

Abstract
Rare diseases are very difficult to study mechanistically and to develop therapies for because of the scarcity of patients. Here, the rare neuro-metabolic disorder Rett syndrome (RTT) is discussed as a prototype for precision medicine, demonstrating how mouse models have led to an understanding of the development of symptoms. RTT is caused by mutations in the X-linked gene methyl-CpG-binding protein 2 (MECP2). Mecp2-mutant mice are being used in preclinical studies that target the MECP2 gene directly, or its downstream pathways. Importantly, this work may improve the health of RTT patients. Clinical presentation may vary widely among individuals based on their mutation, but also because of the degree of X chromosome inactivation and the presence of modifier genes. Because it is a complex disorder involving many organ systems, it is likely that recovery of RTT patients will involve a combination of treatments. Precision medicine is warranted to provide the best efficacy to individually treat RTT patients.
Understanding and developing therapies for rare disorders
Rett syndrome (RTT) is a rare disorder that occurs in 1 in 10,000 females (Rett 1966; Haas 1988). It is characterized by seemingly normal neurological and physical development during the early postnatal period, followed by symptom manifestation between 6 and 18 months of age (Hagberg 2002). Symptoms progress over several stages (Table 1): stagnation, rapid regression, plateau, and late motor deterioration. The stagnation stage is characterized by subtle developmental delays in motor and language skills, and possible decreased alertness. This stage is often overlooked and leads to a delayed diagnosis, as parents and doctors may not notice these subtle changes. During the rapid regression stage, the child loses purposeful hand skills and spoken language, experiences motor impairments, and develops breathing abnormalities. Children may develop autistic-like features such as loss of interest in social interaction, and seizures may occur. This is followed by the plateau stage, during which motor problems and seizures become more common, but communication skills may improve. Lastly, children enter the late motor deterioration stage during which severe physical disability is common, and many patients become wheelchair dependent.
Table 1.
Rett syndrome progresses over several stages
| Stage | Age | Symptoms |
|---|---|---|
| 1. Stagnation | 6–18 months |
• Developmental delays (postural control, motor, language) • Reduced eye contact • Hand-wringing may occur • Microcephaly may occur |
| 2. Rapid regression | 1–4 years |
• Loss of purposeful hand skills • Stereotypical hand movements (wringing, washing, tapping) • Loss of spoken language • Walking may be unsteady • Breathing irregularities may occur • Autistic-like features • Microcephaly progression • Seizures may occur |
| 3. Plateau/pseudo-stationary | 2–potentially life |
• Hand apraxia/dyspraxia • Motor coordination difficulties and/or loss of motor skills • Improvement of communication skills may occur • Seizures are common |
| 4. Late motor deterioration | 10–life |
• Severe physical disability • Muscle weakness, rigidity, or spasticity • Wheelchair dependency may occur |
Independent of disease stage, subsets of patients also experience gastrointestinal problems (Motil et al. 2012), abnormal cardiorespiratory coupling (Kumar et al. 2017), decreased bone density (Shapiro et al. 2010), early-onset osteoporosis (Haas et al. 1997), bruxism (Alpoz et al. 1999), dyslipidaemia (Justice et al. 2013; Segatto et al. 2014), inflammation of the gallbladder (Anderson et al. 2014), scoliosis (Anderson et al. 2014), urological dysfunction (Ward et al. 2016), and sleep disturbances (Young et al. 2007). Additionally, RTT patients have an increased incidence of unexpected death, and often die due to respiratory infection, cardiac instability, and respiratory failure (Laurvick et al. 2006; Anderson et al. 2014). Current treatment options are limited to symptom control.
Rett syndrome is caused by mutations in methyl-CpG-binding protein 2 (MECP2)
Although a genetic basis for RTT was hypothesized as early as 1983 based on the preferential involvement of females (Hagberg et al. 1983), it was not until 1999 that a causative gene for the disorder was identified. Using a systematic gene screening approach, Amir et al. identified mutations in the gene methyl-CpG-binding protein 2 (MECP2) as the cause of some cases of RTT (Amir et al. 1999). It is now known that de novo mutations in MECP2 account for 95% of typical RTT cases (Bienvenu et al. 2000). Although nearly 600 RTT-causing mutations have been identified in MECP2, only eight missense and nonsense mutations account for approximately 70% of mutations in RTT (R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C) (Neul et al. 2008). Large deletions in MECP2 account for another 15% of RTT-causing mutations. Interestingly, additional mutations in MECP2 have been associated with autism (Xi et al. 2011), intellectual disability (Bianciardi et al. 2016), and lupus erythematosus (Liu et al. 2013).
MECP2 encodes a nuclear protein (MECP2), which is especially abundant within neurons but is expressed at varying levels in every human tissue (Lewis et al. 1992; Tate et al. 1996; Shahbazian et al. 2002b). It is comprised of four domains and is host to several posttranslational modifications (Reviewed in Kyle et al. 2018). MECP2 functions as a global transcriptional regulator by binding specifically to methylated DNA, recruiting protein partners and regulatory complexes to modify transcriptional activity (Nan et al. 1997; Chandler et al. 1999). Although MECP2 is thought to primarily repress gene transcription, its role in transcriptional activation (Chahrour et al. 2008), chromatin remodeling, and mRNA splicing has also been described (reviewed in Lyst and Bird 2015). MECP2 expression correlates with the postnatal maturation of the central nervous system (CNS) and neuronal differentiation, suggesting a role in CNS function and maintenance (Kishi and Macklis 2004). Within the brain, MECP2 is seven times higher in neurons than in glia; however, MECP2 has important roles in glia as well (Ballas et al. 2009; Skene et al. 2010; Lioy et al. 2011).
Clinical studies have highlighted the degree of phenotypic variation in Rett syndrome patients (Fig. 1a). Genotype–phenotype correlation studies demonstrate that early truncating mutations in MECP2 (R168X, R255X, and R270X) and large INDELs cause the most severe phenotype, whereas most missense mutations (R133C and R306C) and late truncating mutations (R294X) are the mildest (Neul et al. 2008; Cuddapah et al. 2014). Thus, MECP2 mutation status is a predictor of disease severity. Despite this, phenotypic variation is also reported in familial cases of RTT where affected sisters present with the same mutation (Zhang et al. 2017); these differences may be due to differences in X chromosome inactivation (XCI). Because MECP2 is inherited on the X chromosome, female heterozygous RTT patients are mosaic carriers of normal and mutated MECP2. XCI is a random process by which one X chromosome is silenced in each cell. XCI can be skewed where the X chromosome carrying the mutated MECP2 is more or less expressed throughout the brain and body, influencing the clinical presentation of RTT (Fig. 1b) (Ishii et al. 2001; Knudsen et al. 2006). Tests for skewed XCI are possible in the clinic. In cases where XCI is not skewed toward one allele, phenotypic variation may be due to the presence of modifier mutations. Modifiers are genes whose function has phenotypic outcomes on the effect of another gene. Mutations in modifier genes may alleviate or enhance clinical symptoms in patients as well (Fig. 1c).
Fig. 1.

Symptom severity in RTT is influenced by mutation status, XCI pattern, and modifier genes. a Of the 8 most common RTT-causing MECP2 mutations, R133C and R306C cause the least severe clinical presentation, whereas the missense mutations R106W and T158M, and nonsense mutations R168X, R255X, R270X, and R294X cause the most severe phenotype. Large deletions in the MECP2 gene also cause a severe phenotype, whereas smaller C-terminal truncations are less severe. b Differences in XCI skewing patterns can influence clinical presentation, where patients with fewer cells expressing the mutant MECP2 gene will have less severe symptoms. c Individuals who have modifier mutations in genes that suppress the RTT phenotype have a more favorable clinical presentation than individuals with mutations in genes that enhance detrimental symptoms
Mouse models recapitulate key symptoms of RTT
Mecp2 is found in all vertebrates, but not in non-vertebrate genetic model organisms, including the fruit fly or the worm (Hendrich and Tweedie 2003). Therefore, developing mouse models of the disorder was needed for a mechanistic understanding of the onset and severity of clinical signs. Shortly after the identification of MECP2 as the causative gene in RTT, two Mecp2-null mouse models were generated, which are now the primary models used to study the disease (Table 2). The Mecp2tm1.1Bird mouse line completely lacks MECP2 protein product (Guy et al. 2001), whereas the Mecp2tm1.1Jae line expresses small MECP2 protein fragments (Chen et al. 2001). However, both null models display a similar phenotype that recapitulates symptoms of RTT and both have been used extensively to study the mechanistic basis for disease.
Table 2.
Many mouse models have been created to study Rett syndrome
| Allele type | Allele | Description | Male phenotype | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RB | BW | BR | AX | M | PD | Age of death (weeks) | ||||
| Null | ||||||||||
| Mecp2 tm1.1Bird | Null | Exon 3–4 deletion | X | X^ | X | X | X | X | 6–12 | Guy et al. (2001) |
| Mecp2 tm1.1Jae | Null; some protein product retained | Exon 3 deletion | X | X* | X | X | X | X | 10 | Chen et al. (2001) |
| Mecp2 tm1Pplt | Null | MBD deletion | X | X | NT | X | X | X | 8 | Pelka et al. (2006) |
| Human point mutations | ||||||||||
| Mecp2 tm4.1Joez | R106W | Missense mutation | X | NT | NT | NT | NT | X | 10 | Unpublished; MGI submission |
| Mecp2 tm1Nlnd | Y120D | Missense mutation | X | X | NT | – | X | X | 14–17 | Gandaglia et al. (2018) |
| Mecp2 tm6.1Bird | R133C | Missense mutation | X | X | NT | X | X | X | 42 | Brown et al. (2016) |
| Mecp2 tm1.1Joez | T158A | Missense mutation | X | – | NT | X | X | X | 16 | Goffin et al. (2011) |
| Mecp2 tm4.1Bird | T158M | Missense mutation | X | X | NT | X | X | X | 13 | Lyst et al. (2013), Brown et al. (2016) |
| Mecp2 tm3.1Joez | T158M | Missense mutation | X | NT | NT | NT | NT | X | 14 | Unpublished; MGI submission |
| Mecp2 tm1.1Jtc | R168X | Stop mutation; truncation | X | X | X | X | X | X | 12–14 | Lawson-Yuen et al. (2007), Schaevitz et al. (2013), Wegener et al. (2014) |
| Mecp2 tm1.1Irsf | R255X | Stop mutation; truncation | X | X | X | X | X | X | 8–10 | Pitcher et al. (2015) |
| Mecp2 tm5.1Bird | R306C | Missense mutation | X | X | NT | X | X | X | 30 | Lyst et al. (2013), Brown et al. (2016) |
| Other mutations | ||||||||||
| Mecp2 tm2.1Jae | S80A | Missense mutation | NT | X* | NT | NT | X | NT | NT | Tao et al. (2009) |
| Mecp2 tm1Vnar | A140V | Missense mutation | – | – | – | – | – | – | Normal lifespan | Jentarra et al. (2010) |
| Mecp2 tm3Meg | T308A | Missense mutation | X | NT | NT | NT | X | NT | > 16 | Ebert et al. (2013) |
| Mecp2 tm1Hzo | R308X | Stop mutation; truncation | X | – | NT | X* | X | X | 6–12 | Shahbazian et al. (2002a) |
| Mecp2 tm1.1Meg | S421A | Missense mutation | – | – | – | – | – | – | Normal lifespan | Cohen et al. (2011) |
| Mecp2 tm1.1Mitoh | Deletion | Isoform 2 deletion | – | – | – | – | – | – | Normal lifespan | Itoh et al. (2012) |
| Mecp2 tm1.1Dhy | Deletion | Isoform 1 deletion | X | – | NT | X | X | X | 7–31 | Yasui et al. (2014) |
| Conditional alleles | ||||||||||
| Mecp2 tm1Bird | – | Exons 3–4 floxed | X* | – | X | X | X | – | Normal lifespan | Guy et al. (2001), Samaco et al. (2008) |
| Mecp2 tm1Jae | – | Exon 3 floxed | – | – | – | – | – | – | Normal lifespan | Chen et al. (2001) |
| Mecp2 tm2Bird | – | Floxed-stop upstream of exon 3 | X | X | X | X | X | X | 8–10 weeks | Guy et al. (2007) |
These mouse models include null alleles, point mutations designed to recapitulate mutations observed in human RTT patients, whole exon deletions, and conditional alleles used in combination with targeted Cre mice to achieve temporal deletion of Mecp2. In BW category, alleles marked with *which have an increased body weight, and alleles marked with ^show either increase or decrease depending on mouse background. In the AX category, alleles marked with *show increased anxiety
RB reduced brain size, BW body weight reduction, BR breathing abnormalities, AX reduced anxiety, MA motor abnormalities, PD premature death In categories, X present, – not present, NT not tested
RTT predominantly affects females, so female Mecp2-mutant mice are the most clinically relevant model. However, female Mecp2-mutant mice display deviations in phenotype presentation due to XCI skewing making it difficult to discern which phenotypes arise through cell autonomous versus non-autonomous pathways. Therefore, the majority of RTT mouse studies take place in hemizygous Mecp2-mutant male mice due to their consistent phenotype and their complete lack of MECP2 protein, which is advantageous for mechanistic research.
