

Abstract
Advances in cardiovascular research have identified oxidative stress as an important pathophysiological pathway in the development and progression of heart failure. Oxidative stress is defined as the imbalance between the production of reactive oxygen species (ROS) and the endogenous antioxidant defence system. Under physiological conditions, small quantities of ROS are produced intracellularly, which function in cell signalling, and can be readily reduced by the antioxidant defence system. However, under pathophysiological conditions, the production of ROS exceeds the buffering capacity of the antioxidant defence system, resulting in cell damage and death. Over the last decades several studies have tried to target oxidative stress with the aim to improve outcome in patients with heart failure, with very limited success. The reasons as to why these studies failed to demonstrate any beneficial effects remain unclear. However, one plausible explanation might be that currently employed strategies, which target oxidative stress by exogenous inhibition of ROS production or supplementation of exogenous antioxidants, are not effective enough, while bolstering the endogenous antioxidant capacity might be a far more potent avenue for therapeutic intervention. In this review, we provide an overview of oxidative stress in the pathophysiology of heart failure and the strategies utilized to date to target this pathway. We provide novel insights into modulation of endogenous antioxidants, which may lead to novel therapeutic strategies to improve outcome in patients with heart failure.
Keywords: Heart failure, Oxidative stress, γ‐Glutamyl cycle, Glutathione, Nicotinamide adenine dinucleotide
Introduction
Oxidative stress is involved in the development and progression of clinical and experimental heart failure.1, 2, 3, 4 Oxidative stress is defined as a dysregulation between the production of reactive oxygen species (ROS) and the endogenous antioxidant defence mechanisms, the so called ‘redox state’. When present in low concentrations, ROS plays a critical function in cell homeostasis. However, excess ROS causes cellular dysfunction, protein and lipid peroxidation, DNA damage, and eventually leads to irreversible cell damage and death. This is also evident in the heart where high sensitive troponin assays have demonstrated an increase in troponin release during heart failure progression, suggesting a gradual loss in cardiomyocytes.5
In the heart, an overabundance of ROS can lead to the development and progression of maladaptive myocardial remodelling and heart failure (Figure 1). ROS directly impairs the electrophysiology and the contractile machinery of cardiomyocytes by modifying proteins central to excitation–contraction coupling, including L‐type calcium channels, sodium channels, potassium channels, and the sodium–calcium exchanger.6 ROS can also alter the activity of the sarcoplasmic reticulum Ca2+‐adenosine triphosphatase (SERCA) as well as reduce myofilament calcium sensitivity.6 Furthermore, ROS induces an energy deficit by affecting the function of proteins involved in energy metabolism.6 Finally, ROS has a pro‐fibrotic function, by inducing cardiac fibroblast proliferation and matrix metalloproteinases resulting in extracellular remodelling.6
Figure 1.
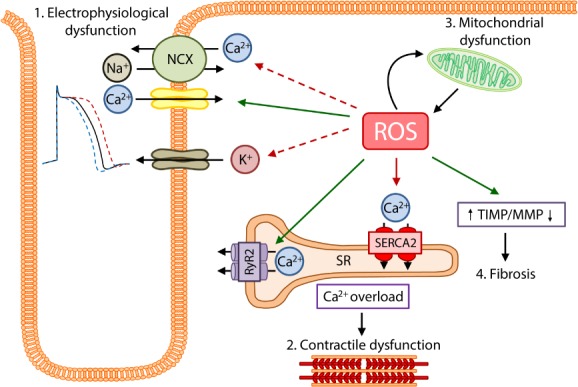
The effects of excessive oxidative stress on the myocardium. As a result of cardiac injury there is a severe accumulation of oxidative stress (reactive oxygen species, ROS), which has several detrimental effects on the myocardium. 1. Cardiomyocyte electrophysiology is severely affected by increased ROS. ROS reverses the function of the Na+/Ca2+ exchanger (NCX), leading to Ca2+ influx and Na+ efflux. ROS also increases the influx of Ca2+ via the L‐type calcium channels. Increased ROS also increases sarcKATP currents, leading to action potential duration shortening, while also reducing KV currents and increasing late sodium currents leading to prolonged action potential durations. 2. Excessive ROS promotes ryanodine receptor 2 (RyR2) activity and inhibits sarcoplasmic reticulum Ca2+‐adenosine triphosphatase 2 (SERCA2) activity, resulting in calcium overload and reduced myofilament calcium sensitivity, eventually leading to contractile dysfunction. 3. The mitochondria react to ischaemic injury by producing increased levels of ROS, however the overabundance of ROS inversely results in further mitochondrial and energy metabolism dysfunction. 4. The increase in ROS is also responsible for increased fibrosis resulting from an increase in tissue inhibitors of metalloproteinases (TIMP) and reduction in matrix metalloproteinase (MMP) expression.
This review will summarize the current knowledge regarding oxidative stress production and the antioxidant defence mechanism in the heart, under physiological and pathophysiological conditions. Furthermore, we recapitulate the current knowledge, failures and successes, regarding the treatment of heart failure by targeting oxidative stress. Finally, we discuss the future potential of targeting endogenous oxidative stress defence mechanisms, to improve clinical outcome in patients with heart failure.
Reactive oxygen species in heart failure and antioxidant mechanisms: a brief summary
Reactive oxygen species production in the heart is primarily achieved by the mitochondria, NADPH oxidases, xanthine oxidase, and uncoupled nitric oxide synthase (NOS) (Figure 2). Under pathological conditions, the electron transport chain of the mitochondria induces the formation of large quantities of superoxide. This increase has been shown to contribute to cardiomyocyte damage and larger myocardial injury after an acute myocardial infarction.7, 8 ROS production is also enhanced due to an increased expression and activity of NADPH oxidase, resulting from several pathological stimuli, including mechanical stretch, angiotensin II, endothelin‐1, and tumour necrosis factor (TNF)‐α.9, 10, 11 Similarly, xanthine oxidase expression and activity is also increased in the failing heart, again leading to an increased production of ROS.11 Finally, as a result of cardiac injury, NOS becomes uncoupled and structurally unstable leading to an increased generation of ROS. In mice, increased generation of ROS leads to left ventricular (LV) dilatation, contractile dysfunction, and LV remodelling.12
Figure 2.
