

Short abstract
Review on the evidence for the neutrophil as a mediator of tissue injury in seven conditions making it a potential therapeutic target.
Keywords: adhesion molecules, integrin, inflammation, cytokine, reactive oxygen species, tissue injury
Abstract
The neutrophil is an essential component of the innate immune system, and its function is vital to human life. Its production increases in response to virtually all forms of inflammation, and subsequently, it can accumulate in blood and tissue to varying degrees. Although its participation in the inflammatory response is often salutary by nature of its normal interaction with vascular endothelium and its capability to enter tissues and respond to chemotactic gradients and to phagocytize and kill microrganisms, it can contribute to processes that impair vascular integrity and blood flow. The mechanisms that the neutrophil uses to kill microorganisms also have the potential to injure normal tissue under special circumstances. Its paradoxical role in the pathophysiology of disease is particularly, but not exclusively, notable in seven circumstances: 1) diabetic retinopathy, 2) sickle cell disease, 3) TRALI, 4) ARDS, 5) renal microvasculopathy, 6) stroke, and 7) acute coronary artery syndrome. The activated neutrophilˈs capability to become adhesive to endothelium, to generate highly ROS, and to secrete proteases gives it the potential to induce local vascular and tissue injury. In this review, we summarize the evidence for its role as a mediator of tissue injury in these seven conditions, making it or its products potential therapeutic targets.
Abbreviations
- AGE
advanced glycation end‐products
- ANCA
antineutrophil cytoplasmic antibodies
- ARDS
acute respiratory distress syndrome
- BALF
BAL fluid
- ELAM‐1
endothelial leukocyte adhesion molecule‐1
- HNA
human neutrophil antigens
- IL‐1Rα
IL‐1R antagonist
- KDR
kinase domain receptor
- MIF
macrophage inhibitory factor
- PR3
proteinase 3
- SLPI
secretory leukocyte protease inhibitor
- tPA
tissue plasminogen activator
- TRALI
transfusion‐related lung injury
Introduction
The human neutrophil, as part of the innate immune system, evolved principally to destroy invading microrganisms. It does so by bringing the organisms into contact with several microbial tissue‐destroying chemicals generated by neutrophils. The two most notable are highly reactive oxygen‐derived compounds (e.g., hypochlorous acid) and defensins, proteins that act as microbicidal antibiotics. The chemistry of the mechanisms involved results in the death of the microorganism but also the neutrophil. This cell loss is compensated for by the ability to produce neutrophils at a prodigious rate and to have them accumulate in tissue in response to need. In some cases, the inflammatory process is not generated by invading microorganisms, and the accumulation of neutrophils and their activation may result in injury to host tissues. We review the role of the neutrophil in seven conditions in which evidence has accumulated that indicates it contributes to vascular injury. In six of the seven conditions, normal or increased numbers of neutrophils are part of the pathogenetic pathway. In one, TRALI, neutropenia occurs [1]. In circumstances in which there is no associated tissue or organ injury, neutrophilia, per se, is asymptomatic. In the seven circumstances described here, the process is triggered by an incident that results in a tissue‐specific inflammatory response in which the neutrophil is one of several participants in the resultant tissue injury. The critical role of the neutrophil, per se, has, in some cases, been demonstrated experimentally by decreasing its numbers or preventing its action, resulting in amelioration of the disease process. In this review article, we discuss the potentially tissue‐damaging effect of tissue neutrophils in seven noninfectious, inflammatory disorders and describe circumstances in humans or experimental animal models in which antineutrophil therapy is effective. These therapeutic observations and experiments add weight to the argument that there are selected circumstances in which intervention to lower the neutrophil count or to nullify the effects of its products may be beneficial.
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF DIABETIC RETINOPATHY
Diabetic retinopathy is initiated by aberrant carbohydrate pathways induced during hyperglycemia; these include increased polyol flux, leading to increased sorbitol accumulation, activation of diacylglycerol, and the resultant increase in PKC, principally, the β isoform [2, –, 4], which leads to damage to retinal endothelium [1] ( Fig. 1 ). AGE further increase PKC‐β activity in vascular endothelial cells and also induce smooth muscle proliferation [5]. Increased sorbitol levels are associated with increased superoxide production. An elevated neutrophil count is associated with the vascular complications of type 2 diabetes [7], and the associated inflammatory processes are presented (see Table 1 ). The inflammatory response may be initiated or heightened by the C677T mutation in the methylene tetrahydrofolate reductase gene and by hyperhomocysteinemia. AGE also bind to receptors on endothelial cells and macrophages, resulting in ROS production and the release of inflammatory cytokines (e.g., IL‐1, TNF‐ α, and TGF‐β). The release of cytokines, such as VEGF, results in activation of PKC‐β and to the inflammatory response [6, 8, 9]. The effect of VEGF is initiated by binding to the endothelial cell membrane‐bound KDR, which activates PI3K, resulting in phosphorylation of PLCγ and increasing diacylgycerol levels and activation of PKC‐β [6]. Also, the release of prosclerotic cytokines alters the ECM and enhances fibrosis [5]. Under these circumstances, increased expression of neutrophil adhesion molecules results in their adhesion to endothelium [10, 11]. This is facilitated by hyperglycemia‐induced up‐regulation of endothelial cell ICAM‐1, ELAM‐1, selectins, and other cell adhesion molecules [12, –, 14, 15, 16, 17], through a pathway related to NF‐κB and PKC [12]. These effects are evident in the neutrophil shedding of L‐selectin with poor glycemic control and heightened levels of serum L‐selectin that correlate with the hemoglobin A1C levels [11]. The neutrophils of diabetics are less deformable in micropipette measurements [18], and leukostasis develops in the retinal capillaries, leading to capillary obstruction, intraretinal hemorrhages, exudates, and microaneurisms [19].
Figure 1.
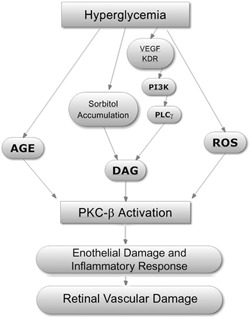
Metabolic alterations in diabetes mellitus, type II, leading to retinal vascular damage. The activation of isomers of PKC, particularly PKC‐β, appears central to the pathophysiological changes resulting from hyperglycemia, leading to the development of diabetic retinopathy [2, 4]. Although atypical PKCs also play a role in glucose metabolism, their role in the genesis of diabetes is unclear [3]. There are multiple pathways for the initiation of PKC‐β activity, examples of which are shown. Hyperglycemia results in the formation of AGE, which can lead to activation of PKC [2, 5]. Increased polyol flux and the accumulation of sorbitol, likewise, result in increased DAG production and PKC activation [2]. Heightened levels of growth factors, such as VEGF, are found in the eyes of patients with diabetic retinopathy, and VEGF binds to an endothelial membrane KDR, which activates PI3K [6]. The latter phosphorylates PLCγ‐generating DAG. Hyperglycemia‐induced generation of ROS, such as superoxide, also produces PKC activation. The activation of PKC‐β is key to the development of retinal vascular endothelial damage. Important in the pathological events and the development of microaneurisms is the concomitant induction of neutrophil adhesion, the release of cytokines and chemokines [4]. Inhibitors of PKC‐β, such as ruboxistaurin, prevent endothelial damage and loss of vision in diabetic patients. This figure is adapted from a figure in ref. [2].
Table 1.
Citations Supporting the Relationship of the Initiating Agent or Pathophysiologic Finding with the Disorder Listed
| Diabetic retinopathy | Pulmonary TRALI | Renal vasculopathy | Sickle cell disease | ARDS | Cerebral vasculopathy (stroke) | Acute coronary artery disease | |
| Initiating agents | |||||||
| Hyperglycemia | [2, 10] | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant |
| Antineutrophil antibody | Not relevant | Antibodies to human neutrophil antigens or HLA [83, 86, 87, 89, 99] | [120] | Not relevant | Not relevant | Not relevant | Not relevant |
| Leukocytosis, neutrophillia | [7] | Not relevant | [121, 134] | [34, –, 36, 37, 38, 39] | [116] | [142, 154] | [189, 191, 195, 219] |
| Neutropenia | Not relevant | [84, 87] | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant |
| Predisposing Inflamation | Not found | Not found | Not found | Not found | [102, 103, 108] | Not found | Not found |
| Reperfusion | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant | [137] | [210] |
| Trigger factors in transfused blood | Not relevant | [87] | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant |
| Pathophysiology | |||||||
| Expression of neutrophil adhesion molecules (selectins and integrins) | [10, 11] | [91, –, 93] | [123] | [42, 43, 75] | [94, 106, 117] | [138, 155, 156] | [195] |
| Expression of endothelial cell adhesion molecules (ICAM‐1, ELAM‐1, E‐selectin, CD18) | [12, –, 14, 15, 16, 17, 22, –, 24] | [63, 83, 91] | [120] | [41, 43,45, 60, 63, 76, 77] | [108] | [138, 155] | [189, 195] |
| Soluble Intercellular adhesion molecules (sICAM‐1, sVCAM‐1, SE‐selectin) | [25, 26] | Not found | Not found | [44, 45, 78] | Not found | Not found | [208] |
| Leukocyte adhesion to endothelium | [10, 12, 24, 27] | [83, 87] | [123] | [50, –, 52] | [104, 105] | [137, 157] | [191, 220] |
| Red cell adhesion to endothelium | Not relevant | Not relevant | Not relevant | [32, 33, 43, 48] | Not relevant | Not relevant | Not relevant |
| Decreased neutrophil deformability and capillary trapping | [18] | [1, 90] | Not found | Not found | [103, 106, 111] | [146] | Not found |
| Leukostasis | [10, 19] | [83, 94, 100] | [120] | [75] | [103, 105] | [138, 156, 158] Aggregation | [191, 194] |
| Transepithelial migration | Not Reported | [83, 87, 94, 100] | [120] | [51] | [119] | Not found | [194] |
| Interaction of monocytes and Tlymphocytes | [10] | [101] | [120, 128, 131, 135] | [45, 52] | [100, 108] | [137, 138] | [189] |
| Cytokines | [8, 9] | [1, 83] | TNF, TGF‐β [129, –, 131, 132] | [53, –, 55, 56, 57, 79] | [100, 105, 108, –, 110] | [137, 160] | [189, 191, 195] |
| Release of oxidants, proteases, and cationic proteins | [10, 20, 21] | [87, 91, 94] | [120] | [49, 58, 60, 80, 81] | [112, –, 114] | [157, 160] | [189, –, 191, 200] |
| Inflammation | [10, 25] | [89] | [120, 131] | [59, 60] | [108] | [157, 160] | [199, 221] |
| Capillary changes | [10] | [90, 95, –, 97] | [120] | [60] | [118] | [138] | [206] |
| Microaneurism formation | [19] | Not found | Not found | Not found | Not found | Nof found | Notably Kawasaki disease |
| Hemorrhage | Retinal [19] | Not found | Not found | Not found | Not found | [157] | Not found |
| Fibrosis | [28] | Not found | (Glomerular) [131, 132] | [82] | [108, 115] | Not found | Myocardial |
| Macrophage MIF | [9, 29] | Not found | [126, 136] | Not found | Not found | Not found | Not found |
| Cardiac myocyte damage | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant | Not relevant | [191] |
| Decreased neutrophil apoptosis | Not found | [100] | Not found | Not found | [100] | Not found | Increased telomerase [189] |
| P‐selectirr‐induced platelet increase | Not found | Not found | [123] | Not found | Not found | Not found | Not found |
There are increased numbers of neutrophils throughout the retina in experimental diabetes in monkeys, and they are observed frequently within microaneurisms. There also is production of superoxide from activated neutrophils [20] and monocytes [21], along with a release of neutrophilic cationic proteins and proteases, which are key factors in producing endothelial damage, contributing to microaneurism formation and hemorrhage [19]. The several pathophysiological mechanisms relating neutrophils to the progression of diabetic retinopathy are listed (see Table 1) [22, –, 24, 25, 26, 27, 28, 29]. Other atypical forms of PKC have been proposed as being activated by hyperglycemia and contributing to tissue injury by inducing apoptosis of retinal vascular cells [30, 31].
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF SICKLE CELL DISEASE
A vaso‐occlusive sickle cell crisis results from a complex set of cellular interactions leading to the obstruction of blood flow to various organs, including muscle, bone, viscera, and brain. This process is initiated by adherence of sickle cells to vascular endothelium mediated by the interaction of integrin α4β1 with VCAM and fibronectin [32, 33], coupled with the red cell distortion and rigidity that result from sickling. The latter change is the result of lower oxygen tension in the microvasculature, which causes polymerization of sickle hemoglobin. The rate of this process is dependent on the concentration of intraerythrocytic sickle hemoglobin and the solubility state of the hemoglobin. Thus, rapid polymerization occurs when the red cells are dehydrated, and sickle hemoglobin concentration is high. Concurrent interactions of the sickle cell with neutrophils, platelets, and the endothelium of the vasculature significantly decrease the flow of blood, leading to pain, infarction, and organ damage.
Neutrophilia is a risk factor for a sickle crisis [34, –, 36, 37]. For example, the higher the white cell count, the greater the frequency of acute chest syndrome [38] and stroke [39]. Conversely, reduction of the count in patients with sickle cell disease is beneficial, as illustrated by the positive therapeutic effect of hydroxyurea in association with a decrease in neutrophil count but not an increase in red cell fetal hemoglobin [40].
The rolling and tethering of sickle red cells, as that of neutrophils loosely adherent to endothelium, involve the expression of endothelial P‐selectin [41]. The surface expression of the integrin α subunits, CD11a and CD11b, on neutrophils is not increased, but these integrins may have increased affinity for adhesion molecules on endothelial cells [42]. Reticulocytes in sickle cell patients have increased expression of α4β1 integrin and CD36, making them particularly adherent to endothelium [43]. CD36 also serves as a ligand for platelet thrombospondin, generated by activated platelets. Endothelial adhesion molecules, VCAM‐1, and soluble VCAM‐1, complementary to CD36, are increased in most sickle cell patients and are reduced by treatment with hydroxyurea [44].