Hemizygous male Mecp2-null mice are phenotypically normal until 4 weeks of age when they develop a RTT-like phenotype consisting of hind limb clasping, tremors, breathing irregularities, loss of muscle tone, and hypoactivity (Chen et al. 2001; Guy et al. 2001). These mice also display a reduced brain weight and body weight, experience a rapid phenotypic regression, and die between 6 and 12 weeks of age. Female mice heterogeneous for Mecp2 deletions develop the same features at 4–6 months of age and typically live a normal lifespan.
Mecp2-mutant mouse models recapitulate a broad spectrum of phenotypes seen in human RTT patients (Fig. 2). The most profound symptoms in human patients and most Mecp2-mutant mice are motor abnormalities, including reduced mobility, impaired motor coordination, ataxic gait, and tremors (Hagberg and Witt-Engerström 1986). While human patients replace purposeful hand use with stereotypic movements, mouse models clasp their hindlimbs (Chen et al. 2001; Guy et al. 2001); this may resemble the hallmark human symptom, but it is not a phenotype specific to RTT mouse models (Lalonde and Strazielle 2011). Morphologically, both human patients and Mecp2 mouse models exhibit a reduced brain volume and neuronal hypotrophy (Chahrour and Zoghbi 2007). Behaviorally, RTT patients experience a neurological regression and loss of speech, two traits which cannot be measured in mouse models. However, both may experience some learning deficits (Elefant and Wigram 2005; Moretti 2006). Interestingly, while human RTT patients typically exhibit increased anxiety and social avoidance (Mount et al. 2002), Mecp2-mutant mice are less anxious and more social, as assessed by phenotypic tests available for rodents (Samaco et al. 2012; Orefice et al. 2016; Wu et al. 2016). Metabolic disturbances in both human patients and mice include abnormalities in neurometabolites (Goffin and Zhou 2012), increased serum cholesterol and triglycerides (Buchovecky et al. 2013; Justice et al. 2013; Segatto et al. 2014; Kyle et al. 2018), abnormal mitochondrial structure (Shulyakova et al. 2017), and increased oxidative stress (De Felice et al. 2009; Janc et al. 2016). Finally, both experience breathing irregularities (Ramirez et al. 2013), cardiac abnormalities (prolonged QTc intervals) (McCauley et al. 2011; Hara et al. 2015), seizures, and a shortened lifespan (Guy et al. 2001; Anderson et al. 2014). Overall, Mecp2-mutant mice exhibit a broad range of phenotypes that recapitulate symptoms of human RTT patients, making them excellent models to study the disorder.
Fig. 2.
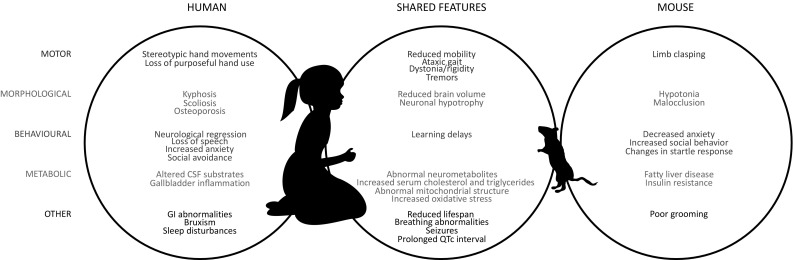
RTT patients and Mecp2-mutant mouse models share many features
Despite their face validity, null mouse models do not always represent human cases molecularly since many RTT patients carry missense mutations that result in a less-efficient or unstable MECP2 protein rather than a complete loss of it. To circumvent this caveat, several mouse lines with point mutations and deletions in MECP2 have been engineered to recapitulate clinically relevant mutations observed in human patients (Table 2). Of the eight most common missense mutations in RTT patients, six have an established mouse model which recapitulates symptoms of the disease. These mice have been instrumental in understanding the functional consequences of RTT-causing mutations and their correlation to symptom severity. For example, RTT patients carrying MECP2 T158M mutations display severe RTT symptoms, while motor and speech preservation is observed in patients with a R133C mutation (Cuddapah et al. 2014). Consistently, male mice carrying these missense mutations have a lifespan of 13 weeks and 42 weeks, respectively. Functional studies in mice determined that while both the T158M and R133C mutations create MECP2 proteins with reduced DNA-binding ability, the T158M mutation also leads to protein instability, likely resulting in the unique and more severe phenotype presentation (Brown et al. 2016).
Further, mice with Mecp2 mutations have been designed to study specific biological questions of functional relevance. For example, while MECP2 is expressed as two isoforms, their expression levels vary among tissues, with MECP2_e1 being more abundant than e2 in the brain. Studies that deleted either MECP2_e1 or e2 throughout the body found that only deletion of MECP2_e1 led to RTT-like phenotypes and a shortened lifespan, while deletion of e2 was required for placental development (Itoh et al. 2012; Yasui et al. 2014). Additionally, MECP2 protein is a host to numerous posttranslational modifications (PTMs), including phosphorylation at serine 80 (S80) and 421 (S421). Mutating either of these serine residues in mice prevented this site-dependent phosphorylation and determined that these PTMs are important for the association of MECP2 to chromatin and its role in regulating neuronal gene expression (Tao et al. 2009; Cohen et al. 2011).
Finally, conditional-ready alleles have been constructed by engineering loxP sites flanking a portion of the Mecp2 gene (Floxed) (Chen et al. 2001; Guy et al. 2001). These mice can be crossed with transgenic mice carrying a tissue- or cell type-specific Cre to achieve spatial or temporal deletion of Mecp2. Studies using these mice have greatly informed Mecp2 function both within the CNS and in peripheral tissues. Neuronal dysfunction was once considered the only cause of RTT, despite MECP2’s expression in glial cells. Crossing Mecp2-floxed mice to mice with a glial-specific GFAP-Cre transgene achieved targeted deletion of Mecp2 in astrocytes. These mice developed RTT-like symptoms including decreased body weight, hindlimb clasping, and irregular breathing, demonstrating that glial Mecp2 deficiency could produce some RTT symptoms (Lioy et al. 2011). Additionally, liver-targeted deletion of Mecp2 was achieved by crossing Mecp2-floxed mice to mice expressing Cre under the control of an Albumin promoter (Alb-Cre). The resulting mice develop fatty liver disease and dyslipidemia, implicating MECP2 in the regulation of lipid metabolism in non-CNS tissues (Kyle et al. 2016). Since transgenic mice have been engineered to express Cre from many diverse cell types, continued studies are likely to achieve a thorough understanding of Mecp2’s role in a variety of tissues and cell types.
A significant question in RTT research concerns phenotypic reversibility: Since RTT patients do not display neuronal death, it is possible that Mecp2-deficient cells can be repaired. In a milestone study, a mouse model was engineered to contain a transcriptional STOP cassette flanked by loxP, which was inserted into the endogenous Mecp2 gene (Guy et al. 2007). These ‘FloxedStop’ mice were crossed with mice expressing a ubiquitous Cre-ER transgene. This strategy allowed for Mecp2 to remain silenced in mice until Cre could be activated with injection of tamoxifen. ‘FloxedStop’ male mice were identical to Mecp2-null mice, developed RTT-like symptoms and had a shortened lifespan. Following phenotype onset, tamoxifen was administered systemically, removing the loxP-STOP-loxP cassette and restoring Mecp2 expression to 80% of normal levels. Remarkably, restoring MECP2 expression reversed neurological symptoms and normalized lifespan (Guy et al. 2007). This key study demonstrated that Mecp2-induced pathology is not permanent and symptom reversal may be possible in human patients.
Given MECP2’s high expression levels, its global DNA-binding affinity, and its multitude of proposed binding partners and functions, it is unsurprising that Mecp2-deficiency results in a myriad of dysregulated pathways. MECP2 has been implicated in neuronal maintenance (Kishi and Macklis 2004; Guy et al. 2011), glial cell function (Ballas et al. 2009), neurotransmitter signaling (Goffin and Zhou 2012), growth factor signaling (Itoh et al. 2007), lipid metabolism (Buchovecky et al. 2013), oxidative stress (Großer et al. 2012), and more. Current preclinical research follows two approaches: targeting pathways downstream of MECP2 or directly targeting MECP2 and its protein product (Fig. 3).
Fig. 3.

Treatment options in RTT either directly target MECP2 mutation or target pathways downstream of MECP2. Treatment options are further divided within these two groups
Using mouse models to develop therapeutics for RTT targeting pathways downstream of Mecp2
The first approach of recognizing components that lie in pathways downstream of MECP2 has identified many targets with the potential for treatment. Pharmacological therapeutics for targets downstream of MECP2 can be repurposed to improve neurotransmitter signaling, growth factor signaling, and metabolism (Table 3). Importantly, several of these treatments ameliorated symptoms in Mecp2-mutant mice and have spawned clinical trials in patients. However, these strategies revealed several limitations. First, because the precise functions of MECP2 are still unknown, identifying the pathways it regulates, especially those which may be subtly changed in the absence of MECP2, is difficult. Another challenge is specificity: MECP2 is recruited to DNA methylation signals, which are highly cell-specific (Cedar 1988; Deaton et al. 2011). Therefore, the genes MECP2 regulates will likely vary based on methylation status in different cell types. Consistently, a recent study found that gene misregulation within subtypes of neurons in Mecp2 mutant mice is highly dependent on cell type-specific epigenetic marks (Renthal et al. 2018). Finally, it is difficult to discern which pathways are affected as the primary result of MECP2 loss and which are secondary effects farther downstream that will likely have less value as therapeutic targets. Nevertheless, advances in molecular biology techniques are making it easier to circumvent these issues. For example, single-cell RNA sequencing can identify genes misregulated in individual Mecp2-deficient cells and chromatin immunoprecipitation of MECP2’s known binding partners could identify its direct transcriptional targets in individual cells. Here, we highlight preclinical treatments targeting pathways downstream of Mecp2 which have led to clinical trials in RTT patients.