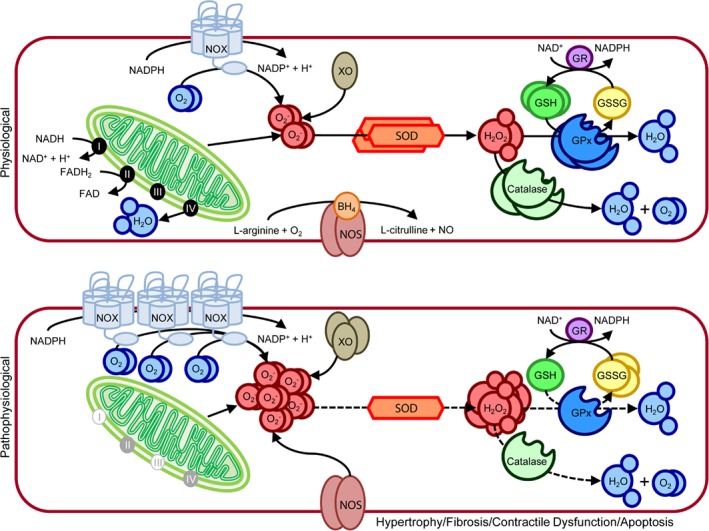
Oxidative stress production and scavenging in cardiomyocytes under physiological and pathophysiological conditions. (Top) Under physiological conditions oxidative stress in the form of reactive oxygen species (ROS) is produced in small quantities by the mitochondrial electron chain, NADPH oxidase (NOX), xanthine oxidase (XO), and nitric oxide synthase (NOS). Mitochondrial respiration converts oxygen to water, resulting in the production of small quantities of superoxide (O2 ‐) as a by‐product. The process starts with electrons derived from NADH2 and FADH2 moving along the respiratory transport chain through a series of cytochrome‐based complexes (I, III, and IV). These complexes eventually transport electrons to molecular oxygen. The high free energy of the electrons is gradually extracted and converted into adenosine triphosphate. NOX is a multimeric complex composed of a plasma membrane spanning cytochrome b558 (NOX2) and cytosolic components (Rac1, p47phox, p67phox, p40phox). Under physiological conditions this complex is in a resting state, producing minimal O2 ‐, by transferring an electron from NADPH to molecular oxygen. XO, which is a cytoplasmic enzyme that catalyzes the oxidation of hypoxanthine and xanthine to uric acid using molecular oxygen as an electron receptor, produces O2 ‐ and hydrogen peroxide (H2O2) in the process. NOS oxidizes the NOS cofactor BH4 utilizing NADPH to generate nitric oxide and L‐citrulline from L‐arginine and oxygen. Superoxide dismutase (SOD) initiates the detoxification of ROS, by scavenging O2 ‐ and converting it to H2O2. Both catalase and glutathione peroxidase (GPx) further detoxify the H2O2 to water and oxygen. GPx utilizes two glutathione (GSH) molecules as electron donors in the reduction of H2O2 to water, producing oxidized glutathione (GSSG) in the process. Once GPx oxidizes GSH to GSSG, GSH reductase (GR) can reduce GSSG back to GSH at the expense of NADPH, forming the GSH redox cycle. The ratio of GSH to GSSG largely determines the intracellular redox potential. (Bottom) Under pathophysiological conditions, oxidative stress production is increased as a result of increased NOX and XO expression, coupled to blockage of the mitochondrial electron chain and uncoupling of NOS. Furthermore, the expression and activity (dotted lines) of SOD, catalase, and GPx are reduced. The levels of GSH are also reduced, while the levels of GSSG are increased. This severe increase in oxidative stress eventually leads to hypertrophy, fibrosis, apoptosis, and contractile dysfunction in the myocardium.
Besides the drastic increase in oxidative stress production, heart failure is also characterized by an exhaustion of the innate antioxidant defence mechanism. In cardiomyocytes, as in most cell types, the major endogenous components of the antioxidant defence mechanism responsible for the inactivation of ROS are superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), nicotinamide adenine dinucleotide (NAD+) and glutathione (GSH) (Figure 2). Several studies have observed a significant decrease in the activities of SOD, catalase, and GPx in animal models for heart failure.13, 14, 15 Furthermore, mice lacking SOD or GPx exposed to cardiac injury have demonstrated worse outcomes when compared to their wild‐type littermates.16, 17, 18, 19, 20 NAD+, together with its reduced dinucleotide NADH, are pivotal in driving oxidation–reduction reactions involved in energy production.21, 22 Besides its role in regulating cellular energy metabolism, NAD+ is a precursor for the phosphorylated dinucleotide pair NADP+/NADPH, which plays a major role in the detoxification of ROS.21, 22 In several murine models of heart failure, a reduction in myocardial NAD+ levels has been observed.23, 24, 25 Interestingly, a recent study demonstrated that nicotinamide mononucleotide adenylyl transferase (Nmnat), an enzyme responsible for the production of NAD+, is significantly repressed in both models of murine heart failure and in heart failure patients.26 This observation suggests that NAD+ reduction also occurs in the human setting. Furthermore GSH, like NAD+, is another major antioxidant of mammalian cells, by scavenging radicals and the elimination of lipid peroxidation products.27 Interestingly, a reduction in total GSH has been observed in animals post‐cardiac injury.28, 29 Furthermore, depletion of GSH was highly correlated with serum TNF‐α levels.29 In LV tissue of end‐stage dilated or ischaemic cardiomyopathy patients, total GSH was decreased by 54% when compared to controls.28 In another study, serum GSH levels highly correlated with the symptom severity in heart failure patients.30
Lessons from previous oxidative stress treatments in heart failure
Based on the observation that the redox state is in disarray during heart failure,31 several experimental and clinical studies targeted oxidative stress producers (i.e. NADPH oxidases, xanthine oxidase, and uncoupled NOS) or scavengers [i.e. SOD, catalase, exogenous antioxidant (vitamin E, or folic acid), and GPx] to treat heart failure (Table 1).11, 12, 18, 19, 26, 28, 29, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54
Table 1.
