

Short abstract
Review on the expression patterns and functions of long noncoding RNAs in the T cell compartment of the adaptive immune system.
Keywords: epigenetics, lncRNA, adaptive immunity
Abstract
Long noncoding RNAs are recently discovered regulatory RNA molecules that do not code for proteins but influence a vast array of biologic processes. In vertebrates, the number of long noncoding RNA genes is thought to greatly exceed the number of protein‐coding genes. It is also thought that long noncoding RNAs drive the biologic complexity observed in vertebrates compared with that in invertebrates. Evidence of this complexity has been found in the T‐lymphocyte compartment of the adaptive immune system. In the present review, we describe our current level of understanding of the expression of specific long or large intergenic or intervening long noncoding RNAs during T‐lymphocyte development in the thymus and differentiation in the periphery and highlight the mechanisms of action that specific long noncoding RNAs employ to regulate T‐lymphocyte function, both in vitro and in vivo.
Abbreviations
- DN
double negative (CD4−, CD8− thymocyte)
- DP
double positive (CD4+, CD8+ thymocyte)
- eQTL
expression quantitative trait loci
- FasL
Fas ligand
- GWASs
genome‐wide association studies
- ILC
innate lymphocyte
- LCR
locus control region
- lncRNA
long noncoding RNA
- lincRNA
long or large intergenic or intervening lncRNA
- NFAT
nuclear factor of activated T cells
- NRON
noncoding RNA repressor of NFAT
- pre‐mRNA
premature RNA
- RA
rheumatoid arthritis
- RBM5
RNA binding motif protein 5
- RNA‐seq
RNA sequencing
- sFas
soluble form of Fas
- SNP
single nucleotide polymorphism
- TB
tuberculosis
- Tcm
central memory T cell
- Tem
effector memory T cell
- Th2LCC
Th2 locus control region
- Tregs
T regulatory cells
Introduction
Most would agree that humans possess greater biologic complexity than worms ( Fig. 1A ). However, worms, jawless vertebrates, vertebrates, and humans all have about the same number of protein‐coding genes (∼20,000), which is not much greater than that of yeast (∼5000; Fig. 1B). Most proteins expressed by worms and humans have similar functions. However, humans possess far greater numbers of discrete cell lineages, organ systems, and physical and intellectual capacity than worms. Therefore, one must ask where is the information that programs this increased complexity? In contrast to the numbers of protein‐coding genes, the genome size between lower organisms, such as worms, and higher organisms, such as humans, is markedly different. The human genome contains about 3,000,000,000 nucleotides, and a worm genome is about 100,000,000 nucleotides; thus, the human genome is ∼30 times larger than that of the worm genome (Fig. 1C) [1, 2].
Figure 1.
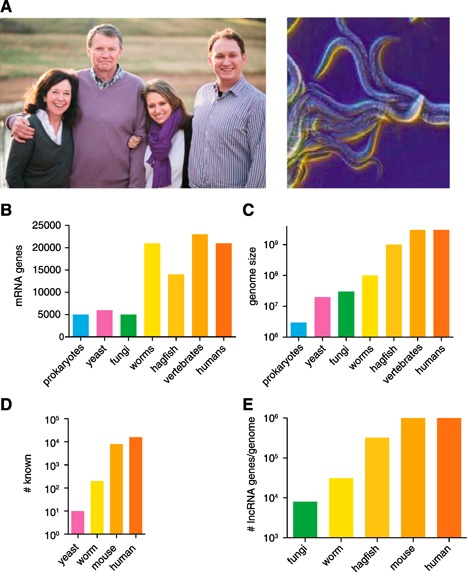
Biologic complexity and expansion of genomes. (A) Illustration of humans and worms (Caenorhabditis elegans). (B) Numbers of mRNA genes in genomes of different organisms from different phyla. (C) Genome size in different organisms from different phyla. (D) Organisms from many different phyla produce lncRNAs. (E) Potential number of lncRNAs produced by organisms from different phyla.
A few years after release of the first draft sequence of the human genome [3, 4], the Encyclopedia of DNA Elements consortium was founded in 2003 with the goal of identifying and defining functions of all DNA elements in the human genome [5, 6, 7, 8–9]. One of the surprising discoveries was that the vast majority of the human genome (current estimates are ∼75%) is transcribed in some cell lineages during some stage of development [2, 6, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31–32]. This contrasts with the amount of genome space used to encode protein‐coding genes, ∼2%. These studies have led to the discovery of a new class of RNA species termed “long noncoding RNA” or “lncRNA” [32, 33, 34, 35, 36, 37, 38–39]. Genes that encode lncRNAs might have an exon–intron structure or might exist as a single exon similar to protein‐coding genes. Most lncRNAs discovered thus far have 5′ cap structures and are polyadenylated. Similar to protein‐coding genes, when undergoing active transcription, lncRNA genes exhibit characteristic H3K4‐trimethylation and H3K36‐trimethylation marks at transcription initiation sights and gene bodies, respectively. The major difference between lncRNAs and mRNAs is that lncRNAs are littered with translational stop codons and have little, if any, protein‐coding potential. This has been demonstrated computationally using programs that predict translational potential and experimentally by approaches such as ribosome footprinting, lncRNA association with polysomes, and/or in vitro transcription/translation. However, just as with any negative result, these must be approached with a level of caution, and methods are being actively sought to positively identify lncRNAs [40, 41, 42–43]. To date, lncRNAs have been identified in multiple species (Fig. 1D). About 20,000, 8000, 100, and 10 unique lncRNA genes have been identified in humans, mice, worms, and yeast, respectively. Thus, lncRNAs are even produced by organisms with very compact genomes. lncRNAs are operationally defined as >200 nucleotides in length to distinguish them from smaller RNAs such as microRNAs. If one assumes the average lncRNA gene is 3000 nucleotides in length and that most of the genome is transcribed, and we know the size of the genome of different species, it would be possible to predict the numbers of unique lncRNAs that might be produced. For humans and other vertebrates, this number could be as high as 1 million unique lncRNA genes; for hagfish, worms, and fungi, the number of lncRNA genes might be as high as 300,000, 30,000, and 8000, respectively (Fig. 1E). Thus, the number of lncRNA genes might vastly exceed the number discovered to date. It is likely that lncRNAs provide the biologic programs that confer increased complexity on higher organisms.
lncRNA genes are often named according to their location in genomes relative to protein‐coding genes and whether they are transcribed in sense or antisense orientations (e.g., IFNG and IFNG‐AS1) or other similar naming conventions (i.e., lincR‐Ccr2‐5′AS) [44, 45]. Investigators and organizations are striving for a common naming convention, such as we have for protein‐coding genes; however, this remains a goal rather than a reality. lncRNA genes with no overlap between 2 protein‐coding genes are referred to as long or large intergenic or intervening lncRNAs and abbreviated lincRNAs. lncRNA genes can totally reside within an intron of a protein‐coding gene, and these are referred to as intronic lncRNA genes. Antisense lncRNA genes are typically difficult to unravel, because they reside within both exons and introns of protein‐coding genes and also because they use novel exons not located within the body of the protein‐coding gene and are often transcribed from the opposite DNA strand. The Th2LCRR lncRNA gene is an example of this category [46]. Divergent lncRNA genes are another class of genes. They are termed divergent because they initiate transcription from the opposite strand of a protein‐coding gene; however, the initiation sites of the 2 genes are typically within a few hundred nucleotides of each other [47]. GATA3 and GATA3‐AS1 represent a protein‐coding gene/lncRNA gene pair that fit this category [48]. Undoubtedly, additional classes of lncRNAs await discovery such that new rules and naming conventions will emerge as this field moves forward.
Another general rule is that lncRNAs, either directly or indirectly, often regulate transcription of protein‐coding genes but use an array of unique mechanisms. First, intracellular signaling paths and combinations of signaling‐responsive and basal transcription factors govern lncRNA expression, similar to mRNA expression. Second, certain lncRNAs regulate the expression of a single gene, often a neighboring protein‐coding gene in the genome to the lncRNA gene, such as IFNG and IFNG‐AS1. However, lncRNAs can regulate transcription of an array of protein‐coding genes positioned throughout the genome. A single lncRNA might also stimulate transcription of certain protein‐coding genes and inhibit transcription of other protein‐coding genes. Exactly how this is achieved is incompletely understood. From mechanistic studies of a few lncRNAs, a few general themes have emerged. One is that lncRNAs associate with chromatin‐modifying enzymes and target gene loci to write the epigenetic code. A second is that lncRNAs form ribonucleoprotein complexes, notably with heterogeneous ribonucleoproteins or hnRNPs, and this complex positively or negatively regulates the transcription of the target genes. A third mechanism is that lncRNAs might associate with transcription factors and titrate them away from target gene loci or stabilize transcription factors at target gene loci or multiple gene loci to negatively or positively regulate mRNA expression. Therefore, common mechanistic themes of lncRNA actions have emerged.