Table 3.
Preclinical treatments targeting pathways downstream of Mecp2
| Treatment | Mechanism | Mouse model | Prolong lifespan | Improved phenotype | References | Clinical trial |
|---|---|---|---|---|---|---|
| Neurotransmitter signaling | ||||||
| Citalopram | Serotonin reuptake blocker | Mecp2 tm1.1Bird | NT | Improved sensitivity to carbon dioxide exposure | Toward et al. (2013) | – |
| 8-OH-DPAT | Serotonin 1a agonist | Mecp2 tm1.1Bird | NT | Reduced apneas | Abdala et al. (2010) | – |
| F15599 | Serotonin 1a agonist | Mecp2 tm1.1Bird | NT | Reduced apneas and improves breathing irregularity | Levitt et al. (2013) | – |
| Sarizotan | Serotonin 1a agonist & dopamine D2-like receptor |
Mecp2 tm1.1Bird Mecp2 tm1.1Jae Mecp2 tm1.1Jtc |
NT | Reduced apneas and improved breathing irregularity | Abdala et al. (2014) | Y |
| LP-211 | Serotonin 7 receptor agonist | Mecp2 tm1Hzo | NT | Improved overall health, memory and anxiety | De Filippis et al. (2014) | – |
| Levodopa | Dopaminergic stimulation | Mecp2 tm1.1Bird | Y | Improved motor activity | Szczesna et al. (2014) | – |
| Ketamine | NMDA receptor antagonist | Mecp2 tm1.1Jae | NT | Improved startle response | Kron et al. (2012) | Y |
| Ketamine | NMDA receptor antagonist | Mecp2 tm1.1Bird | Y | Improved limb clasping, motor coordination and reduced apneas | Patrizi et al. (2016) | Y |
| NO-711 | GABA reuptake blocker | Mecp2 tm1.1Bird | NT | Reduced apneas | Abdala et al. (2010) | – |
| Benzodiazepine diazepam | GABA reuptake blocker | Mecp2 tm1.1Bird | NT | Reduced apneas | Abdala et al. (2010) | – |
| L-838,417 | GABA reuptake blocker | Mecp2 tm1.1Bird | NT | Reduced apneas | (Abdala et al. 2010) | – |
| Tiagabine | GABA reuptake blocker | Mecp2 tm1.1Bird | Y | No improvement | El-Khoury et al. (2014) | – |
| THIP | GABA receptor agonist | Mecp2 tm1.1Bird | Y | Improved motor function, social behavior, and reduced apneas | Zhong et al. (2016) | – |
| Mirtazapine | GABA release promoter | Mecp2 tm1.1Bird | NT | Improved overall healthy, neuronal morphology, dendritic spine number, anxiety | Bittolo et al. (2016) | – |
| VUO462807 | mGlu5 positive allosteric modulator | Mecp2 tm1.1Bird | N | Improved motor function and limb clasping | Gogliotti et al. (2016) | – |
| VU0422288 | mGlu7 positive allosteric modulator | Mecp2 tm1.1Bird | NT | Reduced apneas, improves learning and memory | Gogliotti et al. (2017) | – |
| CTEP | mGluR5 negative allosteric modulator | Mecp2 tm1.1Bird | Y | Reduced apneas, improved memory | Tao et al. (2016) | – |
| Acetyl-L-carnitine | Acetyl group donor | Mecp2 tm1.1Jae | NT | Improved weight gain, motor activity and memory | Schaevitz et al. (2012) | – |
| Acetyl-L-carnitine | Acetyl group donor | Mecp2 tm1.1Jae | NT | Improved weight gain, motor activity and memory | Schaevitz et al. (2012) | – |
| Choline | ACh | Mecp2 tm1.1Jae | NT | Improved motor coordination and activity | Nag and Berger-Sweeney (2007) | – |
| Choline | ACh | Mecp2 tm1Hzo | NT | Improved motor activity | Ricceri et al. (2011) | – |
| Choline | ACh | Mecp2 tm1.1Bird | NT | Improved motor coordination, anxiety and social behavior | Chin et al. (2018) | – |
| D-NAC | Dendrimer-conjugated N-acetyl cysteine | Mecp2 tm1.1Bird | NT | Improved overall health, limb clasping | Nance et al. (2017) | – |
| Desipramine | Norepinephrine reuptake inhibitor | Mecp2 tm1.1Bird | Y | Improved breathing irregularities | Roux et al. (2007) | Y |
| Desipramine | Norepinephrine reuptake inhibitor | Mecp2 tm1.1Bird | Y | Reduced apneas | Zanella et al. (2008) | Y |
| Clenbuterol | B2-adrenergic receptor agonist | Mecp2 tm1.1Bird | Y | Improved motor coordination and breathing irregularities | Mellios et al. (2014) | – |
| D-cycloserine | D-alanine analog | Mecp2 tm1.1Jae | NT | No improvement | Na et al. (2017) | – |
| Growth factor signaling | ||||||
| CX546 | Ampakine (BDNF) | Mecp2 tm1.1Jae | NT | Improved breathing irregularity | Ogier et al. (2007) | – |
| Fingolimod | Sphingosine-1 phosphate receptor (BDNF) | Mecp2 tm1.1Bird | NT | Improved motor activity | Deogracias et al. (2012) | Y |
| CPT157633 | PTP1B inhibitor (BDNF) | Mecp2 tm1.1Bird | Y | Reduced limb clasping, partially improved motor activity | Krishnan et al. (2015) | – |
| LM22A-4 | TrkB agonist (BDNF) | Mecp2 tm1.1Jae | NT | Improved breathing irregularity | Schmid et al. (2012) | – |
| 7,8-DHF | TrKB agonist (BDNF) | Mecp2 tm1.1Jae | Y | Improved motor activity and breathing irregularities | Johnson et al. (2012) | – |
| LM22A-4 | TrkB agonist (BDNF) | Mecp2 tm1.1Jae | NT | Reduced apneas | Kron et al. (2014) | – |
| LM22A-4 | TrkB agonist (BDNF) | Mecp2 tm1.1Jae | NT | Improved memory | Li et al. (2017) | – |
| IGF-1 | IGF-1 | Mecp2 tm1.1Jae | Y | Improves motor activity, breathing irregularities, increased brain size | Tropea et al. (2009) | Y |
| PEG-IGF-1 | Slow release IGF-1 | Mecp2 tm1.1Bird | Y | No improvement | Pitcher et al. (2013) | – |
| RhIGF01 | Recombinant human IGF1-1 | Mecp2 tm1.1Bird | Y | Improves motor activity, breathing irregularities, social behavior and anxiety | Castro et al. (2014) | Y |
| Metabolism | ||||||
| Diet restriction | Caloric deficit | Mecp2 tm1Hzo | NT | Improved motor activity and anxiety | Mantis et al. (2009) | – |
| Statins | Cholesterol-lowering medication |
Mecp2 tm1.1Bird Mecp2 tm1.1Jae |
Y | Improved overall health, motor coordination, motor activity, serum lipids and liver lipids | Buchovecky et al. (2013) | Y |
| Dietary triheptanoin | Energy use (mitochondria) | Mecp2 tm1.1Jae | Y | Improved motor coordination, social behavior, insulin sensitivity, metabolic homeostasis | Park et al. (2014) | Y |
| Trolox | Vitamin E derivative | Mecp2 tm1.1Bird | NT | Blood glucose levels normalized, improved response to hypoxia | Janc et al. (2016) | – |
| Corticosterone | Glucocorticoid activation | Mecp2 tm1.1Bird | Decreased lifespan | Worsened motor activity | Braun et al. (2012) | – |
| Corticosterone | Glucocorticoid activation | Mecp2 tm1Hzo | NT | Improved motor activity | De Filippis et al. (2013) | – |
| RU486 | Glucocorticoid repression | Mecp2 tm1.1Bird | No | Delayed progression of symptoms, improved motor activity | Braun et al. (2012) | – |
| Curcumin | Anti-oxidant, anti-inflammatory | Mecp2 tm1.1Jae | NT | NT | Panighini et al. (2013) | – |
| Insulin | Glucose signaling | Mecp2 tm1.1Bird | Decreased lifespan | No improvement | Pitcher et al. (2013) | – |
| Other | ||||||
| Zoledronic acid | Anti-osteoclastic | Mecp2 tm1.1Bird | NT | Increased bone volume and connectivity | Shapiro et al. (2010) | – |
| Cannabidivarin | Phytocannabinoid | Mecp2 tm1Hzo | NT | Improved overall health, motor activity and social behavior | Vigli et al. (2018) | – |
| CNF1 | RhoGTPase | Mecp2 tm1Hzo | NT | Improved motor activity | De Filippis et al. (2012) | – |
| CNF1 | Rho GTPase | Mecp2 tm1Hzo | NT | Improved mitochondrial dysfunction and memory | De Filippis et al. (2015) | – |
| Non-pharmacological | ||||||
| Enriched environment | Environmental modulation | Mecp2 tm1Pplt | NT | Improved motor coordination | Kondo et al. (2008) | – |
| Enriched environment | Environmental modulation | Mecp2 tm1.1Jae | NT | Improved motor activity | Nag et al. (2009) | – |
| Enriched environment | Environmental modulation | Mecp2 tm1.1Jae | N | Improved motor coordination and activity | Lonetti et al. (2010) | – |
| Enriched environment | Environmental modulation | Mecp2 tm1Pplt | NT | Reduced anxiety | Kondo et al. (2016) | – |
| Forniceal deep brain stimulation | Neural circuit stimulation | Mecp2 tm1.1Bird | NT | Improved memory | Hao et al. (2015) | – |
| Bone marrow transplantation | Brain microglia repopulation | Mecp2 tm1.1Bird | Y | Reduced apneas, improved breathing irregularities, improved locomotor activity | Derecki et al. (2012) | – |
Treatment strategies are divided into those influencing neurotransmitter signaling, growth factor signaling, metabolism, other pharmacological treatments, and non-pharmacological treatments. In prolong lifespan column, Y yes, N no, NT not tested. In clinical trial column, Y yes, –: not yet tested
Potential treatments targeting neurotransmitter signaling
A prominent feature of RTT and Mecp2-mutant mouse models is the reduction in the number and length of dendrites (Armstrong 1992). Since dendrites receive electrical signals and neurotransmitters from pre-synaptic neurons, a neurotransmitter signaling defect was proposed in RTT. In support of this idea, dysfunction in dopamine (Wenk 1995), serotonin (Paterson et al. 2005; Ohno et al. 2016), norepinephrine (Viemari 2005; Santos et al. 2010), glutamate (Chao et al. 2010; Abdala et al. 2016), and NMDA signaling (Katz et al. 2016; Patrizi et al. 2016) has been observed in Mecp2-mutant mice. Treatments that target these pathways have shown varied effects in mice, and a few have been tested in clinical trials. Desipramine, a norepinephrine reuptake inhibitor, improved breathing irregularities and apneas in Mecp2-mutant mice (Roux et al. 2007; Zanella et al. 2008). However, a clinical trial found no clinical improvements in RTT patients treated with this drug (Mancini et al. 2018). In contrast, sarizotan, a serotonin 1a agonist and dopamine D2-like receptor, reduced breathing apneas by 15–30% in Mecp2-mutant mice but had no effect on motor activity (Abdala et al. 2014). As a result, it is currently being tested for its efficacy in improving respiratory symptoms in RTT (NCT02790034).