Summary of pre‐clinical and clinical trials using anti‐oxidative stress treatments
| Experimental animal studies | Target | Treatment | Model | Results | Ref. |
|---|---|---|---|---|---|
| Inhibition of oxidative stress producers | NADPH oxidase | Cytosolic NADPH oxidase component p47phox knock‐out | MI mice | Protected the heart from LV remodelling and dysfunction post‐MI | 32 |
| Xantine oxidase | Oxypurinol administration | Spontaneous hypertensive/HF (SHHF) rat | Improved LV contractility and myocardial efficiency | 33 | |
| Allopurinol administration | Exercise‐induced HF in dogs | In pacing‐induced CHF, allopurinol improved LV systolic function | 34 | ||
| NOS uncoupling | Sapropterin administration | Chronic transverse aortic constriction mice | Improved cardiac function | 12 | |
| Improving endogenous antioxidant capacity | SOD | SOD overexpression | Ischaemia/reperfusion injury in mice | Reduced oxidative stress production, improved contractility, and reduced infarct size | 35 |
| Catalase | Catalase overexpression | Myocyte‐specific overexpression of G(alpha)q mice (a model for dilated cardiomyopathy) crossbred with myocyte‐specific overexpression of catalase | Reduced myocyte hypertrophy, myocyte apoptosis, and fibrosis | 36 | |
| GPx | GPx overexpression | MI mice | Prevention of adverse LV remodelling | 18 | |
| Ischaemia/reperfusion injury in mice | Improved contractility and reduced infarct size | ||||
| GSH | N‐acetylcysteine administration | MI rats | Improved LV GSH levels, improved contractility, and reduced LV remodelling | 28 | |
| Hypertensive rat model (induced by NOS inhibitor N(G)‐nitro‐L‐arginine methyl ester and high‐salt diet) | Improved cardiac GSH levels, reduced LV remodelling and dysfunction, improved TNF‐α levels, and reduced cardiac fibrosis | 29 | |||
| NAD+ | Nicotamide riboside administration | Mouse model of dilated cardiomyopathy | Improved cardiac function and redox state | 26 | |
| Supplementation of exogenous antioxidants | ROS | Vitamin E supplementation | Ascending aortic banding in guinea pigs (cardiac hypertrophy) | Improved myocardial redox state and cardiac function | 37 |
| Diabetic rat model by injection of streptozotocin | Improved myocardial redox state and cardiac function | 38 | |||
| Volume overload dog model | Reduced oxidative stress and improved myocardial contractility | 39 | |||
| Folic acid supplementation | Mouse model of high‐fat diet‐induced obesity | Reduced cardiac dysfunction, oxidative stress, and myocardial fibrosis | 40 | ||
| Inhibition of oxidative stress producers | Xanthine oxidase | Oxypurinol administration | Chronic HF (n = 60) | Improved LV ejection fraction | 41 |
| Symptomatic HF (n = 405) | No improved clinical outcome | 42, 43 | |||
| Allopurinol administration | Idiopathic dilated cardiomyopathy (n = 9) | Improved myocardial efficiency | 11 | ||
| Chronic HF (n = 50) | Reduced plasma BNP levels | 44 | |||
| Primary percutaneous transluminal coronary angioplasty in patients with acute MI (n = 38) | Reduced oxidative stress and improved LV function | 45 | |||
| Hyperuricaemic chronic HF (n = 19) | Improved peripheral vasodilator capacity and blood flow locally and systemically | 46 | |||
| Chronic HF (n = 11) | Improved endothelial dysfunction | 47 | |||
| NOS uncoupling | Sapropterin administration | Coronary artery disease (n = 49) | No effect on vascular function or redox state | 48 | |
| Improving endogenous antioxidant capacity | GSH | N‐acetylcysteine administration | Acute MI (n = 30) | Improved cardiac function | 49 |
| N‐acetylcysteine and streptokinase administration | Acute MI (n = 1, case study) | Improved cardiac function | 50 | ||
| N‐acetylcysteine, nitroglycerin and streptokinase administration | Acute MI (n = 27) | Reduced oxidative stress and improved LV function | 51 | ||
| Supplementation of exogenous antioxidants | ROS | Vitamin E supplementation | Ischaemic heart disease (n = 2002) | Reduced rate of non‐fatal MI | 52 |
| Combined vitamin A, C, E, and β‐carotene | Suspected acute MI (n = 125) | Reduced cardiac necrosis and oxidative stress | 53 | ||
| Meta‐analysis of randomized controlled trials | Cardiovascular diseases (50 studies, n = 294 478), including coronary heart disease, acute MI, unstable angina, TIA, stroke, and angiographically proved coronary atherosclerosis | No beneficial effects of vitamin supplementation on preventing cardiovascular disease | 54 |
BNP, B‐type natriuretic peptide; CHF, congestive heart failure; GPx, glutathione peroxidase; GSH, glutathione; HF, heart failure; LV, left ventricle; MI, myocardial infarction; NAD+, nicotinamide adenine dinucleotide; NOS, nitric oxide synthase; SOD, superoxide dismutase; ROS, reactive oxygen species; TIA, transient ischaemic attack; TNF, tumour necrosis factor.
Pre‐clinical animal models for anti‐oxidative stress therapies
Targeting oxidative stress in the pre‐clinical setting has been extensively studied and demonstrated highly promising results. These experimental studies have focused on three distinct approaches to target oxidative stress in heart failure: (i) inhibition of oxidative stress producers, (ii) improving endogenous antioxidant capacity, and (iii) improving antioxidant capacity by supplementation of exogenous antioxidants.
Initial experimental animal studies focused on inhibiting oxidative stress production by targeting NADPH oxidases, xanthine oxidase, or NOS uncoupling. NADPH oxidase inhibition in mice lacking the cytosolic NADPH oxidase component p47phox, protected the heart from LV remodelling and dysfunction post‐myocardial infarction.32 Inhibition of xanthine oxidase, by means of oxypurinol (rats) or allopurinol (dogs), protected the heart from LV remodelling, improved LV contractile function and myocardial efficiency post‐cardiac injury.33, 34 The production of ROS by the uncoupling of NOS has also been studied as a possible target for heart failure. Mice, with a knock‐out for NOS3, exposed to transverse aortic constriction (TAC), demonstrated a reduction in fibrosis and myocyte hypertrophy.12 Similarly, inhibiting NOS by means of BH4 (sapropterin) treatment also protected mice from TAC‐induced cardiac injury.12 These observations suggested that directly inhibiting ROS producers, thereby reducing oxidative stress, can result in improved survival and cardiac function following cardiac injury.
Besides targeting oxidative stress production, early experimental studies demonstrated that increasing the endogenous antioxidant capacity leads to improved cardiac function in rodent models for heart failure. Specifically, these studies focused on the primary antioxidant enzymes (SOD, catalase, and GPx) and antioxidants (NAD+, GSH, vitamin E, and folic acid). Mice overexpressing SOD when exposed to ischaemia/reperfusion injury were found to have severely decreased levels of superoxide production, improved contractile function, and a decrease in infarct size.35 Similarly, mice with a cardiomyocyte specific overexpression of catalase in a transgenic model for dilated cardiomyopathy were found to have a significant reduction in adverse remodelling (i.e. myocyte hypertrophy, myocyte apoptosis, and interstitial fibrosis) and the progression of heart failure.36 Finally, mice with an overexpression of GPx exposed to ischaemia/reperfusion injury were also found to have improved cardiac tissue survival and function, resulting from an inhibition of LV remodelling.18, 19
Following these positive results obtained by increasing the endogenous expression of antioxidant enzymes, several experimental studies tried to improve the endogenous antioxidant capacity. Early studies focused on increasing the endogenous levels of GSH, by the administration of N‐acetylcysteine (NAC), a precursor of GSH. NAC is readily absorbed into cells, where it is converted into cysteine, the rate‐limiting amino acid in the synthesis of GSH. These studies demonstrated that NAC can improve GSH levels, reduce oxidative stress, and improve cardiac function in rat models of cardiac injury.28, 29 To further address the importance of the antioxidant capacity, several experimental studies demonstrated that supplementation of vitamin E and folic acid leads to improved cardiac function in models for heart failure.37, 38, 39, 40 Recently, a study has demonstrated that by increasing the levels of NAD+, by supplementation of nicotinamide riboside (NR), a precursor of NAD+, leads to improved cardiac function and redox state in a murine heart failure model.26 These findings suggest that by increasing the antioxidant capacity of the heart, cardiomyocyte survival is improved, and the myocardium is better able to cope with injury.
