

Abstract
Leukocyte recruitment in inflammation is a multistep, sequential cascade where the initial step is the selectin‐dependent tethering, followed by the formation of firmer integrin‐mediated adhesive forces leading to extravasation. The α(1,3)‐fucose‐containing sialyl‐Lewis X (sLeX) is the archetypical ligand on leukocyte surfaces mediating selectin interactions. Canonically, disruption of α(1,3)‐fucose formation ablates selectin‐mediated adhesion, dramatically reducing trafficking. We report a paradoxical response to α(1,3)‐fucose deficiency in which the loss exacerbated rather than attenuated leukocyte recruitment in a murine model of acute airway inflammation. The architecture of the capillary‐dominated vasculature in the lung minimized the importance of the selectin dependent step, and we observed that α(1,3)‐fucose deficiency augmented CXCR2‐mediated Rap1‐GTP signaling to enhance the β2‐integrin‐ICAM‐1‐binding axis. The data disclose a previously unknown function for α(1,3)‐fucose, in which this structure negatively regulates the integrin activation step in leukocyte recruitment.
Keywords: fucosyltransferase, neutrophil, ICAM‐1, CXCR2, NTHI
Abbreviations
- BAL
bronchial alveolar lavage fluid
- Fut
fucosyltransferase
- Mac‐1
macrophage‐1 Ag
- NTHI
nontypeable Haemophilus influenzae
- PMN
polymorphonuclear neutrophil
- PSGL
P‐selectin glycoprotein‐ligand‐1
- sLeX
sialyl Lewis‐X
- WT
wild‐type
Introduction
Leukocyte recruitment is a central component of acute inflammation. Although normally a beneficial process, excessive or unresolved inflammation is deleterious and has been implicated in a wide variety of chronic medical conditions, such as cardiovascular and rheumatic diseases, and inflammatory bowel conditions, such as Crohn’s and celiac diseases. Inflammatory cell recruitment, culminating in the extravasation of the recruited cells to inflamed sites, is a multistep, sequential process beginning with selectin–selectin ligand–mediated tethering and rolling [1]. Upon selectin‐mediated tethering, activation by chemokine signaling triggers leukocyte integrins to transition from bent, low‐affinity states to open, higher affinity states that allow for stronger adhesive interactions with their binding partners, VCAM‐1, and ICAM‐1 [2]. The transition from low‐ to high‐affinity states triggers firm leukocyte arrest, transmigration, and the emergence of the recruited cells in the inflamed tissue. The selectin ligands on the surface of leukocytes are sialofucosylated carbohydrates, the principal being the tetrasaccharide sLeX containing an essential α(1,3)‐fucose moiety [3, 4]. The only documented physiologic function for α(1,3)‐fucose is within the context of the selectin ligands. Disruption of the glycosyltransferases responsible for the synthesis of sLeX, particularly the α(1,3)‐Futs, attenuates neutrophil tethering to activated endothelium and mitigates selectin‐dependent recruitment in both human [5, 6] and murine neutrophils [7, 8, 9–10]. Given the position of selectin–selectin ligands in the initial stage of inflammatory cell recruitment, there is significant ongoing interest in disrupting these interactions to control leukocyte recruitment in peritonitis, sickle cell disease, stroke, myocardial infarction, and some forms of cancers in rodent models and, more important, in the clinic [11, 12, 13–14].
In airway inflammation, acute exacerbations are instances where leukocytes are recruited to the sites of inflammation in response to bacterial infections, allergens, or other noxious stimuli [15, 16–17]. At worst, these episodes are directly life threatening; at best, repeated episodes promote long‐term destruction of the airway leading to permanently diminished function [16]. Based on the conventional thinking of adhesion cascade, leukocyte recruitment in the acute airway may be managed by disrupting sLex ligands of selectins. However, there may be important differences between the commonly understood vascular leukocyte adhesion cascade and those that govern recruitment to the lung. Capillaries dominate the lung microvasculature, where small diameters coupled with lower leukocyte transit rates may diminish the requirement for selectin‐mediated braking to slow the leukocyte sufficiently to engage the later steps of the recruitment cascade [18]. Therefore, targeting the selectin ligands may not be an effective approach to manage inflammation of the airways.
To assess the contribution of the selectin axis in lung inflammation, we examined mouse models defective in Fut4 and ‐7. In the mouse, only these 2 Futs construct the α(1,3)‐fucosyl linkage in sLeX. Neutrophils from Fut4−/−Fut7−/− animals have no sLex, do not bind to selectin‐bearing substrates, and have severely attenuated recruitment in acute peritonitis, a well‐studied selectin‐mediated inflammation event [5, 8]. We observed that the loss of α(1,3) fucosylation, instead of blocking cell recruitment related to the absence of sLex, actually worsened pulmonary neutrophilic infiltration. These data disclosed an unexpected role for α(1,3)‐fucose, in addition and unrelated to the formation of the canonical selectin ligand sLeX. The data showed that the lack of α(1,3)‐fucose enhanced the Rap1‐GTP levels downstream of CXCR2, which in turn promoted greater ICAM‐1 affinity by both neutrophil‐borne β2‐integrins: LFA‐1 (or CD11a/CD18) and MAC‐1 r (CD11b/CD18). This unexpected and novel role for α(1,3)‐fucose, unrelated to the formation of the canonical selectin ligand sLeX, was uniquely visualized in a capillary‐dominated vasculature of the lung where low leukocyte transit rates de‐emphasize the role for selectin–selectin ligand–mediated tethering and rolling.
MATERIALS AND METHODS
Animal and inflammation models
The St6gal1−/− mouse, which has a globally inactivated ST6Gal‐1 gene, was obtained from the consortium for functional glycomics. The St3gal4−/− mouse was from Jackson Laboratories (Bar Harbor, ME, USA). Both strains were backcrossed for >10 generations on the C57BL/6 background. The heterozygous Fut4+/− and Fut7+/− mice on a C57BL/6 background were obtained from Jackson Laboratories. Homozygous Fut7−/− mice were generated by breeding homozygous Fut7+/− pairs. Homozygous Fut4−/−Fut7−/− double‐knockout mice were generated by breeding Fut4+/− and Fut7+/− pairs. All deletions were confirmed by genotyping.
The NTHI model of acute pulmonary inflammation was performed as described earlier [15, 19, 20]. In brief, mice received an oropharyngeal instillation of 1 × 106 colony‐forming units of live NTHI strain 1479. After either 4 or 18 h, BALF was analyzed. Alternatively, entire lungs were excised and digested in 2 mg/ml collagenase D (Sigma‐Aldrich, St. Louis, MO, USA) and 80 U/ml DNAse I (New England BioLabs, Ipswich, MA, USA) for 1 h at 37°C at 300 rpm in a Shaking Incubator Model 1585 (VWR, Radnor, PA, USA) to collect the immune cells [21]. A WBC count of BALF was assessed with a TC20 automated cell counter (BioRad, Hercules, CA, USA). After lavage, the lungs were excised and fixed in 10% formaldehyde in PBS, embedded in paraffin, sectioned, and stained with H&E. Lung pathology was evaluated by a pulmonary pathologist blinded to the identity of the slides.
In some experiments, 50 μg of blocking Abs against specific receptors were injected via tail vein immediately before instillation of NTHI. These Abs include purified YN1/1.7.4 (anti‐ICAM‐1), 3C4 (anti‐ICAM‐2), 429 (anti‐VCAM‐1), M17/4 (anti‐CD11a), M1/70 (anti‐CD11b), M18/2 (anti‐CD18), or SA044G4 (anti‐CXCR2). Unless otherwise stated, all immunoreagents for flow cytometry were from Biolegend (San Diego, CA, USA). Also, an sLeX analog, TBC1269 (a gift from Peter Vanderslice, Texas Heart Institute), was used in a similar fashion, as an inhibitor of selectin‐mediated adhesion.
To elicit an inflammatory response and leukocyte emigration to the peritoneum, 1 ml of 4% wt/vol sterile Brewer’s yeast thioglycollate solution (BD Microbiology, Baltimore, MD, USA) was administered intraperitoneally into each recipient animal [22, 23]. At various time points after thioglycollate challenge, the animals were euthanized by CO2 asphyxiation, and peritoneal cells were recovered by peritoneal lavage with 6 ml ice‐cold PBS.
All animal protocols were approved by the Institutional Animal Care and Use Committee of the Roswell Park Cancer Institute.
Ex vivo labeling of cells and reintroduction into recipients
Mice aged 10 to 11 wk were euthanized by CO2 asphyxiation, and bone marrow cells were recovered in cold sterile PBS. To label with PKH26‐red or PKH67‐green (Sigma‐Aldrich, St. Louis, MO, USA), cells were washed in RPMI medium (without serum), and 107 cells were resuspended in 1 ml Diluent C (Sigma‐Aldrich) and rapidly added to 1 ml of 4 μM PKH26‐red. The cells were incubated at 25°C for 5 min, terminated by the addition of 2.5% FCS. Differentially labeled donor cells were recombined immediately before infusion into recipient animals, WT C57BL/6 or Fut4−/−Fut7−/−. Each mouse received intravenous injection of pooled cells consisting of 3 × 106 cells from each labeled group.
Flow cytometric profiling of inflammatory cells
Immunofluorescent staining and flow cytometric analysis of inflammatory cell subsets were performed as follows. Cells (0.5 × 106–2 × 106) were washed in PBS containing 0.5% BSA and 0.02% sodium azide, with combinations of fluorescently labeled Abs 1A8 (anti‐Ly6G), M1/70 (anti–CD11b/Mac‐1), Gr‐1 (anti‐Ly6G, anti‐Ly6C, clone RB6‐8C5), 17A2 (anti‐CD3), 6D5 (anti‐CD19), 30‐F11 (anti‐CD45), G8.8 (anti‐CD326/Ep‐CAM), N418 (anti‐CD11c), BM8 (anti‐F4/80), FA‐11 (anti‐CD68), M5/114.15.2 (anti‐1‐A/1‐E/MHC‐class II), and Sambucus nigra lectin (SNA; Vector Laboratories, Peterborough, UK). To monitor static ligand binding by flow cytometry, we coincubated 5 μg/ml of recombinant E‐selectin Fc Chimera (R&D Systems, Minneapolis, MN, USA) or recombinant ICAM‐1 Fc chimera with 6 μg/ml goat anti‐human F(ab′)2 FITC‐conjugated secondary Ab (Jackson ImmunoResearch, West Grove, PA, USA) in HEPES buffer supplemented with 1.5 mM Ca2+ for 10 min, followed by 15 min incubation with cells to be analyzed. Flow cytometric analysis was performed using either LSR‐II or Fortessa flow cytometer (BD Immunocytometry Systems, Franklin Lakes, NJ, USA) and the FACS Diva software package (BD Biosciences).