Finally, ketamine, an NMDA receptor agonist, was tested in two different laboratories for its therapeutic potential in RTT (Kron et al. 2012; Patrizi et al. 2016). These preclinical studies showed that low-dose ketamine could increase activity in the cortical network while decreasing synaptic excitability in the brainstem network, targeting a possible imbalance in neuronal activity throughout the Mecp2-mutant brain. Treatment with ketamine improved limb clasping, motor coordination, and breathing apneas in Mecp2-mutant mice. The safety of ketamine is currently being assessed in RTT patients (NCT03633058).
Growth factor signaling as a treatment for RTT
Early studies of MECP2 identified BDNF as one of its transcriptional targets (Chen et al. 2003). BDNF is a member of the neurotrophin family of growth factors, which binds to tropomyosin-related kinase B (TrkB) to stimulate signaling cascades involved in neurite outgrowth, synaptic function, and neuronal differentiation (Amaral and Pozzo-Miller 2007). In RTT patients and Mecp2-mutant mice, brain Bdnf expression is drastically reduced (Chang et al. 2006; Deng et al. 2007). However, direct administration of BDNF is not a feasible treatment option because it is unable to cross the blood–brain barrier (BBB). However, fingolimod (FTY720), a sphingosine-1 phosphate analog, has been shown to partially increase BDNF levels and improve motor activity in Mecp2-mutant mice (Deogracias et al. 2012). Fingolimod is currently being assessed for its safety and efficacy in patients (NCT02061137).
Insulin-like growth factor-1 (IGF) binds to the IGF-1 receptor (IGF-1R), activating a signaling cascade similar to the BDNF receptor TrkB. IGF-1 is also transcriptionally regulated by MECP2 (Itoh et al. 2007), but unlike BDNF, it can cross the BBB, increasing its therapeutic potential (Baker et al. 2005). IGF-1 signaling is involved in neuronal survival, neural outgrowth, and synapse formation (D’Ercole et al. 1996). It has been implicated as a promising therapeutic in cancer, diabetes, and ALS.
Mecp2-mutant mice treated with IGF-1 showed improved locomotor function, breathing irregularities, and extended lifespan. IGF-1 also increased the brain weight of mutant mice while restoring neural spine density in the motor cortex and improving excitatory synaptic transmission in sensorimotor cortex neurons (Tropea et al. 2009). Importantly, full-length IGF-1 is already an approved treatment in North America used to treat growth failure in children. A phase 1 clinical trial in RTT patients found no adverse effects of recombinant human IGF-1 (rhIGF-1) (Khwaja et al. 2014). However, subsequent clinical trials found it produced no significant improvement of RTT symptoms (O’Leary et al. 2018).
Recently, a synthetic analog derived from IGF-1, NNZ-2566 (Trofinetide) was developed which inhibits neuroinflammation, restores glial function, corrects synaptic deficits, and regulates oxidative stress response (Deacon et al. 2015). When administered to mice with Fragile X syndrome, NNZ-2566 drastically improved their aberrant symptoms (Ligsay and Hagerman 2016). A phase 2 clinical trial of NNZ-2566 in RTT patients showed that treatment improved overall health, neurobehavioral and motor symptoms. These promising results enabled a phase 3 clinical trial that is currently recruiting patients (NCT02715115).
Metabolic defects in RTT can be therapeutically targeted
Modifiers of MECP2 have implicated neurite outgrowth and energy homeostasis in altering phenotypic outcome in human patients (Artuso et al. 2011; Pizzo et al. 2018). Modifiers are also being identified and validated by carrying out modifier screens in a mouse model (Buchovecky et al. 2013). In a modifier screen, unbiased random mutation identifies key genes that can change a phenotype of interest, allowing the model to reveal important pathways of interest. Such an approach was applied to identify novel therapeutic targets in RTT; ethylnitrosourea (ENU), a mouse supermutagen, was used to induce mutations in male mice, which were then bred to females carrying a mutant Mecp2. Male mice inheriting mutant Mecp2 would normally become very ill and die. Dominant second site mutations that could improve the health and neurological phenotype of the Mecp2 mutant males were then identified molecularly (Buchovecky et al. 2013). One suppressor of disease was a nonsense mutation in a rate-limiting enzyme in cholesterol synthesis—a pathway that had not previously been associated with RTT.
The identification of this modifier locus led to additional studies that revealed lipid metabolism was severely perturbed in Mecp2-mutant mice. Prior to symptom onset, brain cholesterol was already markedly elevated in Mecp2-mutant mice, and cholesterol and triglycerides were elevated in the serum and liver. Remarkably, lipid-lowering statin drugs regulated lipid levels, ameliorated motor symptoms, and extended lifespan in mice (Buchovecky et al. 2013). Furthermore, elevated lipids have also been observed in a subset of RTT patients, indicating that repurposing of statin drugs may be a viable treatment option to benefit patients (Justice et al. 2013; Segatto et al. 2014). Abnormalities in cholesterol homeostasis are associated with many neurological diseases (Tint et al. 1994; Puglielli et al. 2003; Bi and Liao 2010; Berry-Kravis et al. 2015), and therefore RTT is no exception. Importantly, these findings led to a clinical trial testing the efficacy and safety of statins in RTT patients (NCT02563860).
Metabolic defects in RTT are not limited to cholesterol synthesis. Both RTT patients and Mecp2-mutant mouse models display abnormal mitochondrial structure and function (Eeg-Olofsson et al. 1988; Dotti et al. 1993; Kriaucionis et al. 2006; Gold et al. 2014). Mitochondria are organelles within cells that convert glucose into adenosine triphosphate (ATP), the energy currency of the cell. Importantly, compromised mitochondrial function can greatly affect cellular energy production. While mitochondria are necessary in all cells of the body, their role is especially important in tissues with high energy demands, such as nerves and muscle. RTT shares many features of mitochondrial diseases, including early symptomatic onset, developmental delay, neurological regression, poor muscle tone, seizures, and gastrointestinal issues (Schon and Manfredi 2003). Consistently, both RTT patients and Mecp2-mutant mice present with increased oxidative stress and decreased levels of mitochondrial enzymes (Haas et al. 1995; Kriaucionis et al. 2006; De Felice et al. 2009; Leoncini et al. 2011). In Mecp2-mutant mice, markers of oxidative stress increase with age, suggesting a progressive dysfunction in mitochondrial function (De Felice et al. 2014). Altogether, this suggests defects in mitochondrial energy production may be present in RTT. Importantly, mitochondrial energy production is tightly linked with cholesterol synthesis. Several steps in the biosynthesis of cholesterol require mitochondrial sources of ATP as an electron donor in oxygenation reactions. Thus, cholesterol perturbations may be linked with mitochondrial dysfunction in RTT.
Interestingly, anaplerotic substances can replenish intermediate compounds in the energy production pathway, enhancing mitochondrial energy production. A diet supplemented with the anaplerotic triheptanoin reversed symptoms of some metabolic disorders by correcting energy production (Roe et al. 2002; Mochel et al. 2005). This strategy was adapted for use in mouse models of RTT. Strikingly, Mecp2-mutant mice fed a diet with triheptanoin supplementation showed improved mitochondrial morphology and improved energy production. This translated to improved motor coordination and increased lifespan in these mice. Two clinical trials investigating the efficacy of triheptanoin in improving seizures, muscle tone, and symptom improvement in RTT patients are currently in their early stages (NCT02696044 and NCT03059160).
Using mouse models to develop therapies for RTT that directly target MECP2
Directly restoring MECP2 is an attractive therapeutic strategy (Fig. 3) since all pathways downstream of MECP2 could subsequently recover as well. However, the largest concern of these approaches is dosage. While lack of MECP2 causes RTT, an abundance of MECP2 causes MECP2 duplication syndrome, a disorder characterized by neurological dysfunction, intellectual disability, and some RTT-like features (Van Esch et al. 2005). In mice, MECP2 overexpression causes hypoactivity and seizures (Collins et al. 2004; Bodda et al. 2013). The amount of MECP2 required to cause toxicity associated with overexpression has been debated, with some studies finding that 1.6 × normal levels of MECP2 cause behavioral impairments in mice (Jugloff, 2008) and others finding that 2.4 × normal MECP2 can be tolerated (Koerner et al. 2018). Despite this discrepancy, small deviations in MECP2 levels in humans have been linked to milder neurological and psychiatric conditions including autism, intellectual disability, and lupus erythematosus. Thus, caution must be taken to provide enough MECP2 per cell to impart a therapeutic benefit while reducing the risk of MECP2 overexpression.
Read-through compounds may facilitate the recovery of MECP2 product
One strategy involves directly targeting the MECP2 mutation with small molecules known as translational read-through-inducing drugs (TRIDs). This is an attractive strategy for 35–40% of Rett syndrome patients who have nonsense mutations in MECP2 that result in premature stop codons, which inhibit normal full-length protein expression (Neul et al. 2008). TRIDs permit read-through of premature stop codons by binding to ribosomes and impairing codon/anticodon recognition, allowing for the insertion of another amino acid in place of the stop codon (Nagel-Wolfrum et al. 2016). Notably, this causes a missense mutation rather than a nonsense mutation, which may impart less of a functional consequence on the protein product. Importantly, this strategy would increase MECP2 in cells with the nonsense mutation, while avoiding increasing dosage in cells with normal MECP2. However, because TRIDs would create a mutated MECP2 rather than a fully functional protein, this treatment strategy may only modestly improve symptoms in patients. An additional concern is nonsense mediated decay (NMD), a cellular process that facilitates the degradation of mRNA transcripts with premature stop codons because their translation could lead to mutated proteins with deleterious gain-of-function or dominant-negative effects (Conti and Izaurralde 2005). In mammalian cells, when a premature stop codon is located greater than 50–55 nucleotides away from an exon–exon junction, NMD is activated (Nagy and Maquat 1998; Kuzmiak and Maquat 2006). NMD naturally reduces the number of transcripts available for TRIDs (Linde et al. 2007). Therefore, TRIDs may not be a suitable treatment option for RTT patients with certain nonsense mutations which are prone to NMD as the best responders will have high levels of target transcript. However, the efficacy of NMD varies naturally between individuals (Nguyen et al. 2014) and patient transcript levels could be assayed to determine if they are candidates for TRID therapy.
One class of TRIDs is aminoglycoside antibiotics, of which gentamicin is the most commonly studied. Gentamicin has been shown to suppress nonsense mutations and restore function protein in mouse models of Duchenne muscular dystrophy (DMD) (Barton-Davis et al. 1999), cystic fibrosis (CF) (Du et al. 2008), retinal degeneration (Guerin et al. 2008), and several other diseases. Importantly, clinical trials in DMD and CF patients have demonstrated gentamicin’s ability to restore protein products in a subset of patients, giving hope for the effectiveness of TRIDs in other diseases (Nagel-Wolfrum et al. 2016).