Diminished L‐selectin expression and increased expression of β2 integrin on the neutrophil surface in sickle cell patients are associated with heightened shedding and elevated serum levels of L‐selectin [43, 45]. Neutrophil adhesion molecules interact with complementary endothelial molecules such as ICAM‐1, VCAM‐1, E‐selectin, and P‐selectin during vaso‐occlusive crisis [37, 46, –, 48]. Increased adhesiveness of neutrophils to fibronectin, sickle cells, and endothelium promotes vascular occlusion. Monocytes from patients with severe sickle syndromes are activated, possibly as a result of phagocytosis of senescent sickle red cells, other debris, or cellular microparticles [45]. These activated monocytes, in turn, activate endothelial cells via the production of TNF‐α, IL‐1 β, or both [45]. The endothelial cell activation results in increased expression of endothelial adhesion molecules for neutrophils, e.g., E‐selectin, P‐selectin, and ICAM‐1.
Although the precise temporal sequence of adhesion events of sickle cells, activated neutrophils, activated platelets, and altered endothelium is unclear, it appears that neutrophils adhere to endothelium, trap, and adhere to circulating and adherent sickle cells and platelets [49], which is a principal factor in vaso‐occlusion [50]. Adhesion assays using immobilized fibronectin [51] and TNF‐α‐activated endothelial cells [52] confirm the heightened adhesive properties of neutrophils from sickle cell patients. Neutrophil adhesion is augmented by exposure to IL‐8 by an increase in cAMP, and this effect is diminished when cAMP is inhibited [51].
Cytokines play a role in the genesis of vascular and cellular activation and thus, in the vaso‐occlusive crises. They contribute to the vaso‐occlusive event, initiated by sickle cell adherence, neutrophil and platelet adherence, and endothelial activation. Activated endothelial cells produce IL‐1, IL‐6, IL‐8, and TNF‐α; activated platelets elaborate IL‐1 and TNF‐α; and monocytes or macrophages secrete IL‐1 and TNF‐α. Plasma TNF‐α and sometimes IL‐1 are increased in patients with sickle cell disease [53, 54]. They induce further red cell and neutrophil adhesion to the vascular endothelium, activate the endothelium, and activate platelets [55]. The inflammatory cytokines, TNF‐α, IL‐1, and IL‐8, heighten the interaction of reticulocyte surface integrins with endothelial fibronectin and VCAM‐1. IL‐8 regulates the production of TNF‐α and IL‐1, which in turn, stimulates the production of chemokines that heighten the adhesion of leukocytes via their integrins, complementary to endothelial adhesion molecules, such as fibronectin [55, 56].
Endothelin‐1, a potent vasoconstricting agent, is increased in sickle cell patients during vaso‐occlusive crisis and may remain elevated for weeks thereafter [57]. This cytokine may be responsible for prolongation of symptoms secondary to vaso‐occlusion. Activated neutrophils produce increased amounts of ROS. Increased ROS and consequent reduced levels of the antioxidant glutathione are found in neutrophils and platelets from sickle cell patients [58]. In addition, COX is increased significantly in sickle cell neutrophils. As shown in transgenic sickle cell mice, hypoxia‐reoxygenation results in ROS generation in endothelial cells [59]. These processes coupled with reduced glutathione can exacerbate the inflammatory reaction. Endothelial and tissue damage as well as platelet activation and a coagulopathy may be related to generation of ROS ( Fig. 2 ).
Figure 2.
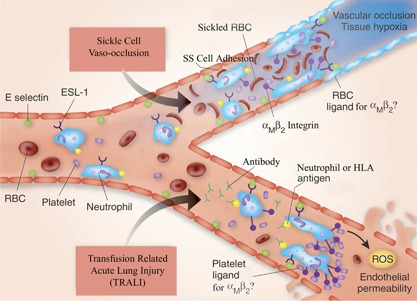
A schematic representation of the major pathophysiological events in the development of sickle cell vaso‐occlusion and TRALI. In sickle cell vaso‐occlusion, there is adhesion of neutrophils to the endothelial cell surface, mediated by E‐selectin ligand‐1 (ESL‐1) on the neutrophil and E‐selectin on the endothelial cell surface. Sickle cells (SS) also adhere to the endothelium via ligands (e.g., CD36, CD47, α4β1) interacting with endothelial cell vascular cell adhesion molecule‐1, αVβ3, fibronectin, thrombospondin, and others, in some cases, mediated by von Willebrand factor. Adhesion of red cells and platelets to neutrophils occurs and may be mediated by αMβ2 neutrophil integrins. The red cell and platelet ligands are uncertain. This intercellular reaction involving red cells, neutrophils, and platelets (and possibly monocytes) results in vascular occlusion and tissue hypoxia [60]. In TRALI, antibodies present in transfusion products with specificity against HNAs (e.g., HNA‐1a, ‐2a, or ‐3a, HLA‐A2) react with their requisite antigen and initiate neutrophil activation. A two‐event model is proposed for the pathophysiology of TRALI, in which the first is the underlying inflammatory disease, inducing activation of the vascular endothelium of the lung and consequent neutrophil sequestration. The second event is the antibody antigen‐induced activation of neutrophils, which adhere to E‐selectin on the endothelial surface via their surface E‐selectin ligand‐1, similar to the process in sickle cell vaso‐occlusion. Platelets bind to neutrophil αMβ2 by an as‐yet unknown, complementary molecule. The resultant production of ROS in conjunction with inflammatory cytokines and chemokines leads to capillary permeability and fluid leak and the clinical syndrome. The inflammatory cytokines are released from the endothelial and inflammatory cells, and this contributes to the vascular effects. TRALI, induced by HNA‐3a, is particularly severe, and this antigen is encoded by the gene for the choline transporter‐like protein‐2 (SLC44A2), in which a single nucleotide polymorphism substitutes arginine for glutamine at position 154 [61, 62]. An animal model of TRALI has been used to identify a possible alternative mechanism of the disease involving the transfusion of an antibody to MHC I antigen on endothelial cells in the lung and activation of neutrophils via their FcγR [63]. Antineutrophil antibodies are, however, the principal pathogenetic mechanism in humans [64]. SS, homozygous sickle hemoglobin. This figure is adapted from a figure in ref. [63].
Several observations support the inference that the neutrophil has a notable place in the pathogenetic sequence of sickle vaso‐occlusive episodes. The findings to support the singular role of the neutrophil include: first, data from several studies that indicate a correlation of the degree of leukocytosis (neutrophilia) with the morbidity of sickle disease patients [35, –, 37, 65]. In a study of sickle cell patients, the absolute neutrophil count increased step‐wise in three groups of sickle cell patients categorized by increasing severity of symptoms [66]. A higher white cell count early in pregnancy predicts a higher likelihood of subsequent sickle cell‐related, clinical problems, including painful crises and acute chest syndrome [34]. In a prospective study of 3764 sickle cell patients, those with a white cell count greater than 15.1 × 109/L had a higher risk of death [67]. Neutrophil growth factors, such as G‐CSFs, cause life‐threatening complications in sickle cell patients, presumably in large measure, as a result of neutrophilia [68]. Second, reduction of the blood neutrophil concentration diminishes the frequency and severity of vaso‐occlusive events [37, 40, 69]. Hydroxurea, the principal drug used to decrease the frequency of sickle vaso‐occlusive events, may have its major effect by decreasing blood neutrophil concentration. Other agents, such as 5‐azacytidine, reported to heighten fetal hemoglobin, also decrease the absolute neutrophil count [70]. The increase in fetal hemoglobin related to hydroxyurea or 5‐azacytidine treatment is unlikely to play a principal role in their salutary effects, as the increase resides primarily in a clonal cohort of hemoglobin F cells in insufficient proportion to influence sickle symptoms. Third, hydroxyurea increases L‐selectin shedding by neutrophils in patients with sickle cell disease and may, thereby, decrease their firm adhesion to endothelium and also decreases neutrophil H2O2 production [47]. Fourth, hydroxyurea diminishes the heightened adhesion of sickle red cells to endothelium, in part, as a result of its action to decrease the concentration of neutrophils in the circulation. In addition, the down‐regulation of various neutrophil adhesion molecules, particularly αMβ2 integrin, by hydroxyurea, may decrease the risk of vaso‐occlusion [71]. In confirmation of this effect, i.v. Ig administered to sickle cell mice inhibits neutrophil adhesion to endothelium and reverses vaso‐occlusion [50]. In addition, GMI‐1070, an investigational agent, studied in a humanized model of sickle cell vaso‐occlusion in mice and analyzed by intravital microscopy, principally blocks E‐selectin but also, P‐selectin and L‐selectin, resulting in decreased adhesion of sickle red cells to endothelial cells and to neutrophils and decreased adhesion of neutrophils to endothelium [72]. These effects were associated with improved microcirculatory blood flow and improved survival of the mice. Last, activated neutrophils in patients with sickle cell disease increase the exposure of red cell phosphatidylserine, which facilitates sickle red cell adhesion to endothelium [73], and hydroxyurea attenuates this process. These observations have led some to conclude that a decrease in blood neutrophil concentration and a deactivation of the adhesive interaction of neutrophils and sickle red cells comprise the primary mechanism of the beneficial effect of hydroxyurea treatment [47]. Hydroxyurea also may have a direct effect on other adhesion molecules on the red cell surface [48] and increase NO to enhance production of hemoglobin F, decrease sickle cell adhesion, or augment vasodilatation [74]. Further documentation of the pathophysiology of vaso‐occlusion in sickle disease is provided (see Table 1) [60, 75, –, 77, 78, 79, 80, 81, 82].
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF TRANSFUSION‐RELATED ACUTE LUNG INJURY
Acute respiratory distress with new bilateral lung infiltrates within 6 h of receipt of a blood transfusion in a recipient, not volume‐overloaded or in heart failure, is the essential finding of TRALI [83]. This complication occurs approximately once in every 5000 blood transfusions [83]. The prior use of G‐CSF may heighten this risk, and a transient decrease in the white cell count may be an early feature of the syndrome [84, 85]. The presence of antineutrophil antibodies is central to the development of most cases of TRALI. There are in vivo and ex vivo animal models to support the antibody‐mediated disease hypothesis [86]. The antibodies may react with HNAs, e.g., HNA‐3a, or HLAs, e.g., HLA‐A2, which have been associated with fatalities, or to HNA‐1a and ‐2a, which have been associated with less‐severe disease [87]. An animal model supports the requirement for neutrophils in this process [88]. Mice made neutropenic were resistant to antibody‐induced lung injury. Neutrophilia is not a feature of this transfusion reaction in humans and is not essential, as approximately one‐quarter of human blood neutrophils is in the pulmonary vascular bed at any time [87]. A “two‐event model” has been suggested in the pathophysiology of this syndrome. The initial event is the underlying illness, which induces activation of the vascular endothelium of the lung and subsequent neutrophil sequestration [87]. The second event involves antineutrophil antibody from a transfused blood product that primes and activates the adherent neutrophils and results in endothelial damage and capillary leak. An illustration of this process and a comparison with the vaso‐occlusive events in sickle cell disease are shown in Fig. 2 [63].
The adhesion of neutrophils in the pulmonary capillary bed may be aggravated by the anatomy. The neutrophil diameter is >50% of that of the capillaries connecting the pulmonary arterioles to the venules, requiring neutrophil deformation for passage [83, 89]. Thus, neutrophils do not tether, roll, and adhere as in other vessels. There is decreased deformability of the neutrophils from polymerization of their cytoplasmic actin filaments, which may be complement‐mediated [1], and this change contributes to neutrophil sequestration in the lung [83, 90]. Adhesion molecules, such as β2 integrins (αMβ2), undergo a conformational change from nonadherent to adherent, and these bind to ICAM‐1 on the endothelial surface [63, 83, 91], immobilizing neutrophils. There is consequent trapping of platelets, but the platelet adhesion molecules involved are not known [63]. The trapped neutrophils are activated by circulating cytokines (e.g., TNF‐α, IL‐6), chemokines, bioactive lipids from the transfusion product, platelet‐derived CD40 ligand, and antineutrophil antibodies. The cross‐linking of integrin or selectin adherence molecules by an antibody leads to an increased oxidative burst in stimulated neutrophils [92, 93]. Activation results in release of ROS and proteases [87, 91, 94], which further damage the endothelium [95, 96] and cause capillary leak and pulmonary edema. Blockage of ROS in an animal model prevented vascular leak, edema, and lung injury [90, 91]. Plasma lipids from platelet concentrates in the absence of antineutrophil antibodies also can precipitate TRALI in an animal model [97]. The animals were pretreated with LPS as the first event, and lysophosphatidylcholine served in lieu of antibody as the second event, resulting in endothelial damage and oxidase activity. This may explain the development of TRALI in ∼10% of patients in whom antibody cannot be demonstrated and is consistent with data indicating that lysophosphatidylcholine is a biologically active lipid that accumulates in stored blood components [98]. Additional data that support the pathophysiology described in regard to TRAILI are included (see Table 1) [99, –, 101].