Lung histology
Excised lungs were fixed in 10% formaldehyde (Polysciences, Washington, PA, USA) in PBS, embedded in paraffin, sectioned, and stained with H&E by the Roswell Park Cancer Institute Histopathology Core Facility. Images were obtained on a light microscope (Olympus America, Center Valley, PA, USA) equipped with a CCD camera and Spot image analysis software (ver. 25.4; Diagnostics Instruments, Sterling, MI, USA). Lung pathology was evaluated by a pathologist (P.N.B.). Identity of the slides remained blinded during 2 independent scoring sessions by the pathologist.
Rap‐1 activation assay
To assess the levels of activated Rap1 in WT and Fut4−/− Fut7−/− neutrophils, we used a Rap‐1 activation kit (Cell Signaling Technology, Danvers, MA, USA), according to the manufacturer’s instructions. In brief, 4 × 106 murine neutrophils were incubated, either with PBS or CXCL1 (KC, 5 μg/ml) from Biolegend (San Diego, CA, USA) for 5 min at 37°C in complete RPMI medium. GST‐RalGDS‐RBD (20 μg; Cell Signaling Technology) was added to each sample and incubated for 1 h at 4°C with gentle rocking. Precipitates were washed, resuspended in 50 μl of 2× sample buffer with DTT, and boiled for 5 min at 95°C. Western blot analysis was performed on a 10% SDS‐PAGE gel with 1 μg/ml rabbit Rap‐1 Ab (Cell Signaling Technology). Whole‐cell lysates (5 μl) were run on a parallel gel to detect total Rap‐1. The band intensity was quantified by using Image J software (National Institutes of Health, Bethesda, MD, USA) [24].
Slide preparation and flow chamber assembly and assay
Plastic microscope slides (1 × 3 in; Thermo Fisher Scientific) were coated with 2 μg/ml protein A/G (Biovision, Milpitas, CA, USA) for 1 h at 4°C (and 5 μg/ml CXCL1, if necessary). After the slides were washed 3 times with 1× PBS, the surfaces were treated with 0.2% pluronic acid F127 (Sigma‐Aldrich) for 1 h to block nonspecific binding to the surface. After 3 more PBS washes, the surfaces were then coated with 5 μg/ml of recombinant human ICAM‐1 Fc for 1 h. A parallel‐plate flow chamber (GlycoTech, Wilmington, NC, USA) was used with the prepared plastic slide coated with either ICAM‐1 Fc or ICAM‐1 Fc and 5 μg/ml recombinant murine CXCL1 (Biolegend). The channel template was cut from 0.01 in. thick Duralastic sheeting (Allied Biomedical, Woodbridge, NJ, USA). For each flow experiment, the template was placed over the prepared slide. The template and slide were placed in the bottom well of the flow chamber, and the top was secured with screws. The chamber was assembled underwater to minimize the introduction of air. It was then mounted on the microscope in a 5% CO2 and 37°C environment for 10 min to allow for equilibration. Before introduction of either WT or Fut4−/−Fut7−/− neutrophils, the chamber was flushed with running medium consisting of Iscove’s modified Dulbecco’s medium. A volume of 2 ml containing 1 × 106 cells in running medium was introduced into the chamber and cells were allowed to attach for 30 min. Fluid flow was initiated with a syringe pump (11 Plus; Harvard Apparatus, Holliston, MA, USA), and volumetric flow rates were adjusted accordingly to correspond to desired shear rates. Shear rate was calculated as τw = (6μQ)/(h 2 w) where μ is the fluid viscosity, Q is the volumetric flow rate, h is the channel height, and w is the channel width. For this chamber, height = 0.023 cm; width = 0.1 cm. Images were captured every minute on a motorized stage and observed with an Eclipse TE300 phase contrast microscope (Nikon, Melville, NY, USA). Images were captured with a ×10 objective. Migrating cells had a polarized morphology consisting of a lamellipodia at the front and a uropod at the rear. The number of cells per square millimeter was calculated by using the average of the number of cells migrating at 5 unique positions in the chamber and normalizing this average per square millimeter. Spherical and nonadherent cells were either washed away upon application of flow or demonstrated no motility and were not included in analysis.
Ex vivo fucosylation of mouse neutrophils
Exofucosylation was performed as published [25], except the cells were mouse neutrophils instead of LSK cells. In brief, neutrophils isolated from the Fut4−/−Fut7−/− mouse were washed with HBSS, counted, and resuspended in 60 mU/ml human FUT7 (R&D Systems) in HBSS (without Ca2+ or Mg2+) containing 20 mM HEPES, 0.1% human serum albumin, and 1 mM GDP‐fucose for 1 h at 37°C. These FUT7‐treated cells were referred to as FUT7 EXO. For control, the WT and Fut4−/−Fut7−/− neutrophils were resuspended in HBSS, 20 mM HEPES, 0.1% human serum albumin, and 1 mM GDP‐fucose without human FUT7 for 1 h at 37°C.
Statistics
Testing for differences between means was determined with either Student’s t test or 2‐way ANOVA with post hoc comparisons in Prism 6 software (GraphPad, San Diego, CA, USA). P < 0.05 was considered significant.
RESULTS
Fut‐deficient animals exhibit more severe neutrophilic airway inflammation
The contribution of sLeX bearing selectin ligands during an acute airway episode was assessed using 2 global Fut‐knockout models lacking Fut7, alone or lacking both Fut4 and Fut7. The single Fut4‐deficient mouse was not studied because of data have shown a compensatory role in sLeX synthesis [9], resulting in only a relatively minor increase in rolling velocity in neutrophils from the Fut4−/− animals [26], with minor attenuation of neutrophil recruitment in thioglycollate‐induced peritonitis [8]. Together Fut4 and ‐7 form the α(1,3)‐fucose linkage in sLeX on murine neutrophils [8, 9]. To this end, WT, Fut7−/−, and Fut4−/−Fut7−/− mice were exposed to NTHI. The BALF was collected either 4 or 18 h after instillation of the bacteria and assessed for cell composition with multicolor flow cytometry. In NTHI‐elicited acute airway inflammation, ∼90% of the immune cells recovered in the BALF were neutrophils, with the remaining cells being composed of mostly macrophages with some dendritic, B, and T cells (Supplemental Fig. 1).
We expected that sLeX deficiency by Fut inactivation would significantly hamper overall inflammatory cell recruitment into the airway. However, the opposite was observed, in that Fut7−/− animals had similar to slightly elevated levels of total immune cell infiltration recoverable in the BALF compared to WT ( Fig. 1A ). The Fut4−/−Fut7−/− animals, which are completely sLeX deficient, had a pronounced (1.65‐fold) increase in cell infiltrates. In neutrophil infiltration, the Fut7−/− and Fut4−/−Fut7−/− animals had 1.25‐ and 1.9‐fold increased BALF neutrophil counts, respectively, over WT controls (Fig. 1B). The T‐cells followed our initially expected outcome in which the Fut7−/− and Fut4−/−Fut7−/− deficiencies drastically reduced their recruitment to the airway (Supplemental Fig. 1), most likely because of a difference in the recruitment mechanism of T cells compared to neutrophils, as T cells must encounter the Ag in the secondary lymphoid organs and acquire effector functions before trafficking to the lung [27]. Why the B cells recruit significantly more in the lung in the Fut7−/− mice and then return to WT levels in the Fut4−/−Fut7−/− is unknown and warrants further investigation. Blinded histopathologic evaluation of the inflamed lungs confirmed that the severity of pulmonary inflammation followed the sequence WT < Fut7−/− < Fut4−/−Fut7−/−, which was the reverse of what was initially expected, based on the defective selectin ligands in the knockouts (Fig. 1C–E). The histopathology thus confirmed an exaggerated rather than a mitigated inflammatory response in the α(1,3) Fut‐deficient animals. More severe inflammation manifested as larger, more frequent parenchymal neutrophilic aggregates and greater infiltration of alveolar walls by individual myeloid cells. In some areas of the Fut4−/−Fut7−/− lung, the inflammation completely obscured the underlying pulmonary architecture.
Figure 1.
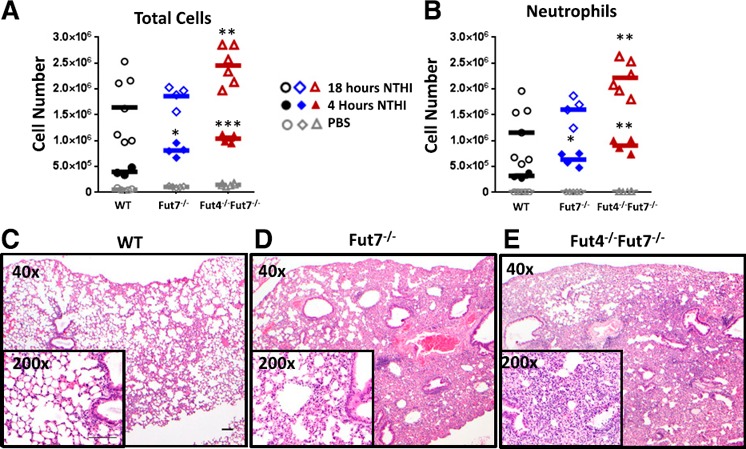
Fut‐deficient mice have increased cell recruitment and inflammation upon NTHI instillation. Colony‐forming units (1 × 106) of NTHI were instilled intratracheally into WT, Fut7−/−, and Fut4−/−Fut7−/− mice. The BALF was collected 4 and 18 h after instillation by flushing twice with 800 μl PBS. (A) Total immune cells were counted by cell counter immediately after hypotonic lysis of erythrocytes. (B) Total neutrophil counts were determined by flow cytometric staining with anti‐CD11b and anti‐Ly6G Abs and multiplying percentages by the total counts in (A). Images of H&E‐stained lung sections of WT (C), Fut7−/− (D), and Fut4−/−Fut7−/− (E) animals treated with NTHI. Magnification, ×40 and ×200 (inset). Overall, neutrophil recruitment was greater in Fut7−/− and Fut4−/−Fut7−/− compared with WT mice (n = 8 mice for each subgroup). Scale bar, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.005 vs. WT.