Clinical anti‐oxidative stress therapies
Due to these highly promising results in animal models, several studies have assessed the potential of anti‐oxidative stress therapies in the clinical setting. Similar to the animal studies, clinical trials have taken three approaches to targeting oxidative stress in heart failure patients: (i) inhibition of oxidative stress producers (xanthine oxidase and NOS uncoupling), (ii) improving endogenous antioxidant capacity (NAC), and (iii) improving antioxidant capacity by supplementation of exogenous antioxidants (vitamin A, vitamin C, vitamin E, and folic acid).
At present, the best studied therapy in patients with heart failure is the inhibition of xanthine oxidase by the administration of allopurinol or oxypurinol.11, 41, 42, 43, 44, 45, 46, 47 The initial clinical trials were small studies (n = 9–60) in patients with dilated cardiomyopathy and chronic heart failure. These trials all demonstrated that treatment with allopurinol or oxypurinol improved myocardial function, peripheral vasodilatation capacity, blood flow, endothelial dysfunction, reduced plasma B‐type natriuretic peptide levels, and increased LV ejection fraction.11, 41, 44, 45, 46, 47 However, in a larger randomized controlled trial in 405 patients with heart failure, oxypurinol did not improve clinical outcome.42, 43 The primary endpoint of the study was a combined clinical endpoint that classified the patient's clinical status as improved, worsened, or unchanged 24 weeks after the initiation of the study. Compared to the placebo group, patients demonstrated no improvement in clinical status following oxypurinol treatment.42, 43 Similarly, inhibition of NOS uncoupling, by means of sapropterin treatment, has also been studied in the clinical setting. Several small clinical trials have been performed with oral sapropterin administration in patients (n = 18–49) with systemic or pulmonary hypertension.48, 55, 56 However, these trials all failed to demonstrate significant differences in nitric oxide synthesis, oxidative stress, systemic haemodynamics, vascular redox state, or endothelial function.
Following these disappointing results by inhibiting oxidative stress production in patients, clinical studies went on to assess the potential of increasing the antioxidant capacity in heart failure patients. The majority of the trials performed to date have involved the supplementation of exogenous antioxidants (vitamin A, vitamin C, vitamin E, and folic acid). Initial studies found that the supplementation of exogenous antioxidants leads to a reduction in cardiovascular events, infarct sizes, and oxidative stress.52, 53 However, a recent meta‐analysis of 50 randomized controlled trials studying the effects of vitamin and antioxidant supplementation, including 294 478 participants, concluded that supplementation with exogenous vitamins and antioxidants was not associated with reductions in the risk of major cardiovascular diseases.54 More interestingly, several clinical trials have demonstrated that improving the antioxidant capacity, by bolstering endogenous GSH levels, does show some promise in heart failure patients. These trials demonstrated that supplementation of NAC in patients resulted in a reduction in oxidative stress, as measured by an increase in the GSH/oxidized GSH (GSSG) ratio, infarct size and an improved cardiac function in patients with heart failure and acute myocardial infarction.49, 50, 51, 57
Why have clinical anti‐oxidative stress therapies largely failed?
Taken together, these findings suggested that although targeting oxidative stress is theoretically logical, the majority of the strategies currently employed in the clinical setting have failed to improve patient prognosis. Furthermore, the exact reasons and mechanisms as to why these studies have failed to produce the expected beneficial effects, remain largely unknown. One reason could be that in the experimental setting, the majority of the studies utilized heart failure models to test the efficacy of anti‐oxidative stress treatments, while in the clinical setting anti‐oxidative stress treatments were primarily tested in patients with acute myocardial infarction and not heart failure. Although it is well documented that following myocardial infarction there is a surge in oxidative stress,58 it maybe that at this stage anti‐oxidative stress therapies are not capable of limiting the production of oxidative stress. Rather, these strategies may serve to improve outcome in heart failure patients, where there is less oxidative stress production. Another reason for this discrepancy could be that only specific patient populations benefit from anti‐oxidative stress treatments. Or else the inadequate understanding of the mechanistic mode of action of the administered antioxidant.
With regard to targeting oxidative stress production in human heart failure, it has been speculated that these therapies may be beneficial for a specific subset of patient. Oxypurinol has been shown to improve heart failure symptoms in a specific subset of patients with elevated uric acid, the product of xanthine oxidase.43, 59 Thus, identifying patients with increases in oxidative stress production resulting from xanthine oxidase activity may still benefit from this therapeutic approach. Several current anti‐oxidative stress therapies have also been found to result in some off‐target effects. Sapropterin administration was found to result in an unexpected increase in oxidized BH4, BH2, a competitive inhibitor of BH4 that promotes NOS uncoupling.48, 60 Similarly, supplementation with α‐tocopherol (vitamin E) has been shown to drastically suppresses the levels of γ‐tocopherol (the more potent antioxidant, found primarily in the diet), thereby possibly reducing rather than increasing total antioxidant capacity.61
Although the findings of clinical trials aimed at reducing ROS production and increasing exogenous antioxidants have been disappointing, targeting oxidative stress, specifically the endogenous antioxidant capacity, in heart failure should not be entirely disregarded. Noteworthy has been the observation that increasing the antioxidant capacity, by bolstering endogenous GSH levels via NAC supplementation, results in improved patient outcome, without any adverse side effects.49, 50, 51, 57 Therefore, future oxidative stress therapies should focus on improving the endogenous antioxidant capacity, rather than inhibiting oxidative stress production or supplementation of exogenous antioxidants.
The future of oxidative stress as a therapeutic target in heart failure
The major endogenous antioxidants in mammalian cells are NAD+ and GSH, the latter produced via the γ‐glutamyl cycle. NAD+ has a multitude of cellular functions, and more recently it has become evident that it plays an important role in the detoxification of cellular ROS. Similarly, GSH protects cells against oxidative stress, and both NAD+ and GSH levels have been shown to be associated with heart failure in the experimental and clinical setting.23, 24, 25, 26, 27, 28, 29, 30 Furthermore, supplementation of NAD+ or GSH precursors has been found to improve cardiac function and redox state in models for heart failure.26, 28, 29 Thus, we propose two approaches for future anti‐oxidative stress therapies in heart failure patients: (i) increasing the endogenous antioxidant capacity, and (ii) increasing the expression/activity of antioxidant producing enzymes.
