bagni C. Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: what a fly and mouse model can teach us. Learn Mem. 2014;21:543–555. doi: 10.1101/lm.035956.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PH, Tassone F, Greco CM, Nethercott HE, Ziaeian B, Hagerman RJ, Hagerman PJ. Neural progenitor cells from an adult patient with fragile X syndrome. BMC Med Genet. 2005;6:2. doi: 10.1186/1471-2350-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Bassell GJ. Dendritic protein synthesis in the normal and diseased brain. Neuroscience. 2012;232:106–127. doi: 10.1016/j.neuroscience.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telias M, Ben-Yosef D. Modeling neurodevelopmental disorders using human pluripotent stem cells. Stem Cell Rev. 2014;10:494–511. doi: 10.1007/s12015-014-9507-2. [DOI] [PubMed] [Google Scholar]
- Telias M, Segal M, Ben-Yosef D. Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol. 2013;374:32–45. doi: 10.1016/j.ydbio.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Telias M, Segal M, Ben-Yosef D. Electrical maturation of neurons derived from human embryonic stem cells. F1000Research. 2014;3:196. doi: 10.12688/f1000research.4943.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CC, Combe CL, Dyar KA, Inglis FM. Modest alterations in patterns of motor neuron dendrite morphology in the Fmr1 knockout mouse model for fragile X. Int J Dev Neurosci. 2008;26:805–811. doi: 10.1016/j.ijdevneu.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Doering LC. Induced pluripotent stem cells to model and treat neurogenetic disorders. Neural Plast. 2012;2012:346053. doi: 10.1155/2012/346053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev. 2012;22:256–263. doi: 10.1016/j.gde.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XS, Peng CZ, Cai WJ, Xia J, Jin D, Dai Y, Luo XG, Klyachko VA, Deng PY. Activity-dependent regulation of release probability at excitatory hippocampal synapses: a crucial role of fragile X mental retardation protein in neurotransmission. Eur J Neurosci. 2014;39:1602–1612. doi: 10.1111/ejn.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. 2002;110:601–605. doi: 10.1007/s00439-002-0723-5. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Oostra BA, Bassell GJ, Dictenberg J. The fragile X syndrome: from molecular genetics to neurobiology. Ment Retard Dev Disabil Res Rev. 2004;10:60–67. doi: 10.1002/mrdd.20010. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, Issner R, Gifford CA, Goren A, Xing J, Gu H, Cacchiarelli D, Tsankov AM, Epstein C, Rinn JL, Mikkelsen TS, Kohlbacher O, Gnirke A, Bernstein BE, Elkabetz Y, Meissner A. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature. 2015;518:355–359. doi: 10.1038/nature13990. [DOI] [PMC free article] [PubMed] [Google Scholar]