To determine whether the paradoxical observation of more severe lung inflammation upon reduced α(1,3)‐fucosylation is unique to the NTHI model and live bacteria in general, we substituted NTHI with the sterile agent LPS (Supplemental Fig. 2). The relative severity and immune cell infiltration during acute airway inflammation was identical to the NTHI‐challenged animals with WT < Fut7−/− < Fut4−/−Fut7−/−. Thus, the observations were not NTHI specific.
α(1,3)‐Fucose deficient neutrophils are recruited to lung and peritoneum at different efficiencies
The more pronounced pulmonary airway inflammation in the Fut‐deficient animals may be caused by a known leukocytosis phenotype in the Fut7−/− and Fut7−/−Fut4−/− mice (Supplemental Table S1 [8]) or by α(1,3)‐fucose alterations in other nonleukocyte cell types. Thus, a surplus of available circulatory neutrophils could be passively driving excessive infiltration. To assess the total peripheral neutrophil pool available for recruitment in the different animal models, marginated cells were released into circulation by intravenous epinephrine. This process reduced the baseline ratio of circulatory neutrophils available from ∼8 to ∼3.5 in both the Fut7−/− and Fut4−/−Fut7−/− animals, as compared to the WT animals (Supplemental Fig. 3). After accounting for the marginated cells, Fut deficiency still conferred a surplus of neutrophils in the periphery.
To examine specifically the effects of fucose ligand deficiency on neutrophil recruitment, we compared inflammation in the lung and peritoneum ( Fig. 2 ). Neutrophils were isolated from WT and either Fut7−/− or Fut4−/−Fut7−/− donor mice, differentially labeled with PKH26‐red or PKH67‐green dyes, pooled at known red:green ratios, and reintroduced into the systemic circulation of the recipient mice, whereupon an acute inflammatory response was elicited 1 h before cell injection. In the NTHI‐induced acute airway inflammation, α(1,3)‐fucose‐deficient neutrophils were preferentially recruited into the lung, as evidenced by a ratio of colored cells from the BALF 18 h after instillation that was skewed toward the Fut‐deficient cells (Fig. 2A, B). A preference for airway recruitment of α(1,3)‐fucose‐deficient neutrophils was observed, regardless of the fucosylation genotype of the recipient animals, but the preferential recruitment was more than twice as pronounced in WT recipients than in the Fut4−/−Fut7−/− recipients (Fig. 2A). Significantly more (40%) total cells and neutrophils alike were recovered in the Fut4−/−Fut7−/− recipients as compared to WT (Fig. 2A, B, F, G) [28].
Figure 2.
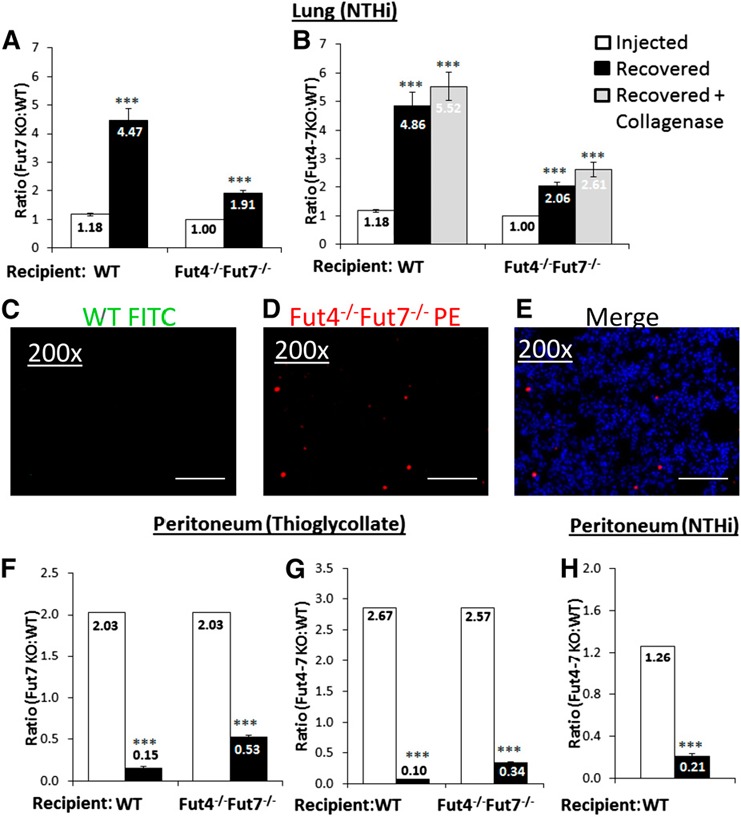
Injecting an equal number of WT and Fut‐deficient neutrophils reveals trafficking preferences to the lung and peritoneum. WT and Fut7−/− (A, F) or WT and Fut4−/−Fut7−/− (B, G, H) neutrophils (2 × 106) were injected at a fixed initial ratio into either WT or Fut4−/−Fut7−/− animals. Intratracheal NTHI (A, B), intraperitoneal thioglycollate (F, G), or intraperitoneal NTHI (H) 1 h was instilled before neutrophil injection. (C, D, E) Images show WT cells (green), Fut4−/−Fut7−/− (red) cells, and a merged image from the lungs of WT recipient mice from the experiment in (B). Open bars: injected ratio of Fut4−/−Fut7−/− PMN to WT neutrophils. Solid bars: ratio of cells recovered from peritoneum or BALF. Shaded bars: ratio of neutrophils recovered after collagenase digestion of lung tissue and BALF (n = 8 mice for each subgroup). Scale bars, 100 μm. ***P < 0.001 vs. injection ratio.
We addressed the possibility that the skewed ratios favoring α(1,3)‐fucose‐deficient cells in the BALF were caused by the preferential retention of WT neutrophils within the lung tissue. Total lung PMNs were collected by collagenase digestion of the lungs, resulting in the recovery of 10‐fold more PMNs than the collection of BALF alone (data not shown). The collagenase‐released cell ratios were even more skewed, favoring Fut4−/−Fut7−/− over WT neutrophils, with ratios of 5.5 and 2.6 when the pooled cells were injected in WT and Fut4−/−Fut7−/− recipients, respectively (Fig. 2B). The preferential recruitment of α(1,3)‐fucose‐deficient cells into the airway is supported by fluorescent imaging of NTHI‐challenged WT recipient lungs, where Fut4−/−Fut7−/− (red‐dyed) and WT (green‐dyed) donor cells from a ∼1:1 premixed pool resulted in very few visible green but strikingly more abundant red cells in the inflamed lung (Fig. 2C–E).
To comparatively contrast these observations on the acute airway against a well‐documented selectin‐dependent model, we examined leukocyte recruitment in thioglycollate‐elicited peritonitis [7, 8, 9–10, 29]. A differentially labeled pool of Fut4−/−Fut7−/− and WT neutrophils with initial ratios of 2 or 2.6 were introduced into WT or Fut4−/−Fut7−/− recipients; peritoneum neutrophilic recruitment heavily favored WT over the Fut‐deficient cells (Fig. 2F, G). To address the caveat that the discordant neutrophil recruitment patterns between NTHI lung inflammation and thioglycollate peritonitis are related to differences between the 2 eliciting agents (e.g., NTHI and thioglycollate), peritonitis was induced by intraperitoneal instillation of NTHI. Here again, neutrophil recruitment in peritonitis markedly favored WT neutrophils, regardless of the eliciting agent (Fig. 2H). NTHI‐induced peritonitis in this case experimentally replicates a clinical condition known as spontaneous bacterial peritonitis, which is bacterial colonization of the ascites fluid that can be a severe complication in patients with cirrhosis of the liver [30].
It was noted further that in both NTHI lung inflammation and thioglycollate peritonitis, the skewed ratios of recruited WT and Fut4−/−Fut7−/− PMNs were more pronounced in WT recipients and less so in α(1,3)‐fucose deficient recipients, perhaps because of an additional consequence of α(1,3)‐fucose insufficiency unrelated to the absence of fucose ligands on the PMNs, which was not explored further. We restricted consideration only to the α(1,3)‐fucose ligands on PMN surfaces; all subsequent recipients were of the WT genotype. Overall, the data illuminated an additional role for α(1,3)‐fucosylated structures on inflammatory cell surfaces, which influenced cell recruitment in a paradoxical manner to the well‐established role of α(1,3)‐fucoses as selectin ligands. Although α(1,3)‐fucose deficiency is traditionally thought to prevent sLeX biosynthesis and neutrophil recruitment by preventing selectin–selectin ligand interactions, the deficiency paradoxically promotes neutrophil migration into the lung.