COMPLEXITY OF THE T CELL LINEAGE
Arguably, the immune and nervous systems are 2 of the most complex systems in vertebrates. The immune system can be divided into innate and adaptive arms, in which the innate immune system provides both initial protection against pathogens and responds to the adaptive arm to provide various effector functions. The adaptive arms are provided by T and B lymphocytes that possess the ability to clonally express a vast array of antigen receptors via either TCRs or Igs, respectively. All T cells possess a similar morphology and many common features. All lymphocytes develop from a common lymphoid progenitor ( Fig. 2 ). In the thymus, T cells that will become TCR‐αβ+ develop through a series of clearly identifiable cell stages: DN1 → DN2 → DN3 → DN4 → DP (CD4+, CD8+) → single positive (either single CD4+ or CD8+) before emigrating to the periphery as naïve CD4+, TCR‐αβ+ or CD8+, TCR‐αβ+ cells. CD4−, CD8− TCR‐γδ T cells also develop from DN stages in the thymus [49, 50, 51–52]. Natural CD4+ and CD8+ TCR‐αβ+ Tregs develop in the thymus from T cell progenitors. NKT cells, both type 1 and type 2, develop in the thymus from the DP stage. In the periphery, naïve CD4+ αβ+ T cells can differentiate in response to environmental cues, mostly cytokines, into a number of cell lineages defined by their effector functions, Th1 (IFN‐γ), Th2 (IL‐4, IL‐13, IL‐5), Th17 (IL‐17), Th9 (IL‐9), Th22 (IL‐22), T follicular helper cells, and induced Tregs. The Th cells are defined by the cytokines they produce, and T follicular helper cells provide help to B cells for antibody production, and induced Tregs possess regulatory function. Although CD8+ T cells are mostly defined by their cytotoxic functions, CD8+ T cells also differentiate into effector cells with the ability to selectively express the same cytokines as Th cells, TC1 and TC2 and, by extension, TC17, TC9, and TC22. Similar to the type 1 and type 2 NKT cells (type 1 NKT cells express an invariant TCR, and type 2 NKT cells express a more diverse pattern of TCRs), the CD4−, CD8−, TCR‐γδ+ cells appear to emerge from the thymus with a preprogrammed array of effector functions, including cytotoxicity and release of cytokines and a pattern of receptors to enable specific responses to environmental cues. The cytokines expressed by both NKT cells and γδ‐T cells include IFN‐γ, IL‐4 IL‐13, and IL‐17, among others. Both these cell types are thought to represent a bridge between the more primitive innate immune system and the adaptive immune system.
Figure 2.
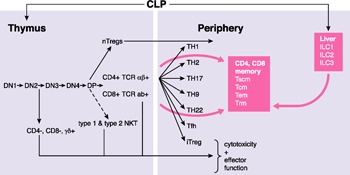
Complexity of T‐lymphocyte development and differentiation. iTregs, induced Tregs; Tfh, T follicular helper (cell).
In the periphery, memory T cells develop from naïve precursors in response to antigen recognition, which is one hallmark of adaptive immunity and are of both CD4+ and CD8+ TCR‐αβ+ lineages [53, 54–55]. The precise developmental pathways of memory cells are incompletely understood, but these cells also exist as discrete lineages, including Tcm and Tem cells. In general, it is thought that Tcm cells have the capacity for self‐renewal but lack or have reduced effector function and that Tem cells lack the ability for self‐renewal and are short‐lived but express effector functions. In humans, it has been argued that a discrete stem cell memory T cell exists that gives rise to the Tcm lineage; however, this has not been seen in the mouse. In addition, studies have suggested distinct Tcm or Tem cell lineages might exist that permanently reside in specific tissues [e.g., skin, gut, lung (resident memory T cells)] and do not recirculate throughout the host.
Another recently discovered class of lymphocytes that also arises from common lymphoid progenitors is the innate lymphocyte [8] [56, 57–58]. These lymphocytes develop in the liver and lack TCRs. However, they possess many of the effector functions of Th cells and are subdivided into ILC1, ILC2, and ILC3 lineages according to their ability to express IFN‐γ, IL‐4, IL‐13 and IL‐5, and IL‐17, respectively. These cells also use the same transcriptional “master regulators” used by conventional Th1, Th2, and Th17 cells (e.g., T‐bet, GATA3, and ROR‐γt, respectively) to acquire these unique cytokine expression patterns.
Taken together, these findings indicate that >50 unique cell lineages constitute the T cell compartment of the vertebrate immune system independent of the known TCR diversity. It could be useful to consider this diversity in the context of shared and unique transcriptional modules. Thus, these different cell lineages share common master transcriptional regulators to express key cytokines such as IFN‐γ, IL‐4, and so forth, share common pathways to induce cytotoxicity, and use unique patterns of chemokine receptors to direct their migration to tissues, to name a few. The emergence of this complexity developed with the tremendous expansion of genome size in vertebrates that allowed for the production of many cell type‐specific lncRNAs. Thus, it seems reasonable to postulate that T cell‐specific lncRNAs drive the complexity of the T cell compartment in the adaptive immune system.
The question of T cell stage and lineage‐specific expression of lncRNAs and mRNAs has been addressed in both humans and mice. One of the first comprehensive examinations of lncRNA expression was performed in human CD8+ T cells [59]. The general findings are that most lncRNAs are expressed in a stage‐ or lineage‐specific manner but that only a small percentage of mRNAs exhibit this property. This can be seen across the whole spectrum of T cell development in the thymus to T cell differentiation in the periphery. In the thymus, each stage of differentiation expresses its own unique pattern of lncRNAs, and these lncRNAs are not expressed at the other stages. This is also seen in the periphery, where each lineage expresses a unique pattern of lncRNAs. Furthermore, these T cell‐specific lncRNAs are not expressed, or are expressed at very low levels, in nonlymphoid tissues. Thus, even these highly related cell types can be defined by a unique lncRNA expression profile [45, 46, 60].
Induction of lineage‐specific lncRNAs is also dynamically regulated. lncRNAs that are selectively expressed in Th1 cells are rapidly induced in response to cell‐signaling pathways that direct the Th1 differentiation program, and the induction of these lncRNAs requires the key transcriptional regulators, STAT4 and T‐bet, which drive this program. Furthermore, the gene that encodes the Th1‐specific lncRNA, IFNG‐AS1, uses a unique group of proximal and distal enhancers not used by IFNG, and these enhancers bind the transcription factors T‐bet and NF‐κB to induce transcription of IFNG‐AS1 [61]. Similarly, the induction of Th2‐specific lncRNAs requires the key transcriptional regulators STAT6 and GATA3, which drive the Th2 differentiation program [45]. Thus, the major transcriptional regulators that drive Th cell differentiation programs in the periphery also drive the expression of lineage‐specific lncRNAs.
GUILT BY ASSOCIATION
In general, lncRNAs can regulate the expression of single or multiple genes. In the former case, genes encoding lncRNAs are often in close proximity in the genome to the protein‐coding gene the lncRNA regulates and are coexpressed. This would also be true in the latter case; however, in the latter case, expression of multiple protein‐coding genes would be regulated by the lncRNA via direct or indirect mechanisms, and these genes would likely be scattered throughout the genome. Thus, examination of lncRNA and mRNA coexpression across multiple biologic samples can provide insight into the potential targets of a given lncRNA. For example, effector Th1, Th2, and Th17 cells express multiple lineage‐specific lncRNAs and lineage‐specific mRNAs. Most of the genes that encode these lncRNAs are in the neighborhood of coexpressed lineage‐specific mRNA genes ( Fig. 3 ). Most of these lncRNA‐coding genes are intragenic with lineage‐specific coexpressed protein‐coding genes. Among these lncRNA genes, no clear preference has been found for lncRNA genes to be transcribed in the same (sense) or opposite (antisense) orientation to these coexpressed protein‐coding genes.
Figure 3.
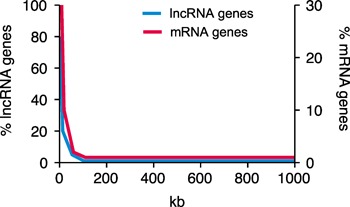
Close proximity of Th cell lineage‐specific mRNA and lncRNA genes to each other in the human genome. Left Y‐axis, percentage of lineage‐specific lncRNA genes the indicated distance from a lineage‐specific mRNA gene; right Y‐axis, percentage of lineage‐specific mRNA genes the indicated distance from a lineage‐specific lncRNA gene.
It is also useful to perform the opposite comparison and determine the distances between all Th1, Th2, and Th17 lineage‐specific mRNA genes and all lineage‐specific lncRNA genes. This analysis has demonstrated that most lineage‐specific mRNA genes are not randomly distributed in the genome but are, in fact, enriched in the genome in regions containing lineage‐specific lncRNA genes (Fig. 3). From this type of analysis, it is possible to produce an overview of lncRNA genes that might be candidates for regulating adjacent protein‐coding genes in the genome and of lncRNA genes that might be candidates for regulating multiple protein‐coding genes distributed throughout the genome ( Table 1 ). These data also suggest that lncRNAs shape the transcriptional response to Th1, Th2, and Th17 differentiation signals
Table 1.
Overview of T cell lncRNAs
| lncRNA | Expression | Targets | +/− | Mechanism | In vivo | References |
|---|---|---|---|---|---|---|
| lincRNA‐p21 | DNA damage | Multiple genes | − | Binds hnRNP | 66 | |
| Multiple (also in T cells) | RelA, JunB | − | Binds target mRNA and inhibits translation | 64, 65 | ||
| Hypoxia | HIF1α | + | Prevents VHL directed biquitination | 62 | ||
| NRON | T cells | NFAT protein | − | Sequesters NFAT in cytoplasm | 72, 73 | |
| IFNG‐AS1 | CD4 Th1, CD8 | IFNG | + | Epigenetic | Yes | 44, 76 |
| linc‐MAF‐4 | CD4 Th1 | MAF | − | Epigenetic | 60 | |
| Th2LCRR | CD4 Th2 | IL4, IL13, IL5 | + | Epigenetic | 46 | |
| Linc‐Ccr2‐5′AS | CD4 Th2 | Ccr1, Ccr3, Ccr2, Ccr5 | + | ? | Yes | 45 |
| GATA3‐AS1 | CD4 Th2 | ? | ? | ? | 48 | |
| Fas‐AS1 | T and B cells | FAS/sFAS | − | Splicing | 82 | |
| LncRNA‐CD244 | CD8 | IFNG/TNF | − | Epigenetic | Yes | 81 |
| hTR | CD4+, others | Apoptosis | − | ? | 85 |
, positive regulator; −, negative regulator; HIF1α, hypoxia‐inducing factor‐1α; hnRNP, heterogeneous ribonucleoprotein.