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF THE ARDS
Acute lung injury or ARDS is defined as severe arterial hypoxemia reflected by a partial arterial pressure of oxygen divided by the fractional concentration of oxygen in the inspired air below 200 [102, 103]. It occurs directly, as a complication of pneumonia or aspiration, or indirectly, as a consequence of sepsis or trauma. The common feature of ARDS is injury to the alveolar capillary membrane, resulting in a failure to permit the transfer of oxygen from the alveolar sac to the pulmonary capillary circulation [103]. Activated neutrophils and cytokines from macrophages play a critical role in the failure of oxygen transport [100]. With lung injury, an early response is the release of TNF‐α and IL‐ 1β from macrophages [103]. Neutrophils and endothelial cells are activated. Neutrophil sequestration in the alveolar capillaries and interaction with other inflammatory cells, such as monocytes, macrophages, and lymphocytes, result [100]. The neutrophilic response, as measured in the fluid from BAL, is correlated directly with the severity of the impaired gas exchange and protein leak [104]; this response is correlated closely with neutrophil activation and adhesion [105]. Platelet activation of neutrophils via release of their stored mediators, including von Willebrand factor, tissue factor, chemokines, and cytokines, is part of the response [106, 107]. In addition, cytokines, chemokines, acute‐phase reactants, and coagulation factors are released from injured tissues, including TNF‐α, IL‐1, IL‐6, and IL‐8 [105, 108, –, 110]. Neutrophils exhibit decreased deformability, which augments their trapping in capillaries [106, 111]. Tethering, rolling, activation, and adhesion of neutrophils occur prior to transendothelial migration [94]. Stimulated neutrophils migrate along chemotactic gradients into the lung parenchyma [106]. Neutrophil migration across the epithelium is more complex than its transendothelial migration. The pathway appears to be longer (20 μm or more), takes an intercellular route, and proceeds from the basolateral to the apical surface [94]. Selectins are not involved in transepithelial migration of neutrophils, but Β1 integrins may mediate adhesion to fibroblasts or the interstitial matrix [94]. Neutrophils are retained at the apical epithelial surface by adhering to ICAM‐1 and by Fc interactions [94]; there is a decrease in neutrophil apoptosis proportional to the severity of sepsis [100]. Neutrophils release neutrophil elastase, metalloproteinases, defensins, and ROS locally, and if in sufficient numbers, neutrophils, thereby, contribute to tissue injury. Pro‐oxidant substrates, such as hypoxanthine, are increased in BALF from patients with ARDS, reflecting the heightened activity of oxidative reactions [112], whereas antioxidant mechanisms are impaired [113]. These toxic substances damage the epithelium so that there is heightened paracellular permeability and fluid leakage characteristic of the acute lung injury in ARDS [114]. Neutrophils are the principal cell type in BALF early in the development of ARDS; the fluid contains TNF, IL‐1, IL‐6, and IL‐8 [115]. The liberation of neutrophil‐derived proteases inhibits surfactant activity, thereby further diminishing lung function. Within 2 weeks of onset, a fibrinoproliferative process begins with the neutrophils in lung fluid replaced by alveolar macrophages, the proliferation of type 2 epithelial cells, and collagen deposition with evolution of lung fibrosis [115]. Other supporting data for the role of leukocytosis [116], neutrophil adhesion molecules [117], capillary leakage [118], and transepithelial migration of neutrophils [119] in the ARDS shown (see Table 1). Neutrophilia, a normal neutrophil count, or neutropenia may be observed in patients with ARDS. The circulatory neutrophil pool size is, however, less relevant than the intrapulmonary pool, which is characteristically engorged with neutrophils, as indicated by the lavage cellular content. There may be some limitation of the pulmonary neutrophil pool size in patients with neutropenia, and this may be associated with somewhat less‐severe manifestations.
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF IMMUNE RENAL VASCULITIS AND GLOMERULONEPHRITIS
ANCA lead to neutrophil activation, neutrophil sequestration in the glomerulus, and glomerular capillaritis [120]. This sequence of events results in the clinical manifestation of glomerulonephritis, manifested by proteinuria, hematuria, hypertension, and ultimately, renal failure. If the creatinine is above 4 mg/dl, the greater the neutrophilia, the poorer the clinical outcome [121]. This process is observed primarily in the proliferative forms of glomerulonephritis, characterized by glomerular hypercellularity, composed of neutrophils, macrophages, and T lymphocytes accumulating in the glomerular tuft and producing crescent formation. ANCA‐related glomerulonephritis, antiglomerulo‐basement membrane nephritis, and nephritis associated with systemic lupus erythematosis are the three major diseases that produce severe proliferative, crescentic glomerulonephritis [120]. In ANCA‐related glomerulonephritis, a loss of tolerance to MPO and PR3 released from neutrophil granules results in antibodies to these proteins [121, 122]. Neutrophils do not initially roll prior to adhesion in glomeruli [123], rather, they undergo “immediate arrest” within the glomerular capillaries. It has been proposed that P‐selectin, derived from platelets, serves as the endothelial adhesion molecule interacting with ICAM‐1 and CD11b/CD18 on neutrophils, which are up‐regulated by the activation of neutrophils by ANCA and possibly, C5a in an activation loop [124]. In confirmation of this pathogenesis, inhibition of ICAM‐1 and CD11b/CD18 or P‐selectin reduces leukocyte recruitment and the severity of experimental glomerulonephritis [123, 125]. Cytokines, including IFN‐γ from CD4 lymphocytes, interact with CD40 to activate macrophages in concert with macrophage MIF, the latter involved in macrophage recruitment and activation [126]. TNF‐α plays a role in ANCA‐induced glomerulonephritis, as leukocyte recruitment is decreased after anti‐TNF antibody treatment in experimental animals [127]. An interaction of neutrophils and macrophages with Th1‐type Th cells plays a prominent role in producing the proliferative and crescentic form of glomerulonephritis [128]. The Th1‐type Th cells produce IFN‐γ, IL‐2, lymphotoxin‐α, and TNF‐β, which can induce neutrophil, macrophage, and cytotoxic T cell activation [128]. Inflammatory cytokines, such as IL‐1, IL‐6, and TNF‐α, are also produced by macrophages and to a lesser extent, by resident glomerular cells [129, 130]. Macrophages, tubular cells, and myofibroblasts produce and secrete fibrosis‐promoting molecules, including TGF‐β, and there are reduced levels of antifibrotic factors, leading ultimately to renal fibrosis and chronic kidney disease [131]. Attenuation of renal fibrosis occurs if there is a reduction of TGF‐β [132]. The PDGF group of cytokines also contributes to myofibroblast proliferation and renal scarring [133]. Additional evidence for the involvement of leukocytosis [134], neutrophil adhesion molecules [134], leukocytes [135], and MIF [136] in ANCA‐mediated glomerulonephritis is shown (see Table 1).
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF STROKE
The accumulation of neutrophils in ischemic brain regions has been appreciated for years, and mechanisms investigating their potential cytotoxic effects in this context remain an area of active investigation [137, 138]. Epidemiological studies indicate patients with neutrophil counts >8.2 × 109 cells/L have a higher risk of stroke compared with controls. In these studies, neutrophilia also predicted poor outcome after stroke, and these associations were not influenced by the use of aspirin or clopidogrel [139]. Neutrophil accumulation within brain territories begins within 6 h of ischemia onset, lasting up to 6 days, and in many studies, the degree of infiltration correlates with the severity of the neurological injury [140, 141] Results from diffusion‐weighted MRI indicate that early neutrophilia, but not elevated lymphocyte counts, is associated with increased infarct volumes after stroke [142]. Similar trends have been reported using serum markers of neutrophil activation, including gelatinase‐associated lipocalin, PR4, and TNF‐α, which were elevated after acute ischemic stroke [143, 144]. The expression of leukocyte adhesion molecules is also increased in patients admitted to the hospital after a stroke [145].
Preclinical modeling supports the notion that neutrophils alter microcirculatory homeostasis, causing what has been termed the “no‐reflow” phenomenon [146]. Central to these events is the interaction between neutrophil activation and adherence to endothelium and transendothelial migration; the contributing factors include MCP‐1 and the intercellular adhesion molecules, ICAM‐1, integrin‐β2 (CD18), PECAM‐1, and others [147]. Their expression is causally related to circulatory defects after stroke. For example, anti‐CD18 mAb inhibit neutrophil adherence to the vasculature and suppressed the no‐reflow phenomenon in a baboon model of focal cerebral ischemia [148]. Also, loss of CD47 expression reduced neutrophil accumulation, matrix metalloproteinase‐9 activity, and cerebral edema after transient middle cerebral artery occlusion in mice [149]. Targeted disruption of P‐selectin (an early‐phase endothelial marker involved in neutrophil recruitment) is protective against cerebral ischemia in mouse models [150]. It is also clear that ischemia induces rapid changes in the expression of genes, induction of cell surface molecules, and release of soluble markers associated with neutrophil activation. Two such targets induced by hypoxic vascular endothelia include ICAM‐5 and MCP‐1 [151]. The extent to which neutrophils participate in tissue damage depends on the type and severity of ischemic injury, which in turn, define the range of neutrophil‐specific, interacting molecules activated. For example, although blockade with CD18‐specific antibodies protects against transient focal ischemia, there is no effect after middle cerebral artery occlusion [152]. These data highlight the particularly important role of neutrophils in the pathological response associated with reperfusion injury. In this regard, although inhibiting neutrophil function with anti‐CD18 antisera alone was not beneficial in a preclinical model of focal stroke, when given in combination with tPA, it augmented protection and extended the therapeutic window for thrombolytic administration [153]. Other data supporting the role of leukocytosis [154], neutrophil adhesion [155, –, 157], leukostasis [156, 158, 159], and resultant inflammation [160] in the development of a stroke are shown (see Table 1).
Results from a number of genetic and pharmacological studies support a pathological role for PMNs after stroke. ROS generated through the NADPH oxidase respiratory burst produce direct injury to the vascular endothelium and brain parenchyma. Pharmacological inhibition or genetic ablation of NADPH oxidase function is neuroprotective by blocking neutrophil superoxide generation [161]. Other factors, including peroxisome proliferator‐activated receptor‐γ, C/EBP‐β, and PKC‐δ, have been targeted, resulting in reductions in neutrophil accumulation and neuronal injury after cerebral ischemia [162, 163]. Loss of PKC‐δ appears particularly important after stroke‐reperfusion injury, as gene loss‐of‐function was associated with impaired adhesion, migration, ROS generation, and degranulation of blood neutrophils [164]. Also, pretreatment with the platelet‐activating factor receptor antagonist LAU‐0901 reduced infarct volumes via its antineutrophil and anti‐inflammatory properties [165]. Elastases secreted by neutrophils are elevated after stroke and contribute to tissue degradation [166]. Consequently, the small molecule elastase inhibitors, ONO‐5046 and sivelestat, decrease neuronal damage following transient global and focal ischemia, respectively [167, 168]. The SLPI (antileukoproteinase), a highly cationic, single‐chain protein with eight intramolecular disulfide bonds, is an endogenous inhibitor of elastase and other leukocyte serine proteases and exhibits potent anti‐inflammatory properties [169]. As predicted, adenoviral‐mediated expression of SLPI in the cortex prior to ischemia protects against tissue injury [170]. Although nonspecific in nature, ischemic preconditioning limits brain injury after stroke by reducing inflammatory gene expression (e.g., cytokines, chemokines, adhesion molecules) and the net infiltration of neutrophils into the ischemic cortex [171].
Several risk factors commonly linked with stroke are also associated with neutrophil activation. For example, the AGE products associated with the diabetic state reduce the deformability and enhance the intrinsic respiratory burst activity of neutrophils [172, 173]. Similarly, although smoking and hypertension are associated with neutrophilia, tobacco use further impairs neutrophil membrane compliance [174, 175] Elevated plasma homocysteine, which is associated with accelerated rates of atherosclerosis and small vessel stroke, enhances neutrophil‐endothelial interactions in vivo [176]. Conversely, moderate alcohol use, which reduces the risk of stroke, also reduces the expression of the neutrophil adhesion molecule CD18 [177]. Given the common occurrence of diabetes, hypertension, and tobacco use, it is plausible that when combined, these variables may produce synergistic and deleterious effects on the cerebral microcirculation during stroke, in part, as a result of their effects on neutrophil function.
Neutrophilia plays a central role in several systemic disorders in which stroke is frequently encountered as a complication. For example, in Wegenerˈs granulomatosis, ANCA titers correlate with disease activity and cause cerebrovascular damage through antibody‐mediated release of proteolytic enzymes and the priming of neutrophil reactivity against the endothelium [178]. Surface expression of neutrophil adhesion molecules, including β‐2 integrin and L‐selectin, is increased in pregnant women who progress to eclampsia before the clinical features of disorder are manifest [179]. In view of these observations, whether a result of local hypoxia‐ischemia or in response to a distinct stimulus, neutrophilia and stroke coexist in a number of non‐neurological, systemic conditions.
In addition to the risk of aspiration‐related pneumonia, inflammatory cytokines released after ischemic stroke impair immune defenses, leaving the host particularly susceptible to nosocomial infection [180]. Despite this risk, antineutrophil therapies are starting to make their debut. Based on the ability of the neutrophil inhibitory factor UK‐279‐276 to reduce infarct volume after reperfusion in animal models, the safety and efficacy of the drug have been tested in combination with rtPA, administered for acute stroke [181]. Although the trial was stopped early for futility, the experimental drug was generally well‐tolerated and did not appear to increase the incidence of infectious complications. The endogenous cytokine IL‐1Rα also protects against cerebral injury in a range of neuropathological conditions including stroke. i.v. delivery of IL‐1Rα, followed by a 2‐mg/kg/h infusion over 72 h, produced no adverse events and lowered neutrophil, C‐reactive protein, and IL‐6 levels in treated patients [182]. Data regarding imaging and clinical outcomes supported improvements in the treatment group compared with controls receiving placebo.
Although most studies cast the neutrophil in primarily pathological roles, some reports indicate that they play only a limited role in this regard and may in fact provide some benefit after stroke [183, –, 185]. Studies primarily done in rodent systems suggest that the rheological and pathological changes that occur early after reperfusion cannot be explained by the actions of the neutrophil, given the delayed profile of their accumulation. In this context, neutrophilia may simply act as a marker rather than a participant in the pathological response. Neutrophil depletion decreases levels of several growth factors, including brain‐derived neurotrophic factor and VEGF, attenuating induced focal angiogenesis in the mature mouse brain [186]. Moreover, G‐CSF treatment, which produces neutrophilia in WT and G‐CSF knockout mice, is, in fact, protective against transient focal ischemia [187]. Thus, taking these observations into consideration, it may be possible that PMNs can have protective and harmful effects in the ischemic brain depending on additional cues that influence the highly regulated process of neutrophil activation.