Sialic acid ligand deficiency does not affect neutrophil trafficking to the inflamed lung
To investigate further the note of sLeX in airway inflammation, ST3gal4 deficiency was also examined. α(2,3)‐Sialylation by the sialyltransferase ST3Gal4 is also a critical enzyme in the formation of sLeX type selectin ligands [6, 7, 10], and ST3Gal4 inactivation results in diminished E‐selectin binding in mice and humans. Consistent with the loss of the sLeX ligand, ST3Gal4−/− neutrophils had diminished recruitment in the thioglycollate‐elicited peritoneum when compared to WT neutrophils ( Fig. 3C ). In the NTHI‐challenged airway, the ST3Gal4−/− neutrophils behaved unlike the Fut4−/−Fut7−/− neutrophils; the ST3Gal4−/− neutrophils were not preferentially recruited (Fig. 3A). In additional controls, neutrophils from another sialyltransferase‐deficient mouse, ST6Gal1−/−, which forms α(2,6)‐sialic acid linkages that are not part of sLeX [20, 23, 31, 32], also did not alter neutrophil recruitment efficiency to the lung or to the peritoneum (Fig. 3B, D). Thus, the data revealed that the exaggerated recruitment of the Fut4−/−Fut7−/− neutrophils into the acute lung was because of deficiency in α(1,3)‐fucose structures that were not the traditional α(2,3)‐sialylated sLeX selectin‐ligands or those involving α(2,6)‐sialylated structures.
Figure 3.
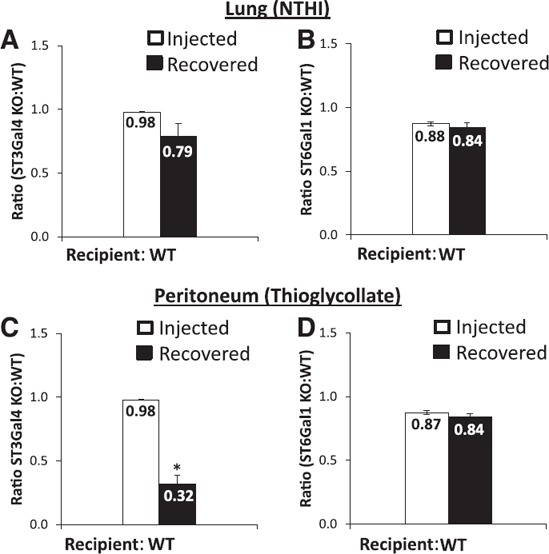
Sialyltransferase deficiency affects neutrophil trafficking to the peritoneum but not the lung. WT and ST3Gal4−/− (A, C) or WT and ST6Gal1−/− (2 × 106) (B, D) neutrophils (2 × 106) were injected in a 1:1 ratio into WT‐recipient animals. Animals were given intratracheal NTHI (C, D) or intraperitoneal thioglycollate (A, B) 1 h before neutrophil injection. Open bars: injected ratio of sialyltransferase KO neutrophils to WT neutrophils. Solid bars: ratio of cells recovered from the peritoneum or BALF. In general, sialyltransferase deficiency did not affect neutrophil trafficking to the lung. ST3Gal4 deficiency reduced neutrophil migration to peritoneum (n = 8 mice for each subgroup). *P < 0.01 vs. injection ratio.
The β2‐integrin–ICAM‐1 axis, not the selectin–selectin ligand axis, controls the exaggerated pulmonary recruitment of Fut4−/−Fut7−/− neutrophils
To determine whether selectin ligands contribute to pulmonary neutrophil recruitment, we used the sLeX‐mimetic panselectin inhibitor TBC1269 [33, 34] to inhibit PMN migration into the lung upon NTHI challenge. TBC1269 treatment impeded inflammatory cell accumulation in the NTHI‐induced acute airway [50 and 25% reductions in total accumulated cells ( Fig. 4A ), or 75 and 50% reductions specifically to PMNs (Fig. 4B) in the BALF of WT and Fut4−/−Fut7−/− mice, respectively]. On the one hand, the contribution of the selectin–selectin ligand axis in the acute airway is supported by the ability of the selectin mimetic to diminish cell migration. On the other hand, the inability of the selectin mimetic to normalize the observed differences in recruitment efficiencies between WT and Fut4−/−Fut7−/− PMNs implies another role for α(1,3)‐fucose structures, independent of the canonical sLeX‐selectin axis in airway recruitment.
Figure 4.
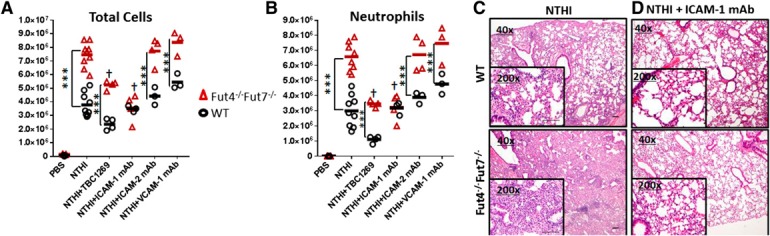
Blocking the ICAM‐1 binding axis but not the selectin axis normalizes the exaggerated airway recruitment of Fut‐deficient neutrophils. Colony‐forming units of NTHI (1 × 106) were instilled intratracheally into WT and Fut4−/−Fut7−/− mice immediately after the injection of 50 μg of the sLeX analog TBC1269, anti‐ICAM‐1, anti‐ICAM‐2, or anti‐VCAM‐1 mAbs. BALF was collected 18 h after instillation by flushing twice with 800 μl PBS. Total immune cells were counted by cell counter immediately after hypotonic lysis of erythrocytes (A). Total neutrophil counts were determined by flow cytometric staining with anti‐CD11b and anti‐Ly6G Abs and multiplying percentages by the total counts. (B) Images of H&E‐stained lung sections of WT or Fut4−/−Fut7−/− animals treated with NTHI and either (C) no mAb or (D) ICAM‐1. Magnification, ×40 and ×200 (inset) (n = 6 mice for each subgroup). Scale bars in NTHI conditions, 100 μm. ***P < 0.001 vs. WT of each subgroup. †P < 0.01 vs. Fut4−/−Fut7−/−+NTHI sample.
To interrogate how the leukocyte adhesion cascade was affected by the α(1,3)‐fucose deficiency, blocking mAbs against the endothelial ligands ICAM‐1, ICAM‐2, and VCAM‐1 of the selectin‐independent steps of the adhesion cascade were used to determine which mAb could return the exaggerated Fut4−/−Fut7−/− neutrophil recruitment to WT levels. Among these, only blocking ICAM‐1 was effective in normalizing total cell or neutrophil infiltration into the BALF (Fig. 4A, B, respectively). The effectiveness of the ICAM‐1 blockade in normalizing the difference was reaffirmed by histopathologic examination (Fig. 4C, D). Although there was marked reduction in solid aggregates of neutrophils and alveolar inflammatory infiltration upon ICAM‐1 mAb treatment of the Fut4−/−Fut7−/− animals, there was little appreciable reduction in inflammation in the WT lung. Thus, the ability to normalize excessive inflammation by a blockade of ICAM‐1 suggests a role for α(1,3) fucosylation in altering how endothelial ICAM‐1 can interact with its neutrophil‐borne binding partner—namely, the β2‐integrin family.
To test the hypothesis excessive inflammatory cell recruitment in α(1,3)‐fucose deficiency is caused by the altered β2‐integrin function on the neutrophils, Abs against the αL‐chain of LFA‐1 (CD11a), the αM‐chain of Mac‐1 (CD11b), and the common β2‐chain (CD18) of β2‐integrin were tested for their ability to normalize inflammation to WT levels. Blockades of CD11a, CD11b, and CD18, similar to the blockade of ICAM‐1, were all equally effective in reducing excessive inflammatory cell recruitment in Fut4−/−Fut7−/− to WT levels (Fig. 5A, B), which was confirmed by histopathologic evaluation (Fig. 5C, D).
Figure 5.
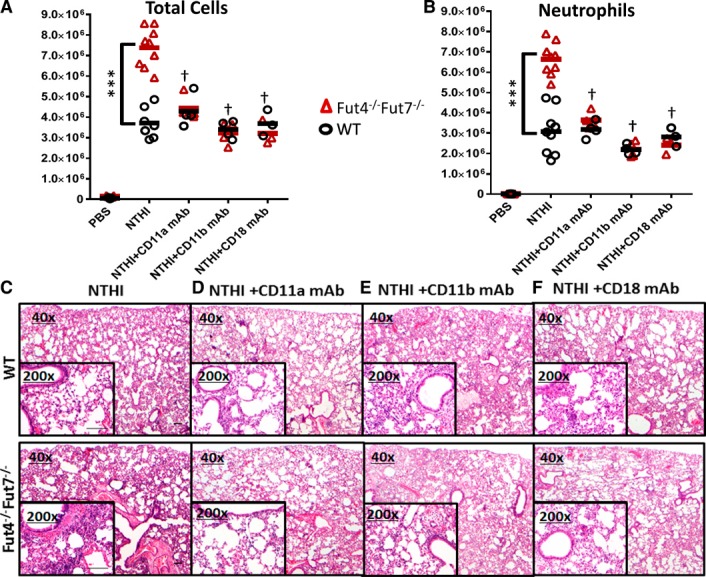
Blocking the α and β subunits of the β2‐integrins normalizes the excess ICAM‐1‐mediated recruitment of the Fut4−/−Fut7−/− neutrophils. Colony‐forming units of NTHI (1 × 106) were instilled intratracheally into WT and Fut4−/−Fut7−/− mice immediately after the injection of 50 μg of anti‐CD11a mAb M17/4, anti‐CD11b mAb M1/70, or anti‐CD18 mAb M18/2. BALF was collected, and total cell counts were determined as described in Fig. 4A. Total neutrophil counts were determined as in Fig. 4B. (C–F) H&E‐stained lung sections of WT or Fut4−/−Fut7−/− animals treated with NTHI and no mAb (C), CD11a (D), CD11b (E), or CD18 (F) showed a normalization of the inflammation and inflammatory cell numbers between WT and Fut4−/−Fut7−/− lungs upon blocking with any of the β2‐integrin mAbs (n = 8 mice for each subgroup). Magnification, ×40 and ×200 (inset). Scale bar, 100 μm. ***P < 0.001 vs. WT of each subgroup. † P < 0.01 vs. Fut4−/−Fut7−/−+NTHI sample.