LincRNA‐p21
lincRNA‐p21 has been included in the present overview because its levels are reduced in inflammatory diseases, such as RA, and in coronary artery disease, and these diseases are thought to critically depend on T cells. A second corollary is that RA is a major risk factor for developing coronary artery disease, suggesting that lincRNA‐p21 might play important roles in these diseases, which affect a large percentage of the human population. The prevalence of RA in the general population is a little <1% and the prevalence of coronary artery disease is ∼10–20%. Reduced levels of lincRNA‐p21 have also been associated with various cancers and their prognoses. Taken together, these studies suggest that lincRNA‐p21 could be an important therapeutic target for a number of diseases that affect a large percentage of the human population. Furthermore, lincRNA‐p21 has been extensively studied; thus, a review of its roles and mechanisms of action in different biologic systems should be instructive [62, 63, 64, 65, 66–67].
lincRNA‐p21 was first discovered as a critical regulator of the cellular response to DNA damage induced by the tumor suppressor, p53. In turn, lincRNA‐p21 associates with 1 of the heterogeneous ribonucleoproteins, hnRNP‐K, to inhibit expression, either directly or indirectly, of a number of genes that encode proteins with proapoptotic functions, thus contributing to the induction of apoptosis. In addition to p53, lincRNA‐p21 expression is also induced 1) by concentrations of methotrexate similar to those used to treat RA via a DNA‐protein kinase catalytic subunit–dependent pathway, resulting in inhibition of the proinflammatory transcription factor, NF‐κB; 2) by the tumor suppressor ING1b; and 3) by the transcription factor HIF1α (hypoxia‐inducing factor‐1α), resulting in increased glycolysis. The other actions of lincRNA‐p21 include 1) inhibition of MDM2‐mediated ubiquitination of p53, resulting in increased p53 activity; 2) induction of a second tumor suppressor, p21; 3) inhibition of the Wnt/β‐catenin signaling pathway; and 4) inhibition of translation of mRNAs encoding JunB, β‐catenin, and RelA. To some degree, these results might seem paradoxical. Many of the known functions of lincRNA‐p21 are both anti‐inflammatory and tumor suppressive. However, activation of glycolysis is thought to be both tumor promoting and proinflammatory, because the shift to glycolysis is thought to represent a key mechanism allowing tumors to survive and grow under conditions of low oxygen. This shift is a key element in the activation and growth of T lymphocytes [62, 68, 69, 70–71]. Also, the mechanisms of action of lincRNA‐p21 are quite diverse, ranging from transcriptional regulation of target gene expression via epigenetic mechanisms, control of mRNA translation by forming sequence‐specific RNA–RNA duplexes, to association with proteins to alter their functions or stability. Many of the other known lncRNAs have not been studied in this detail; thus, it is not known whether other lncRNAs also possess multiple modes of action capable of altering a variety of cellular processes or whether multiple lincRNA‐p21 isoforms might exist, each with unique functions.
SPECIFIC lncRNAs EXPRESSED BY T CELLS
NRON
Most lncRNAs reside in and perform their functions in the nucleus. An example of a cytoplasmic lncRNA is NRON [72, 73]. The transcription factor NFAT was originally discovered as a transcriptional activator of the IL2 gene in activated T cells. We now know that multiple isoforms of NFAT exist and that NFAT isoforms are critical transcriptional regulators of many genes in cells of many different lineages and organ systems. To maintain NFAT in the inactive state, it is sequestered in the cytoplasm by phosphorylation. In response to increased intracellular Ca2+, calcineurin dephosphorylates NFAT, allowing it to translocate to the nucleus and bind to specific target DNA sequences in promoters/enhancers of NFAT‐responsive genes, such as IL2, resulting in transcriptional activation. NFAT is retained in the cytoplasm by a ribonucleoprotein complex that includes NRON and a group of NFAT kinases that keep NFAT phosphorylated. Additional proteins are also present in this complex that contribute to maintaining NFAT in the cytoplasm. NRON acts as a scaffold to maintain the structure of this ribonucleoprotein complex. Depletion of NRON disrupts this complex, leading to dephosphorylation of NFAT and translocation to the nucleus, allowing NFAT to act as a transcriptional activator of target genes.
Ets‐1 is a transcription factor essential for optimal IL‐2 production by T cells. Ets‐1 moves rapidly from the nucleus to the cytoplasm in response to Ca2+ signaling. Ets‐1 does not directly interfere with the dephosphorylation of NFAT by calcineurin. Rather, Ets‐1 interferes with the binding of NFAT to the NRON complex, and this frees NFAT from its cytoplasmic trap, permitting rapid translocation of NFAT to the nucleus, where it can fulfill its transactivating functions [74].
lncRNAs and Th cell differentiation
The “master transcriptional regulators” of Th1, Th2, and Th17 differentiation programs include T‐bet, GATA3, and Ror‐γT and BATF, respectively. The corresponding major cytokines selectively produced by these lineages are IFN‐γ, IL‐4, IL‐13 and IL5, and IL‐17. As described in previous sections, the searches for lncRNAs expressed selectively by these lineages have been performed in both human and mouse models [45, 46, 60]. If one considers the “guilt by association” idea, it might be worthwhile to ask whether the genes encoding lineage‐specific lncRNAs exist in the genome near to genes that encode these “master regulators” or the cytokines that define these lineages. In large part, in both mouse and human studies, lineage‐specific lncRNAs have been identified that are transcribed from genes in the genome adjacent or from nearby genes that encode these “master regulators” or lineage‐specific cytokines. Clear rules have not emerged regarding the locations of these lncRNA genes in the genome relative to the relevant mRNA genes. Certain lncRNA genes, such as IFNG‐AS1 and GATA3‐AS1, are near to IFNG and GATA3, respectively, intergenic, and transcribed in an antisense direction. A Th17 lineage‐specific lncRNA gene near to RORC (encodes ROR‐γT) is transcribed in the sense orientation, is several genes away from RORC, and is partially embedded within another gene in the genome, OAZ3. Similarly, a lineage‐specific lncRNA gene near BATF is partially embedded within the neighboring FLVCR2 gene and is transcribed in the antisense orientation to BATF. Neither OAZ3 nor FLVCR2 gene is a Th17 lineage‐specific gene. A Th1 lineage‐specific lncRNA gene is also located near Tbx21 in the mouse and transcribed in the sense orientation. Th2 lineage‐specific lncRNA genes are also found near the IL4, IL13, and IL5 genes in both mice and humans. In humans, these genes are transcribed in the antisense orientation relative to the IL4 and IL13 genes and are partially embedded within the RAD50 gene. Finally, a Th17 lineage‐specific lncRNA gene has been identified in close proximity to Il17f in the mouse genome. These whole genome RNA‐seq analyses have also described lineage‐specific lncRNAs unique to Tregs and both CD4+ and CD8+ naïve T cells and CD4+ and CD8+ Tcm and Tem cells. The functions of most of these lncRNAs have yet to be explored.
Th1‐specific lncRNAs
IFNG‐AS1.
IFNG‐AS1, originally named TMEVPG1 or Theiler's murine encephalomyelitis virus persistence candidate gene 1 and also named NeST or Nettoie Salmonella pas Theiler's (cleanup Salmonella not Theiler's), was originally discovered by studies to identify the genetic factors controlling Theiler's viral persistence in the brain [75]. IFNG‐AS1 is transcribed in the antisense orientation relative to IFNG in both mouse and human genomes. In the human genome, the transcriptional start sites for IFNG and IFNG‐AS1 are separated by ∼200 kb. IFNG‐AS1 is composed of multiple isoforms with unique and shared exons and introns. Most of these isoforms span ∼20 kb of genome space; however, 1 isoform spans ∼250 kb, with the 3′ end located between IL26 and IL22.
IFNG‐AS1 is induced in CD4+ T cells in response to Th1 differentiation signals that requires both Stat4 and T‐bet [44]. The responses in effector Th1 cells to secondary stimulation results in marked increases in IFNG‐AS1 expression, which is also dependent on non–lineage‐specific transcription factors, NF‐κB and Ets‐1. A number of distal transcriptional enhancers exist across the IFNG–IFNG‐AS1 genomic locus, and it appears that these enhancers are used by either IFNG or IFNG‐AS1 but are not shared between both genes [61]. Furthermore, IFNG‐AS1 cooperates with T‐bet to stimulate IFNG transcription by effector Th1 cells. IFNG‐AS1 is also essential for IFNG expression by CD8+ T cells. IFNG‐AS1 associates with the histone H3K4 methyltransferase complex and recruits this complex to the IFNG locus to establish H3K4‐methylation marks at IFNG promoter and intronic regions to facilitate transcription [76]. Other cells that express IFNG include NK cells, NKT cells, and CD4+ and CD8+ Tem cells. At this point, it is not known whether these cells also express IFNG‐AS1 or whether IFNG‐AS1 is required for IFNG expression by these cells.
linc‐MAF‐4.
linc‐MAF‐4 is a second Th1 lineage‐specific lncRNA that has been studied in detail and offers interesting distinctions from other lncRNAs [60]. The linc‐MAF‐4 and MAF genes are ∼100 kb apart in the human genome on chromosome 16, and both genes are transcribed from the same DNA strand. In contrast to linc‐MAF‐4, MAF is expressed in a Th2 lineage‐specific manner and promotes the expression of genes encoding Th2 cytokines. Knockdown of linc‐MAF‐4 in activated CD4+ T cells under nonpolarizing conditions increases expression of MAF, GATA3, IL4, and other Th2 lineage‐specific mRNAs and decreases expression of Th1 lineage‐specific mRNAs. Thus, linc‐MAF‐4 acts by repressing MAF expression, which, in turn, represses the Th2‐differentiation program and thus promoting the Th1‐differentiation program. Chromosome conformation capture assays have indicated that genes encoding linc‐MAF‐4 and MAF are brought into close physical proximity after T cell activation and linc‐MAF‐4 associates with chromatin modifiers LSD1 and EZH2 to facilitate formation of H3K27 trimethylation marks at the MAF gene to produce a transcriptionally repressive environment. This scenario is similar to the mechanisms of other lncRNAs that repress transcription of protein‐coding gene targets. What seems to make it unique is that the biologic function of linc‐MAF‐4 is to make MAF a Th2 lineage‐specific mRNA and transcription factor promoting the Th2‐differentiation program and linc‐MAF‐4 stimulates the Th1‐differentiation program by dampening the overall Th2 transcriptional response, thus playing an important role in shaping overall Th1/Th2 responses.