Four of the most common RTT-causing mutations (R168X, R255X, R270X, R294X) result in premature stop mutations. In vitro studies have shown that read-through efficiency of gentamicin depends heavily on the context of the stop mutation, where gentamicin restored 22% of function MECP2 in cells with R294X mutations and only 10% in cells with R168X (Brendel et al. 2011), possibly due to variable NMD. Increasing the concentration of gentamicin increased read-through efficiency and amounts of MECP2 protein. However, these high levels of aminoglycosides could cause toxic side effects if administered over a long period of time (Popescu et al. 2010). Novel aminoglycosides and different TRIDs may impart higher read-through activity and lower toxicity (Brendel et al. 2011; Pitcher et al. 2015), though no in vivo tests with these novel compounds have performed in Mecp2-mutant mice.
One new promising TRID is Ataluren, an orally bioavailable oxadiazole with no antibacterial activity and minimal off-target effects (Keeling et al. 2014). Ataluren restored 20% of normal functional protein in mouse models of DMD and CF, leading to clinical trials for both diseases. However, in both cases, Ataluren did not produce enough protein to see a significant benefit in patients (Kerem et al. 2014; McDonald et al. 2017). This may be a worthwhile avenue to pursue for RTT since even small increases in MECP2 are associated with improved symptoms. Nonetheless, efforts are underway to improve the efficacy of TRIDs for suppression therapy, making them more likely to assist RTT patients.
Reactivating the silent X chromosome could restore endogenous MECP2 to cells
Through XCI, mammalian cells silence one X chromosome in each cell, allowing for dosage compensation of X-linked genes (Bhatnagar et al. 2014). XCI is initiated by the non-coding RNA Xist, which coats the inactive X chromosome (Xi) from which it is produced. On the active X (Xa), Tsix RNA blocks Xist upregulation. As MECP2 is expressed on the X chromosome, one copy is silenced by XCI. Therefore, in cells of RTT patients where the mutated MECP2 is on the Xa, a normal copy of MECP2 lies dormant on the Xi.
Interestingly, the silent state of the Xi is temporarily reversed during development and stem cell remodeling. In the case of X-linked diseases, identification of key molecular players involved in reactivation of the Xi could be repurposed to facilitate the re-expression of its genes. Using this strategy to target MECP2 could provide a viable treatment option for RTT patients (Vacca et al. 2016). This is an attractive strategy for RTT because patient cells could use their own regulatory functions to control MECP2 expression. However, sex chromosome dosage is an important developmental process and reactivating the second X chromosome may lead to a pathological level of expression for other X chromosome loci. Ideally, these strategies will aim to target the MECP2 gene or its local vicinity alone. Additionally, the Xi can be difficult to reactivate due to multiple mechanisms of epigenetic silencing and attempting to disengage these processes could disrupt epigenetic patterns throughout the genome which could cause adverse long-term effects. Finally, because RTT patients are heterozygous mosaics for MECP2 mutation, approximately half of their cells already express a normal copy of MECP2. Thus, Xi reactivation will lead to 2x the normal amount of MECP2 in these cells. In patients carrying mutations that only reduce efficiency of MECP2 rather than destroy its function, Xi reactivation will lead to anywhere from 1 to 2x the normal amount of MECP2 in some cells. This could lead to symptoms associated with overexpression of MECP2. Therefore, Xi reactivation will need to be adapted to target only MECP2 or its local vicinity, and may only be a practical treatment option for patients who have loss-of-function mutations in MECP2.
One group reactivated the Xi using an antisense oligonucleotide to 5-aza-2′-deoxycytidine (Aza), a DNA methylation inhibitor, in a mouse with an Xist deletion on one chromosome and Mecp2-GFP on the other. This ensured that Mecp2-GFP was silenced in almost all cells. Treatment resulted in a great increase in GFP expression, indicating that Mecp2 could be targeted by Xi reactivation tools (Carrette et al. 2017). Recently, a heterozygous female mouse was engineered with a mutation of Tsix on one chromosome and a null mutation of Mecp2 on the other, directing the preferential expression of the chromosome with the mutated Mecp2 (Carrette et al. 2018). This strategy achieved a Mecp2-null female mouse with all cells expressing the same Xa. Additionally, this group found that while Mecp2-null females had a shortened lifespan, a 5% increase in MECP2 levels increased the lifespan by 50%, and a 10–20% increase in MECP2 restored a normal lifespan (Carrette et al. 2018). Thus, small increases in MECP2 can have great positive impacts on life expectancy. However, as a global inhibitor of methylation, Aza is toxic when administered over a long time course. Ongoing screens aim to identify new molecules to induce Xi reactivation that can be further tested and optimized for treatment (Bhatnagar et al. 2014; Minkovsky et al. 2015; Sripathy et al. 2017). Importantly, the unique mice designed in these studies will provide an excellent avenue to test candidate Xi-reactivating drugs specifically for the treatment of RTT.
Gene therapy
A final potential avenue of treatment for RTT involves introducing a normal copy of MECP2 into cells by gene therapy. Gene therapy is a promising approach for the treatment of many disorders and has been successful in reversing symptoms in mouse models of CF, hemophilia, Hunter syndrome, diabetes, obesity, ALS, and more. Recently, the first targeted gene therapy treatment was approved in North America to treat patients with Lever’s congenital amaurosis, a rare inherited eye disease (Kumaran et al. 2018).
To treat RTT, gene therapy approaches must utilize an appropriate vector able to cross the BBB and transduce many cells in the CNS, and able to maintain stable long-term expression of the exogenously derived MECP2. Additionally, strategies must be developed to avoid transgene repression and avoid overexpression of MECP2. Like Xi reactivation, gene therapy targets all cells regardless of MECP2 mutation status. Thus, cells expressing normal MECP2 will have 2x the normal level of the protein leading to potential toxic effects of MECP2 overexpression. An additional concern of gene therapy is that high viral titres are often needed to infect a large proportion of cells, but cells that inadvertently receive more than one viral particle, and hence more than one copy of MECP2, would be overburdened. Strategies to circumvent these issues could supply a factor such as an siRNA to suppress endogenous MECP2 so that only the transgenic MECP2 is expressed (Gadalla et al. 2011). However, this would require the transgenic MECP2 to possess a seamless promoter to completely mimic endogenous gene expression. MicroRNAS (miRNAs) have been used to control exogenous transgene expression by mediating the degradation of transgene mRNAs in a tissue-specific manner and may be beneficial in RTT gene therapy approaches to limit off-target toxicity (Geisler and Fechner 2016).
Recombinant adeno-associated virus (AAV) vectors have been used in preclinical gene therapy studies for their ability to cross the BBB, infect neurons, and mediate stable long-term expression of the transgene without inflammation or toxicity (Gonçalves 2005; Foust et al. 2009). AAV has a 4.7-kb ssDNA genome from which 4.4 kb of the viral DNA can be removed and replaced with a human transgene. Self-complementary (sc) AAV vectors have a 10–100-fold higher transduction efficiency, but their drawback is that their packaging capacity is cut in half to only 2.2 kb, making it difficult to package large genes (McCarty et al. 2001). When AAV9-MECP2 under the promoter of chicken β-actin was injected into the brains of neonatal Mecp2-null mice, transduction efficiency varied between brain regions from 7 to 42% of cells infected, with the highest infection rate in the hypothalamus and lowest in the striatum (Gadalla et al. 2013). However, this low efficiency of infection was sufficient to increase lifespan of male mice to 16.6 weeks and improve motor impairments, but it did not have any effect on respiratory symptoms. In contrast, scAAV9-MECP2 under a truncated Mecp2 promoter injected into mice systemically had a very low transduction efficiency in the brain of 2–4%. Despite this, mice survived to 15 weeks, indicating that low levels of MECP2 re-expression in the brain and/or re-expression in non-CNS tissues could modestly improve lifespan. Notably, AAV9 preferentially targeted the liver and spleen, with some cells in these tissues receiving ten copies of the vector and causing liver damage. Therefore, future studies must increase the specificity of AAV9 therapy before moving to the clinic.
A subsequent study delivered scAAV9-MECP2 under a truncated Mecp2 promoter systemically and found a range of transduction efficiencies in the brain from 10% in the cerebellum to 25% in the cortex and brainstem (Garg et al. 2013). This produced some behavioral improvements but did not rescue breathing symptoms. A second-generation AAV9 vector with a modified 3′UTR and a panel of miRNA binding sites was developed with the goal of biasing transgene expression away from the liver (Sinnett et al. 2017). This vector, injected in the cisterna magna, was better tolerated and improved the lifespan of Mecp2 mutant mice but could not improve behavioral traits without being used at very high doses. However, direct cerebroventricular injection of this vector into neonatal Mecp2-null mice resulted in a high brain transduction efficiency, increased survival, and the amelioration of RTT-like phenotypes, highlighting the importance of endogenous regulatory elements in the gene expression cassette (Gadalla et al. 2017).
Finally, a recent study designed a minimal-MECP2 protein lacking all amino acids except those encoding two functional domains: the methyl-binding domain, and the NCOR-interaction domain (Tillotson et al. 2017). When neonatal mice were injected intracranially with a scAAV9 vector encoding this minimal protein, they showed improved phenotypes and survival in the absence of toxic effects, indicating that the primary role of MECP2 is to physically connect the NCOR-containing complex to DNA. This study provides a new avenue to pursue in gene therapy studies as a functional minimal MECP2 protein creates room for additional regulatory sequences to be packaged into the limited capacity of scAAV9 vectors. Importantly, this would allow for more precise temporal control of Mecp2 expression. Future studies should aim to introduce additional regulatory elements into the gene therapy vector while also controlling for timing of treatment to best represent therapeutic utility in human patients.
An additional obstacle of gene therapy treatment for any disorder involves scaling the dosage for humans. Due to their small size, mice cannot reliably inform effective dosages for clinical applications. Therefore, gene therapy treatment is being tested in other animal models including canines and non-human primates. The brains of non-human primates are similar to humans with regard to neural circuitry, physiology, and behavioral characteristics, making them an ideal model to test gene therapy for neurological diseases (Gopinath et al. 2015). However, studies in larger models are expensive and require longer experimental periods. Importantly, primate models lacking MECP2 have been generated which can be utilized to accelerate gene therapy treatment for RTT (Liu et al. 2014; Chen et al. 2017). Recently, systemic administration of high-dose AAV9 was found to cause severe liver and neuronal toxicity in three non-human primates indicating that efforts in dosage optimization will be imperative (Hinderer et al. 2018).
The need for precision medicine for a monogenic disease
Rare diseases have limited statistical power in clinical trials making animal models our greatest opportunity at developing, adapting, and enhancing therapeutics to be clinical trial-ready. Importantly, treatments that are rigorously tested and streamlined for efficiency before advancing to human trials achieve higher levels of success than those for which multiple preclinical trials are not conducted (Morgan et al. 2018). To do this, studies should address age- and method-dependent effects of treatments. For example, in the case of gene therapy, treatment is often more effective in neonatal mice compared to adult mice, or through direct brain injection compared to systemic administration. It is likely that most treatment options will be more effective if delivered to patients early, but as this is not always feasible, preliminary studies should aim to recapitulate treatment methods available in human patients. This would involve testing treatments in Mecp2 mutant mice after symptom onset. Additionally, identifying and publishing negative effects of potential therapeutics is beneficial as they inform about off-target effects and toxicity. This ultimately provides better criteria to assess the safety of treatments that will lead to the development of more effective therapeutic strategies.