In WT animals, blocking either subunit did not drastically alter the total number of infiltrated cells (Fig. 5A) or neutrophils (Fig. 5B) compared with non–Ab‐treated controls. However, blocking either the α‐ or β‐chain reduced the number of recruited cells in the Fut4−/−Fut7−/− mice to levels identical with those observed in WT animals in exactly the same manner as blocking ICAM‐1. Histopathologic evaluation of the lungs (Fig. 5C–F) confirmed the abilities of CD11a, CD11b, and CD18 Abs to reduce the level of cell infiltrates in Fut4−/−Fut7−/− animals to WT levels. Histopatholgic evaluation also confirmed the effectiveness of CD11a, CD11b, or CD18 blockade in markedly reducing the solid aggregates of neutrophils and alveolar inflammatory infiltration in Fut4−/−Fut7−/− animals to WT levels, but without appreciable reduction to the baseline pulmonary inflammation in WT animals. This result is consistent with the notion that α(1,3)‐fucosylation deficiency promotes excess inflammation by enhancing the β2‐integrin–ICAM‐1 axis, and this mechanism is independent of the requirement for fucosylation in selectin ligand biosynthesis.
Enhanced signaling through CXCR2 causes greater β2‐integrin–ICAM‐1 adhesive interactions in Fut4−/−Fut7−/− neutrophils
In neutrophils, β2‐integrin conformation and binding affinity are controlled via engagement of CXCR2, the murine IL‐8Rβ homolog, and acting via the intracellular signaling intermediary Rap1‐GTP [35, 36]. To test whether α(1,3)‐fucosylation affects the availability of active β2‐integrins on neutrophil surfaces, a blocking mAb against CXCR2 was used. We observed that CXCR2 blockade not only completely eliminated excess cell recruitment in the Fut4−/−Fut7−/− animals, it also reduced overall airway inflammation both in the Fut4−/−Fut7−/− and WT animals after NTHI exposure, as assessed by recovery of inflammatory cells in the BALF (Fig. 6A, B) and by histopathologic evaluations of the lungs (Fig. 6C, D). Together, the data are consistent with the idea that CXCR2 signaling is a major component in inflammatory cell recruitment during pulmonary inflammation. We hypothesize further that excess recruitment in the Fut4−/−Fut7−/− animals is related to hyperactivated CXCR2 signaling.
Figure 6.
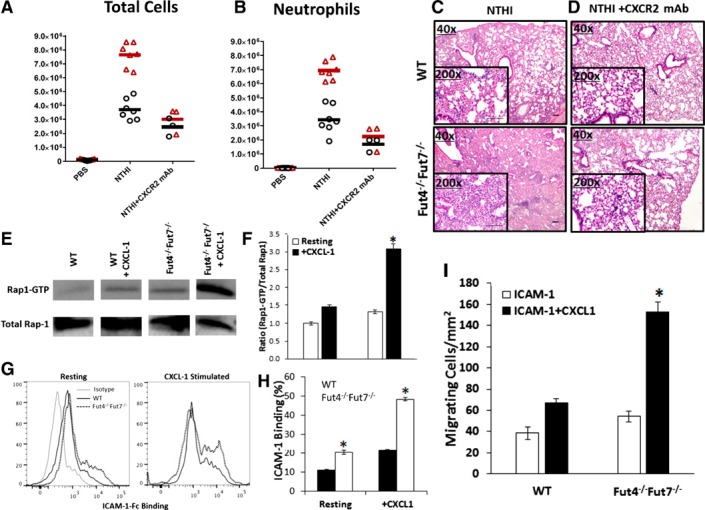
Blocking the CXCL‐1 CXCR2 axis normalizes and reduces recruitment to the lung by attenuating the excess Rap‐1 GTP signaling and ICAM‐1 binding in Fut‐deficient neutrophils. BALF was collected 18 h after instillation of NTHI and total cells (A), and neutrophil counts (B) were taken (n = 6 mice for each subgroup). *P < 0.05 vs. WT of each subgroup. ***P < 0.001 vs. WT of each subgroup. ‡ P < 0.01 vs. WT+NTHI sample. Images of lung sections of WT or Fut4−/−Fut7−/− animals treated with NTHI and no mAb (C) or anti‐CXCR2 (D). Magnification, ×40 and ×200 (inset). Scale bar, 100 μm. Representative immunoblots (E) and density blots (F) are shown for total Rap1 and Rap1‐GTP from murine neutrophils stimulated with PBS or recombinant CXCL1. Density plots were calculated using image‐analysis software. Data are presented as means ± sem. *P < 0.05 vs. WT ratio. (G, H) Percentage of resting or CXCL1 stimulated neutrophils binding to recombinant ICAM‐1 Fc. (I) Number of migrating neutrophils on ICAM‐1 and ICAM‐1+CXCL‐1 surfaces at a shear flow rate of 800 s−1. *P < 0.05 vs. the no‐CXCL1 subtype.
Next, we assessed the active intermediate of the neutrophil inside‐out signaling pathway Rap1‐GTP. Mouse neutrophils were stimulated for 5 min at 37°C in the presence of CXCL1, a known CXCR2 ligand, or with PBS as a control. The addition of recombinant CXCL1 (KC) to both WT and Fut4−/−Fut7−/− neutrophils ex vivo resulted in a 1.5‐fold up‐regulation in WT and a 2.4‐fold elevation of Rap1‐GTP levels (Fig. 6E, F) in Fut4−/−Fut7−/− neutrophils. CXCL1 stimulation also significantly increased the binding to ICAM‐1 Fc in both types of neutrophils (1.8‐fold in WT and 2.5‐fold in Fut4−/−Fut7−/−), as compared to resting, in which greater than 2‐fold more ICAM‐1 Fc were bound by the α(1,3)‐fucose‐deficient Fut4−/−Fut7−/− neutrophils as compared to WT neutrophils (Fig. 6G, H). We then studied whether more Fut4−/−Fut7−/− neutrophils would migrate on ICAM‐1 surfaces under shear flow (Fig. 6I), and the data showed that 45% more Fut4−/−Fut7−/− neutrophils migrated on ICAM‐1, as compared to WT. Furthermore, on ICAM‐1+ surface‐bound CXCL1‐treated surfaces, the number of Fut4−/−Fut7−/− neutrophils migrating under shear flow was increased 2.3‐fold, as compared to that in the WT. The number of cells migrating was also 2.8‐fold more, as compared to Fut4−/−Fut7−/− neutrophils on ICAM‐1‐treated surfaces (WT was up‐regulated only 1.7‐fold).
Together, these data clearly support a model where α(1,3)‐fucose‐deficient neutrophils have enhanced CXCR2 signaling, and acting via the intermediary Rap1‐GTP, results in a greater proportion of active β2‐integrins that can support ICAM‐1 engagement and migration, and culminating in enhanced leukocyte recruitment in the lung.
α(1,3)‐Fucosylation of Fut4−/−Fut7−/− neutrophils with rFUT7 partially reverses recruitment to WT levels in both lung and peritoneal models of inflammation
To test directly the hypothesis that defective α(1,3)‐fucosylation leads to the excessive infiltration of the Fut4−/−Fut7−/− neutrophils in the inflamed airway, we used rFUT7 to re‐engineer the α(1,3)‐fucose linkages onto the Fut4−/−Fut7−/− PMNs. This fucosylation strategy partially restored E‐selectin binding activity on the Fut4−/−Fut7−/− PMNs to ∼2‐fold above their native binding (Fig. 7A). The refucosylated Fut4−/−Fut7−/− neutrophils were introduced into WT recipients. In peritonitis, where defective α(1,3)‐fucosylation resulted in severely attenuated cell recruitment, the refucosylated neutrophils were recruited into the thioglycollate‐elicited peritoneum at a 2.5‐fold greater number than the native Fut4−/−Fut7−/− neutrophils (Fig. 7B), demonstrating the effectiveness of the strategy in partially restoring the α(1,3)‐fucosylated selectin‐ligand sLeX. In the inflamed airway where defective α(1,3)‐fucosylation resulted in exaggerated inflammation, the refucosylated Fut4−/−Fut7−/− neutrophils had a ∼50% reduction in binding to ICAM‐1‐Fc (Fig. 7C) and, in turn, a reduction in Rap1‐GTP signaling back to almost WT levels (Fig. 7D, E). The refucosylated neutrophils also had more than a 2‐fold reduced recruitment to the acute airway when compared to the native Fut4−/−Fut7−/− neutrophils (Fig. 7F). Thus, refucosylation is an effective strategy for the repairing the altered function of Fut4−/−Fut7−/− neutrophils.
Figure 7.
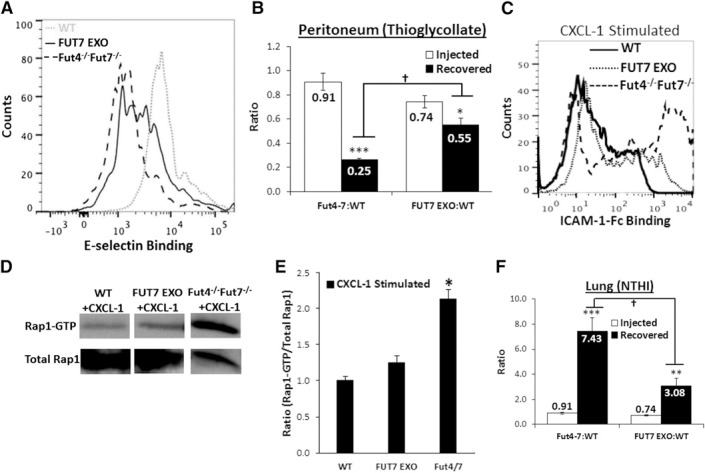
Refucosylating the Fut4−/−Fut7−/− neutrophils restores native trafficking to lung and peritoneum. Neutrophils isolated from Fut4−/−Fut7−/− mice were ex vivo fucosylated, resulting in a 2‐fold increase in binding of Fut4−/−Fut7−/− neutrophils to E‐selectin Fc (A) and a 2‐fold reduction in ICAM‐1 Fc binding (C). Immunoblot (D) and density blot (E) profiles showed that refucosylation also reduced Rap1‐GTP signaling ∼40%. Data are presented as means ± sem. *P < 0.05 vs. WT ratio; 2×106 WT, Fut4−/−Fut7−/− and FUT7 EXO (B, E) neutrophils were injected into WT recipient animals in an ∼1:1 ratio. Animals received intraperitoneal thioglycollate (B) or intratracheal NTHI (F) 1 h before PMN injection. Open bars: initial injected ratio of FUT7 EXO PMN or Fut4−/−Fut7−/− to WT PMN. Solid bars: ratio of cells recovered from peritoneum or BALF (n = 8 mice for each subgroup). *P < 0.05, **P < 0.01, and ***P < 0.005 vs. injection ratio.