NEUTROPHILS IN THE PATHOPHYSIOLOGY OF THE ACUTE CORONARY SYNDROME
Myocardial infarction is most often caused by the acute thrombotic occlusion of patent coronary arteries rather than from chronic, progressive luminal narrowing from atherosclerosis. Progression of the intimal fatty streak (composed of lipid droplets and foam cells) to the unstable plaque involves the accumulation of, and signaling between, immune components, including DCs, mast cells, B cells, and NK cells [188]. These cellular elements also contribute to plaque instability through the induction of proteases, free radicals, and vasoactive molecules [189, –, 191]. Rupture of the unstable fibrous cap and release of the tissue factor‐rich lipid core precipitate focal platelet activation and intraluminal thrombosis. Neutrophil leukostasis occurs at sites of plaque rupture and is associated with endothelial dysfunction and the expression of neutrophil and vascular endothelial adhesion molecules [188, 191, –, 193, 194, 195]. Acute coronary syndrome is also associated with several markers of intrinsic neutrophil activation. Compared with blood neutrophils, those associated with coronary plaques, harvested during percutaneous coronary intervention, exhibit increased telomerase activity, which is a marker reflecting a prolonged neutrophil lifespan [196]. Premature telomere shortening promotes apoptosis in postmitotic cells, whereas enhanced telomerase activity promotes survival [197, 198]. Local enrichment of activated “survivor” plaque neutrophils could exaggerate local inflammatory responses and promote stent restenosis [199]. In addition, neutrophils obtained from patients with acute coronary disease exhibit twice the respiratory burst activity found in patients with stable angina [200]. Analyses of autopsy material from patients who succumbed to acute myocardial infarction indicate that neutrophils are found more frequently in ruptured rather than eroded or stable plaques [201], and analyses using the marker anti‐BP‐30 indicate that neutrophils were present in 72% of ruptures but not in intact fibroatheromas [202]. These data support the assumption that neutrophils participate in plaque rupture and in situ coronary thrombosis.
Various markers related to neutrophil activation have been linked with acute coronary disease. For example, neutrophilia alone is a risk factor for adverse cardiac events, including cardiogenic death, heart failure, and nonfatal myocardial infarction [203, –, 205]. High levels of C‐reactive protein, indicative of systemic inflammation, correlate positively with absolute neutrophil counts but not with increases in other blood cell types [206]. In contrast, T lymphocyte activity is lower in patients with acute coronary syndrome, and a high neutrophil‐to‐lymphocyte ratio may be used to stratify patients by risk of a poor outcome, as it appears to predict in‐hospital and 6‐month mortality [207]. Soluble forms of adhesion molecules, including soluble VCAM‐1 and soluble P‐selectin (markers reflecting endothelial activation), are also elevated in the serum of patients with angina pectoris, referred for coronary angiography [208]. Platelet‐neutrophil interactions are increased fourfold in the circulating venous blood of patients with unstable angina [209].
Aside from the potential pathological association with the unstable plaque, neutrophils may play a particularly important role in promoting myocardial injury after reperfusion [210]. Neutrophil activation in this context could aggravate thrombosis and microvascular dysfunction through the release of vasoactive substances, including platelet activating factor, thromboxane A2, and leukotriene B4 [211]. Similarly, elevated neutrophil counts observed after percutaneous coronary intervention for ST‐segment elevation correlate with the size of the myocardial infarct and resultant left ventricular function [212]. In patients undergoing cardiopulmonary bypass, neutrophil depletion lowered the prevalence of low cardiac index and reperfusion ventricular fibrillation and lowered cardiac enzyme levels postoperatively, suggesting that neutrophils play a pathological role in postrevascularization recovery [213]. However, whereas a single injection of the anti‐CD18 antibody prior to reperfusion in a canine model of acute coronary syndrome limited myocardial infarct size by close to 50% and produced improved global and regional left ventricular function [214], its use in humans was not efficacious [215]. The failure of attempts to block neutrophil function in acute coronary disease in humans has made the concept that neutrophils play a central role in reperfusion injury controversial [216, 217]. However, as β1‐integrin‐VCAM interactions also mediate neutrophil emigration into the heart, achieving a detectable clinical response may require targeting several adhesion molecules [218]. Other evidence supporting a pathophysiologic role for neutrophils in the genesis of acute coronary artery disease is shown in Table 1 [219, 220].
CONCLUDING REMARKS
Neutrophils may contribute to tissue‐specific injury in certain inflammatory states initiated by various incidents. The mechanisms involved include inappropriate microvascular neutrophil adhesion, decreased neutrophil deformability and capillary trapping, release of cytokines, release of oxidants, and release of proteases, each contributing to an exaggerated inflammatory response, which can be more harmful than beneficial. In the pathophysiological processes, the neutrophil can be a key participant, although often not the sole factor in inducing and sustaining detrimental tissue effects. Four compelling observations that neutrophils are key pathogenetic factors in specific tissue injury have been made in sickle cell vaso‐occlusive crisis, in the extent of injury in experimental models of stroke, in the severity of vascular disease in TRALI, and in the amelioration of experimental glomerulonephritis. In human studies and in humanized animal models of sickle cell vaso‐occlusive crisis, reduction of neutrophils or blockade of neutrophil adhesion to sickle cells or endothelium [72] ameliorated the frequency or severity of crisis. In stroke, pharmacological inhibition or genetic ablation of NADPH oxidase function is neuroprotective by blocking neutrophil superoxide generation [161]. Elastases secreted by neutrophils are elevated after stroke and contribute to tissue degradation [166]. Consequently, the small molecule elastase inhibitors, ONO‐5046 and sivelestat, decrease neuronal damage following transient global and focal ischemia, respectively [167, 168]. Adenoviral‐mediated expression of SLPI in the cortex prior to ischemia protects against tissue injury [170]. In experimental animal models of TRALI, blockade of ROS released from neutrophils prevented vascular leak, edema, and lung injury [90, 91]. Inhibition of ICAM‐1 and CD11b/CD18 or P‐selectin reduces leukocyte recruitment and the severity of experimental glomerulonephritis [123, 125]. Thus, the question has been posed, appropriately, whether decreasing neutrophil adhesion, limiting the neutrophil secretory response, and/or decreasing the neutrophil count, if normal or elevated, may be useful in the treatment of these conditions. We anticipate an increase in research focused on methods to accomplish these goals in ways that provide a satisfactory benefit:risk therapeutic relationship.
AUTHORSHIP
G.B.S. and M.A.L. conceived of the review topic and wrote the major portion of the paper. M.W.H. wrote the sections about stroke and acute coronary occlusion.
ACKNOWLEDGMENTS
The authors express their gratitude to Drs. Niels Borregaard (Copenhagen, Denmark) and C. Wayne Smith (Houston, TX, USA) for reading the manuscript and providing helpful suggestions. They bear no responsibility, however, for any errors of fact or interpretation that might be contained herein.
REFERENCES
- 1. Silliman, C. C. , McLaughlin, N. J. (2006) Transfusion‐related acute lung injury. Blood Rev. 20, 139–159. [DOI] [PubMed] [Google Scholar]
- 2. Setter, S. M. , Campbell, R. K. , Cahoon, C. J. (2003) Biochemical pathways for microvascular complications of diabetes mellitus. Ann. Pharmacother. 37, 1858–1866. [DOI] [PubMed] [Google Scholar]
- 3. Liu, X. J. , He, A. B. , Chang, Y. S. , Fang, F. D. (2006) Atypical protein kinase C in glucose metabolism. Cell. Signal. 18, 2071–2076. [DOI] [PubMed] [Google Scholar]
- 4. Das Evcimen, N. , King, G. L. (2007) The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 55, 498–510. [DOI] [PubMed] [Google Scholar]
- 5. Bloomgarden, Z. T. (2002) The epidemiology of complications. Diabetes Care 25, 924–932. [DOI] [PubMed] [Google Scholar]
- 6. Aiello, L. P. , Bursell, S. E. , Clermont, A. , Duh, E. , Ishii, H. , Takagi, C. , Mori, F. , Ciulla, T. A. , Ways, K. , Jirousek, M. , Smith, L. E. , King, G. L. (1997) Vascular endothelial growth factor‐induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective β‐isoform‐selective inhibitor. Diabetes 46, 1473–1480. [DOI] [PubMed] [Google Scholar]
- 7. Tong, P. C. , Lee, K. F. , So, W. Y. , Ng, M. H. , Chan, W. B. , Lo, M. K. , Chan, N. N. , Chan, J. C. (2004) White blood cell count is associated with macro‐ and microvascular complications in Chinese patients with type 2 diabetes. Diabetes Care 27, 216–222. [DOI] [PubMed] [Google Scholar]
- 8. Maeda, M. , Fujio, Y. , Azuma, J. (2006) MTHFR gene polymorphism and diabetic retinopathy. Curr. Diabetes Rev. 2, 467–476. [DOI] [PubMed] [Google Scholar]
- 9. Mitamura, Y. , Harada, C. , Harada, T. (2005) Role of cytokines and trophic factors in the pathogenesis of diabetic retinopathy. Curr. Diabetes Rev. 1, 73–81. [DOI] [PubMed] [Google Scholar]
- 10. Chibber, R. , Ben‐Mahmud, B. M. , Chibber, S. , Kohner, E. M. (2007) Leukocytes in diabetic retinopathy. Curr. Diabetes Rev. 3, 3–14. [DOI] [PubMed] [Google Scholar]
- 11. Karadayi, K. , Top, C. , Gulecek, O. (2003) The relationship between soluble L‐selectin and the development of diabetic retinopathy. Ocul. Immunol. Inflamm. 11, 123–129. [DOI] [PubMed] [Google Scholar]
- 12. Morigi, M. , Angioletti, S. , Imberti, B. , Donadelli, R. , Micheletti, G. , Figliuzzi, M. , Remuzzi, A. , Zoja, C. , Remuzzi, G. (1998) Leukocyte‐endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF‐κB‐dependent fashion. J. Clin. Invest. 101, 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baumgartner‐Parzer, S. M. , Wagner, L. , Pettermann, M. , Gessl, A. , Waldhausl, W. (1995) Modulation by high glucose of adhesion molecule expression in cultured endothelial cells. Diabetologia 38, 1367–1370. [DOI] [PubMed] [Google Scholar]
- 14. Takami, S. , Yamashita, S. , Kihara, S. , Kameda‐Takemura, K. , Matsuzawa, Y. (1998) High concentration of glucose induces the expression of intercellular adhesion molecule‐1 in human umbilical vein endothelial cells. Atherosclerosis 138, 35–41. [DOI] [PubMed] [Google Scholar]
- 15. Kado, S. , Wakatsuki, T. , Yamamoto, M. , Nagata, N. (2001) Expression of intercellular adhesion molecule‐1 induced by high glucose concentrations in human aortic endothelial cells. Life Sci. 68, 727–737. [DOI] [PubMed] [Google Scholar]
- 16. Altannavch, T. S. , Roubalova, K. , Kucera, P. , Andel, M. (2004) Effect of high glucose concentrations on expression of ELAM‐1, VCAM‐1 and ICAM‐1 in HUVEC with and without cytokine activation. Physiol. Res. 53, 77–82. [PubMed] [Google Scholar]
- 17. Miyamoto, K. , Khosrof, S. , Bursell, S. E. , Rohan, R. , Murata, T. , Clermont, A. C. , Aiello, L. P. , Ogura, Y. , Adamis, A. P. (1999) Prevention of leukostasis and vascular leakage in streptozotocin‐induced diabetic retinopathy via intercellular adhesion molecule‐1 inhibition. Proc. Natl. Acad. Si USA 96, 10836–10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Linderkamp, O. , Ruef, P. , Zilow, E. P. , Hoffmann, G. F. (1999) Impaired deformability of erythrocytes and neutrophils in children with newly diagnosed insulin‐dependent diabetes mellitus. Diabetologia 42, 865–869. [DOI] [PubMed] [Google Scholar]