Improving endogenous antioxidant capacity in heart failure
Increasing the endogenous antioxidant capacity can be primarily achieved by supplementation of precursors of the major cellular antioxidants GSH and NAD+. The effectiveness and safety of this approach has previously been demonstrated with the supplementation of NAC, a precursor of GSH, to heart failure patients and acute myocardial infarction patients, resulting in improved patient outcome.49, 50, 51, 57 Thus, increasing the levels of endogenous antioxidants seems to be a promising target for not only treating oxidative stress in myocardial infarction, but also in heart failure patients.
Improving the endogenous levels of GSH can be achieved by supplementation with GSH precursors which can be utilized by the γ‐glutamyl cycle for de novo GSH synthesis (Figure 3). Besides NAC, γ‐glutamylcysteine, another GSH precursor, and 2‐oxothiazolidine‐4‐carboxylate (OTC), an analogue of 5‐oxproline, also seem to have the potential to increase endogenous GSH levels. Both γ‐glutamylcysteine and OTC increased GSH and reduced oxidative stress in experimental and clinical studies.62, 63, 64, 65, 66, 67, 68 Similar to NAC, supplementation of γ‐glutamylcysteine increased the levels of GSH in patients with cancer, with no adverse effects.62 OTC, which is converted to cysteine by 5‐oxoprolinase (OPLAH), increased GSH levels in the experimental setting.63, 64 Interestingly, early experimental studies have shown that OTC improved cardiac function following cardiac injury.65, 67 Furthermore, in several clinical trials OTC treatment had no adverse effects and increased GSH concentrations and decreased oxidative stress in patients with acute respiratory distress syndrome and HIV patients.67, 68
Figure 3.
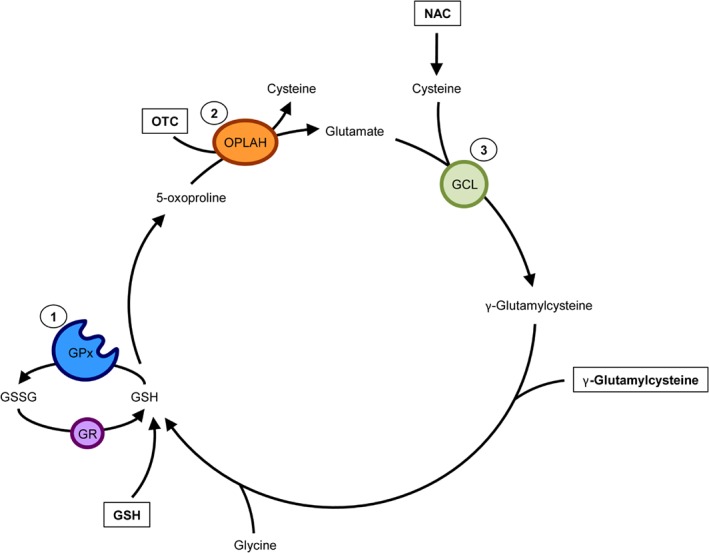
Drug therapies targeting endogenous glutathione (GSH) synthesis. GSH is synthesized from cysteine (the rate‐limiting amino acid), glutamate, and glycine by the γ‐glutamyl cycle. GSH is then utilized by GSH peroxidase (GPx) to reduce oxidative stress, and in the process forming oxidized GSH (GSSG). GSSG is then reduced by action of GSH reductase (GR). Improving the γ‐glutamyl cycle's ability to produce GSH has been characterized as a treatment target in heart failure. N‐acetylcysteine (NAC), γ‐glutamylcysteine, and 2‐oxothiazolidine‐4‐carboxylate (OTC, also known as pro‐cysteine) are compounds which have demonstrated the capacity to increase the endogenous production of GSH. OTC is converted to cysteine, by action of 5‐oxoprolinase (OPLAH), to be used for de novo synthesis of GSH. Similarly, NAC is converted to cysteine intracellularly, and used for GSH synthesis. γ‐Glutamylcysteine is utilized by the γ‐glutamyl cycle to form GSH, by addition of glycine. GCL, glutamate cysteine ligase.
Besides improving the endogenous GSH levels in heart failure, another approach could be to increase the levels of endogenous NAD+. NAD+ can be derived from several precursors, including deamidated precursors, such as tryptophan, nicotinic acid, amidated vitamin B3 nicotinamide and NR.21 The most promising of these precursors to date is NR, which is converted to NAD+ by NR kinase (Nmrk) and nicotinamide mononucleotide adenylyl transferase (Nmnat). It has been recently demonstrated that supplementation of NR in murine models for dilated cardiomyopathy and pressure overload‐induced heart failure, can restore NAD+ levels and preserve cardiac function.26
Although only few studies have focused on increasing the endogenous antioxidant capacity in models for heart failure, the results to date may be promising. Furthermore, the observation that NAC supplementation in heart failure patients leads to increased GSH levels suggests that this might be a potential strategy for reducing the increase in oxidative stress resulting from cardiac injury. Future studies should thus focus on further characterizing the beneficial effects of the supplementation of GSH precursors (γ‐glutamylcysteine and OTC) and NAD+ precursors (NR) on heart failure patient outcome.
Targeting the endogenous production of antioxidants in heart failure
Besides improving endogenous antioxidant capacity by administration of GSH and NAD+ precursors, another avenue for reducing oxidative stress in heart failure is to improve the expression and/or activity of the γ‐glutamyl cycle and NAD+ producers.
Recent experimental studies have demonstrated that several components of the γ‐glutamyl cycle are strongly associated with the development and progression of heart failure (including γ‐glutamylcysteine synthetase, GPx, and OPLAH). Furthermore, modulation of these enzymes, by overexpression, has resulted in cardioprotection.18, 19, 69, 70 Of particular interest is OPLAH, a cytoplasmic enzyme of the GSH cycle whose only function is the conversion of 5‐oxoproline, a degradation product of GSH, into glutamate (Figure 4). Interestingly, studies have demonstrated that excessive 5‐oxoproline accumulation can lead to the induction of intracellular oxidative stress.70, 71, 72 Therefore, OPLAH plays a pivotal role not only in the γ‐glutamyl cycle, by producing glutamate for de novo GSH synthesis, but also as an antioxidant by scavenging 5‐oxoproline.
Figure 4.