DISCUSSION
α(1,3)‐Fucosylated glycans on neutrophils are typically regarded to exist in the context of the tetrasaccharide sLeX glycoform, which is the canonical selectin determinant participating in the initial step of the leukocyte recruitment cascade during inflammation [37]. These fucosylated ligands bind P‐ and E‐selectin on the stimulated endothelial monolayer with rapid on‐and‐off rates, thus facilitating the direct capture or tethering of circulatory leukocytes from free flow onto the vessel wall [1]. In turn, the selectin–selectin ligand–mediated tethering allows cellular activation along a gradient of endothelium‐bound chemokines and promotes the kinetically slower but tighter integrin‐mediated adhesive interactions, resulting ultimately in extravasation of the leukocytes to the inflamed tissue [2]. Owing to its place at the beginning of the cascade, targeting the selectin–selectin ligand axis to attenuate excess immune cell recruitment is of significant clinical interest and the subject of active pharmaceutical clinical trials with generally encouraging outcomes [12, 13]. A physiologic contribution of α(1,3)‐fucosylated glycans outside the context of selectin ligands is not known.
The current work suggests a novel role for α(1,3)‐fucosylated glycans in the leukocyte adhesion cascade, in addition to the sLeX glycoform for selectin binding. This possibility was uniquely visualized in lung inflammation, because leukocyte recruitment to the lung differs from most other vascular sites caused by the unique alveolar capillary beds of pulmonary circulation, portions of which are smaller in diameter than the neutrophils themselves [18]. It requires the neutrophil to deform in these beds [38]. Such close contact between the leukocytes and the capillary endothelium results in slower transit rates. We posit that the slowed transit rate diminishes the importance of selectin‐ligands in the lung compared to other organs [39, 40], which in turn allows visualization of the role of α(1,3)‐fucose in other aspects of the leukocyte recruitment cascade. Consistent with this notion, although the recruitment of Fut4−/−Fut7−/− neutrophils was dramatically reduced, as expected, in selectin–selectin ligand–dominated recruitment in the inflamed peritoneum, α(1,3)‐fucose deficiency had the opposite effect in the inflamed airway.
A role for α(1,3)‐fucose in attenuating airway inflammatory leukocyte recruitment was confirmed in ex vivo fucosylation studies where the partial restoration of cell surface α(1,3)‐fucose on the Fut4−/−Fut7−/−‐deficient neutrophils, using recombinant human FUT7, reduced exaggerated neutrophil recruitment in the airway. Exofucosylation also restored the cell surface sLeX epitope, in that it partially reinstated the selectin‐mediated recruitment of the Fut‐deficient neutrophils in the peritoneum during inflammation. In additional support of the involvement of α(1,3)‐fucose outside of the context of sLeX, the sLeX ‐analog TBC‐1269 [41] could not normalize the exaggerated recruitment of Fut4−/−Fut7−/− neutrophils to WT levels. Although the nature of the α(1,3)‐fucose ligand participating in the selectin‐independent leukocyte recruitment remains to be elucidated, our data are consistent with the idea that this α(1,3)‐fucose does not exist within the context of the canonical selectin ligand, the sLeX tetrasaccharide. As further proof, ST3Gal‐4‐deficient neutrophils, which also lacked sLeX, had the expected attenuation in selectin‐dependent recruitment in peritonitis and in fact had their recruitment into the airway slightly reduced.
The data presented herein disclose a novel role for α(1,3) fucosylation in controlling CD18/β2‐integrin‐dependent firm arrest to ICAM‐1. In the current work, we focused on whether the binding between neutrophil borne β2‐integrins Mac‐1 (CD11b/CD18) and LFA‐1 (CD11a/CD18) and their principal binding partner ICAM‐1 [1, 42] were affected by the loss of α(1,3)‐fucose. Both LFA‐1 and Mac‐1 have been shown to be important during neutrophil recruitment to the lungs in response to specific inflammatory insults [42, 43, 44–45]. Consistent with this, the same exaggerated airway recruitment was observed when the gram‐negative NTHI was replaced with the sterile agent LPS (Supplemental Fig. 1). With this said, it cannot be discounted that the utility of this α(1,3) fucose to alter neutrophil recruitment could be different based on the type of inflammatory insult, as some gram‐positive bacteria such as Streptococcus pneumoniae have been shown to use a β2‐integrin‐independent axis of recruitment [46]. The binding of both LFA‐1 and MAC‐1 to their partners is also known to be distinct from the selectin–selectin ligand mechanism. In our studies, Fut4−/−Fut7−/− neutrophil migration to the lung was normalized or reduced to WT levels upon blocking either the unique α‐chains or the common β2‐chain. Because the recruitment was only normalized and not completely abrogated (WT levels were barely affected by treatment with β2‐integrin blocking mAbs), the α(1,3)‐fucose axis was responsible only for the augmented trafficking observed in the Fut4−/−Fut7−/− neutrophils.
The blocking efficacy of the anti‐integrin mAbs suggests that the lack of α(1,3)‐fucose increases the number of active β2‐integrins by affecting mechanistic steps upstream of the β2‐integrin–ICAM‐1 axis. In this regard, human neutrophils have 2 neutrophil‐bound receptors for the endothelium‐bound cytokines CXCR1 and CXCR2, whereas murine neutrophils have only CXCR2 [47]. CXCR2 has been thought to have 2 N‐glycan sites [48], to be sensitive to enzymatic deglycosylation, which affects its normal function [49] and has been hypothesized to carry either α(1,2)‐ or α(1,3)‐fucose residues [50], which lends credence to our theory that the α(1,3) fucose may affect CXCR2 function. Also, endothelial CXCR2 has been implicated in the regulation of lung endothelial leakiness upon LPS stimulation [28] which may serve to explain why the ratio of Fut4−/−Fut7−/−:WT cells recovered was higher in the WT recipients (Fig. 2), given that loss of α(1,3)‐fucose may modify these sites as well.
The engagement of the chemokine by these cognate receptors initiates an inside‐out signaling cascade in which the chemokine receptor participates in a series of interactions culminating in the phosphorylation of Rap1‐GDP into Rap1‐GTP [51]. Rap1‐GTP is the immediate upstream step of talin‐1 mediated β2‐integrin opening to the low‐affinity conformation and also kindlin‐3‐mediated opening to the high‐affinity state [52]. Together, these processes facilitate β2‐integrin’s binding to endothelial ICAM‐1 and the firm arrest step of the multistep adhesion cascade. The talin‐1‐mediated transition from the bent to the low‐affinity state has been associated with PSGL‐1 binding to E‐/P‐selectin [36, 53], which would be absent in the Fut4−/−Fut7−/− neutrophils because of their lack of sLeX. On the other hand, kindlin‐3 transitions to the high‐affinity state are mediated through CXCR2 stimulation. Therefore, monitoring the levels of Rap1‐GTP upon chemokine activation of CXCR2 should be a direct indicator of the amount of kindlin‐3‐mediated transition of the β2‐integrins to high affinity, especially because no PSGL‐1‐selectin interactions occur in the Fut4−/−Fut7−/− neutrophils. Indeed, our data show increased Rap1‐GTP levels in both resting and CXCL1‐stimulated Fut4−/−Fut7−/− neutrophils, as compared to WT leukocytes.
Furthermore, the binding and migration on recombinant ICAM‐1 is significantly increased in knockout neutrophils compared to WT cells. An interesting side point is that our data suggest that a small fraction of β2‐integrins remain in the active (intermediate or high‐affinity) form, even in the resting state. In contrast to the classic model on integrin activation, increasing evidence has demonstrated that neutrophils maintain a small number of active integrins in circulation [54, 55], and this is seen in both the WT and Fut4−/−Fut7−/− neutrophils, albeit to various degrees (10% vs. 20%). Thus α(1,3)‐fucose may be a method for increasing the levels of circulating leukocytes in the immediate state of integrin affinity and supports our model of increased emigration to the lung. In addition, high Rap1‐GTP levels, ICAM‐1 Fc binding, and excessive leukocyte emigration to the lung could be either partially or fully reversed by refucosylating the Fut4−/−Fut7−/− neutrophils, using recombinant FUT7. Thus, the loss of the α(1,3)‐fucose in neutrophils may increase leukocyte trafficking to the lung because of a hyperactive CXCR2 receptor that up‐regulates Rap1‐GTP, leading to more active β2‐integrins.
The observations support the idea that enhancement of integrin‐mediated interactions in the α(1,3)‐fucose‐deficient cells have been overlooked because of the dominance of the selectin‐mediated interactions in most tissues and models of inflammation. The severe requirement for α(1,3)‐fucose for selectin interactions does not allow for chemokine signaling and integrin binding to occur downstream in its absence in traditional inflammatory models. To this end, lung biology has identified an additional previously overlooked α(1,3)‐fucose‐dependent, selectin‐independent role in inflammatory cell recruitment.
The data support a mechanism, depicted in Fig. 8 , where an α(1,3)‐fucose structure, distinct from the sLeX participating in the selectin axis, can modulate the function of the chemokine receptor CXCR2 and is mediated via the inside‐out signaling pathway intermediate Rap1‐GTP, to control β2‐integrin conformation and binding affinity. Thus, a condition of α(1,3)‐fucose deficit would drive enhanced recruitment of the affected leukocytes by enhancing the binding efficacy of the β2‐integrin axis. Moreover, these observations caution that targeting the selectin–selectin ligand axis may have limited effectiveness when the diameter of the normal vasculature has been compromised, such as during atherosclerosis. Finally, the existence of a novel α(1,3)‐fucosylated glycan affecting inflammatory cell trafficking and acting on the level of chemokine–chemokine receptor engagement may present an additional exploitable target for therapeutic intervention in immune cell recruitment.