While many potential treatments for RTT have been identified in model organisms, many have not shown extreme efficacy in human trials. Clinical trial design for rare diseases is especially important due to the sparsity of patients. In diseases like RTT where there is substantial variability in clinical presentation, patient stratification should be considered to reduce the variability in the sample and maximize the potential to detect efficacy (Modi and Sahin 2018). Natural history studies assessing genotype–phenotype correlations, symptom progression, and biomarkers of disease may provide tools to identify patients likely to respond to new treatments. For example, there may be critical windows in which specific interventions will have a maximal effect. Additionally, clinical trials should be designed with enough statistical power for the endpoints being tested, with consideration given to placebo effects.
Given the drastic variation in phenotype presentation, it is likely that every RTT patient is unique and will not respond to the same treatment. In this respect, precision medicine for RTT is warranted (Fig. 4). Precision medicine approaches are taken because patients carrying different mutations do not always respond to treatments in the same way. Ideally, RTT patient age, their specific mutation and individual level of XCI skewing should be taken into consideration when developing a personalized treatment regimen. It is likely that some patients will make excellent candidates for gene therapy, mutation read-through, or reactivation of the silent X chromosome, while others may not benefit greatly from any of these treatments.
Fig. 4.
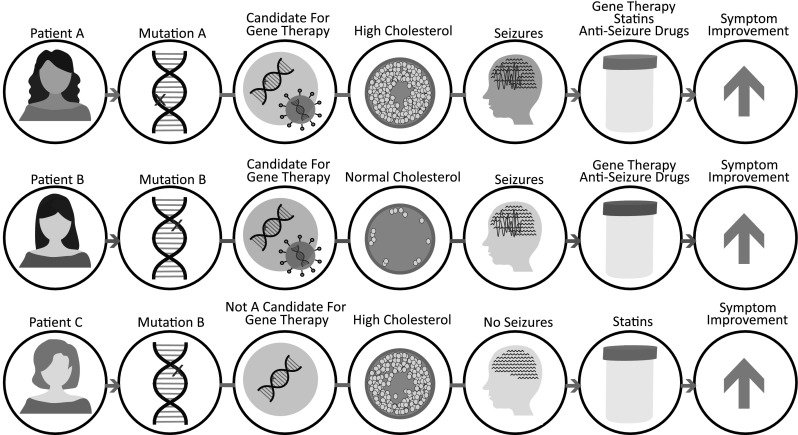
Precision medicine for RTT. Different RTT patients will likely benefit from different combinations of treatment. When treating patients for RTT, their mutation status and XCI skewing should be taken into consideration. It is likely some patients will not be ideal candidates for gene therapy. Biomarkers, such as serum cholesterol, can be used to determine which patients will benefit from pharmacological intervention, such as statins. Management of other symptoms (seizures, scoliosis, lung infection, etc.) should also be considered. Together, RTT patients should receive individualized treatment to maximize their symptom improvement
As CNS-targeted gene therapy becomes a more realistic therapeutic approach, peripheral deficiency of MECP2 also needs to be considered. Loss of MECP2 in non-CNS tissues is associated with cardiac defects (Hara et al. 2015), decreased bone density (Haas et al. 1997), urological dysfunction (Ward et al. 2016), elevated lipid metabolism (Justice et al. 2013; Segatto et al. 2014), and increased oxidative stress (Leoncini et al. 2011). It is likely that when MECP2 is genetically treated exclusively in the brain, these peripheral symptoms will persist. For this reason, biomarkers of non-CNS perturbations in RTT are needed to ensure adequate treatment. For example, high serum cholesterol or increased oxidative stress can be used as an indicator for statin or triheptanoin treatment, respectively. As more perturbed pathways downstream of Mecp2 mutation are discovered, it is likely that the list of biomarkers for pharmacological intervention will increase. Importantly, patients should continue to be assessed for seizures, scoliosis, lung infection, and other features of RTT that present differently in each patient. Ultimately, because small increases in MECP2 can lead to drastic improvements (Carrette et al. 2018), it is probable that a combination of gene therapy paired with personalized pharmacological treatments will provide the best efficacy to treat most RTT patients. Treating each patient individually is expensive which can reduce profit for drug companies. However, the NIH instituted an orphan drug program to provide incentives for pharma companies to develop treatments for rare diseases such as RTT (Engstrom 1985).
Although it is a rare disorder, RTT has served as a paradigm for the understanding and developing treatments for rare diseases because of the early development of mouse models, intensive mechanistic studies, a NIH-funded natural history study (NCT02738281), and funding by focused patient groups (International Rett Syndrome Foundation and Rett Syndrome Research Trust). Because it is a complex disorder involving many organ systems, it is likely that recovery of RTT patients will involve a combination of treatments. Model organisms will continue to serve as an indispensable tool in accelerating drug discovery and adapting therapeutics, leading to better treatment strategies for many rare disorders.
Acknowledgements
We thank all the girls with Rett syndrome and their families who provide the motivation for our group to try to alleviate symptoms. We thank Julie Ruston, Adebola Enikanolaiye, and Rebekah Tillotson for their critical comments on the manuscript.
Funding
MJJ and NV were supported by grants from the Canadian Institute for Health Research (CIHR) and the Angel Ava Fund while writing this review.
Compliance with ethical standards
Conflict of interest
All authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abdala APL, Dutschmann M, Bissonnette JM, Paton JFR. Correction of respiratory disorders in a mouse model of Rett syndrome. Proc Natl Acad Sci. 2010 doi: 10.1073/pnas.1012104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Lioy DT, Garg SK, et al. Effect of sarizotan, a 5-HT1aand D2-like receptor agonist, on respiration in three mouse models of rett syndrome. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2013-0372OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Toward MA, Dutschmann M, et al. Deficiency of GABAergic synaptic inhibition in the Kölliker-Fuse area underlies respiratory dysrhythmia in a mouse model of Rett syndrome. J Physiol. 2016;594:223–237. doi: 10.1113/JP270966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpoz AR, Ergul N, Oncag O. Bruxism in Rett syndrome: a case report. J Clin Pediatr Dent. 1999;23:161–163. [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to Induce dendritic spine formation. J Neurosci. 2007;27:5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Anderson A, Wong K, Jacoby P, et al. Twenty years of surveillance in Rett syndrome: what does this tell us? Orphanet J Rare Dis. 2014;9:87. doi: 10.1186/1750-1172-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DD. The neuropathology of the Rett syndrome. Brain Dev. 1992;14:S89–S98. [PubMed] [Google Scholar]
- Artuso R, Papa FT, Grillo E, et al. Investigation of modifier genes within copy number variations in Rett syndrome. J Hum Genet. 2011;56:508–515. doi: 10.1038/jhg.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AM, Batchelor DC, Thomas GB, et al. Central penetration and stability of N-terminal tripeptide of insulin-like growth factor-I, glycine-proline-glutamate in adult rat. Neuropeptides. 2005;39:81–87. doi: 10.1016/j.npep.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non–cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Levin R, Shah H, et al. Cholesterol levels in Fragile X syndrome. Am J Med Genet A. 2015;167:379–384. doi: 10.1002/ajmg.a.36850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Zhu X, Ou J, et al. Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc Natl Acad Sci. 2014;111:12591–12598. doi: 10.1073/pnas.1413620111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Liao G. Cholesterol in niemann–pick type C disease. Subcell Biochem. 2010;51:319–335. doi: 10.1007/978-90-481-8622-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianciardi L, Fichera M, Failla P, et al. MECP2 missense mutations outside the canonical MBD and TRD domains in males with intellectual disability. J Hum Genet. 2016;61:95–101. doi: 10.1038/jhg.2015.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, Carrié A, de Roux N, et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9:1377–1384. doi: 10.1093/hmg/9.9.1377. [DOI] [PubMed] [Google Scholar]
- Bittolo T, Raminelli CA, Deiana C, et al. Pharmacological treatment with mirtazapine rescues cortical atrophy and respiratory deficits in MeCP2 null mice. Sci Rep. 2016 doi: 10.1038/srep19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodda C, Tantra M, Mollajew R, et al. Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am J Pathol. 2013;183:195–210. doi: 10.1016/j.ajpath.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Braun S, Kottwitz D, Nuber UA. Pharmacological interference with the glucocorticoid system influences symptoms and lifespan in a mouse model of Rett syndrome. Hum Mol Genet. 2012 doi: 10.1093/hmg/ddr602. [DOI] [PubMed] [Google Scholar]
- Brendel C, Belakhov V, Werner H, et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med. 2011;89:389–398. doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Selfridge J, Lagger S, et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet. 2016;25:558–570. doi: 10.1093/hmg/ddv496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchovecky CM, Turley SD, Brown HM, et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet. 2013;45:1013–1020. doi: 10.1038/ng.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrette LLG, Wang C-Y, Wei C, et al. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc Natl Acad Sci. 2017 doi: 10.1073/pnas.1715124115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrette LLG, Blum R, Ma W, et al. Tsix–Mecp2 female mouse model for Rett syndrome reveals that low-level MECP2 expression extends life and improves neuromotor function. Proc Natl Acad Sci. 2018 doi: 10.1073/pnas.1800931115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J, Garcia RI, Kwok S, et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett syndrome. Proc Natl Acad Sci. 2014;111:9941–9946. doi: 10.1073/pnas.1311685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H. DNA methylation and gene activity. Cell. 1988;53:3–4. doi: 10.1016/0092-8674(88)90479-5. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SYY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler SP, Guschin D, Landsberger N, Wolffe AP. The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry. 1999;38:7008–7018. doi: 10.1021/bi990224y. [DOI] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, et al. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao H-TT, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu J, Niu Y, et al. Modeling Rett syndrome ising TALEN-Edited MECP2 mutant cynomolgus monkeys. Cell. 2017;169:945–955.e10. doi: 10.1016/j.cell.2017.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin EWM, Lim WM, Ma D, et al. Choline rescues behavioural deficits in a mouse model of Rett syndrome by modulating neuronal plasticity. Mol Neurobiol. 2018;15:1–5. doi: 10.1007/s12035-018-1345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51:152–158. doi: 10.1136/jmedgenet-2013-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- De Felice C, Ciccoli L, Leoncini S, et al. Systemic oxidative stress in classic Rett syndrome. Free Radic Biol Med. 2009;47:440–448. doi: 10.1016/j.freeradbiomed.2009.05.016. [DOI] [PubMed] [Google Scholar]
- De Filippis B, Fabbri A, Simone D, et al. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of rett syndrome. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis B, Ricceri L, Fuso A, Laviola G. Neonatal exposure to low dose corticosterone persistently modulates hippocampal mineralocorticoid receptor expression and improves locomotor/exploratory behaviour in a mouse model of Rett syndrome. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2012.05.048. [DOI] [PubMed] [Google Scholar]
- De Felice C, Della Ragione F, Signorini C, et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol Dis. 2014;68:66–77. doi: 10.1016/j.nbd.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis B, Nativio P, Fabbri A, et al. Pharmacological stimulation of the brain serotonin receptor 7 as a novel therapeutic approach for rett syndrome. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis B, Valenti D, Chiodi V, et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur Neuropsychopharmacol. 2015 doi: 10.1016/j.euroneuro.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Deacon RMJ, Glass L, Snape M, et al. NNZ-2566, a novel analog of (1–3) IGF-1, as a potential therapeutic agent for Fragile X syndrome. NeuroMol Med. 2015;17:71–82. doi: 10.1007/s12017-015-8341-2. [DOI] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr ARW, et al. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21:1074–1086. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng V, Matagne V, Banine F, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- Deogracias R, Yazdani M, Dekkers MP, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012 doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti MT, Manneschi L, Malandrini A, et al. Mitochondrial dysfunction in Rett syndrome an ultrastructural and biochemical study. Brain Dev. 1993;15:103–106. doi: 10.1016/0387-7604(93)90045-A. [DOI] [PubMed] [Google Scholar]