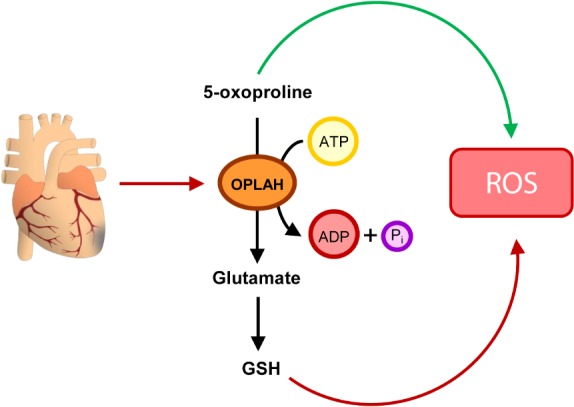
Targeting 5‐oxoprolinase (OPLAH) to reduce oxidative stress in heart failure. Following cardiac injury, OPLAH expression is reduced, leading to the accumulation of 5‐oxoproline. 5‐Oxoproline then leads to drastic increase in oxidative stress (reactive oxygen species, ROS). To help reduce the insult of 5‐oxoproline to the injured myocardium, two strategies could be developed: (i) to pharmacologically improve the remaining OPLAH's ability to reduce 5‐oxoproline, or (ii) to increase OPLAH expression by means of gene therapy. ADP, adenosine diphosphate; ATP, adenosine triphosphate; GSH, glutathione.
OPLAH expression is suppressed in heart failure, both in the experimental and clinical setting.70, 73 In the murine setting, reduction in OPLAH increased plasma 5‐oxoproline levels in cardiac tissue and plasma, which coincided with an increase in oxidative stress.70 Interestingly, elevated levels of plasma 5‐oxoproline in chronic heart failure patients were associated with higher N‐terminal pro‐B‐type natriuretic peptide, incidence of atrial fibrillation, and all‐cause mortality.70 In a recent study, OPLAH overexpression in mice exposed to ischaemia/reperfusion injury or permanent myocardial ischaemia improved cardiac function, reduced infarct size and fibrosis, when compared to wild‐type littermates.70 Improved cardiac function in the OPLAH overexpression mice was coupled to reduced 5‐oxoproline levels and improved GSH/GSSG ratio post‐cardiac injury.70 Thus, stimulating the expression and/or activity of OPLAH could lead to novel therapeutic strategies for patients with heart failure.
Similar to the γ‐glutamyl cycle genes, there are several NAD+ producers, most noteworthy is Nampt. Nampt has been shown to be repressed in both model for heart failure and the clinical setting.26 Furthermore, overexpression of Nampt in mice was found to protect these animals against ischaemia/reperfusion injury and isoproterenol‐induced hypertrophy.23, 24 Therefore, increasing the expression and/or activity of Nampt might be a potential target for the treatment of heart failure.
To date there are no known pharmacological agents (i.e. drugs or small molecules) that have the capacity to induce OPLAH activity. There are however several Nampt activators, although limited information is currently available about these compounds in patients.74, 75 Future studies should therefore focus on (i) identifying novel pharmacological agents that specifically target OPLAH, and (ii) characterize Nampt activators in the clinical setting. Similarly, the development of an OPLAH or Nampt gene therapy, as recently described for SERCA2, could also serve as a viable therapeutic strategy.76 Furthermore, besides OPLAH and Nampt, other members of the γ‐glutamyl cycle and NAD+ producers should also be screened for their potential use as therapeutic targets in heart failure.
Conclusion
The role of oxidative stress in the onset and progression of heart failure has been extensively studied. Pre‐clinical studies showed promising results with various anti‐oxidative strategies, but these beneficial effects did not translate into positive results in clinical studies in patients with heart failure. This might be caused by inadequate patient inclusion criteria or off target effects of currently employed therapies. Therefore, we believe there is still room for novel antioxidant approaches in heart failure. In particular, targeting the endogenous antioxidant capacity might be interesting new targets in the treatment of heart failure. Of interest would be the development of medications capable of interacting with the components of the γ‐glutamyl cycle or NAD+ production, which may lead to novel treatment options for heart failure in the future.
Conflict of interest: none declared.
References
- 1. Karimi Galougahi K, Antoniades C, Nicholls SJ, Channon KM, Figtree GA. Redox biomarkers in cardiovascular medicine. Eur Heart J 2015;36:1576–1582. [DOI] [PubMed] [Google Scholar]
- 2. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 2011;301:H2181‐2190. [DOI] [PubMed] [Google Scholar]
- 3. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) . Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 4. Wolfram R, Oguogho A, Palumbo B, Sinzinger H. Enhanced oxidative stress in coronary heart disease and chronic heart failure as indicated by an increased 8‐epi‐PGF 2α. Eur J Heart Fail 2005;7:167–172. [DOI] [PubMed] [Google Scholar]
- 5. Sato Y, Fujiwara H, Takatsu Y. Cardiac troponin and heart failure in the era of high‐sensitivity assays. J Cardiol 2012;60:160–167. [DOI] [PubMed] [Google Scholar]
- 6. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007;49:241–248. [DOI] [PubMed] [Google Scholar]
- 7. Sawyer DB, Colucci WS. Mitochondrial oxidative stress in heart failure : “oxygen wastage” revisited. Circ Res 2000;86:119–120. [DOI] [PubMed] [Google Scholar]
- 8. Perrelli M, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol 2011;3:186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II‐mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 2008;102:488–496. [DOI] [PubMed] [Google Scholar]
- 10. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 2003;41:2164–2171. [DOI] [PubMed] [Google Scholar]
- 11. Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 2001;104:2407–2411. [DOI] [PubMed] [Google Scholar]
- 12. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase‐3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 2005;115:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 1996;148:291–300. [PMC free article] [PubMed] [Google Scholar]
- 14. Khaper N, Singal PK. Effects of afterload‐reducing drugs on pathogenesis of antioxidant changes and congestive heart failure in rats. J Am Coll Cardiol 1997;29:856–861. [DOI] [PubMed] [Google Scholar]
- 15. Khaper N, Kaur K, Li T, Farahmand F, Singal PK. Antioxidant enzyme gene expression in congestive heart failure following mycardial infarction. Mol Cell Biochem 2003;251:9–15. [PubMed] [Google Scholar]
- 16. van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, Duncker DJ, Chen Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic Biol Med 2008;44:1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding Y, Li YL, Zimmerman MC, Davisson RL, Schultz HD. Role of CuZn superoxide dismutase on carotid body function in heart failure rabbits. Cardiovasc Res 2009;81:678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004;109:544–549. [DOI] [PubMed] [Google Scholar]