Th2‐specific lncRNAs
linc‐Ccr2‐5′AS.
Genes encoding a cluster of chemokine receptors, CCR1, CCR3, CCR2, and CCR5, are located on human chromosome 3 and mouse chromosome 9. In the mouse, a lncRNA gene has been identified between the Ccr3 and Ccr2 genes named linc‐Ccr2‐5′AS, because it is transcribed in the antisense orientation to Ccr2 [45]. linc‐Ccr2‐5′AS fulfills the criterion of an intergenic lncRNA gene. This lncRNA gene has not been identified in the human genome. linc‐Ccr2‐5′AS is preferentially expressed by the Th2 lineage and is dependent on GATA3. This lncRNA is also coexpressed with these chemokine receptor genes, and depletion of linc‐Ccr2‐5′AS results in loss of Ccr1, Ccr3, Ccr2, and Ccr5 expression, indicating that linc‐Ccr2‐5′AS positively regulates the expression of these genes encoding chemokine receptors. In contrast to many other enhancer lncRNAs, linc‐Ccr2‐5′AS does not appear to modulate the H3K4 di‐ and trimethylation marks at these gene loci. However, it is noteworthy that the regulatory properties of linc‐Ccr2‐5′AS appear not to be limited to just the Ccr1, Ccr3, Ccr2, and Ccr5 genes, because depletion of linc‐Ccr2‐5′AS alters the expression of a number of Th2 lineage‐specific mRNAs. Thus, the precise mechanisms of action by which linc‐Ccr2‐5′AS regulates expression of Ccr1, Ccr3, Ccr2, and Ccr5 genes is unknown but will most certainly by the subject of future investigations.
Th2LCRR.
A second Th2‐specific lncRNA, originally termed Th2‐LCR lncRNA, has been identified from studies examining lncRNA expression during differentiation of human effector Th1, Th2, and Th17 cells [46, 77]. Effector Th2 cells selectively express this lncRNA, and this lncRNA is coexpressed with IL4, IL5, and IL13. Th2‐LCR lncRNA actually consists of ≥4 unique isoforms that contain both shared and unique RNA sequences. Using these definitions, Th2‐LCR lncRNA can be considered an antisense lncRNA and is transcribed antisense to the RAD50 gene and partially shares RAD50 exons and introns and exons between the RAD50 and IL13 genes and IL13 and IL4 genes. This is approximately the same genomic region identified in mice that functions as a LCR and controls the expression of genes encoding Th2 cytokines [78]. The suggested Human Genome Organization gene symbol for this lncRNA gene is Th2LCRR‐associated RNA. Depletion of all Th2LCRR isoforms by small interfering RNA targeting of a common sequence abrogates expression of IL4, IL13, and IL5 in human T cell cultures. Depletion of individual Th2LCRR isoforms does not effectively reduce the expression of IL4, IL13, or IL5, suggesting that individual isoforms do not target individual genes encoding these Th2 cytokines. Just as with certain other activating lncRNAs, Th2LCRR lncRNA binds to the WDR5 component of the H3K4 methyltransferase enzyme complex and depletion of Th2LCRR lncRNA inhibits recruitment of WDR5 to IL4 and IL13 gene promoters and enhancer elements and the formation of H3K4 methylation marks at these sites.
LCRs are defined as cis‐regulatory DNA elements that act over genomic distances to regulate the expression of linked genes, often gene families. The prototypical LCR is the β‐globin gene LCR that regulates coordinated expression of globin genes during development [79]. These LCRs function by recruiting chromatin‐modifying complexes, coactivators, and transcriptional complexes to target genes. These are some of the same properties that are also now ascribed to lncRNAs. Studies of Th2LCRR lncRNA suggest that this lncRNA might actually mediate the activities of the Th2 LCR. Thus, it is tempting to speculate that a general mechanism of how LCRs function might be to encode lncRNAs capable of directly regulating and coordinating the expression of linked gene families.
Closer examination of the Th2LCRR family of lncRNAs also illustrates certain problems or hurdles inherent to the analysis of lncRNAs ( Fig. 4 ). In general, investigators have used a variety of approaches to identify lncRNAs of interest in a given biologic system. Whole genome RNA‐seq is the most encompassing. Even with the sophisticated computational programs currently available, it would be very difficult to determine by RNA‐seq alone that Th2LCRR actually consists of 4 unique RNA sequences. The analysis is further complicated because certain Th2LCRR‐transcribed sequences are shared with RAD50 exons, which are transcribed at much higher levels than Th2LCRR family isoforms. However, Th2LCRR family isoforms were already annotated in the human genome assembly as spliced ESTs. Thus, when RNA‐seq data identified this region as a Th2 lineage‐specific lncRNA, it was obvious to first question whether all Th2LCRR isoforms are Th2 lineage‐specific lncRNAs, which they were. Sequencing of each isoform made it possible to identify the shared and unique sequences within the Th2LCRR family. With this information, it was possible to design a small interfering RNA knockdown strategy to target all members of the Th2LCRR family or individual family members. This was critical information that made it possible to identify the gene targets of Th2LCRR and underlying mechanisms of action. The field of lncRNA biology is relatively new, and many fundamental aspects are poorly understood. For example, we do not know how common lncRNA isoform families are and whether the individual isoforms within these families have shared or unique functions. We also do not have a complete understanding of how lncRNA sequences recognize their target genes and proteins required to perform their biologic functions. In the case of the Th2LCRR isoform family, it would have been very difficult to assemble the individual isoform family members into unique sequences using sequencing data alone. It would have also been very difficult to uncover the functions of the Th2LCRR isoform family without previous knowledge of the Th2 LCRR isoform sequences available from the available spliced EST sequence information.
Figure 4.
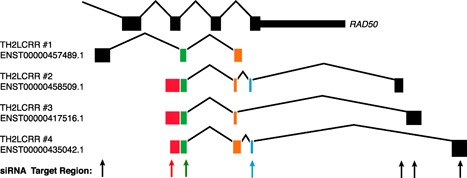
Illustration of RAD50 and Th2LCRR alternatively spliced exon boundaries. Rectangles represent spliced exons for the indicated transcripts. Black rectangle sequences do not match with other Th2LCRR transcripts; the green rectangle sequence is the shared sequence among all Th2LCRR isoforms; the blue rectangle sequence is a shared sequence among 2 Th2LCRR isoforms; the red rectangle sequence is a shared sequence among 3 Th2LCRR isoforms; orange is shared sequence among each Th2LCRR isoform.
GATA3‐AS1.
Human effector Th2 cells also selectively express the lncRNA, GATA3‐AS1 [48]. Transcriptional start sites for GATA3 and GATA3‐AS1 are within a few hundred base pairs of each other, and these genes are transcribed in opposite orientations. Thus, GATA3‐AS1 can be considered a divergent lncRNA gene. Although the functions of GATA3‐AS1 lncRNA are unknown, GATA3‐AS1 is expressed at higher levels by CD4+ T cells from allergic individuals in response to challenge by allergens. In addition, genetic studies have identified SNPs around the GATA3‐AS1 gene that confer greater risk and/or a poorer prognosis for certain forms of cancers, including certain forms of breast cancer and acute lymphoblastic leukemia in children [80]. Taken together, these results suggest that GATA3‐AS1 might positively or negatively influence diverse human diseases. In addition, other lncRNA genes several hundred kilobytes from GATA3 and GATA3‐AS1 genes identified in the mouse genome are also expressed selectively by murine effector Th2 cells compared with other T cell lineages [45].
Additional T cell–associated lncRNAs
LncRNA‐CD244.
The costimulatory receptor, CD244 (2B4) is expressed by virus‐specific CD8+ T cells, and CD244 signaling is associated with the persistence of certain viral infections in humans. TB infection also induces expression of certain T cell‐inhibitory molecules by CD8+ T cells, and this is thought to inhibit CD8+ T cell immunity. CD244 mediates the inhibition of the expression of IFN‐γ and TNF‐α, key cytokines that protect against tuberculosis pathogenesis, by inducing expression of a lncRNA termed lncRNA‐CD244 [81]. CD244 stimulates expression of lncRNA‐CD244 by creating a permissive transcriptional environment at the lncRNA‐CD244 gene locus by causing the loss of repressive histone marks, H3K27Me3. In turn, lncRNA‐CD244 represses transcription of IFNG and TNF genes by recruiting the EZH2 enzyme complex to IFNG and TNF promoters to facilitate H3K27 methylation, creating a repressive transcriptional environment. This is also an example of failure of the “guilt by association” model. The CD244 gene is on human chromosome 1, the lncRNA‐CD244 gene is on chromosome 22, and the genes regulated by lncRNA‐CD244 are on chromosomes 12 (IFNG) and 6 (TNF).