- 19. Kim, S. Y. , Johnson, M. A. , McLeod, D. S. , Alexander, T. , Hansen, B. C. , Lutty, G. A. (2005) Neutrophils are associated with capillary closure in spontaneously diabetic monkey retinas. Diabetes 54, 1534–1542. [DOI] [PubMed] [Google Scholar]
- 20. Ohmori, M. , Harada, K. , Kitoh, Y. , Nagasaka, S. , Saito, T. , Fujimura, A. (2000) The functions of circulatory polymorphonuclear leukocytes in diabetic patients with and without diabetic triopathy. Life Sci. 66, 1861–1870. [DOI] [PubMed] [Google Scholar]
- 21. Hiramatsu, K. , Arimori, S. (1988) Increased superoxide production by mononuclear cells of patients with hypertriglyceridemia and diabetes. Diabetes 37, 832–837. [DOI] [PubMed] [Google Scholar]
- 22. Song, H. , Wang, L. , Hui, Y. (2007) Expression of CD18 on the neutrophils of patients with diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 245, 24–31. [DOI] [PubMed] [Google Scholar]
- 23. Moore, T. C. , Moore, J. E. , Kaji, Y. , Frizzell, N. , Usui, T. , Poulaki, V. , Campbell, I. L. , Stitt, A. W. , Gardiner, T. A. , Archer, D. B. , Adamis, A. P. (2003) The role of advanced glycation end products in retinal microvascular leukostasis. Invest. Ophthalmol. Vis. Sci. 44, 4457–4464. [DOI] [PubMed] [Google Scholar]
- 24. Barouch, F. C. , Miyamoto, K. , Allport, J. R. , Fujita, K. , Bursell, S. E. , Aiello, L. P. , Luscinskas, F. W. , Adamis, A. P. (2000) Integrin‐mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest. Ophthalmol. Vis. Sci. 41, 1153–1158. [PubMed] [Google Scholar]
- 25. Van Hecke, M. V. , Dekker, J. M. , Nijpels, G. , Moll, A. C. , Heine, R. J. , Bouter, L. M. , Polak, B. C. , Stehouwer, C. D. (2005) Inflammation and endothelial dysfunction are associated with retinopathy: the Hoorn Study. Diabetologia 48, 1300–1306. [DOI] [PubMed] [Google Scholar]
- 26. Mastej, K. , Adamiec, R. (2008) Neutrophil surface expression of CD11b and CD62L in diabetic microangiopathy. Acta Diabetol. 45, 183–190. [DOI] [PubMed] [Google Scholar]
- 27. Hirata, F. , Yoshida, M. , Ogura, Y. (2006) High glucose exacerbates neutrophil adhesion to human retinal endothelial cells. Exp. Eye Res. 82, 179–182. [DOI] [PubMed] [Google Scholar]
- 28. Ban, C. R. , Twigg, S. M. (2008) Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 4, 575–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tashimo, A. , Mitamura, Y. , Nagai, S. , Nakamura, Y. , Ohtsuka, K. , Mizue, Y. , Nishihira, J. (2004) Aqueous levels of macrophage migration inhibitory factor and monocyte chemotactic protein‐1 in patients with diabetic retinopathy. Diabet. Med. 21, 1292–1297. [DOI] [PubMed] [Google Scholar]
- 30. Liu, X. J. , He, A. B. , Chang, Y. S. , Fang, F. D. (2006) Atypical protein kinase C in glucose metabolism. Cell. Signal. 18, 2071–2076. [DOI] [PubMed] [Google Scholar]
- 31. Geraldes, P. , Hiraoka‐Yamamoto, J. , Matsumoto, M. , Clermont, A. , Leitges, M. , Marette, A. , Aiello, L. P. , Kern, T. S. , King, G. L. (2009) Activation of PKC‐δ and SHP‐1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 15, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoover, R. , Rubin, R. , Wise, G. , Warren, R. (1979) Adhesion of normal and sickle erythrocytes to endothelial monolayer cultures. Blood 54, 872–876. [PubMed] [Google Scholar]
- 33. Hebbel, R. P. , Yamada, O. , Moldow, C. F. , Jacob, H. S. , White, J. G. , Eaton, J. W. (1980) Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J. Clin. Invest. 65, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Litos, M. , Sarris, I. , Bewley, S. , Seed, P. , Okpala, I. , Oteng‐Ntim, E. (2007) White blood cell count as a predictor of the severity of sickle cell disease during pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 133, 169–172. [DOI] [PubMed] [Google Scholar]
- 35. Quinn, C. T. , Lee, N. J. , Shull, E. P. , Ahmad, N. , Rogers, Z. R. , Buchanan, G. R. (2008) Prediction of adverse outcomes in children with sickle cell anemia: a study of the Dallas Newborn Cohort. Blood 111, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller, S. T. , Sleeper, L. A. , Pegelow, C. H. , Enos, L. E. , Wang, W. C. , Weiner, S. J. , Wethers, D. L. , Smith, J. , Kinney, T. R. (2000) Prediction of adverse outcomes in children with sickle cell disease. N. Engl. J. Med. 342, 83–89. [DOI] [PubMed] [Google Scholar]
- 37. Okpala, I. (2004) The intriguing contribution of white blood cells to sickle cell disease—a red cell disorder. Blood Rev. 18, 65–73. [DOI] [PubMed] [Google Scholar]
- 38. Castro, O. , Brambilla, D. J. , Thorington, B. , Reindorf, C. A. , Scott, R. B. , Gillette, P. , Vera, J. C. , Levy, P. S. (1994) The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 84, 643–649. [PubMed] [Google Scholar]
- 39. Ohene‐Frempong, K. , Weiner, S. J. , Sleeper, L. A. , Miller, S. T. , Embury, S. , Moohr, J. W. , Wethers, D. L. , Pegelow, C. H. , Gill, F. M. (1998) Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91, 288–294. [PubMed] [Google Scholar]
- 40. Charache, S. , Terrin, M. L. , Moore, R. D. , Dover, G. J. , Barton, F. B. , Eckert, S. V. , McMahon, R. P. , Bonds, D. R. (1995) Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 332, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 41. Matsui, N. M. , Borsig, L. , Rosen, S. D. , Yaghmai, M. , Varki, A. , Embury, S. H. (2001) P‐selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood 98, 1955–1962. [DOI] [PubMed] [Google Scholar]
- 42. Canalli, A. A. , Franco‐Penteado, C. F. , Saad, S. T. , Conran, N. , Costa, F. F. (2008) Increased adhesive properties of neutrophils in sickle cell disease may be reversed by pharmacological nitric oxide donation. Haematologica 93, 605–609. [DOI] [PubMed] [Google Scholar]
- 43. Harlan, J. M. (2000) Introduction: anti‐adhesion therapy in sickle cell disease. Blood 95, 365–367. [PubMed] [Google Scholar]
- 44. Saleh, A. W. , Hillen, H. F. , Duits, A. J. (1999) Levels of endothelial, neutrophil and platelet‐specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 102, 31–37. [DOI] [PubMed] [Google Scholar]
- 45. Belcher, J. D. , Marker, P. H. , Weber, J. P. , Hebbel, R. P. , Vercellotti, G. M. (2000) Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso‐occlusion. Blood 96, 2451–2459. [PubMed] [Google Scholar]
- 46. Fadlon, E. , Vordermeier, S. , Pearson, T. C. , Mire‐Sluis, A. R. , Dumonde, D. C. , Phillips, J. , Fishlock, K. , Brown, K. A. (1998) Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood 91, 266–274. [PubMed] [Google Scholar]
- 47. Benkerrou, M. , Delarche, C. , Brahimi, L. , Fay, M. , Vilmer, E. , Elion, J. , Gougerot‐Pocidalo, M. A. , Elbim, C. (2002) Hydroxyurea corrects the dysregulated L‐selectin expression and increased H(2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood 99, 2297–2303. [DOI] [PubMed] [Google Scholar]
- 48. Johnson, C. , Telen, M. J. (2008) Adhesion molecules and hydroxyurea in the pathophysiology of sickle cell disease. Haematologica 93, 481–485. [DOI] [PubMed] [Google Scholar]
- 49. Hofstra, T. C. , Kalra, V. K. , Meiselman, H. J. , Coates, T. D. (1996) Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood 87, 4440–4447. [PubMed] [Google Scholar]
- 50. Chang, J. , Shi, P. A. , Chiang, E. Y. , Frenette, P. S. (2008) Intravenous immunoglobulins reverse acute vaso‐occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood 111, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Canalli, A. A. , Franco‐Penteado, C. F. , Traina, F. , Saad, S. T. , Costa, F. F. , Conran, N. (2007) Role for cAMP‐protein kinase A signaling in augmented neutrophil adhesion and chemotaxis in sickle cell disease. Eur. J. Haematol. 79, 330–337. [DOI] [PubMed] [Google Scholar]
- 52. Finnegan, E. M. , Turhan, A. , Golan, D. E. , Barabino, G. A. (2007) Adherent leukocytes capture sickle erythrocytes in an in vitro flow model of vaso‐occlusion. Am. J. Hematol. 82, 266–275. [DOI] [PubMed] [Google Scholar]
- 53. Francis Jr., R. B. , Haywood, L. J. (1992) Elevated immunoreactive tumor necrosis factor and interleukin‐1 in sickle cell disease. J. Natl. Med. Assoc. 84, 611–615. [PMC free article] [PubMed] [Google Scholar]
- 54. Croizat, H. (1994) Circulating cytokines in sickle cell patients during steady state. Br. J. Haematol. 87, 592–597. [DOI] [PubMed] [Google Scholar]
- 55. Makis, A. C. , Hatzimichael, E. C. , Bourantas, K. L. (2000) The role of cytokines in sickle cell disease. Ann. Hematol. 79, 407–413. [DOI] [PubMed] [Google Scholar]
- 56. Assis, A. , Conran, N. , Canalli, A. A. , Lorand‐Metze, I. , Saad, S. T. , Costa, F. F. (2005) Effect of cytokines and chemokines on sickle neutrophil adhesion to fibronectin. Acta Haematol. 113, 130–136. [DOI] [PubMed] [Google Scholar]
- 57. Graido‐Gonzalez, E. , Doherty, J. C. , Bergreen, E. W. , Organ, G. , Telfer, M. , McMillen, M. A. (1998) Plasma endothelin‐1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso‐occlusive sickle crisis. Blood 92, 2551–2555. [PubMed] [Google Scholar]
- 58. Amer, J. , Ghoti, H. , Rachmilewitz, E. , Koren, A. , Levin, C. , Fibach, E. (2006) Red blood cells, platelets and polymorphonuclear neutrophils of patients with sickle cell disease exhibit oxidative stress that can be ameliorated by antioxidants. Br. J. Haematol. 132, 108–113. [DOI] [PubMed] [Google Scholar]
- 59. Kaul, D. K. , Hebbel, R. P. (2000) Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J. Clin. Invest. 106, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frenette, P. S. (2002) Sickle cell vaso‐occlusion: multistep and multicellular paradigm. Curr. Opin. Hematol. 9, 101–106. [DOI] [PubMed] [Google Scholar]
- 61. Greinacher, A. , Wesche, J. , Hammer, E. , Fürll, B. , Völker, U. , Reil, A. , Bux, J. (2010) Characterization of the human neutrophil alloantigen‐3a. Nat. Med. 16, 45–48. [DOI] [PubMed] [Google Scholar]
- 62. Curtis, B. R. , Cox, N. J. , Sullivan, M. J. , Konkashbaev, A. , Bowens, K. , Hansen, K. , Aster, R. H. (2010) The neutrophil alloantigen HNA‐3a (5b) is located on choline transporter‐like protein 2 and appears to be encoded by an R>Q154 amino acid substitution. Blood 115, 2073–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Looney, M. R. , Matthay, M. A. (2009) Neutrophil sandwiches injure the microcirculation. Nat. Med. 15, 364–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Frenette, P. S. , Mohandas, N. (2010) Bad blood: a trigger for TRALI. Nat. Med. 16, 382–383. [DOI] [PubMed] [Google Scholar]
- 65. Coller, B. S. (2005) Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler. Thromb. Vasc. Biol. 25, 658–670. [DOI] [PubMed] [Google Scholar]
- 66. Anyaegbu, C. C. , Okpala, I. E. , Akren'Ova, Y. A. , Salimonu, L. S. (1998) Peripheral blood neutrophil count and candidacidal activity correlate with the clinical severity of sickle cell anaemia (SCA). Eur. J. Haematol. 60, 267–268. [DOI] [PubMed] [Google Scholar]
- 67. Platt, O. S. , Brambilla, D. J. , Rosse, W. F. , Milner, P. F. , Castro, O. , Steinberg, M. H. , Klug, P. P. (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 330, 1639–1644. [DOI] [PubMed] [Google Scholar]
- 68. Adler, B. K. , Salzman, D. E. , Carabasi, M. H. , Vaughan, W. P. , Reddy, V. V. , Prchal, J. T. (2001) Fatal sickle cell crisis after granulocyte colony‐stimulating factor administration. Blood 97, 3313–3314. [DOI] [PubMed] [Google Scholar]
- 69. Charache, S. (1997) Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin. Hematol. 34 (Suppl. 3), 15–21. [PubMed] [Google Scholar]
- 70. Koshy, M. , Dorn, L. , Bressler, L. , Molokie, R. , Lavelle, D. , Talischy, N. , Hoffman, R. , van Overveld, W. , DeSimone, J. (2000) 2‐Deoxy 5‐azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood 96, 2379–2384. [PubMed] [Google Scholar]
- 71. Okpala, I. (2006) Leukocyte adhesion and the pathophysiology of sickle cell disease. Curr. Opin. Hematol. 13, 40–44. [DOI] [PubMed] [Google Scholar]