Figure 8.
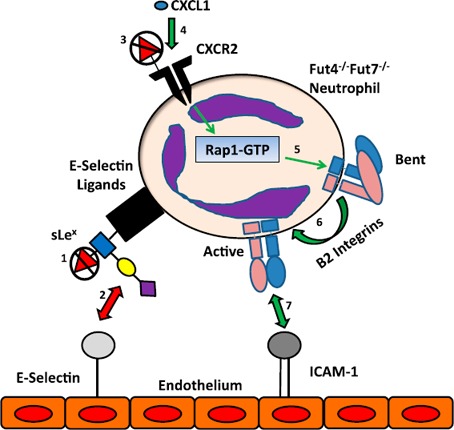
Altered binding pathways in Fut4−/−Fut7−/− neutrophils. The different binding mechanisms affected by the loss of α(1,3) fucose in Fut4−/−Fut7−/− deficient mice during recruitment of neutrophils to the acutely inflamed lungs. Previous studies have demonstrated that Fut4−/−Fut7−/− neutrophils have lost the fucose residue of sLeX (1) on their selectin ligands and therefore do not interact with endothelial bound E‐selectin (2). On the other hand, the loss of fucose on CXCR2 (3) leads to enhanced chemokine signaling (CXCL1 was tested but other cytokines can signal through CXCR2, as well) through the receptor (4), which in turn up‐regulates Rap1‐GTP (5). This process leads to more β2‐integrins in their high‐affinity state (6). More active β2‐integrins (LFA‐1/Mac‐1) lead to a greater interaction with ICAM‐1 on the endothelial surface and therefore more migration on the endothelium and emigration into the lung (7). Red arrows: interactions that are down‐regulated, whereas green arrows indicate those that are up‐regulated upon α(1,3) fucose deficiency.
AUTHOR CONTRIBUTIONS
A.B.J. designed and performed experiments and wrote the manuscript. M.N., C.M., and A.L. designed and performed the experiments. P.N.B. analyzed lung sections and wrote the paper. R.S., Y.T., S.N., and J.T.Y.L. designed research, provided critical methods, coordinated project activities, and wrote the paper.
DISCLOSURE
The authors declare no conflicts of interest.
Supporting information
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported by the U. S. National Institutes of Health (NIH), National Heart, Lung, and Blood Institute Grants HL63014 and HL103411 (to S.N.), and a Program of Excellence in Glycosciences grant HL107146 (J.T.Y.L.). The core facilities of Roswell Park Cancer Institute used in this work were supported in part by NIH National Cancer Institute Cancer Center Support Grant CA15056. The authors thank Melinda Haarmeyer for help with the lung sections, Prof. Peter Vanderslice for the TBC1269, and Dr. Daniel Hammer for his guidance on the ICAM‐1 migration assays.
REFERENCES
- 1. Ley, K. , Laudanna, C. , Cybulsky, M. I. , Nourshargh, S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. [DOI] [PubMed] [Google Scholar]
- 2. Springer, T. A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314. [DOI] [PubMed] [Google Scholar]
- 3. Phillips, M. L. , Nudelman, E. , Gaeta, F. C. , Perez, M. , Singhal, A. K. , Hakomori, S. , Paulson, J. C. (1990) ELAM‐1 mediates cell adhesion by recognition of a carbohydrate ligand, sialyl‐Lex. Science 250, 1130–1132. [DOI] [PubMed] [Google Scholar]
- 4. Polley, M. J. , Phillips, M. L. , Wayner, E. , Nudelman, E. , Singhal, A. K. , Hakomori, S. , Paulson, J. C. (1991) CD62 and endothelial cell‐leukocyte adhesion molecule 1 (ELAM‐1) recognize the same carbohydrate ligand, sialyl‐Lewis x. Proc. Natl. Acad. Sci. USA 88, 6224–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buffone, A., Jr. , Mondal, N. , Gupta, R. , McHugh, K. P. , Lau, J. T. Y. , Neelamegham, S. (2013) Silencing a1,3‐fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E‐selectin‐mediated cell adhesion. J. Biol. Chem. 288, 1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mondal, N. , Buffone, A., Jr. , Stolfa, G. , Antonopoulos, A. , Lau, J. T. Y. , Haslam, S. M. , Dell, A. , Neelamegham, S. (2015) ST3Gal‐4 is the primary sialyltransferase regulating the synthesis of E‐, P‐, and L‐selectin ligands on human myeloid leukocytes. Blood 125, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellies, L. G. , Sperandio, M. , Underhill, G. H. , Yousif, J. , Smith, M. , Priatel, J. J. , Kansas, G. S. , Ley, K. , Marth, J. D. (2002) Sialyltransferase specificity in selectin ligand formation. Blood 100, 3618–3625. [DOI] [PubMed] [Google Scholar]
- 8. Homeister, J. W. , Thall, A. D. , Petryniak, B. , Malý, P. , Rogers, C. E. , Smith, P. L. , Kelly, R. J. , Gersten, K. M. , Askari, S. W. , Cheng, G. , Smithson, G. , Marks, R. M. , Misra, A. K. , Hindsgaul, O. , von Andrian, U. H. , Lowe, J. B. (2001) The α(1,3)fucosyltransferases FucT‐IV and FucT‐VII exert collaborative control over selectin‐dependent leukocyte recruitment and lymphocyte homing. Immunity 15, 115–126. [DOI] [PubMed] [Google Scholar]
- 9. Malý, P. , Thall, A. , Petryniak, B. , Rogers, C. E. , Smith, P. L. , Marks, R. M. , Kelly, R. J. , Gersten, K. M. , Cheng, G. , Saunders, T. L. , Camper, S. A. , Camphausen, R. T. , Sullivan, F. X. , Isogai, Y. , Hindsgaul, O. , von Andrian, U. H. , Lowe, J. B. (1996) The α(1,3)fucosyltransferase Fuc‐TVII controls leukocyte trafficking through an essential role in L‐, E‐, and P‐selectin ligand biosynthesis. Cell 86, 643–653. [DOI] [PubMed] [Google Scholar]
- 10. Yang, W. H. , Nussbaum, C. , Grewal, P. K. , Marth, J. D. , Sperandio, M. (2012) Coordinated roles of ST3Gal‐VI and ST3Gal‐IV sialyltransferases in the synthesis of selectin ligands. Blood 120, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Broderick, J. , Brott, T. , Kothari, R. , Miller, R. , Khoury, J. , Pancioli, A. , Gebel, J. , Mills, D. , Minneci, L. , Shukla, R. (1998) The Greater Cincinnati/Northern Kentucky Stroke Study: preliminary first‐ever and total incidence rates of stroke among blacks. Stroke 29, 415–421. [DOI] [PubMed] [Google Scholar]
- 12. Chang, J. , Patton, J. T. , Sarkar, A. , Ernst, B. , Magnani, J. L. , Frenette, P. S. (2010) GMI‐1070, a novel pan‐selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 116, 1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tardif, J.‐C. , Tanguay, J.‐F. , Wright, R. S. , Duchatelle, V. , Petroni, T. , Grégoire, J. C. , Ibrahim, R. , Heinonen, T. M. , Robb, S. , Bertrand, O. F. , Cournoyer, D. , Johnson, D. , Mann, J. , Guertin, M.‐C. , L'Allier, P. L. (2013) Effects of the P‐selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non‐ST‐segment elevation myocardial infarction: results of the SELECT‐ACS trial. J. Am. Coll. Cardiol. 61, 2048–2055. [DOI] [PubMed] [Google Scholar]
- 14. Barbier, V. , Nutt, H. L. , Hasnain, S. Z. , Levesque, J.‐P. , Magnani, J. L. , McGuckin, M. A. (2013) Administration of E‐selectin antagonist GMI‐1271 improves survival after high‐dose chemotherapy by alleviating mucositis and accelerating neutrophil recovery. Blood 122, 2266. [Google Scholar]
- 15. Lugade, A. A. , Bogner, P. N. , Murphy, T. F. , Thanavala, Y. (2011) The role of TLR2 and bacterial lipoprotein in enhancing airway inflammation and immunity. Front. Immunol. 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grommes, J. , Soehnlein, O. (2011) Contribution of neutrophils to acute lung injury. Mol. Med. 17, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang, M. , Kumar, R. K. , Hansbro, P. M. , Foster, P. S. (2012) Emerging roles of pulmonary macrophages in driving the development of severe asthma. J. Leukoc. Biol. 91, 557–569. [DOI] [PubMed] [Google Scholar]
- 18. Doerschuk, C. M. , Beyers, N. , Coxson, H. O. , Wiggs, B. , Hogg, J. C. (1993) Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J. Appl. Physiol. 74, 3040–3045. [DOI] [PubMed] [Google Scholar]
- 19. Lugade, A. A. , Vethanayagam, R. R. , Nasirikenari, M. , Bogner, P. N. , Segal, B. H. , Thanavala, Y. (2011) Nrf2 regulates chronic lung inflammation and B‐cell responses to nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol. 45, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nasirikenari, M. , Chandrasekaran, E. V. , Matta, K. L. , Segal, B. H. , Bogner, P. N. , Lugade, A. A. , Thanavala, Y. , Lee, J. J. , Lau, J. T. Y. (2010) Altered eosinophil profile in mice with ST6Gal‐1 deficiency: an additional role for ST6Gal‐1 generated by the P1 promoter in regulating allergic inflammation. J. Leukoc. Biol. 87, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jungblut, M. , Oeltze, K. , Zehnter, I. , Hasselmann, D. , Bosio, A. (2009) Standardized preparation of single‐cell suspensions from mouse lung tissue using the gentle MACS dissociator. J. Vis. Exp. 29, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marathe, D. D. , Buffone, A., Jr. , Chandrasekaran, E. V. , Xue, J. , Locke, R. D. , Nasirikenari, M. , Lau, J. T. Y. , Matta, K. L. , Neelamegham, S. (2010) Fluorinated per‐acetylated GalNAc metabolically alters glycan structures on leukocyte PSGL‐1 and reduces cell binding to selectins. Blood 115, 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nasirikenari, M. , Segal, B. H. , Ostberg, J. R. , Urbasic, A. , Lau, J. T. (2006) Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal I. Blood 108, 3397–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , Preibisch, S. , Rueden, C. , Saalfeld, S. , Schmid, B. , Tinevez, J.‐Y. , White, D. J. , Hartenstein, V. , Eliceiri, K. , Tomancak, P. , Cardona, A. (2012) Fiji: an open‐source platform for biological‐image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merzaban, J. S. , Burdick, M. M. , Gadhoum, S. Z. , Dagia, N. M. , Chu, J. T. , Fuhlbrigge, R. C. , Sackstein, R. (2011) Analysis of glycoprotein E‐selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood 118, 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weninger, W. , Ulfman, L. H. , Cheng, G. , Souchkova, N. , Quackenbush, E. J. , Lowe, J. B. , von Andrian, U. H. (2000) Specialized contributions by α(1,3)‐fucosyltransferase‐IV and FucT‐VII during leukocyte rolling in dermal microvessels. Immunity 12, 665–676. [DOI] [PubMed] [Google Scholar]