Fas‐AS1.
Fas‐AS1 is a lncRNA involved in the Fas‐mediated extrinsic pathway of apoptosis, which is critical for both T cell development and activation [82]. The Fas protein is a membrane‐bound protein. The elimination of 1 exon by alternative splicing produces a soluble form of Fas, termed sFas. The Fas ligand, FasL, binds to Fas to trigger apoptosis. sFas also binds to FasL, which inhibits Fas‐FasL–mediated apoptosis. Synthesis of Fas and sFas is controlled by the antisense lncRNA termed Fas‐AS1. The RBM5 protein is a component of one of the spliceosome complexes and regulates pre‐mRNA splicing of many target genes. Fas‐AS1 binds to RBM5 and inhibits exon skipping during splicing of Fas pre‐mRNA to mature mRNA. This increases the Fas:sFas ratios and causes an increase in FasL‐dependent apoptosis. Certain primary B‐cell lymphomas exhibit depressed levels of Fas‐AS1 and corresponding increases in sFas, making them more resistant to Fas‐FasL–mediated apoptosis. This is thought to promote resistance to apoptosis and lymphoma survival. Fas‐AS1 is also induced in T lymphocytes in response to ionizing radiation by an ataxia telangiectasia, mutated–dependent mechanism, a critical mediator of the DNA damage response and a sentinel of genomic integrity [83]. Importantly, the RBM5 protein is involved in pre‐mRNA to mature mRNA splicing of a number of tumor suppressor genes, including p53. It is not known whether Fas‐AS1 also regulates splicing of these pre‐mRNAs or whether its actions are limited to the gene that encodes Fas and sFas. Furthermore, the extrinsic pathway of apoptosis plays important roles in lymphocyte development and in the elimination of lymphocytes during immune responses. The results from these initial studies suggest that Fas‐AS1 might play important roles in these critical processes.
hTR.
Telomerase is a ribonucleoprotein complex consisting of a reverse transcriptase protein component named hTERT and an essential RNA component named hTR. Telomerase adds DNA to the ends of chromosomes, which is essential for long‐term maintenance of telomeres. Because hTR is >200 bp in length and is transcribed from the TERC gene, it fulfills the criteria of a lncRNA. The loss of telomerase activity in general has been associated with a variety of disease risk factors, and it is thought that appropriate levels of telomerase activity are important for human health and maintaining immune function. The loss of telomerase activity in CD4+ T cells occurs in RA, and it is thought that this contributes to both accelerated immunosenescence and inflammation seen in RA [84]. However, it has never been completely clear why the loss of telomerase activity and shortened telomeres should be associated with this type of pathogenesis. Recent studies have shed some light on this question. In addition to its role in telomerase activity, the hTR RNA molecule also protects against apoptosis in CD4+ T cells and thus is important for the short‐term survival of T lymphocytes [85]. Specifically, depletion of hTR is sufficient to activate Bim‐mediated apoptosis via activation of caspase‐9 and caspase 3/7. Thus, normal levels of hTR inhibit the intrinsic pathway of apoptosis by suppressing Bim levels, which, in turn, suppresses caspase‐9 activity. The biochemical mechanisms underlying hTR regulation of the intrinsic pathway of apoptosis are not understood. Whether hTR acts to influence the expression of target genes or can influence the activity of pro‐ or anti‐apoptotic proteins or other mechanisms will undoubtedly be the subject of future investigations.
Activities of known lncRNAs in immune responses in vivo.
Although large numbers of tissue culture correlates of immune responses exist, the study of immune responses to antigens, autoantigens, and pathogens in animal models has greatly increased our understanding of the immune system. Thus, it is critical to understand whether and how immune‐specific lncRNAs regulate the immune responses in vivo. Of the lncRNAs we have discussed, 3, IFNG‐AS1, linc‐Ccr2‐5′AS, and lncRNA‐CD244, have been studied in vivo.
IFNG‐AS1: Because of a genetic deletion, B10.S mice do not express IFNG‐AS1. However, this was corrected in B10.S mice by the introduction of a transgene, allowing for expression of IFNG‐AS1 under the control of a T cell‐specific promoter. Wild‐type B10.S mice succumb to Salmonella infection, but B10.S‐IFNG‐AS1 transgenic mice are relatively resistant to infection by Salmonella. The converse is true for infection with Theiler's virus. Theiler's virus does not persist in the brain long term in B10.S mice but does in B10.S‐IFNG‐AS1 transgenic mice. Thus, the persistence of Theiler's virus in the brain originally observed in Tmevp3SJL/J congenic mice is recapitulated by transgenic expression of IFNG‐AS1 in T cells [76].
linc‐Ccr2‐5′AS: The migration of effector Th2 cells to the lung is chemokine dependent. Depletion of linc‐CCR2‐5′AS greatly impairs the migration of effector Th2 cells into the lung of C57BL/6 mice. This indicates that the lncRNA, linc‐CCR2‐5′AS, is an important regulator of migration of effector Th2 cells to peripheral tissues in vivo [45].
lncRNA‐CD244: lncRNA‐CD244 was originally discovered as a lncRNA highly expressed in CD244+ CD8+ T cells during active infection with TB [81]. This lncRNA‐CD244 gene has not been discovered in mice, so functions of lncRNA‐CD244, in vivo, were investigated in a cell transfer and tuberculosis infection model in SCID mice. Depletion of lncRNA‐CD244 via lentiviral‐shRNA transduction results in a marked increase in production of IFN‐γ and TNF‐α, in vivo, and markedly reduced TB bacterial burdens in lungs and blood. Lung necrosis, hemorrhage, and damage to pulmonary structures were also significantly reduced. Taken together, these studies indicate that lncRNA‐CD244 plays an important role in TB pathogenesis.
As observed in other experimental systems, several of these lncRNAs function by establishing activating or repressive histone marks at target gene loci ( Fig. 5 ). Furthermore, these studies make it clear that lncRNAs within the T cell compartment of the immune system play important roles in regulating immune responses in vivo. These studies also suggest that lncRNAs expressed by T cells might be important therapeutic targets for infections and, perhaps, autoimmune diseases.
Figure 5.

Cartoon model illustrating mechanisms of action of T cell lncRNAs. LncRNA and mRNA genes are depicted by speckled green and red, respectively. Black arrows below indicate orientation of transcription. lncRNAs are depicted by green squiggly line with cargo, either WDR5 or EZH2/LSD1 (blue). Green arrows connect lncRNA gene to the lncRNA or the target of the lncRNA to the mRNA gene (green, activating; red, inhibiting). Yellow stars show locations of H3K4Me marks that favor transcriptional activation, and black “explosions” show locations of H3K27Me marks that suppress transcription of gene targets.
lncRNAs, GWASs, AND AUTOIMMUNE DISEASE
As a group, diseases of the immune system, including autoimmune and related diseases, are a heterogeneous group of disorders affecting ∼10% of the worldwide population. Both genetic and environmental factors contribute to the onset and severity of these diseases. GWASs have identified numerous SNPs that confer the risk of developing individual autoimmune diseases [86]. The mechanistic understanding of how these genetic variants confer disease risk is complicated because <10% are in protein‐coding regions of the genome and existing linkage disequilibrium across the genome makes it difficult to identify the causative genetic variants that actually confer the disease risk. The identification of associations between mRNA expression levels and the presence or absence of a given genetic variant is one avenue pursued to better understand how a given SNP might confer disease risk. Thus, mRNA expression levels that vary depending on a given disease‐associated SNP are termed “expression quantitative trait loci” or eQTL and are considered cis‐eQTL if the SNP and gene encoding the associated mRNA are on the same chromosome or trans‐eQTL if the SNP and gene encoding the associated mRNA are on different chromosomes.
With the recognition that many lncRNAs are transcribed from regions of the genome that do not contain protein‐coding genes, investigators have begun to question whether lncRNA genes map to SNP loci (identified by GWASs) that confer the risk of developing an autoimmune disease and the relationship between lncRNA eQTL and mRNA eQTL [87, 88]. These studies are early in their development but are showing that lncRNA genes expressed predominantly in immune cells, including T cells, are significantly enriched in autoimmune disease genetic loci. Analysis of the coexpression between these lncRNA eQTL and mRNA eQTL has also predicted the signaling pathways in which these lncRNAs might contribute. Thus, it might be possible to infer defective biologic processes that might contribute to autoimmunity and inflammatory disease. It is likely that this type of approach will contribute greatly to our understanding of the genetic risk of an individual to develop a given complex disease, including an autoimmune disease.
BIOINFORMATICS PIPELINES
A significant hurdle for scientists studying lncRNAs is the necessary computational framework. Next‐generation sequencing technologies, such as RNA‐seq, enables not only quantification of annotated lncRNAs, but also the discovery of novel lncRNAs through de novo sequence assembly. Strand‐specific RNA sequencing permits identification of both sense and anti‐sense transcripts. The read depth across sequencing platforms is also important, because many lncRNAs are weakly expressed [64, 77]. As a general rule, mRNA measurements require ∼30 million reads per sample, but lncRNAs are ideally quantified with ∼40 million reads per sample. The detection of novel transcripts, spliced isoforms, and de novo transcript assembly is possible using paired end sequencing.