- 72. Chang J., Patton J. T., Sarkar A., Ernst B., Magnani J. L., Frennette P. S. (2010) GMI‐1070, a novel pan selectin antagonist, reverses acute vascular occlusion in sickle cell mice. Blood 116, 1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Haynes Jr., J. , Obiako, B. , Hester, R. B. , Baliga, B. S. , Stevens, T. (2008) Hydroxyurea attenuates activated neutrophil‐mediated sickle erythrocyte membrane phosphatidylserine exposure and adhesion to pulmonary vascular endothelium. Am. J. Physiol. Heart Circ. Physiol. 294, H379‐H385. [DOI] [PubMed] [Google Scholar]
- 74. Morris, C. R. , Vichinsky, E. P. , van Warmerdam, J. , Machado, L. , Kepka‐Lenhart, D. , Morris Jr., S. M. , Kuypers, F. A. (2003) Hydroxyurea and arginine therapy: impact on nitric oxide production in sickle cell disease. J. Pediatr. Hematol. Oncol. 25, 629–634. [DOI] [PubMed] [Google Scholar]
- 75. Turhan, A. , Weiss, L. A. , Mohandas, N. , Coller, B. S. , Frenette, P. S. (2002) Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc. Natl. Acad. Sci. USA 99, 3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Swerlick, R A. , Eckman, J. R. , Kumar, A. , Jeitler, M. , Wick, T. M. (1993) α 4 β 1‐Integrin expression on sickle reticulocytes: vascular cell adhesion molecule‐1‐dependent binding to endothelium. Blood 82, 1891–1899. [PubMed] [Google Scholar]
- 77. Gee, B. E. , Platt, O. S. (1995) Sickle reticulocytes adhere to VCAM‐1. Blood 85, 268–274. [PubMed] [Google Scholar]
- 78. Lard, L. R. , Mul, F. P. , de Haas, M. , Roos, D. , Duits, A. J. (1999) Neutrophil activation in sickle cell disease. J. Leukoc. Biol. 66, 411–415. [DOI] [PubMed] [Google Scholar]
- 79. Croizat, H. (1994) Circulating cytokines in sickle cell patients during steady state. Br. J. Haematol. 87, 592–597. [DOI] [PubMed] [Google Scholar]
- 80. Aslan, M. , Canatan, D. (2008) Modulation of redox pathways in neutrophils from sickle cell disease patients. Exp. Hematol. 36, 1535–1544. [DOI] [PubMed] [Google Scholar]
- 81. Lanaro, C. , Franco‐Penteado, C. F. , Albuqueque, D. M. , Saad, S. T. , Conran, N. , Costa, F. F. (2009) Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J. Leukoc. Biol. 85, 235–242. [DOI] [PubMed] [Google Scholar]
- 82. Minter, K. R. , Gladwin, M. T. (2001) Pulmonary complications of sickle cell anemia. A need for increased recognition, treatment, and research. Am. J. Respir. Crit. Care Med. 164, 2016–2019. [DOI] [PubMed] [Google Scholar]
- 83. Bux, J. , Sachs, U. J. (2007) The pathogenesis of transfusion‐related acute lung injury (TRALI). Br. J. Haematol. 136, 788–799. [DOI] [PubMed] [Google Scholar]
- 84. Nakagawa, M. , Toy, P. (2004) Acute and transient decrease in neutrophil count in transfusion‐related acute lung injury: cases at one hospital. Transfusion 44, 1689–1694. [DOI] [PubMed] [Google Scholar]
- 85. Fadeyi, E. A. , De Los Angeles Muniz, M. , Wayne, A. S. , Klein, H. G. , Leitman, S. F. , Stroncek, D. F. (2007) The transfusion of neutrophil‐specific antibodies causes leukopenia and a broad spectrum of pulmonary reactions. Transfusion 47, 545–550. [DOI] [PubMed] [Google Scholar]
- 86. Kleinman, S. , Caulfield, T. , Chan, P. , Davenport, R. , McFarland, J. , McPhedran, S. , Meade, M. , Morrison, D. , Pinsent, T. , Robillard, P. , Slinger, P. (2004) Toward an understanding of transfusion‐related acute lung injury: statement of a consensus panel. Transfusion 44, 1774–1789. [DOI] [PubMed] [Google Scholar]
- 87. Fung, Y. L. , Silliman, C. C. (2009) The role of neutrophils in the pathogenesis of transfusion‐related acute lung injury. Transfus. Med. Rev. 23, 266–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Looney, M. R. , Su, X. , Van Ziffle, J. A. , Lowell, C. A. , Matthay, M. A. (2006) Neutrophils and their Fc γ receptors are essential in a mouse model of transfusion‐related acute lung injury. J. Clin. Invest. 116, 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Triulzi, D. J. (2009) Transfusion‐related acute lung injury: current concepts for the clinician. Anesth. Analg. 108, 770–776. [DOI] [PubMed] [Google Scholar]
- 90. Sachs, U. J. (2007) The pathogenesis of transfusion‐related acute lung injury and how to avoid this serious adverse reaction of transfusion. Transfus. Apher. Sci. 37, 273–282. [DOI] [PubMed] [Google Scholar]
- 91. Mulligan, M. S. , Varani, J. , Warren, J. S. , Till, G. O. , Smith, C. W. , Anderson, D. C. , Todd, R. F. III , Ward, P. A. (1992) Roles of β 2 integrins of rat neutrophils in complement‐ and oxygen radical‐mediated acute inflammatory injury. J. Immunol. 148, 1847–1857. [PubMed] [Google Scholar]
- 92. Waddell, T. K. , Fialkow, L. , Chan, C. K. , Kishimoto, T. K. , Downey, G. P. (1994) Potentiation of the oxidative burst of human neutrophils. A signaling role for L‐selectin. J. Biol. Chem. 269, 18485–18491. [PubMed] [Google Scholar]
- 93. Liles, W. C. , Ledbetter, J. A. , Waltersdorph, A. W. , Klebanoff, S. J. (1995) Cross‐linking of CD18 primes human neutrophils for activation of the respiratory burst in response to specific stimuli: implications for adhesion‐dependent physiological responses in neutrophils. J. Leukoc. Biol. 58, 690–697. [DOI] [PubMed] [Google Scholar]
- 94. Zemans, R. L. , Colgan, S. P. , Downey, G. P. (2009) Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am. J. Respir. Cell Mol. Biol. 40, 519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Silliman, C. C. , Kelher, M. (2005) The role of endothelial activation in the pathogenesis of transfusion‐related acute lung injury. Transfusion 45 (Suppl.), 109S–116S. [DOI] [PubMed] [Google Scholar]
- 96. Sachs, U. J. , Hattar, K. , Weissmann, N. , Bohle, R. M. , Weiss, T. , Sibelius, U. , Bux, J. (2006) Antibody‐induced neutrophil activation as a trigger for transfusion‐related acute lung injury in an ex vivo rat lung model. Blood 107, 1217–1219. [DOI] [PubMed] [Google Scholar]
- 97. Silliman, C. C. , Bjornsen, A. J. , Wyman, T. H. , Kelher, M. , Allard, J. , Bieber, S. , Voelkel, N. F. (2003) Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion 43, 633–640. [DOI] [PubMed] [Google Scholar]
- 98. Cherry, T. , Steciuk, M. , Reddy, V. V. , Marques, M. B. (2008) Transfusion‐related acute lung injury: past, present, and future. Am. J. Clin. Pathol. 129, 287–297. [DOI] [PubMed] [Google Scholar]
- 99. Fung, Y. L. , Goodison, K. A. , Wong, J. K. , Minchinton, R. M. (2003) Investigating transfusion‐related acute lung injury (TRALI). Intern. Med. J. 33, 286–290. [DOI] [PubMed] [Google Scholar]
- 100. Chopra, M. , Reuben, J. S. , Sharma, A. C. (2009) Acute lung injury: apoptosis and signaling mechanisms. Exp. Biol. Med. (Maywood) 234, 361–371. [DOI] [PubMed] [Google Scholar]
- 101. Nishimura, M. , Hashimoto, S. , Takanashi, M. , Okazaki, H. , Satake, M. , Nakajima, K. (2007) Role of anti‐human leucocyte antigen class II alloantibody and monocytes in development of transfusion‐related acute lung injury. Transfus. Med. 17, 129–134. [DOI] [PubMed] [Google Scholar]
- 102. Atabai, K. , Matthay, M. A. (2002) The pulmonary physician in critical care. 5: Acute lung injury and the acute respiratory distress syndrome: definitions and epidemiology. Thorax 57, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Suratt, B. T. , Parsons, P. E. (2006) Mechanisms of acute lung injury/acute respiratory distress syndrome. Clin. Chest Med. 27, 579–589. [DOI] [PubMed] [Google Scholar]
- 104. Weiland, J. E. , Davis, W. B. , Holter, J. F. , Mohammed, J. R. , Dorinsky, P. M. , Gadek, J. E. (1986) Lung neutrophils in the adult respiratory distress syndrome. Clinical and pathophysiologic significance. Am. Rev. Respir. Dis. 133, 218–225. [DOI] [PubMed] [Google Scholar]
- 105. Chollet‐Martin, S. , Jourdain, B. , Gibert, C. , Elbim, C. , Chastre, J. , Gougerot‐Pocidalo, M. A. (1996) Interactions between neutrophils and cytokines in blood and alveolar spaces during ARDS. Am. J. Respir. Crit. Care Med. 154, 594–601. [DOI] [PubMed] [Google Scholar]
- 106. Maniatis, N. A. , Kotanidou, A. , Catravas, J. D. , Orfanos, S. E. (2008) Endothelial pathomechanisms in acute lung injury. Vascul. Pharmacol. 49, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bozza, F. A. , Shah, A. M. , Weyrich, A. S. , Zimmerman, G. A. (2009) Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am. J. Respir. Cell Mol. Biol. 40, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bellingan, G. J. (2002) The pulmonary physician in critical care: the pathogenesis of ALI/ARDS. Thorax 57, 540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pugin, J. , Ricou, B. , Steinberg, K. P. , Suter, P. M. , Martin, T. R. (1996) Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin‐1. Am. J. Respir. Crit. Care Med. 153, 1850–1856. [DOI] [PubMed] [Google Scholar]
- 110. Donnelly, S. C. , Strieter, R. M. , Kunkel, S. L. , Walz, A. , Robertson, C. R. , Carter, D. C. , Grant, I. S. , Pollok, A. J. , Haslett, C. (1993) Interleukin‐8 and development of adult respiratory distress syndrome in at‐risk patient groups. Lancet 341, 643–647. [DOI] [PubMed] [Google Scholar]
- 111. Worthen, G. S. , Schwab, B. III , Elson, E. L. , Downey, G. P. (1989) Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science 245, 183–186. [DOI] [PubMed] [Google Scholar]
- 112. Zhang, H. , Slutsky, A. S. , Vincent, J. L. (2000) Oxygen free radicals in ARDS, septic shock and organ dysfunction. Intensive Care Med. 26, 474–476. [DOI] [PubMed] [Google Scholar]
- 113. Ware, L. B. (2006) Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 27, 337–349. [DOI] [PubMed] [Google Scholar]
- 114. Hogg, J. C. , (1994) Felix Fleischner Lecture. The traffic of polymorphonuclear leukocytes through pulmonary microvessels in health and disease. AJR Am. J. Roentgenol. 163, 769–775. [DOI] [PubMed] [Google Scholar]
- 115. Redding, G. J. (2001) Current concepts in adult respiratory distress syndrome in children. Curr. Opin. Pediatr. 13, 261–266. [DOI] [PubMed] [Google Scholar]
- 116. Lien, T. C. , Sung, C. S. , Lee, C. H. , Kao, H. K. , Huang, Y. C. , Liu, C. Y. , Perng, R. P. , Wang, J. H. (2008) Characteristic features and outcomes of severe acute respiratory syndrome found in severe acute respiratory syndrome intensive care unit patients. J. Crit. Care 23, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Doerschuk, C. M. , Winn, R. K. , Coxson, H. O. , Harlan, J. M. (1990) CD18‐dependent and ‐independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J. Immunol. 144, 2327–2333. [PubMed] [Google Scholar]
- 118. Rocco, P. R. , Dos Santos, C. , Pelosi, P. (2009) Lung parenchyma remodeling in acute respiratory distress syndrome. Minerva Anestesiol. 75, 730–740. [PubMed] [Google Scholar]
- 119. Reutershan, J. , Basit, A. , Galkina, E. V. , Ley, K. (2005) Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS‐induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L807–L815. [DOI] [PubMed] [Google Scholar]
- 120. Kitching, A. R. , Holdsworth, S. R. , Hickey, M. J. (2008) Targeting leukocytes in immune glomerular diseases. Curr. Med. Chem. 15, 448–458. [DOI] [PubMed] [Google Scholar]
- 121. Morgan, M. D. , Harper, L. , Williams, J. , Savage, C. (2006) Anti‐neutrophil cytoplasm‐associated glomerulonephritis. J. Am. Soc. Nephrol. 17, 1224–1234. [DOI] [PubMed] [Google Scholar]
- 122. Jennette, J. C. , Xiao, H. , Falk, R. J. (2006) Pathogenesis of vascular inflammation by anti‐neutrophil cytoplasmic antibodies. J. Am. Soc. Nephrol. 17, 1235–1242. [DOI] [PubMed] [Google Scholar]
- 123. Kuligowski, M. P. , Kitching, A. R. , Hickey, M. J. (2006) Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet‐derived P‐selectin in the absence of rolling. J. Immunol. 176, 6991–6999. [DOI] [PubMed] [Google Scholar]