- Du M, Liu X, Welch EM, et al. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Gabel HW, Robinson ND, et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeg-Olofsson O, Al-Zuhair AG, Teebi AS, Al-Essa MM. Abnormal mitochondria in the Rett syndrome. Brain Dev. 1988;10:260–262. doi: 10.1016/S0387-7604(88)80010-X. [DOI] [PubMed] [Google Scholar]
- Elefant C, Wigram T. Learning ability in children with Rett syndrome. Brain Dev. 2005;27:S97–S101. doi: 10.1016/j.braindev.2005.03.020. [DOI] [PubMed] [Google Scholar]
- El-Khoury R, Panayotis N, Matagne V, et al. GABA and glutamate pathways are spatially and developmentally affected in the brain of Mecp2-deficient mice. PLoS One. 2014 doi: 10.1371/journal.pone.0092169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom LO. The National Institutes of Health Small Business Innovation Research (SBIR) Program: opportunities for orphan drug development. Prog Clin Biol Res. 1985;192:25–30. [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KKE, Bailey MES, Cobb SR. MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J. 2011;439:1–14. doi: 10.1042/BJ20110648. [DOI] [PubMed] [Google Scholar]
- Gadalla KK, Bailey ME, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21:18–30. doi: 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KKE, Vudhironarit T, Hector RD, et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther-Methods Clin Dev. 2017 doi: 10.1016/j.omtm.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandaglia A, Brivio E, Carli S, et al. A novel Mecp2 Y120D knock-in model displays similar behavioral traits but distinct molecular features compared to the Mecp2-null mouse implying precision medicine for the treatment of Rett syndrome. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1412-2. [DOI] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J Exp Med. 2016 doi: 10.5493/wjem.v6.i2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Allen M, Zhang L, et al. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat Neurosci. 2011;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Zhou Z, Joe The neural circuit basis of Rett syndrome. Front Biol (Beijing) 2012;7:428–435. doi: 10.1007/s11515-012-1248-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Senter RK, Rook JM, et al. MGlu5positive allosteric modulation normalizes synaptic plasticity defects and motor phenotypes in a mouse model of Rett syndrome. Hum Mol Genet doi. 2016 doi: 10.1093/hmg/ddw074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Senter RK, Fisher NM, et al. MGlu7potentiation rescues cognitive, social, and respiratory phenotypes in a mouse model of Rett syndrome. Sci Transl Med. 2017 doi: 10.1126/scitranslmed.aai7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold WA, Williamson SL, Kaur S, et al. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): implications for the disease phenotype. Mitochondrion. 2014;15:10–17. doi: 10.1016/j.mito.2014.02.012. [DOI] [PubMed] [Google Scholar]
- Gonçalves MA, et al. Adeno-associated virus: from defective virus to effective vector. Virol J. 2005;2:43–43. doi: 10.1186/1743-422X-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath C, Nathar T, Ghosh A, et al. Contemporary animal models for human gene therapy applications. Curr Gene Ther. 2015 doi: 10.2174/1566523215666150929110424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großer E, Hirt U, Janc OA, et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol Dis. 2012;48:102–114. doi: 10.1016/j.nbd.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Guerin K, Gregory-Evans CY, Hodges MD, et al. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp Eye Res. 2008;87:197–207. doi: 10.1016/j.exer.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, et al. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Haas RH. The history and challenge of Rett syndrome. J Child Neurol. 1988;3:S3–S5. doi: 10.1177/0883073888003001S02. [DOI] [PubMed] [Google Scholar]
- Haas RH, Nasirian F, Hua X, et al. Oxidative metabolism in Rett syndrome: 2—biochemical and molecular studies. Neuropediatrics. 1995;26:95–99. doi: 10.1055/s-2007-979735. [DOI] [PubMed] [Google Scholar]
- Haas RH, Dixon SD, Sartoris DJ, Hannessy MJ. Osteopenia in Rett syndrome. J Pediatr. 1997;131:771–774. doi: 10.1016/S0022-3476(97)70113-6. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Witt-Engerström I. Rett syndrome: a suggested staging system for describing impairment profile with increasing age towards adolescence. Am J Med Genet. 1986;1:47–59. doi: 10.1002/ajmg.1320250506. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Hao S, Tang B, Wu Z, et al. Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature. 2015 doi: 10.1038/nature15694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Takahashi T, Mitsumasu C, et al. Disturbance of cardiac gene expression and cardiomyocyte structure predisposes Mecp2-null mice to arrhythmias. Sci Rep. 2015;5:1–17. doi: 10.1038/srep11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an AAV vector expressing human SMN. Hum Gene Ther. 2018 doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Makita Y, Ogawa A, et al. Brain and development. Amsterdam: Elsevier; 2001. The role of different X-inactivation pattern on the variable clinical phenotype with Rett syndrome. [DOI] [PubMed] [Google Scholar]
- Itoh M, Ide S, Takashima S, et al. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J Neuropathol Exp Neurol. 2007;66:117–123. doi: 10.1097/nen.0b013e3180302078. [DOI] [PubMed] [Google Scholar]
- Itoh M, Tahimic CGT, Ide S, et al. Methyl CpG-binding protein isoform MeCP2-e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. J Biol Chem. 2012;287:13859–13867. doi: 10.1074/jbc.M111.309864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janc OA, Hüser MA, Dietrich K, et al. Systemic radical scavenger treatment of a mouse model of Rett syndrome: merits and limitations of the vitamin E derivative trolox. Front Cell Neurosci. 2016 doi: 10.3389/fncel.2016.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentarra GM, Olfers SL, Rice SG, et al. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010;11:19. doi: 10.1186/1471-2202-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Lam M, Punzo AM, et al. 7,8-dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J Appl Physiol. 2012 doi: 10.1152/japplphysiol.01361.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice MJ, Buchovecky CM, Kyle SM, Djukic A. A role for metabolism in Rett syndrome pathogenesis: new clinical findings and potential treatment targets. Rare Dis. 2013;1:e27265. doi: 10.4161/rdis.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM, Menniti FS, Mather RJ. N-methyl-D-aspartate receptors, ketamine, and Rett syndrome: something special on the road to treatments? Biol Psychiatry. 2016;79:710–712. doi: 10.1016/j.biopsych.2016.03.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annu Rev Genom Hum Genet. 2014;15:371–394. doi: 10.1146/annurev-genom-091212-153527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja OS, Ho E, Barnes KV, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci. 2014;111:4596–4601. doi: 10.1073/pnas.1311141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Knudsen GPS, Neilson TCS, Pedersen J, et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006;14:1189–1194. doi: 10.1038/sj.ejhg.5201682. [DOI] [PubMed] [Google Scholar]
- Koerner MV, FitzPatrick L, Selfridge J, et al. Toxicity of overexpressed MeCP2 is independent of HDAC3 activity. Genes Dev. 2018;32:1514–1524. doi: 10.1101/gad.320325.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Gray LJ, Pelka GJ, et al. Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome—Mecp2 gene dosage effects and BDNF expression. Eur J Neurosci. 2008 doi: 10.1111/j.1460-9568.2008.06305.x. [DOI] [PubMed] [Google Scholar]
- Kondo MA, Gray LJ, Pelka GJ, et al. Affective dysfunction in a mouse model of Rett syndrome: therapeutic effects of environmental stimulation and physical activity. Dev Neurobiol. 2016 doi: 10.1002/dneu.22308. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Paterson A, Curtis J, et al. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Krishnan K, Connors CR, et al. PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J Clin Invest. 2015;125:3163–3177. doi: 10.1172/JCI80323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 2012;32:13860–13872. doi: 10.1523/JNEUROSCI.2159-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Lang M, Adams IT, et al. A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech. 2014;7:1047–1055. doi: 10.1242/dmm.016030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Jaryal A, Gulati S, et al. Cardiovascular autonomic dysfunction in children and adolescents with Rett syndrome. Pediatr Neurol. 2017;70:61–66. doi: 10.1016/j.pediatrneurol.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Kumaran N, Smith AJ, Michaelides M, et al. Gene therapy for leber congenital amaurosis. Expert Rev Ophthalmol. 2018;13:11–15. doi: 10.1080/17469899.2018.1429916. [DOI] [Google Scholar]
- Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Kyle SM, Saha PK, Brown HM, et al. MeCP2 co-ordinates liver lipid metabolism with the NCoR1/HDAC3 corepressor complex. Hum Mol Genet. 2016;25:3029–3041. doi: 10.1093/hmg/ddw156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle SM, Vashi N, Justice MJ. Rett syndrome: a neurological disorder with metabolic components. Open Biol. 2018;8:170216. doi: 10.1098/rsob.170216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Strazielle C. Brain regions and genes affecting limb-clasping responses. Brain Res. Rev. 2011;67:252259. doi: 10.1016/j.brainresrev.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Laurvick CL, de Klerk N, Bower C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Lawson-Yuen A, Liu D, Han L, et al. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007;1180:1–6. doi: 10.1016/j.brainres.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoncini S, De Felice C, Signorini C, et al. Oxidative stress in Rett syndrome: natural history, genotype, and variants. Redox Rep. 2011;16:145–153. doi: 10.1179/1351000211Y.0000000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt ES, Hunnicutt BJ, Knopp SJ, et al. A selective 5-HT 1a receptor agonist improves respiration in a mouse model of Rett syndrome. J Appl Physiol. 2013 doi: 10.1152/japplphysiol.00889.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-O. [DOI] [PubMed] [Google Scholar]
- Li W, Bellot-Saez A, Phillips ML, et al. A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech. 2017 doi: 10.1242/dmm.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligsay A, Hagerman RJ. Review of targeted treatments in fragile X syndrome. Intractable Rare Dis Res. 2016;5:158–167. doi: 10.5582/irdr.2016.01045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde L, Boelz S, Nissim-Rafinia M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007 doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Zhang L, Chen J, et al. Association of MeCP2 (rs2075596, rs2239464) genetic polymorphisms with systemic lupus erythematosus: a meta-analysis. Lupus. 2013;22:908–918. doi: 10.1177/0961203313496340. [DOI] [PubMed] [Google Scholar]
- Liu H, Chen Y, Niu Y, et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell. 2014;14:323–328. doi: 10.1016/j.stem.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetti G, Angelucci A, Morando L, et al. Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 Null mice. Biol Psychiatry. 2010 doi: 10.1016/j.biopsych.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet. 2015;16:261–275. doi: 10.1038/nrg3897. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16:898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini J, Dubus JC, Jouve E, et al. Effect of desipramine on patients with breathing disorders in RETT syndrome. Ann Clin Transl Neurol. 2018 doi: 10.1002/acn3.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantis JG, Fritz CL, Marsh J, et al. Improvement in motor and exploratory behavior in Rett syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 2009 doi: 10.1016/j.yebeh.2009.02.038. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- McCauley MD, Wang T, Mike E, et al. Pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: implication for therapy in Rett syndrome. Sci Transl Med. 2011;3:113ra125–113ra125. doi: 10.1126/scitranslmed.3002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1489–1498. doi: 10.1016/S0140-6736(17)31611-2. [DOI] [PubMed] [Google Scholar]