The sequencing output is often a demultiplexed FASTQ file containing raw reads ( Fig. 6 ). A quality control step is necessary using packages such as FastQC or QC3 (Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom) to examine the reads from each lane of the flow cell analyzing guanine‐cytosine content, the distribution of nucleotides per cycle, and the total reads per lane looking for consistency across each sample [89]. TopHat2 (The Johns Hopkins University Center for Computational Biology, Bethesda, MD, USA) and Subread (SourceForge, Inc., Mountainview, CA, USA) are available as binary programs, and Rsubread (open source) is a version of Subread written in R language to align the sequencing data to the genome [90, 91]. A number of different computational algorithms (e.g., DESeq2, NBPSeq, baySeq) identify differentially expressed genes between classes [92, 93, 94, 95, 96, 97, 98–99]. Thus, the user has a number of options from which to choose.
Figure 6.
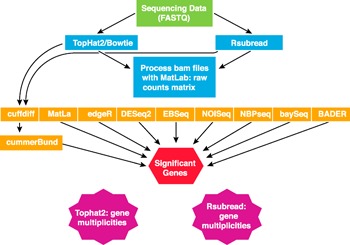
Example of a pipeline for identification and characterization of annotated and novel lncRNAs using whole genome sequencing.
To date, no single piece of software is available to positively or negatively predict a lncRNA from RNA‐seq data. However, different approaches have been developed to build a bioinformatically driven case that a novel gene is, in fact, a lncRNA gene. The use of tools such as BlastX, HMMER/Pfam, PhyloCSF, CPAT, CPC, and GetORF can analyze the sequence and determine the protein‐coding potential of the novel transcript [41, 77, 100, 101]. PhyloCSF, in particular, is a useful tool because it studies sequence conservation across many animal species [42]. A combination of techniques can be applied to each data set to interrogate novel transcripts for protein‐coding potential. High multiplicity gene counts can be checked with software such as the Integrative Genomics Viewer from the Broad Institute (Cambridge, MA, USA) [102, 103].
CONCLUDING REMARKS
Although the Encyclopedia of DNA Elements project was initiated to define and understand DNA elements contributing to transcription of protein‐coding genes, one unexpected discovery was that most of the genome is transcribed into RNA. A debate ensued as to whether this was “noise” or whether these RNA molecules might play important roles in biology. The discovery and study of lncRNAs as a new class of RNA molecule have demonstrated that at least a portion of these newly discovered RNAs have biologic functions. The lncRNAs that have been characterized use a vast array of strategies to regulate the transcription and translation of protein‐coding genes and the functions of proteins. It seems likely that additional mechanisms of action of lncRNAs will emerge. Given the size of the human genome, undoubtedly additional lncRNA genes and other classes of RNA genes are present in the human genome that await discovery.
The adaptive immune system similar to that of humans did not emerge until the evolution of jawed vertebrates. Jawless vertebrates have an adaptive immune system that performs similar functions but uses very different strategies, similar to the concept of our universe and a parallel universe [104]. The development of the adaptive immune system also corresponds in evolution with the vast expansion of the genome and expansion of biologic complexity and cellular diversity between invertebrates and vertebrates. Perhaps >50 unique T cell lineages constitute the adaptive immune system in jawed vertebrates, including both mice and humans. One might predict that this cellular complexity could be achieved by the pattern of expression of both unique and shared lncRNAs. It is easy to see how this is critical to the survival of the host. The adaptive immune system is a very effective protector against infection by pathogens. However, the adaptive immune system also produces a great amount of collateral damage during an immune response. Thus, an ineffective immune response can lead to illness and even death by failure to control an infection and an overexuberant immune response can lead to illness and even death by the collateral damage it produces. lncRNAs expressed by the adaptive immune system can also contribute to the pathogenesis of autoimmune disease. We do not fully understand the genesis of autoimmune diseases or why they persist. Failure to express T cell‐specific lncRNAs in the correct lineages or failure in the function of T cell‐specific lncRNAs might be important contributors to human autoimmune disease. This is especially true if one considers that GWASs show that most genetic loci that confer a risk of developing autoimmune disease are located in regions of the genome that do not code for proteins.
Our understanding of gene regulation has also advanced from our understanding of the contribution of RNA molecules to this process. It was not that long ago that we thought gene regulation was achieved by the combinatorial actions of a few transcription factors acting at conserved DNA enhancers within a few hundred base pairs of promoters and transcriptional start sites. Now, we know that distal enhancers perform very unique developmentally regulated functions, chromatin adopts 3‐dimensional conformations, the epigenetic code is “written” and “read” at both histones and DNA, RNA molecules are transcribed from transcriptional enhancers, and lncRNA molecules acting in cis and trans can regulate all these processes [86, 105, 106, 107, 108, 109, 110, 111–112]. The potential is great that a better understanding of lncRNAs in the adaptive immune system will also lead to identification of new therapeutic strategies to manipulate the immune system to improve the treatment of infectious and autoimmune diseases and cancer.
AUTHORSHIP
T.M.A., P.S.C, and C.F.S. wrote and edited the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported in part by the U.S. National Institutes of Health (Grants R01 AI044924 and R21 AR063846) and the National Science Foundation Graduate Research Fellowship Program (Grant DGE0909667). We apologize for the omission of work by colleagues owing to oversight or space limitations. John Mattick of the Garvin Institute of Medical Research, Sydney, Australia, deserves special acknowledgment for posting his seminar online titled “Most Assumptions in Molecular Biology Are Wrong” as a part of the Australian Nuclear Science and Technology Organization, July 8, 2013, Distinguished Seminar Series, which served as stimulation to highlight the biologic complexity we have presented with a view from the T lymphocyte.
REFERENCES