- 124. Schreiber, A. , Xiao, H. , Jennette, J. C. , Schneider, W. , Luft, F. C. , Kettritz, R. (2009) C5a receptor mediates neutrophil activation and ANCA‐induced glomerulonephritis. J. Am. Soc. Nephrol. 20, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Calderwood, J. W. , Williams, J. M. , Morgan, M. D. , Nash, G. B. , Savage, C. O. (2005) ANCA induces β2 integrin and CXC chemokine‐dependent neutrophil‐endothelial cell interactions that mimic those of highly cytokine‐activated endothelium. J. Leukoc. Biol. 77, 33–43. [DOI] [PubMed] [Google Scholar]
- 126. Lan, H. Y. , Yang, N. , Nikolic‐Paterson, D. J. , Yu, X. Q. , Mu, W. , Isbel, N. M. , Metz, C. N. , Bucala, R. , Atkins, R. C. (2000) Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 57, 499–509. [DOI] [PubMed] [Google Scholar]
- 127. Little, M. A. , Bhangal, G. , Smyth, C. L. , Nakada, M. T. , Cook, H. T. , Nourshargh, S. , Pusey, C. D. (2006) Therapeutic effect of anti‐TNF‐α antibodies in an experimental model of anti‐neutrophil cytoplasm antibody‐associated systemic vasculitis. J. Am. Soc. Nephrol. 17, 160–169. [DOI] [PubMed] [Google Scholar]
- 128. Holdsworth, S. R. , Kitching, A. R. , Tipping, P. G. (1999) Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 55, 1198–1216. [DOI] [PubMed] [Google Scholar]
- 129. Takemura, T. , Yoshioka, K. , Murakami, K. , Akano, N. , Okada, M. , Aya, N. , Maki, S. (1994) Cellular localization of inflammatory cytokines in human glomerulonephritis. Virchows Arch. 424, 459–464. [DOI] [PubMed] [Google Scholar]
- 130. Noronha, I. L. , Krüger, C. , Andrassy, K. , Ritz, E. , Waldherr, R. (1993) In situ production of TNF‐α, IL‐1 β and IL‐2R in ANCA‐positive glomerulonephritis. Kidney Int. 43, 682–692. [DOI] [PubMed] [Google Scholar]
- 131. Eddy, A. A. (2005) Progression in chronic kidney disease. Adv. Chronic Kidney Dis. 12, 353–365. [DOI] [PubMed] [Google Scholar]
- 132. Eitner, F. , Floege, J. (2005) Therapeutic targets for prevention and regression of progressive fibrosing renal diseases. Curr. Opin. Investig. Drugs 6, 255–261. [PubMed] [Google Scholar]
- 133. Bonner, J. C. (2004) Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 15, 255–273. [DOI] [PubMed] [Google Scholar]
- 134. Wilkowski, M. J. , Velosa, J. A. , Holley, K. E. , Offord, K. P. , Chu, C. P. , Torres, V. E. , McCarthy, J. T. , Donadio Jr., J. V. , Wagoner, R. D. (1989) Risk factors in idiopathic renal vasculitis and glomerulonephritis. Kidney Int. 36, 1133–1141. [DOI] [PubMed] [Google Scholar]
- 135. Harris, R. C. , Neilson, E. G. (2006) Toward a unified theory of renal progression. Annu. Rev. Med. 57, 365–380. [DOI] [PubMed] [Google Scholar]
- 136. Bernhagen, J. , Krohn, R. , Lue, H. , Gregory, J. L. , Zernecke, A. , Koenen, R. R. , Dewor, M. , Georgiev, I. , Schober, A. , Leng, L. , Kooistra, T. , Fingerle‐Rowson, G. , Ghezzi, P. , Kleemann, R. , McColl, S. R. , Bucala, R. , Hickey, M. J. , Weber, C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596. [DOI] [PubMed] [Google Scholar]
- 137. Jean, W. C. , Spellman, S. R. , Nussbaum, E. S. , Low, W. C. (1998) Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery 43, 1382–1396, discussion 1396–1397. [DOI] [PubMed] [Google Scholar]
- 138. Kochanek, P. M. , Hallenbeck, J. M. (1992) Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke 23, 1367–1379. [DOI] [PubMed] [Google Scholar]
- 139. Grau, A. J. , Boddy, A. W. , Dukovic, D. A. , Buggle, F. , Lichy, C. , Brandt, T. , Hacke, W. (2004) Leukocyte count as an independent predictor of recurrent ischemic events. Stroke 35, 1147–1152. [DOI] [PubMed] [Google Scholar]
- 140. Akopov, S. E. , Simonian, N. A. , Grigorian, G. S. (1996) Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke 27, 1739–1743. [DOI] [PubMed] [Google Scholar]
- 141. Price, C. J. , Menon, D. K. , Peters, A. M. , Ballinger, J. R. , Barber, R. W. , Balan, K. K. , Lynch, A. , Xuereb, J. H. , Fryer, T. , Guadagno, J. V. , Warburton, E. A. (2004) Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging‐based study. Stroke 35, 1659–1664. [DOI] [PubMed] [Google Scholar]
- 142. Buck, B. H. , Liebeskind, D. S. , Saver, J. L. , Bang, O. Y. , Yun, S. W. , Starkman, S. , Ali, L. K. , Kim, D. , Villablanca, J. P. , Salamon, N. , Razinia, T. , Ovbiagele, B. (2008) Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. [DOI] [PubMed] [Google Scholar]
- 143. Elneihoum, A. M. , Falke, P. , Axelsson, L. , Lundberg, E. , Lindgarde, F. , Ohlsson, K. (1996) Leukocyte activation detected by increased plasma levels of inflammatory mediators in patients with ischemic cerebrovascular diseases. Stroke 27, 1734–1738. [DOI] [PubMed] [Google Scholar]
- 144. Yan, J. , Greer, J. M. , Etherington, K. , Cadigan, G. P. , Cavanagh, H. , Henderson, R. D. , O'Sullivan, J. D. , Pandian, J. D. , Read, S. J. , McCombe, P. A. (2009) Immune activation in the peripheral blood of patients with acute ischemic stroke. J. Neuroimmunol. 206, 112–117. [DOI] [PubMed] [Google Scholar]
- 145. Tsai, N. W. , Chang, W. N. , Shaw, C. F. , Jan, C. R. , Huang, C. R. , Chen, S. D. , Chuang, Y. C. , Lee, L. H. , Lu, C. H. (2009) The value of leukocyte adhesion molecules in patients after ischemic stroke. J. Neurol. 256, 1296–1302. [DOI] [PubMed] [Google Scholar]
- 146. Ames, A. III , Wright, R. L. , Kowada, M. , Thurston, J. M. , Majno, G. (1968) Cerebral ischemia. II. The no‐reflow phenomenon. Am. J. Pathol. 52, 437–453. [PMC free article] [PubMed] [Google Scholar]
- 147. Ley, K. , Laudanna, C. , Cybulsky, M. I. , Nourshargh, S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. [DOI] [PubMed] [Google Scholar]
- 148. Mori, E. , del Zoppo, G. J. , Chambers, J. D. , Copeland, B. R. , Arfors, K. E. (1992) Inhibition of polymorphonuclear leukocyte adherence suppresses no‐reflow after focal cerebral ischemia in baboons. Stroke 23, 712–718. [DOI] [PubMed] [Google Scholar]
- 149. Jin, G. , Tsuji, K. , Xing, C. , Yang, Y. G. , Wang, X. , Lo, E. H. (2009) CD47 gene knockout protects against transient focal cerebral ischemia in mice. Exp. Neurol. 217, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Goussev, A. V. , Zhang, Z. , Anderson, D. C. , Chopp, M. (1998) P‐selectin antibody reduces hemorrhage and infarct volume resulting from MCA occlusion in the rat. J. Neurol. Sci. 161, 16–22. [DOI] [PubMed] [Google Scholar]
- 151. Arnould, T. , Michiels, C. , Remacle, J. (1993) Increased PMN adherence on endothelial cells after hypoxia: involvement of PAF, CD18/CD11b, and ICAM‐1. Am. J. Physiol. 264, C1102–C1110. [DOI] [PubMed] [Google Scholar]
- 152. Prestigiacomo, C. J. , Kim, S. C. , Connolly Jr., E. S. , Liao, H. , Yan, S. F. , Pinsky, D. J. (1999) CD18‐mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 30, 1110–1117. [DOI] [PubMed] [Google Scholar]
- 153. Zhang, L. , Zhang, Z. G. , Zhang, R. L. , Lu, M. , Krams, M. , Chopp, M. (2003) Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 34, 1790–1795. [DOI] [PubMed] [Google Scholar]
- 154. Kazmierski, R. , Guzik, P. , Ambrosius, W. , Ciesielska, A. , Moskal, J. , Kozubski, W. (2004) Predictive value of white blood cell count on admission for in‐hospital mortality in acute stroke patients. Clin. Neurol. Neurosurg. 107, 38–43. [DOI] [PubMed] [Google Scholar]
- 155. Del Zoppo, G. J. (1997) Microvascular responses to cerebral ischemia/inflammation. Ann. N. Y. Acad. Sci. 823, 132–147. [DOI] [PubMed] [Google Scholar]
- 156. Hallenbeck, J. M. (1996) Significance of the inflammatory response in brain ischemia. Acta Neurochir. Suppl. 66, 27–31. [DOI] [PubMed] [Google Scholar]
- 157. Fagan, S. C. , Hess, D. C. , Hohnadel, E. J. , Pollock, D. M. , Ergul, A. (2004) Targets for vascular protection after acute ischemic stroke. Stroke 35, 2220–2225. [DOI] [PubMed] [Google Scholar]
- 158. Fisher, T. C. , Meiselmann, H. J. (1994) Polymorphonuclear leukocytes in ischemic vascular disease. Thromb. Res. 74 (Suppl. 1), S21–S34. [DOI] [PubMed] [Google Scholar]
- 159. Galante, A. , Silvestrini, M. , Stanzione, P. , Pietroiusti, A. , Baldoni, F. , Domenici, B. , Bernardi, G. (1992) Leucocyte aggregation in acute cerebrovascular disease. Acta Neurol. Scand. 86, 446–449. [DOI] [PubMed] [Google Scholar]
- 160. Feuerstein, G. (2006) Inflammation and stroke: therapeutic effects of adenoviral expression of secretory leukocyte protease inhibitor. Front. Biosci. 11, 1750–1757. [DOI] [PubMed] [Google Scholar]
- 161. Chen, H. , Song, Y. S. , Chan, P. H. (2009) Inhibition of NADPH oxidase is neuroprotective after ischemia‐reperfusion. J. Cereb. Blood Flow Metab. 29, 1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kapadia, R. , Tureyen, K. , Bowen, K. K. , Kalluri, H. , Johnson, P. F. , Vemuganti, R. (2006) Decreased brain damage and curtailed inflammation in transcription factor CCAAT/enhancer binding protein β knockout mice following transient focal cerebral ischemia. J. Neurochem. 98, 1718–1731. [DOI] [PubMed] [Google Scholar]
- 163. Luo, Y. , Yin, W. , Signore, A. P. , Zhang, F. , Hong, Z. , Wang, S. , Graham, S. H. , Chen, J. (2006) Neuroprotection against focal ischemic brain injury by the peroxisome proliferator‐activated receptor‐γ agonist rosiglitazone. J. Neurochem. 97, 435–448. [DOI] [PubMed] [Google Scholar]
- 164. Chou, W. H. , Choi, D. S. , Zhang, H. , Mu, D. , McMahon, T. , Kharazia, V. N. , Lowell, C. A. , Ferriero, D. M. , Messing, R. O. (2004) Neutrophil protein kinase Cδ as a mediator of stroke‐reperfusion injury. J. Clin. Invest. 114, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Belayev, L. , Khoutorova, L. , Atkins, K. , Gordon, W. C. , Alvarez‐Builla, J. , Bazan, N. G. (2008) LAU‐0901, a novel platelet‐activating factor antagonist, is highly neuroprotective in cerebral ischemia. Exp. Neurol. 214, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ikegame, Y. , Yamashita, K. , Hayashi, S. , Yoshimura, S. , Nakashima, S. , Iwama, T. (2010) Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens. Res. 33, 703–707. [DOI] [PubMed] [Google Scholar]
- 167. Shimakura, A. , Kamanaka, Y. , Ikeda, Y. , Kondo, K. , Suzuki, Y. , Umemura, K. (2000) Neutrophil elastase inhibition reduces cerebral ischemic damage in the middle cerebral artery occlusion. Brain Res. 858, 55–60. [DOI] [PubMed] [Google Scholar]
- 168. Matayoshi, H. , Hirata, T. , Yamashita, S. , Ishida, K. , Mizukami, Y. , Gondo, T. , Matsumoto, M. , Sakabe, T. (2009) Neutrophil elastase inhibitor attenuates hippocampal neuronal damage after transient forebrain ischemia in rats. Brain Res. 1259, 98–106. [DOI] [PubMed] [Google Scholar]
- 169. Grobmyer, S. R. , Barie, P. S. , Nathan, C. F. , Fuortes, M. , Lin, E. , Lowry, S. F. , Wright, C. D. , Weyant, M. J. , Hydo, L. , Reeves, F. , Shiloh, M. U. , Ding, A. (2000) Secretory leukocyte protease inhibitor, an inhibitor of neutrophil activation, is elevated in serum in human sepsis and experimental endotoxemia. Crit. Care Med. 28, 1276–1282. [DOI] [PubMed] [Google Scholar]
- 170. Wang, X. , Li, X. , Xu, L. , Zhan, Y. , Yaish‐Ohad, S. , Erhardt, J. A. , Barone, F. C. , Feuerstein, G. Z. (2003) Up‐regulation of secretory leukocyte protease inhibitor (SLPI) in the brain after ischemic stroke: adenoviral expression of SLPI protects brain from ischemic injury. Mol. Pharmacol. 64, 833–840. [DOI] [PubMed] [Google Scholar]
- 171. Bowen, K. K. , Naylor, M. , Vemuganti, R. (2006) Prevention of inflammation is a mechanism of preconditioning‐induced neuroprotection against focal cerebral ischemia. Neurochem. Int. 49, 127–135. [DOI] [PubMed] [Google Scholar]
- 172. Wong, R. K. , Pettit, A. I. , Quinn, P. A. , Jennings, S. C. , Davies, J. E. , Ng, L. L. (2003) Advanced glycation end products stimulate an enhanced neutrophil respiratory burst mediated through the activation of cytosolic phospholipase A2 and generation of arachidonic acid. Circulation 108, 1858–1864. [DOI] [PubMed] [Google Scholar]
- 173. Masuda, M. , Murakami, T. , Egawa, H. , Murata, K. (1990) Decreased fluidity of polymorphonuclear leukocyte membrane in streptozocin‐induced diabetic rats. Diabetes 39, 466–470. [DOI] [PubMed] [Google Scholar]
- 174. Pitzer, J. E. , Del Zoppo, G. J. , Schmid‐Schonbein, G. W. (1996) Neutrophil activation in smokers. Biorheology 33, 45–58. [PubMed] [Google Scholar]