- 19. Yoshida T, Watanabe M, Engelman DT, Engelman RM, Schley JA, Maulik N, Ho YS, Oberley TD, Das DK. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J Mol Cell Cardiol 1996;28:1759–1767. [DOI] [PubMed] [Google Scholar]
- 20. Forgione MA, Cap A, Liao R, Moldovan NI, Eberhardt RT, Lim CC, Jones J, Goldschmidt‐Clermont PJ, Loscalzo J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation 2002;106:1154–1158. [DOI] [PubMed] [Google Scholar]
- 21. Mericskay M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis 2016;109:207–215. [DOI] [PubMed] [Google Scholar]
- 22. Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 2017;13:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP‐ribose) polymerase‐1‐dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J Biol Chem 2005;280:43121–43130. [DOI] [PubMed] [Google Scholar]
- 24. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 2009;105:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation‐dependent deactivation of poly(ADP‐ribose) polymerase 1. Mol Cell Biol 2009;29:4116–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, Breton M, Decaux JF, Lavery GG, Baczkó I, Zoll J, Garnier A, Li Z, Brenner C, Mericskay M. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018;137:2256–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother 2003;57:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adamy C, Mulder P, Khouzami L, Andrieu‐abadie N, Defer N, Candiani G, Pavoine C, Caramelle P, Souktani R, Le Corvoisier P, Perier M, Kirsch M, Damy T, Berdeaux A, Levade T, Thuillez C, Hittinger L, Pecker F. Neutral sphingomyelinase inhibition participates to the benefits of N‐acetylcysteine treatment in post‐myocardial infarction failing heart rats. J Mol Cell Cardiol 2007;43:344–353. [DOI] [PubMed] [Google Scholar]
- 29. Bourraindeloup M, Adamy C, Candiani G, Cailleret M, Bourin MC, Badoual T, Su JB, Adubeiro S, Roudot‐Thoraval F, Dubois‐Rande JL, Hittinger L, Pecker F. N‐acetylcysteine treatment normalizes serum tumor necrosis factor‐alpha level and hinders the progression of cardiac injury in hypertensive rats. Circulation 2004;110:2003–2009. [DOI] [PubMed] [Google Scholar]
- 30. Damy T, Kirsch M, Khouzami L, Caramelle P, Le Corvoisier P, Roudot‐Thoraval F, Dubois‐Randé JL, Hittinger L, Pavoine C, Pecker F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS One 2009;4:e4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mohamed BA, Schnelle M, Khadjeh S, Lbik D, Herwig M, Linke WA, Hasenfuss G, Toischer K. Molecular and structural transition mechanisms in long‐term volume overload. Eur J Heart Fail 2016;18:362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doerries C, Grote K, Hilfiker‐Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 2007;100:894–903. [DOI] [PubMed] [Google Scholar]
- 33. Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, Berry CE, Barouch LA, Vandegaer KM, Li D, Hare JM. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res 2006;98:271–279. [DOI] [PubMed] [Google Scholar]
- 34. Ukai T, Cheng CP, Tachibana H, Igawa A, Zhang ZS, Cheng HJ, Little WC. Allopurinol enhances the contractile response to dobutamine and exercise in dogs with pacing‐induced heart failure. Circulation 2001;103:750–755. [DOI] [PubMed] [Google Scholar]
- 35. Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, Zweier JL. Overexpression of human copper, zinc‐superoxide dismutase (SOD1) prevents postischemic injury. Proc Natl Acad Sci U S A 1998;95:4556–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin F, Lennon‐Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ, Colucci WS. Cardiac‐specific overexpression of catalase identifies hydrogen peroxide‐dependent and ‐independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q‐overexpressing transgenic mice. Circ Heart Fail 2010;3:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 1996;28:506–514. [DOI] [PubMed] [Google Scholar]
- 38. Hamblin M, Smith HM, Hill MF. Dietary supplementation with vitamin E ameliorates cardiac failure in type I diabetic cardiomyopathy by suppressing myocardial generation of 8‐iso‐prostaglandin F2alpha and oxidized glutathione. J Card Fail 2007;13:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prasad K, Gupta JB, Kalra J, Lee P, Mantha SV, Bharadwaj B. Oxidative stress as a mechanism of cardiac failure in chronic volume overload in canine model. J Mol Cell Cardiol 1996;28:375–385. [DOI] [PubMed] [Google Scholar]
- 40. Li W, Tang R, Ouyang S, Ma F, Liu Z, Wu J. Folic acid prevents cardiac dysfunction and reduces myocardial fibrosis in a mouse model of high‐fat diet‐induced obesity. Nutr Metab (Lond) 2017;14:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cingolani HE, Plastino JA, Escudero EM, Mangal B, Brown J, Pérez NG. The effect of xanthine oxidase inhibition upon ejection fraction in heart failure patients: La Plata Study. J Card Fail 2006;12:491–498. [DOI] [PubMed] [Google Scholar]
- 42. Freudenberger RS, Schwarz RP, Brown J, Moore A, Mann D, Givertz MM, Colucci WS, Hare JM. Rationale, design and organisation of an efficacy and safety study of oxypurinol added to standard therapy in patients with NYHA class III‐IV congestive heart failure. Expert Opin Investig Drugs 2004;13:1509–1516. [DOI] [PubMed] [Google Scholar]
- 43. Hare JM, Mangal B, Brown J, Fisher C, Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP; Investigators OPT‐CHF. Impact of oxypurinol in patients with symptomatic heart failure. J Am Coll Cardiol 2008;51:2301–2309. [DOI] [PubMed] [Google Scholar]
- 44. Gavin AD, Struthers AD. Allopurinol reduces B‐type natriuretic peptide concentrations and haemoglobin but does not alter exercise capacity in chronic heart failure. Heart 2005;91:749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guan W, Osanai T, Kamada T, Hanada H, Ishizaka H, Onodera H, Iwasa A, Fujita N, Kudo S, Ohkubo T, Okumura K. Effect of allopurinol pretreatment on free radical generation after primary coronary angioplasty for acute myocardial infarction. J Cardiovasc Pharmacol 2003;41:699–705. [DOI] [PubMed] [Google Scholar]
- 46. Doehner W, Schoene N, Rauchhaus M, Leyva‐Leon F, Pavitt D V, Reaveley DA, Schuler G, Coats AJ, Anker SD, Hambrecht R. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo‐controlled studies. Circulation 2002;105:2619–2624. [DOI] [PubMed] [Google Scholar]
- 47. Farquharson CAJ, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 2002;106:221–226. [DOI] [PubMed] [Google Scholar]