- Mellios N, Woodson J, Garcia RI, et al. β2-Adrenergic receptor agonist ameliorates phenotypes and corrects microRNA-mediated IGF1 deficits in a mouse model of Rett syndrome. Proc Natl Acad Sci. 2014 doi: 10.1073/pnas.1309426111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkovsky A, Sahakyan A, Bonora G, et al. A high-throughput screen of inactive X chromosome reactivation identifies the enhancement of DNA demethylation by 5-aza-2′-dC upon inhibition of ribonucleotide reductase. Epigenet Chromatin. 2015 doi: 10.1186/s13072-015-0034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, DeLonlay P, Touati G, et al. Pyruvate carboxylase deficiency: Clinical and biochemical response to anaplerotic diet therapy. Mol Genet Metab. 2005;84:305–312. doi: 10.1016/j.ymgme.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Modi ME, Sahin M. The way forward for mechanism-based therapeutics in genetically defined neurodevelopmental disorders. Clin Pharmacol Ther. 2018;104:60306. doi: 10.1002/cpt.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006 doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan P, Brown DG, Lennard S, et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov. 2018;17:167. doi: 10.1038/nrd.2017.244. [DOI] [PubMed] [Google Scholar]
- Motil KJ, Caeg E, Barrish JO, et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 2012;55:292–298. doi: 10.1097/MPG.0b013e31824b6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount RH, Charman T, Hastings RP, et al. The Rett syndrome behaviour questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry Allied Discip. 2002 doi: 10.1111/1469-7610.00236. [DOI] [PubMed] [Google Scholar]
- Na ES, De Jesús-Cortés H, Martinez-Rivera A, et al. D-cycloserine improves synaptic transmission in an animal mode of Rett syndrome. PLoS ONE. 2017 doi: 10.1371/journal.pone.0183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag N, Berger-Sweeney JE. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Nag N, Moriuchi JM, Peitzman CGK, et al. Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp21lox mice. Behav Brain Res. 2009 doi: 10.1016/j.bbr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Nagel-Wolfrum K, Möller F, Penner I, et al. Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs) BioDrugs. 2016;30:49–74. doi: 10.1007/s40259-016-0157-6. [DOI] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;6:198–199. doi: 10.1016/S0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/S0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nance E, Kambhampati SP, Smith ES, et al. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J Neuroinflamm. 2017 doi: 10.1186/s12974-017-1004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev. 2014;46:175–186. doi: 10.1016/j.neubiorev.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary HM, Kaufmann WE, Barnes KV, et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol. 2018;5:323–332. doi: 10.1002/acn3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, et al. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007 doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Saito Y, Ueda R, et al. Effect of serotonin 1A agonists and selective serotonin reuptake inhibitors on behavioral and nighttime respiratory symptoms in Rett syndrome. Pediatr Neurol. 2016 doi: 10.1016/j.pediatrneurol.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Orefice LLL, Zimmerman ALL, Chirila AMM, et al. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell. 2016 doi: 10.1016/j.cell.2016.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panighini A, Duranti E, Santini F, et al. Vascular dysfunction in a mouse model of Rett syndrome and effects of curcumin treatment. PLoS ONE. 2013 doi: 10.1371/journal.pone.0064863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MJ, Aja S, Li Q, et al. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE. 2014 doi: 10.1371/journal.pone.0109527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson DS, Thompson EG, Belliveau RA, et al. Serotonin transporter abnormality in the dorsal motor nucleus of the vagus in rett syndrome: potential implications for clinical autonomic dysfunction. J Neuropathol Exp Neurol. 2005;64:1018–1027. doi: 10.1097/01.jnen.0000187054.59018.f2. [DOI] [PubMed] [Google Scholar]
- Patrizi A, Picard N, Simon AJ, et al. Chronic administration of the N-methyl-D-aspartate receptor antagonist ketamine improves Rett syndrome phenotype. Biol Psychiatry. 2016;79:755–764. doi: 10.1016/j.biopsych.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelka GJ, Watson CM, Radziewic T, et al. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006 doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- Pitcher MR, Ward CS, Arvide EM, et al. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MR, Herrera JA, Buffington SA, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. 2015;24:2662–2672. doi: 10.1093/hmg/ddv030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo L, Jensen M, Polyak A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. 2018 doi: 10.1038/s41436-018-0266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu AC, Sidorova E, Zhang G, Eubanks JH. Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. J Neurosci Res. 2010;88:2316–2324. doi: 10.1002/jnr.22409. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Ramirez JM, Ward CS, Neul JL. Breathing challenges in Rett Syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 2013;189:280–287. doi: 10.1016/j.resp.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Boxer LD, Hrvatin S, et al. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat Neurosci. 2018;21:1670–1679. doi: 10.1038/s41593-018-0270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr. 1966;1946:723–726. [PubMed] [Google Scholar]
- Ricceri L, De Filippis B, Fuso A, Laviola G. Cholinergic hypofunction in MeCP2-308 mice: beneficial neurobehavioural effects of neonatal choline supplementation. Behav Brain Res. 2011 doi: 10.1016/j.bbr.2011.03.051. [DOI] [PubMed] [Google Scholar]
- Roe CR, Sweetman L, Roe DS, et al. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Investig. 2002;110:259–269. doi: 10.1172/JCI0215311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux JC, Dura E, Moncla A, et al. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007 doi: 10.1111/j.1460-9568.2007.05466.x. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, et al. A partial loss of function allele of Methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, McGraw CM, et al. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet. 2012 doi: 10.1038/ng.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos M, Summavielle T, Teixeira-Castro A, et al. Monoamine deficits in the brain of methyl-CpG binding protein 2 null mice suggest the involvement of the cerebral cortex in early stages of Rett syndrome. Neuroscience. 2010 doi: 10.1016/j.neuroscience.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Schaevitz LR, Nicolai R, Lopez CM, et al. Acetyl-L-carnitine improves behavior and dendritic morphology in a mouse model of Rett syndrome. PLoS ONE. 2012 doi: 10.1371/journal.pone.0051586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaevitz LR, Gómez NB, Zhen DP, Berger-Sweeney JE. MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Genes, Brain Behav. 2013 doi: 10.1111/gbb.12070. [DOI] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, et al. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. 2012 doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Investig. 2003;111:303312. doi: 10.1172/JCI200317741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segatto M, Trapani L, Di Tunno I, et al. Cholesterol metabolism is altered in Rett syndrome: a study on plasma and primary cultured fibroblasts derived from patients. PLoS ONE. 2014;9:e104834. doi: 10.1371/journal.pone.0104834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Young JI, Yuva-Paylor LA, et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/S0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Shapiro JR, Bibat G, Hiremath G, et al. Bone mass in Rett syndrome: association with clinical parameters and MECP2 mutations. Pediatr Res. 2010;68:446–451. doi: 10.1203/PDR.0b013e3181f2edd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulyakova N, Andreazza AC, Mills LR, Eubanks JH. Mitochondrial dysfunction in the pathogenesis of Rett syndrome: implications for mitochondria-targeted therapies. Front Cell Neurosci. 2017 doi: 10.3389/fncel.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett SE, Hector RD, Gadalla KKE, et al. Improved MECP2 gene therapy extends the survival of MeCP2-null mice without apparent Toxicity after intracisternal delivery. Mol Ther-Methods Clin Dev. 2017;5:106–115. doi: 10.1016/j.omtm.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripathy S, Leko V, Adrianse RL, et al. Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-β superfamily as a regulator of XIST expression. Proc Natl Acad Sci. 2017;114:1619–1624. doi: 10.1073/pnas.1621356114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna K, De La Caridad O, Petazzi P, et al. Improvement of the rett syndrome phenotype in a mecp2 mouse model upon treatment with levodopa and a dopa-decarboxylase inhibitor. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Hu K, Chang Q, et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci. 2009;106:4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wu H, Coronado AA, et al. Negative allosteric modulation of mGluR5 partially corrects pathophysiology in a mouse model of Rett syndrome. J Neurosci. 2016 doi: 10.1523/JNEUROSCI.0672-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate P, Skarnes W, Bird A. The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat Genet. 1996;12:205–208. doi: 10.1038/ng0296-205. [DOI] [PubMed] [Google Scholar]
- Tillotson R, Selfridge J, Koerner MV, et al. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature. 2017;550:398–401. doi: 10.1038/nature24058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint GS, Irons M, Elias ER, et al. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- Toward MA, Abdala AP, Knopp SJ, et al. Increasing brain serotonin corrects CO2 chemosensitivity in methyl-CpG-binding protein 2 (Mecp2)-deficient mice. Exp Physiol. 2013 doi: 10.1113/expphysiol.2012.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca M, Della Ragione F, Scalabrì F, D’Esposito M. X inactivation and reactivation in X-linked diseases. Semin Cell Dev Biol. 2016;56:78–87. doi: 10.1016/j.semcdb.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viemari J-C. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigli D, Cosentino L, Raggi C, et al. Chronic treatment with the phytocannabinoid Cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology. 2018 doi: 10.1016/j.neuropharm.2018.07.029. [DOI] [PubMed] [Google Scholar]
- Ward CS, Huang TW, Herrera JA, et al. Loss of MeCP2 causes urological dysfunction and contributes to death by kidney failure in mouse models of rett syndrome. PLoS ONE. 2016 doi: 10.1371/journal.pone.0165550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener E, Brendel C, Fischer A, et al. Characterization of the MeCP2R168X knockin mouse model for Rett syndrome. PLoS ONE. 2014;9:e115444. doi: 10.1371/journal.pone.0115444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL. Alterations in dopaminergic function in Rett syndrome. Neuropediatrics. 1995;26:123–125. doi: 10.1055/s-2007-979741. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhong W, Cui N, et al. Characterization of Rett syndrome-like phenotypes in Mecp2-knockout rats. J Neurodev Disord. 2016;8:23. doi: 10.1186/s11689-016-9156-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi CY, Lu Y, Tan YH, et al. Analysis of MECP2 gene copy number in boys with autism. J Child Neurol. 2011;26:570–573. doi: 10.1177/0883073810387138. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Gonzales ML, Aflatooni JO, et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum Mol Genet. 2014;23:2447–2458. doi: 10.1093/hmg/ddt640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D, Nagarajan L, de Klerk N, et al. Sleep problems in Rett syndrome. Brain Dev. 2007;29:609–616. doi: 10.1016/j.braindev.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella S, Mebarek S, Lajard AM, et al. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008 doi: 10.1016/j.resp.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhao Y, Bao X, et al. Familial cases and male cases with MECP2 mutations. Am J Med Genet B. 2017 doi: 10.1002/ajmg.b.32534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Johnson CM, Wu Y, et al. Effects of early-life exposure to THIP on phenotype development in a mouse model of Rett syndrome. J Neurodev Disord. 2016 doi: 10.1186/s11689-016-9169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]