- 175. Tatsukawa, Y. , Hsu, W. L. , Yamada, M. , Cologne, J. B. , Suzuki, G. , Yamamoto, H. , Yamane, K. , Akahoshi, M. , Fujiwara, S. , Kohno, N. (2008) White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens. Res. 31, 1391–1397. [DOI] [PubMed] [Google Scholar]
- 176. Dudman, N. P. , Temple, S. E. , Guo, X. W. , Fu, W. , Perry, M. A. (1999) Homocysteine enhances neutrophil‐endothelial interactions in both cultured human cells and rats in vivo. Circ. Res. 84, 409–416. [DOI] [PubMed] [Google Scholar]
- 177. Liao, S. L. , Chen, W. Y. , Raung, S. L. , Chen, C. J. (2003) Ethanol attenuates ischemic and hypoxic injury in rat brain and cultured neurons. Neuroreport 14, 2089–2094. [DOI] [PubMed] [Google Scholar]
- 178. Reumaux, D. , Duthilleul, P. , Roos, D. (2004) Pathogenesis of diseases associated with antineutrophil cytoplasm autoantibodies. Hum. Immunol. 65, 1–12. [DOI] [PubMed] [Google Scholar]
- 179. Bar, J. , Bardin, R. , Chen, R. , Pardo, J. , Hod, M. , Peled, Y. , Molad, Y. (2003) Surface expression of neutrophil adhesion molecules in pregnant women at risk for hypertensive complications. Hypertens. Pregnancy 22, 165–172. [DOI] [PubMed] [Google Scholar]
- 180. Haeusler, K. G. , Schmidt, W. U. , Fohring, F. , Meisel, C. , Helms, T. , Jungehulsing, G. J. , Nolte, C. H. , Schmolke, K. , Wegner, B. , Meisel, A. , Dirnagl, U. , Villringer, A. , Volk, H. D. (2008) Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc. Dis. 25, 50–58. [DOI] [PubMed] [Google Scholar]
- 181. Krams, M. , Lees, K. R. , Hacke, W. , Grieve, A. P. , Orgogozo, J. M. , Ford, G. A. (2003) Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose‐response study of UK‐279,276 in acute ischemic stroke. Stroke 34, 2543–2548. [DOI] [PubMed] [Google Scholar]
- 182. Emsley, H. C. , Smith, C. J. , Georgiou, R. F. , Vail, A. , Hopkins, S. J. , Rothwell, N. J. , Tyrrell, P. J. (2005) A randomized phase II study of interleukin‐1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 76, 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Beray‐Berthat, V. , Palmier, B. , Plotkine, M. , Margaill, I. (2003) Neutrophils do not contribute to infarction, oxidative stress, and NO synthase activity in severe brain ischemia. Exp. Neurol. 182, 446–454. [DOI] [PubMed] [Google Scholar]
- 184. Harris, A. K. , Ergul, A. , Kozak, A. , Machado, L. S. , Johnson, M. H. , Fagan, S. C. (2005) Effect of neutrophil depletion on gelatinase expression, edema formation and hemorrhagic transformation after focal ischemic stroke. BMC Neurosci. 6, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Hayward, N. J. , Elliott, P. J. , Sawyer, S. D. , Bronson, R. T. , Bartus, R. T. (1996) Lack of evidence for neutrophil participation during infarct formation following focal cerebral ischemia in the rat. Exp. Neurol. 139, 188–202. [DOI] [PubMed] [Google Scholar]
- 186. Hao, Q. , Chen, Y. , Zhu, Y. , Fan, Y. , Palmer, D. , Su, H. , Young, W. L. , Yang, G. Y. (2007) Neutrophil depletion decreases VEGF‐induced focal angiogenesis in the mature mouse brain. J. Cereb. Blood Flow Metab. 27, 1853–1860. [DOI] [PubMed] [Google Scholar]
- 187. Strecker, J. K. , Sevimli, S. , Schilling, M. , Klocke, R. , Nikol, S. , Schneider, A. , Schabitz, W. R. (2010) Effects of G‐CSF treatment on neutrophil mobilization and neurological outcome after transient focal ischemia. Exp. Neurol. 222, 108–113. [DOI] [PubMed] [Google Scholar]
- 188. Hansson, G. K. , Libby, P. (2006) The immune response in atherosclerosis: a double‐edged sword. Nat. Rev. Immunol. 6, 508–519. [DOI] [PubMed] [Google Scholar]
- 189. Hansson, G. K. (2005) Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695. [DOI] [PubMed] [Google Scholar]
- 190. Goldmann, B. U. , Rudolph, V. , Rudolph, T. K. , Holle, A. K. , Hillebrandt, M. , Meinertz, T. , Baldus, S. (2009) Neutrophil activation precedes myocardial injury in patients with acute myocardial infarction. Free Radic. Biol. Med. 47, 79–83. [DOI] [PubMed] [Google Scholar]
- 191. Ricevuti, G. , Mazzone, A. , Pasotti, D. , de Servi, S. , Specchia, G. (1991) Role of granulocytes in endothelial injury in coronary heart disease in humans. Atherosclerosis 91, 1–14. [DOI] [PubMed] [Google Scholar]
- 192. Viehman, G. E. , Ma, X. L. , Lefer, D. J. , Lefer, A. M. (1991) Time course of endothelial dysfunction and myocardial injury during coronary arterial occlusion. Am. J. Physiol. 261, H874–H881. [DOI] [PubMed] [Google Scholar]
- 193. Sheridan, F. M. , Dauber, I. M. , McMurtry, I. F. , Lesnefsky, E. J. , Horwitz, L. D. (1991) Role of leukocytes in coronary vascular endothelial injury due to ischemia and reperfusion. Circ. Res. 69, 1566–1574. [DOI] [PubMed] [Google Scholar]
- 194. de Servi, S. , Ricevuti, G. , Mazzone, A. , Ghio, S. , Zito, A. , Raffaghello, S. , Specchia, G. (1991) Granulocyte function in coronary artery disease. Am. J. Cardiol. 68, 64B–68B. [DOI] [PubMed] [Google Scholar]
- 195. Biasucci, L. M. , Colizzi, C. , Rizzello, V. , Vitrella, G. , Crea, F. , Liuzzo, G. (1999) Role of inflammation in the pathogenesis of unstable coronary artery diseases. Scand. J. Clin. Lab. Invest. Suppl. 230, 12–22. [PubMed] [Google Scholar]
- 196. Narducci, M. L. , Grasselli, A. , Biasucci, L. M. , Farsetti, A. , Mule, A. , Liuzzo, G. , La Torre, G. , Niccoli, G. , Mongiardo, R. , Pontecorvi, A. , Crea, F. (2007) High telomerase activity in neutrophils from unstable coronary plaques. J. Am. Coll. Cardiol. 50, 2369–2374. [DOI] [PubMed] [Google Scholar]
- 197. Holt, S. E. , Glinsky, V. V. , Ivanova, A. B. , Glinsky, G. V. (1999) Resistance to apoptosis in human cells conferred by telomerase function and telomere stability. Mol. Carcinog. 25, 241–248. [PubMed] [Google Scholar]
- 198. Gupta, M. , Shogreen, M. R. , Braden, G. A. , White, W. L. , Sane, D. C. (2000) Prevalence of telomerase in coronary artery atherosclerosis. J. Anti Aging Med. 3, 5–24. [Google Scholar]
- 199. Sane, D. C. (2008) Telomerase‐positive neutrophils: plaque “survivors” and restenosis. J. Am. Coll. Cardiol. 51, 2443–2444. [DOI] [PubMed] [Google Scholar]
- 200. Takeshita, S. , Isshiki, T. , Ochiai, M. , Ishikawa, T. , Nishiyama, Y. , Fusano, T. , Toyoizumi, H. , Kondo, K. , Ono, Y. , Sato, T. (1997) Systemic inflammatory responses in acute coronary syndrome: increased activity observed in polymorphonuclear leukocytes but not T lymphocytes. Atherosclerosis 135, 187–192. [DOI] [PubMed] [Google Scholar]
- 201. Naruko, T. , Ueda, M. , Haze, K. , van der Wal, A. C. , van der Loos, C. M. , Itoh, A. , Komatsu, R. , Ikura, Y. , Ogami, M. , Shimada, Y. , Ehara, S. , Yoshiyama, M. , Takeuchi, K. , Yoshikawa, J. , Becker, A. E. (2002) Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 106, 2894–2900. [DOI] [PubMed] [Google Scholar]
- 202. Tavora, F. R. , Ripple, M. , Li, L. , Burke, A. P. (2009) Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc. Disord. 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Huang, G. , Zhong, X. N. , Zhong, B. , Chen, Y. Q. , Liu, Z. Z. , Su, L. , Ling, Z. Y. , Cao, H. , Yin, Y. H. (2009) Significance of white blood cell count and its subtypes in patients with acute coronary syndrome. Eur. J. Clin. Invest. 39, 348–358. [DOI] [PubMed] [Google Scholar]
- 204. O'Donoghue, M. , Morrow, D. A. , Cannon, C. P. , Guo, W. , Murphy, S. A. , Gibson, C. M. , Sabatine, M. S. (2008) Association between baseline neutrophil count, clopidogrel therapy, and clinical and angiographic outcomes in patients with ST‐elevation myocardial infarction receiving fibrinolytic therapy. Eur. Heart J. 29, 984–991. [DOI] [PubMed] [Google Scholar]
- 205. Karabinos, I. , Koulouris, S. , Kranidis, A. , Pastromas, S. , Exadaktylos, N. , Kalofoutis, A. (2009) Neutrophil count on admission predicts major inhospital events in patients with a non‐ST‐segment elevation acute coronary syndrome. Clin. Cardiol. 32, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Nijm, J. , Wikby, A. , Tompa, A. , Olsson, A. G. , Jonasson, L. (2005) Circulating levels of proinflammatory cytokines and neutrophil‐platelet aggregates in patients with coronary artery disease. Am. J. Cardiol. 95, 452–456. [DOI] [PubMed] [Google Scholar]
- 207. Tamhane, U. U. , Aneja, S. , Montgomery, D. , Rogers, E. K. , Eagle, K. A. , Gurm, H. S. (2008) Association between admission neutrophil to lymphocyte ratio and outcomes in patients with acute coronary syndrome. Am. J. Cardiol. 102, 653–657. [DOI] [PubMed] [Google Scholar]
- 208. Eschen, O. , Christensen, J. H. , Toft, E. , Schmidt, E. B. (2005) Soluble adhesion molecules and marine n‐3 fatty acids in patients referred for coronary angiography. Atherosclerosis 180, 327–331. [DOI] [PubMed] [Google Scholar]
- 209. Ott, I. , Neumann, F. J. , Gawaz, M. , Schmitt, M. , Schomig, A. (1996) Increased neutrophil‐platelet adhesion in patients with unstable angina. Circulation 94, 1239–1246. [DOI] [PubMed] [Google Scholar]
- 210. Mehta, J. L. , Nichols, W. W. , Mehta, P. (1988) Neutrophils as potential participants in acute myocardial ischemia: relevance to reperfusion. J. Am. Coll. Cardiol. 11, 1309–1316. [DOI] [PubMed] [Google Scholar]
- 211. Huang, Q. , Wu, M. , Meininger, C. , Kelly, K. , Yuan, Y. (1998) Neutrophil‐dependent augmentation of PAF‐induced vasoconstriction and albumin flux in coronary arterioles. Am. J. Physiol. 275, H1138–H1147. [DOI] [PubMed] [Google Scholar]
- 212. Chia, S. , Nagurney, J. T. , Brown, D. F. , Raffel, O. C. , Bamberg, F. , Senatore, F. , Wackers, F. J. , Jang, I. K. (2009) Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST‐elevation myocardial infarction. Am. J. Cardiol. 103, 333–337. [DOI] [PubMed] [Google Scholar]
- 213. Palatianos, G. M. , Balentine, G. , Papadakis, E. G. , Triantafillou, C. D. , Vassili, M. I. , Lidoriki, A. , Dinopoulos, A. , Astras, G. M. (2004) Neutrophil depletion reduces myocardial reperfusion morbidity. Ann. Thorac. Surg. 77, 956–961. [DOI] [PubMed] [Google Scholar]
- 214. Arai, M. , Lefer, D. J. , So, T. , DiPaula, A. , Aversano, T. , Becker, L. C. (1996) An anti‐CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48‐hour reperfusion. J. Am. Coll. Cardiol. 27, 1278–1285. [DOI] [PubMed] [Google Scholar]
- 215. Faxon, D. P. , Gibbons, R. J. , Chronos, N. A. , Gurbel, P. A. , Sheehan, F. (2002) The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT‐MI study. J. Am. Coll. Cardiol. 40, 1199–1204. [DOI] [PubMed] [Google Scholar]
- 216. Rusnak, J. M. , Kopecky, S. L. , Clements, I. P. , Gibbons, R. J. , Holland, A. E. , Peterman, H. S. , Martin, J. S. , Saoud, J. B. , Feldman, R. L. , Breisblatt, W. M. , Simons, M. , Gessler Jr., C. J. , Yu, A. S. (2001) An anti‐CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am. J. Cardiol. 88, 482–487. [DOI] [PubMed] [Google Scholar]
- 217. Baxter, G. F. (2002) The neutrophil as a mediator of myocardial ischemia‐reperfusion injury: time to move on. Basic Res. Cardiol. 97, 268–275. [DOI] [PubMed] [Google Scholar]
- 218. Bowden, R. A. , Ding, Z. M. , Donnachie, E. M. , Petersen, T. K. , Michael, L. H. , Ballantyne, C. M. , Burns, A. R. (2002) Role of α4 integrin and VCAM‐1 in CD18‐independent neutrophil migration across mouse cardiac endothelium. Circ. Res. 90, 562–569. [DOI] [PubMed] [Google Scholar]
- 219. García de Tena, J. (2005) Inflammation, atherosclerosis, and coronary disease. N. Engl. J. Med. 353, 429–430. [DOI] [PubMed] [Google Scholar]
- 220. Harlan, J. M. , Killen, P. D. , Senecal, F. M. , Schwartz, B. R. , Yee, E. K. , Taylor, R. F. , Beatty, P. G. , Price, T. H. , Ochs, H. D. (1985) The role of neutrophil membrane glycoprotein GP‐150 in neutrophil adherence to endothelium in vitro. Blood 66, 167–178. [PubMed] [Google Scholar]
- 221. Tonnesen, M. G. , Smedly, L. A. , Henson, P. M. (1984) Neutrophil‐endothelial cell interactions. Modulation of neutrophil adhesiveness induced by complement fragments C5a and C5a des arg and formyl‐methionylleucyl‐phenylalanine in vitro. J. Clin. Invest. 74, 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]