- 48. Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, Antoniades C, Margaritis M, Lee R, Cerrato R, Crabtree MJ, Francis JM, Sayeed R, Ratnatunga C, Pillai R, Choudhury RP, Neubauer S, Channon KM. Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 2012;125:1356–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Šochman J, Vrbská J, Musilová B, Roček M. Infarct size limitation: acute N‐acetylcysteine defense (ISLAND trial): preliminary analysis and report after the first 30 patients. Clin Cardiol 1996;19:94–100. [DOI] [PubMed] [Google Scholar]
- 50. Sochman J, Peregrin JH. Total recovery of left ventricular function after acute myocardial infarction: comprehensive therapy with streptokinase, N‐acetylcysteine and percutaneous transluminal coronary angioplasty. Int J Cardiol 1992;35:116–118. [DOI] [PubMed] [Google Scholar]
- 51. Arstall MA, Yang J, Stafford I, Betts WH, Horowitz JD. N‐acetylcysteine in combination with nitroglycerin and streptokinase for the treatment of evolving acute myocardial infarction. Circulation 1995;92:2855–2862. [DOI] [PubMed] [Google Scholar]
- 52. Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996;347:781–786. [DOI] [PubMed] [Google Scholar]
- 53. Singh RB, Niaz MA, Rastogi SS, Rastogi S. Usefulness of antioxidant vitamins in suspected acute myocardial infarction (the Indian experiment of infarct survival‐3). Am J Cardiol 1996;77:232–236. [DOI] [PubMed] [Google Scholar]
- 54. Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ; Korean Meta‐Analysis Study Group . Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta‐analysis of randomised controlled trials. BMJ 2013;346:f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reverter E, Mesonero F, Seijo S, Martínez J, Abraldes JG, Peñas B, Berzigotti A, Deulofeu R, Bosch J, Albillos A, García‐Pagán JC. Effects of sapropterin on portal and systemic hemodynamics in patients with cirrhosis and portal hypertension: a bicentric double‐blind placebo‐controlled study. Am J Gastroenterol 2015;110:985–992. [DOI] [PubMed] [Google Scholar]
- 56. Robbins IM, Hemnes AR, Simon Gibbs J, Christman BW, Howard L, Meehan S, Cabrita I, Gonzalez R, Oyler T, Zhao L, Du RH, Mendes LA, Wilkins MR. Safety of sapropterin dihydrochloride (6r–bh4) in patients with pulmonary hypertension. Exp Lung Res 2011;37:26–34. [DOI] [PubMed] [Google Scholar]
- 57. Mehra A, Shotan A, Ostrzega E, Hsueh W, Vasquez‐Johnson J, Elkayam U. Potentiation of isosorbide dinitrate effects with N‐acetylcysteine in patients with chronic heart failure. Circulation 1994;89:2595–2600. [DOI] [PubMed] [Google Scholar]
- 58. Grieve DJ, Byrne JA, Cave AC, Shah AM. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ 2004;13:132–138. [DOI] [PubMed] [Google Scholar]
- 59. von Lueder TG, Girerd N, Atar D, Agewall S, Lamiral Z, Kanbay M, Pitt B, Dickstein K, Zannad F, Rossignol P; High‐Risk Myocardial Infarction Database Initiative Investigators . Serum uric acid is associated with mortality and heart failure hospitalizations in patients with complicated myocardial infarction: findings from the High‐Risk Myocardial Infarction Database Initiative. Eur J Heart Fail 2015;17:1144–1151. [DOI] [PubMed] [Google Scholar]
- 60. Vásquez‐Vivar J, Martásek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J 2002;362(Pt 3):733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gutierrez AD, de Serna DG, Robinson I, Schade DS. The response of γ vitamin E to varying dosages of α vitamin E plus vitamin C. Metabolism 2009;58:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zarka MH, Bridge WJ. Oral administration of γ‐glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study. Redox Biol 2017;11:631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Williamson JM, Meister A. New substrates of 5‐oxo‐L‐prolinase. J Biol Chem 1982;257:12039–12042. [PubMed] [Google Scholar]
- 64. Weitberg AB. The effect of L‐2‐oxothiazolidine on glutathione levels in cultured mammalian cells. Mutat Res 1987;191:189–191. [DOI] [PubMed] [Google Scholar]
- 65. Shug A, Madsen D. Protection of the ischemic rat heart by procysteine and amino acids. J Nutr Biochem 1994;5:356–359. [Google Scholar]
- 66. Poon BY, Goddard CM, Leaf CD, Russell JA, Walley KR. L‐2‐oxothiazolidine‐4‐carboxylic acid prevents endotoxin‐induced cardiac dysfunction. Am J Respir Crit Care Med 1998;158:1109–1113. [DOI] [PubMed] [Google Scholar]
- 67. Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, Wright PE. A trial of antioxidants N‐acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest 1997;112:164–172. [DOI] [PubMed] [Google Scholar]
- 68. Kalayjian RC, Skowron G, Emgushov RT, Chance M, Spell SA, Borum PR, Webb LS, Mayer KH, Jackson JB, Yen‐Lieberman B, Story KO, Rowe WB, Thompson K, Goldberg D, Trimbo S, Lederman MM. A phase I/II trial of intravenous L‐2‐oxothiazolidine‐4‐carboxylic acid (procysteine) in asymptomatic HIV‐infected subjects. J Acquir Immune Defic Syndr 1994;7:369–374. [PubMed] [Google Scholar]
- 69. Schupp N, Schmid U, Heidland A, Stopper H. Rosuvastatin protects against oxidative stress and DNA damage in vitro via upregulation of glutathione synthesis. Atherosclerosis 2008;199:278–287. [DOI] [PubMed] [Google Scholar]
- 70. van der Pol A, Gil A, Silljé HH, Tromp J, Ovchinnikova ES, Vreeswijk‐Baudoin I, Hoes M, Domian IJ, van de Sluis B, van Deursen JM, Voors AA, van Veldhuisen DJ, van Gilst WH, Berezikov E, van der Harst P, de Boer RA, Bischoff R, van der Meer P. Accumulation of 5‐oxoproline in myocardial dysfunction and the protective effects of OPLAH. Sci Transl Med 2017;9:eaam8574. [DOI] [PubMed] [Google Scholar]
- 71. Pederzolli CD, Sgaravatti AM, Braum CA, Prestes CC, Zorzi GK, Sgarbi MB, Wyse AT, Wannmacher CM, Wajner M, Dutra‐Filho CS. 5‐Oxoproline reduces non‐enzymatic antioxidant defenses in vitro in rat brain. Metab Brain Dis 2007;22:51–65. [DOI] [PubMed] [Google Scholar]
- 72. van der Pol A, Gil A, Tromp J, Silljè HHW, van Veldhuisen DJ, Voors AA, Hoendermis ES, Grote Beverborg N, Schouten EM, de Boer RA, Bischoff R, van der Meer P. OPLAH ablation leads to accumulation of 5‐oxoproline, oxidative stress, fibrosis and elevated fillings pressures: a murine model for heart failure with a preserved ejection fraction. Cardiovasc Res 2018;114:1871–1882. [DOI] [PubMed] [Google Scholar]
- 73. Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 2014;129:1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Walker MA, Tian R. Raising NAD in heart failure. Circulation 2018;137:2274–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang P, Miao CY. NAMPT as a therapeutic target against stroke. Trends Pharmacol Sci 2015;36:891–905. [DOI] [PubMed] [Google Scholar]
- 76. Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long‐term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 2014;114:101–108. [DOI] [PubMed] [Google Scholar]