- 27. D'Ambrosio, D. , Mariani, M. , Panina‐Bordignon, P. , Sinigaglia, F. (2001) Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am. J. Respir. Crit. Care Med. 164, 1266–1275. [DOI] [PubMed] [Google Scholar]
- 28. Reutershan, J. , Morris, M. A. , Burcin, T. L. , Smith, D. F. , Chang, D. , Saprito, M. S. , Ley, K. (2006) Critical role of endothelial CXCR2 in LPS‐induced neutrophil migration into the lung. J. Clin. Invest. 116, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forlow, S. B. , Ley, K. (2001) Selectin‐independent leukocyte rolling and adhesion in mice deficient in E‐, P‐, and L‐selectin and ICAM‐1. Am. J. Physiol. Heart Circ. Physiol. 280, H634–H641. [DOI] [PubMed] [Google Scholar]
- 30. Lata, J. , Stiburek, O. , Kopacova, M. (2009) Spontaneous bacterial peritonitis: a severe complication of liver cirrhosis. World J. Gastroenterol. 15, 5505–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones, M. B. , Nasirikenari, M. , Lugade, A. A. , Thanavala, Y. , Lau, J. T. Y. (2012) Anti‐inflammatory IgG production requires functional P1 promoter in β‐galactoside a2,6‐sialyltransferase 1 (ST6Gal‐1) gene. J. Biol. Chem. 287, 15365–15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nasirikenari, M. , Veillon, L. , Collins, C. C. , Azadi, P. , Lau, J. T. Y. (2014) Remodeling of marrow hematopoietic stem and progenitor cells by non‐self ST6Gal‐1 sialyltransferase. J. Biol. Chem. 289, 7178–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davenpeck, K. L. , Berens, K. L. , Dixon, R. A. F. , Dupre, B. , Bochner, B. S. (2000) Inhibition of adhesion of human neutrophils and eosinophils to P‐selectin by the sialyl Lewis antagonist TBC1269: preferential activity against neutrophil adhesion in vitro. J. Allergy Clin. Immunol. 105, 769–775. [DOI] [PubMed] [Google Scholar]
- 34. Hicks, A. E. R. , Abbitt, K. B. , Dodd, P. , Ridger, V. C. , Hellewell, P. G. , Norman, K. E. (2005) The anti‐inflammatory effects of a selectin ligand mimetic, TBC‐1269, are not a result of competitive inhibition of leukocyte rolling in vivo. J. Leukoc. Biol. 77, 59–66. [DOI] [PubMed] [Google Scholar]
- 35. Stadtmann, A. , Brinkhaus, L. , Mueller, H. , Rossaint, J. , Bolomini‐Vittori, M. , Bergmeier, W. , Van Aken, H. , Wagner, D. D. , Laudanna, C. , Ley, K. , Zarbock, A. (2011) Rap1a activation by CalDAG‐GEFI and p38 MAPK is involved in E‐selectin‐dependent slow leukocyte rolling. Eur. J. Immunol. 41, 2074–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pruenster, M. , Kurz, A. R. M. , Chung, K.‐J. , Cao‐Ehlker, X. , Bieber, S. , Nussbaum, C. F. , Bierschenk, S. , Eggersmann, T. K. , Rohwedder, I. , Heinig, K. , Immler, R. , Moser, M. , Koedel, U. , Gran, S. , McEver, R. P. , Vestweber, D. , Verschoor, A. , Leanderson, T. , Chavakis, T. , Roth, J. , Vogl, T. , Sperandio, M. (2015) Extracellular MRP8/14 is a regulator of β2 integrin‐dependent neutrophil slow rolling and adhesion. Nat. Commun. 6, 6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Becker, D. J. , Lowe, J. B. (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R. [DOI] [PubMed] [Google Scholar]
- 38. Huang, Y. , Doerschuk, C. M. , Kamm, R. D. (2001) Computational modeling of RBC and neutrophil transit through the pulmonary capillaries. J. Appl. Physiol. 90, 545–564. [DOI] [PubMed] [Google Scholar]
- 39. Downey, G. P. , Worthen, G. S. , Henson, P. M. , Hyde, D. M. (1993) Neutrophil sequestration and migration in localized pulmonary inflammation: capillary localization and migration across the interalveolar septum. Am. Rev. Respir. Dis. 147, 168–176. [DOI] [PubMed] [Google Scholar]
- 40. Gebb, S. A. , Graham, J. A. , Hanger, C. C. , Godbey, P. S. , Capen, R. L. , Doerschuk, C. M. , Wagner, W. W., Jr. (1995) Sites of leukocyte sequestration in the pulmonary microcirculation. J. Appl. Physiol. 79, 493–497. [DOI] [PubMed] [Google Scholar]
- 41. Watz, H. , Bock, D. , Meyer, M. , Schierhorn, K. , Vollhardt, K. , Woischwill, C. , Pedersen, F. , Kirsten, A. , Beeh, K.‐M. , Meyer‐Sabellek, W. , Magnussen, H. , Beier, J. (2013) Inhaled pan‐selectin antagonist Bimosiamose attenuates airway inflammation in COPD. Pulm. Pharmacol. Ther. 26, 265–270. [DOI] [PubMed] [Google Scholar]
- 42. Smith, C. W. , Marlin, S. D. , Rothlein, R. , Toman, C. , Anderson, D. C. (1989) Cooperative interactions of LFA‐1 and Mac‐1 with intercellular adhesion molecule‐1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 83, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Erzurum, S. C. , Downey, G. P. , Doherty, D. E. , Schwab III, B. , Elson, E. L. , Worthen, G. S. (1992) Mechanisms of lipopolysaccharide‐induced neutrophil retention: relative contributions of adhesive and cellular mechanical properties. J. Immunol. 149, 154–162. [PubMed] [Google Scholar]
- 44. McMillan, S. J. , Sharma, R. S. , McKenzie, E. J. , Richards, H. E. , Zhang, J. , Prescott, A. , Crocker, P. R. (2013) Siglec‐E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b β2‐integrin‐dependent signaling. Blood 121, 2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Asaduzzaman, M. , Zhang, S. , Lavasani, S. , Wang, Y. , Thorlacius, H. (2008) LFA‐1 and MAC‐1 mediate pulmonary recruitment of neutrophils and tissue damage in abdominal sepsis. Shock 30, 254–259. [DOI] [PubMed] [Google Scholar]
- 46. Doerschuk, C. M. , Winn, R. K. , Coxson, H. O. , Harlan, J. M. (1990) CD18‐dependent and ‐independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J. Immunol. 144, 2327–2333. [PubMed] [Google Scholar]
- 47. Olson, T. S. , Ley, K. (2002) Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R7–R28. [DOI] [PubMed] [Google Scholar]
- 48. Ludwig, A. , Ehlert, J. E. , Flad, H.‐D. , Brandt, E. (2000) Identification of distinct surface‐expressed and intracellular CXC‐chemokine receptor 2 glycoforms in neutrophils: N‐glycosylation is essential for maintenance of receptor surface expression. J. Immunol. 165, 1044–1052. [DOI] [PubMed] [Google Scholar]
- 49. Frommhold, D. , Ludwig, A. , Bixel, M. G. , Zarbock, A. , Babushkina, I. , Weissinger, M. , Cauwenberghs, S. , Ellies, L. G. , Marth, J. D. , Beck‐Sickinger, A. G. , Sixt, M. , Lange‐Sperandio, B. , Zernecke, A. , Brandt, E. , Weber, C. , Vestweber, D. , Ley, K. , Sperandio, M. (2008) Sialyltransferase ST3Gal‐IV controls CXCR2‐mediated firm leukocyte arrest during inflammation. J. Exp. Med. 205, 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu, L. H. , Shi, B. Z. , Zhao, Q. L. , Wu, X. Z. (2010) Fucosylated glycan inhibition of human hepatocellular carcinoma cell migration through binding to chemokine receptors. Glycobiology 20, 215–223. [DOI] [PubMed] [Google Scholar]
- 51. Alon, R. , Ley, K. (2008) Cells on the run: shear‐regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr. Opin. Cell Biol. 20, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lefort, C. T. , Ley, K. (2012) Neutrophil arrest by LFA‐1 activation. Front. Immunol. 3, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zarbock, A. , Ley, K. , McEver, R. P. , Hidalgo, A. (2011) Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alon, R. , Dustin, M. L. (2007) Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen‐presenting cells. Immunity 26, 17–27. [DOI] [PubMed] [Google Scholar]
- 55. Johansson, M. W. , Mosher, D. F. (2013) Integrin activation States and eosinophil recruitment in asthma. Front. Pharmacol. 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files