- 1. Liu, G. , Mattick, J.S. , Taft, R.J. (2013) A meta‐analysis of the genomic and transcriptomic composition of complex life. Cell Cycle 12, 2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kapusta, A. , Feschotte, C. (2014) Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet. 30, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lander, E.S.L. , Linton, L.M. , Birren, B. , Nusbaum, C. , Zody, M.C. , Baldwin, J. , Devon, K. , Dewar, K. , Doyle, M. , FitzHugh, W. , Funke, R. , Gage, D. , Harris, K. , Heaford, A. , Howland, J. , Kann, L. , Lehoczky, J. , LeVine, R. , McEwan, P. , McKernan, K. , Meldrim, J. , Mesirov, J.P. , Miranda, C. , Morris, W. , Naylor, J. , Raymond, C. , Rosetti, M. , Santos, R. , Sheridan, A. , Sougnez, C. , Stange‐Thomann, Y. , Stojanovic, N. , Subramanian, A. , Wyman, D. , Rogers, J. , Sulston, J. , Ainscough, R. , Beck, S. , Bentley, D. , Burton, J. , Clee, C. , Carter, N. , Coulson, A. , Deadman, R. , Deloukas, P. , Dunham, A. , Dunham, I. , Durbin, R. , French, L. , Grafham, D. , Gregory, S. , Hubbard, T. , Humphray, S. , Hunt, A. , Jones, M. , Lloyd, C. , McMurray, A. , Matthews, L. , Mercer, S. , Milne, S. , Mullikin, J.C. , Mungall, A. , Plumb, R. , Ross, M. , Shownkeen, R. , Sims, S. , Waterston, R.H. , Wilson, R.K. , Hillier, L.W. , McPherson, J.D. , Marra, M.A. , Mardis, E.R. , Fulton, L.A. , Chinwalla, A.T. , Pepin, K.H. , Gish, W.R. , Chissoe, S.L. , Wendl, M.C. , Delehaunty, K.D. , Miner, T.L. , Delehaunty, A. , Kramer, J.B. , Cook, L.L. , Fulton, R.S. , Johnson, D.L. , Minx, P.J. , Clifton, S.W. , Hawkins, T. , Branscomb, E. , Predki, P. , Richardson, P. , Wenning, S. , Slezak, T. , Doggett, N. , Cheng, J.F. , Olsen, A. , Lucas, S. , Elkin, C. , Uberbacher, E. , Frazier, M. , Gibbs, R.A. , Muzny, D.M. , Scherer, S.E. , Bouck, J.B. , Sodergren, E.J. , Worley, K.C. , Rives, C.M. , Gorrell, J.H. , Metzker, M.L. , Naylor, S.L. , Kucherlapati, R.S. , Nelson, D.L. , Weinstock, G.M. , Sakaki, Y. , Fujiyama, A. , Hattori, M. , Yada, T. , Toyoda, A. , Itoh, T. , Kawagoe, C. , Watanabe, H. , Totoki, Y. , Taylor, T. , Weissenbach, J. , Heilig, R. , Saurin, W. , Artiguenave, F. , Brottier, P. , Bruls, T. , Pelletier, E. , Robert, C. , Wincker, P. , Smith, D.R. , Doucette‐Stamm, L. , Rubenfield, M. , Weinstock, K. , Lee, H.M. , Dubois, J. , Rosenthal, A. , Platzer, M. , Nyakatura, G. , Taudien, S. , Rump, A. , Yang, H. , Yu, J. , Wang, J. , Huang, G. , Gu, J. , Hood, L. , Rowen, L. , Madan, A. , Qin, S. , Davis, R.W. , Federspiel, N.A. , Abola, A.P. , Proctor, M.J. , Myers, R.M. , Schmutz, J. , Dickson, M. , Grimwood, J. , Cox, D.R. , Olson, M.V. , Kaul, R. , Raymond, C. , Shimizu, N. , Kawasaki, K. , Minoshima, S. , Evans, G.A. , Athanasiou, M. , Schultz, R. , Roe, B.A. , Chen, F. , Pan, H. , Ramser, J. , Lehrach, H. , Reinhardt, R. , McCombie, W.R. , de la Bastide, M. , Dedhia, N. , Blöcker, H. , Hornischer, K. , Nordsiek, G. , Agarwala, R. , Aravind, L. , Bailey, J.A. , Bateman, A. , Batzoglou, S. , Birney, E. , Bork, P. , Brown, D.G. , Burge, C.B. , Cerutti, L. , Chen, H.C. , Church, D. , Clamp, M. , Copley, R.R. , Doerks, T. , Eddy, S.R. , Eichler, E.E. , Furey, T.S. , Galagan, J. , Gilbert, J.G. , Harmon, C. , Hayashizaki, Y. , Haussler, D. , Hermjakob, H. , Hokamp, K. , Jang, W. , Johnson, L.S. , Jones, T.A. , Kasif, S. , Kaspryzk, A. , Kennedy, S. , Kent, W.J. , Kitts, P. , Koonin, E.V. , Korf, I. , Kulp, D. , Lancet, D. , Lowe, T.M. , McLysaght, A. , Mikkelsen, T. , Moran, J.V. , Mulder, N. , Pollara, V.J. , Ponting, C.P. , Schuler, G. , Schultz, J. , Slater, G. , Smit, A.F. , Stupka, E. , Szustakowski, J. , Thierry‐Mieg, D. , Thierry‐Mieg, J. , Wagner, L. , Wallis, J. , Wheeler, R. , Williams, A. , Wolf, Y.I. , Wolfe, K.H. , Yang, S.P. , Yeh, R.F. , Collins, F. , Guyer, M.S. , Peterson, J. , Felsenfeld, A. , Wetterstrand, K.A. , Patrinos, A. , Morgan, M.J. , de Jong, P. , Catanese, J.J. , Osoegawa, K. , Shizuya, H. , Choi, S. , Chen, Y.J. , Szustakowki, J.; International Human Genome Sequencing Consortium. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 4. Venter, J.C. , Adams, M.D. , Myers, E.W. , Li, P.W. , Mural, R.J. , Sutton, G.G. , Smith, H.O. , Yandell, M. , Evans, C.A. , Holt, R.A. , Gocayne, J.D. , Amanatides, P. , Ballew, R.M. , Huson, D.H. , Wortman, J.R. , Zhang, Q. , Kodira, C.D. , Zheng, X.H. , Chen, L. , Skupski, M. , Subramanian, G. , Thomas, P.D. , Zhang, J. , Gabor Miklos, G.L. , Nelson, C. , Broder, S. , Clark, A.G. , Nadeau, J. , McKusick, V.A. , Zinder, N. , Levine, A.J. , Roberts, R.J. , Simon, M. , Slayman, C. , Hunkapiller, M. , Bolanos, R. , Delcher, A. , Dew, I. , Fasulo, D. , Flanigan, M. , Florea, L. , Halpern, A. , Hannenhalli, S. , Kravitz, S. , Levy, S. , Mobarry, C. , Reinert, K. , Remington, K. , Abu‐Threideh, J. , Beasley, E. , Biddick, K. , Bonazzi, V. , Brandon, R. , Cargill, M. , Chandramouliswaran, I. , Charlab, R. , Chaturvedi, K. , Deng, Z. , Di Francesco, V. , Dunn, P. , Eilbeck, K. , Evangelista, C. , Gabrielian, A.E. , Gan, W. , Ge, W. , Gong, F. , Gu, Z. , Guan, P. , Heiman, T.J. , Higgins, M.E. , Ji, R.R. , Ke, Z. , Ketchum, K.A. , Lai, Z. , Lei, Y. , Li, Z. , Li, J. , Liang, Y. , Lin, X. , Lu, F. , Merkulov, G.V. , Milshina, N. , Moore, H.M. , Naik, A.K. , Narayan, V.A. , Neelam, B. , Nusskern, D. , Rusch, D.B. , Salzberg, S. , Shao, W. , Shue, B. , Sun, J. , Wang, Z. , Wang, A. , Wang, X. , Wang, J. , Wei, M. , Wides, R. , Xiao, C. , Yan, C. , Yao, A. , Ye, J. , Zhan, M. , Zhang, W. , Zhang, H. , Zhao, Q. , Zheng, L. , Zhong, F. , Zhong, W. , Zhu, S. , Zhao, S. , Gilbert, D. , Baumhueter, S. , Spier, G. , Carter, C. , Cravchik, A. , Woodage, T. , Ali, F. , An, H. , Awe, A. , Baldwin, D. , Baden, H. , Barnstead, M. , Barrow, I. , Beeson, K. , Busam, D. , Carver, A. , Center, A. , Cheng, M.L. , Curry, L. , Danaher, S. , Davenport, L. , Desilets, R. , Dietz, S. , Dodson, K. , Doup, L. , Ferriera, S. , Garg, N. , Gluecksmann, A. , Hart, B. , Haynes, J. , Haynes, C. , Heiner, C. , Hladun, S. , Hostin, D. , Houck, J. , Howland, T. , Ibegwam, C. , Johnson, J. , Kalush, F. , Kline, L. , Koduru, S. , Love, A. , Mann, F. , May, D. , McCawley, S. , McIntosh, T. , McMullen, I. , Moy, M. , Moy, L. , Murphy, B. , Nelson, K. , Pfannkoch, C. , Pratts, E. , Puri, V. , Qureshi, H. , Reardon, M. , Rodriguez, R. , Rogers, Y.H. , Romblad, D. , Ruhfel, B. , Scott, R. , Sitter, C. , Smallwood, M. , Stewart, E. , Strong, R. , Suh, E. , Thomas, R. , Tint, N.N. , Tse, S. , Vech, C. , Wang, G. , Wetter, J. , Williams, S. , Williams, M. , Windsor, S. , Winn‐Deen, E. , Wolfe, K. , Zaveri, J. , Zaveri, K. , Abril, J.F. , Guigó, R. , Campbell, M.J. , Sjolander, K.V. , Karlak, B. , Kejariwal, A. , Mi, H. , Lazareva, B. , Hatton, T. , Narechania, A. , Diemer, K. , Muruganujan, A. , Guo, N. , Sato, S. , Bafna, V. , Istrail, S. , Lippert, R. , Schwartz, R. , Walenz, B. , Yooseph, S. , Allen, D. , Basu, A. , Baxendale, J. , Blick, L. , Caminha, M. , Carnes‐Stine, J. , Caulk, P. , Chiang, Y.H. , Coyne, M. , Dahlke, C. , Mays, A. , Dombroski, M. , Donnelly, M. , Ely, D. , Esparham, S. , Fosler, C. , Gire, H. , Glanowski, S. , Glasser, K. , Glodek, A. , Gorokhov, M. , Graham, K. , Gropman, B. , Harris, M. , Heil, J. , Henderson, S. , Hoover, J. , Jennings, D. , Jordan, C. , Jordan, J. , Kasha, J. , Kagan, L. , Kraft, C. , Levitsky, A. , Lewis, M. , Liu, X. , Lopez, J. , Ma, D. , Majoros, W. , McDaniel, J. , Murphy, S. , Newman, M. , Nguyen, T. , Nguyen, N. , Nodell, M. , Pan, S. , Peck, J. , Peterson, M. , Rowe, W. , Sanders, R. , Scott, J. , Simpson, M. , Smith, T. , Sprague, A. , Stockwell, T. , Turner, R. , Venter, E. , Wang, M. , Wen, M. , Wu, D. , Wu, M. , Xia, A. , Zandieh, A. , Zhu, X. (2001) The sequence of the human genome. Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 5. Harrow, J. , Frankish, A. , Gonzalez, J.M. , Tapanari, E. , Diekhans, M. , Kokocinski, F. , Aken, B.L. , Barrell, D. , Zadissa, A. , Searle, S. , Barnes, I. , Bignell, A. , Boychenko, V. , Hunt, T. , Kay, M. , Mukherjee, G. , Rajan, J. , Despacio‐Reyes, G. , Saunders, G. , Steward, C. , Harte, R. , Lin, M. , Howald, C. , Tanzer, A. , Derrien, T. , Chrast, J. , Walters, N. , Balasubramanian, S. , Pei, B. , Tress, M. , Rodriguez, J.M. , Ezkurdia, I. , van Baren, J. , Brent, M. , Haussler, D. , Kellis, M. , Valencia, A. , Reymond, A. , Gerstein, M. , Guigó, R. , Hubbard, T.J. (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Derrien, T. , Johnson, R. , Bussotti, G. , Tanzer, A. , Djebali, S. , Tilgner, H. , Guernec, G. , Martin, D. , Merkel, A. , Knowles, D.G. , Lagarde, J. , Veeravalli, L. , Ruan, X. , Ruan, Y. , Lassmann, T. , Carninci, P. , Brown, J.B. , Lipovich, L. , Gonzalez, J.M. , Thomas, M. , Davis, C.A. , Shiekhattar, R. , Gingeras, T.R. , Hubbard, T.J. , Notredame, C. , Harrow, J. , Guigó, R. (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rozowsky, J.S. , Newburger, D. , Sayward, F. , Wu, J. , Jordan, G. , Korbel, J.O. , Nagalakshmi, U. , Yang, J. , Zheng, D. , Guigó, R. , Gingeras, T.R. , Weissman, S. , Miller, P. , Snyder, M. , Gerstein, M.B. (2007) The DART classification of unannotated transcription within the ENCODE regions: associating transcription with known and novel loci. Genome Res. 17, 732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. ENCODE Project Consortium; Birney, E. , Stamatoyannopoulos, J.A. , Dutta, A. , Guigó, R. , Gingeras, T.R. , Margulies, E.H. , Weng, Z. , Snyder, M. , Dermitzakis, E.T. , Thurman, R.E. , Kuehn, M.S. , Taylor, C.M. , Neph, S. , Koch, C.M. , Asthana, S. , Malhotra, A. , Adzhubei, I. , Greenbaum, J.A. , Andrews, R.M. , Flicek, P. , Boyle, P.J. , Cao, H. , Carter, N.P. , Clelland, G.K. , Davis, S. , Day, N. , Dhami, P. , Dillon, S.C. , Dorschner, M.O. , Fiegler, H. , Giresi, P.G. , Goldy, J. , Hawrylycz, M. , Haydock, A. , Humbert, R. , James, K.D. , Johnson, B.E. , Johnson, E.M. , Frum, T.T. , Rosenzweig, E.R. , Karnani, N. , Lee, K. , Lefebvre, G.C. , Navas, P.A. , Neri, F. , Parker, S.C. , Sabo, P.J. , Sandstrom, R. , Shafer, A. , Vetrie, D. , Weaver, M. , Wilcox, S. , Yu, M. , Collins, F.S. , Dekker, J. , Lieb, J.D. , Tullius, T.D. , Crawford, G.E. , Sunyaev, S. , Noble, W.S. , Dunham, I. , Denoeud, F. , Reymond, A. , Kapranov, P. , Rozowsky, J. , Zheng, D. , Castelo, R. , Frankish, A. , Harrow, J. , Ghosh, S. , Sandelin, A. , Hofacker, I.L. , Baertsch, R. , Keefe, D. , Dike, S. , Cheng, J. , Hirsch, H.A. , Sekinger, E.A. , Lagarde, J. , Abril, J.F. , Shahab, A. , Flamm, C. , Fried, C. , Hackermüller, J. , Hertel, J. , Lindemeyer, M. , Missal, K. , Tanzer, A. , Washietl, S. , Korbel, J. , Emanuelsson, O. , Pedersen, J.S. , Holroyd, N. , Taylor, R. , Swarbreck, D. , Matthews, N. , Dickson, M.C. , Thomas, D.J. , Weirauch, M.T. , Gilbert, J. , Drenkow, J. , Bell, I. , Zhao, X. , Srinivasan, K.G. , Sung, W.K. , Ooi, H.S. , Chiu, K.P. , Foissac, S. , Alioto, T. , Brent, M. , Pachter, L. , Tress, M.L. , Valencia, A. , Choo, S.W. , Choo, C.Y. , Ucla, C. , Manzano, C. , Wyss, C. , Cheung, E. , Clark, T.G. , Brown, J.B. , Ganesh, M. , Patel, S. , Tammana, H. , Chrast, J. , Henrichsen, C.N. , Kai, C. , Kawai, J. , Nagalakshmi, U. , Wu, J. , Lian, Z. , Lian, J. , Newburger, P. , Zhang, X. , Bickel, P. , Mattick, J.S. , Carninci, P. , Hayashizaki, Y. , Weissman, S. , Hubbard, T. , Myers, R.M. , Rogers, J. , Stadler, P.F. , Lowe, T.M. , Wei, C.L. , Ruan, Y. , Struhl, K. , Gerstein, M. , Antonarakis, S.E. , Fu, Y. , Green, E.D. , Karaöz, U. , Siepel, A. , Taylor, J. , Liefer, L.A. , Wetterstrand, K.A. , Good, P.J. , Feingold, E.A. , Guyer, M.S. , Cooper, G.M. , Asimenos, G. , Dewey, C.N. , Hou, M. , Nikolaev, S. , Montoya‐Burgos, J.I. , Löytynoja, A. , Whelan, S. , Pardi, F. , Massingham, T. , Huang, H. , Zhang, N.R. , Holmes, I. , Mullikin, J.C. , Ureta‐Vidal, A. , Paten, B. , Seringhaus, M. , Church, D. , Rosenbloom, K. , Kent, W.J. , E. A. Stone; S. , Goldman, N. , Hardison, R.C. , Haussler, D. , Miller, W. , Sidow, A. , Trinklein, N.D. , Zhang, Z.D. , Barrera, L. , Stuart, R. , King, D.C. , Ameur, A. , Enroth, S. , Bieda, M.C. , Kim, J. , Bhinge, A.A. , Jiang, N. , Liu, J. , Yao, F. , Vega, V.B. , Lee, C.W. , Ng, P. , Shahab, A. , Yang, A. , Moqtaderi, Z. , Zhu, Z. , Xu, X. , Squazzo, S. , Oberley, M.J. , Inman, D. , Singer, M.A. , Richmond, T.A. , Munn, K.J. , Rada‐Iglesias, A. , Wallerman, O. , Komorowski, J. , Fowler, J.C. , Couttet, P. , Bruce, A.W. , Dovey, O.M. , Ellis, P.D. , Langford, C.F. , Nix, D.A. , Euskirchen, G. , Hartman, S. , Urban, A.E. , Kraus, P. , Van Calcar, S. , Heintzman, N. , Kim, T.H. , Wang, K. , Qu, C. , Hon, G. , Luna, R. , Glass, C.K. , Rosenfeld, M.G. , Aldred, S.F. , Cooper, S.J. , Halees, A. , Lin, J.M. , Shulha, H.P. , Zhang, X. , Xu, M. , Haidar, J.N. , Yu, Y. , Ruan, Y. , Iyer, V.R. , Green, R.D. , Wadelius, C. , Farnham, P.J. , Ren, B. , Harte, R.A. , Hinrichs, A.S. , Trumbower, H. , Clawson, H. , Hillman‐Jackson, J. , Zweig, A.S. , Smith, K. , Thakkapallayil, A. , Barber, G. , Kuhn, R.M. , Karolchik, D. , Armengol, L. , Bird, C.P. , de Bakker, P.I. , Kern, A.D. , Lopez‐Bigas, N. , Martin, J.D. , Stranger, B.E. , Woodroffe, A. , Davydov, E. , Dimas, A. , Eyras, E. , Hallgrímsdóttir, I.B. , Huppert, J. , Zody, M.C. , Abecasis, G.R. , Estivill, X. , Bouffard, G.G. , Guan, X. , Hansen, N.F. , Idol, J.R. , Maduro, V.V. , Maskeri, B. , McDowell, J.C. , Park, M. , Thomas, P.J. , Young, A.C. , Blakesley, R.W. , Muzny, D.M. , Sodergren, E. , Wheeler, D.A. , Worley, K.C. , Jiang, H. , Weinstock, G.M. , Gibbs, R.A. , Graves, T. , Fulton, R. , Mardis, E.R. , Wilson, R.K. , Clamp, M. , Cuff, J. , Gnerre, S. , Jaffe, D.B. , Chang, J.L. , Lindblad‐Toh, K. , Lander, E.S. , Koriabine, M. , Nefedov, M. , Osoegawa, K. , Yoshinaga, Y. , Zhu, B. , de Jong, P.J. (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gerstein, M.B. , Bruce, C. , Rozowsky, J.S. , Zheng, D. , Du, J. , Korbel, J.O. , Emanuelsson, O. , Zhang, Z.D. , Weissman, S. , Snyder, M. (2007) What is a gene, post‐ENCODE? History and updated definition. Genome Res. 17, 669–681. [DOI] [PubMed] [Google Scholar]
- 10. Morris, K.V. , Mattick, J.S. (2014) The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paralkar, V.R. , Mishra, T. , Luan, J. , Yao, Y. , Kossenkov, A.V. , Anderson, S.M. , Dunagin, M. , Pimkin, M. , Gore, M. , Sun, D. , Konuthula, N. , Raj, A. , An, X. , Mohandas, N. , Bodine, D.M. , Hardison, R.C. , Weiss, M.J. (2014) Lineage and species‐specific long noncoding RNAs during erythro‐megakaryocytic development. Blood 123, 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fatica, A. , Bozzoni, I. (2014) Long non‐coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15, 7–21. [DOI] [PubMed] [Google Scholar]
- 13. Alvarez‐Dominguez, J.R. , Hu, W. , Yuan, B. , Shi, J. , Park, S.S. , Gromatzky, A.A. , van Oudenaarden, A. , Lodish, H.F. (2014) Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood 123, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ulitsky, I. , Bartel, D.P. (2013) lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sauvageau, M. , Goff, L.A. , Lodato, S. , Bonev, B. , Groff, A.F. , Gerhardinger, C. , Sanchez‐Gomez, D.B. , Hacisuleyman, E. , Li, E. , Spence, M. , Liapis, S.C. , Mallard, W. , Morse, M. , Swerdel, M.R. , D'Ecclessis, M.F. , Moore, J.C. , Lai, V. , Gong, G. , Yancopoulos, G.D. , Frendewey, D. , Kellis, M. , Hart, R.P. , Valenzuela, D.M. , Arlotta, P. , Rinn, J.L. (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2, e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monnier, P. , Martinet, C. , Pontis, J. , Stancheva, I. , Ait‐Si‐Ali, S. , Dandolo, L. (2013) H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc. Natl. Acad. Sci. USA 110, 20693–20698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hangauer, M.J. , Vaughn, I.W. , McManus, M.T. (2013) Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 9, e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clark, M.B. , Choudhary, A. , Smith, M.A. , Taft, R.J. , Mattick, J.S. (2013) The dark matter rises: the expanding world of regulatory RNAs. Essays Biochem. 54, 1–16. [DOI] [PubMed] [Google Scholar]
- 19. Rinn, J.L. , Chang, H.Y. (2012) Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu, W. , Alvarez‐Dominguez, J.R. , Lodish, H.F. (2012) Regulation of mammalian cell differentiation by long non‐coding RNAs. EMBO Rep. 13, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guttman, M. , Rinn, J.L. (2012) Modular regulatory principles of large non‐coding RNAs. Nature 482, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Djebali, S. , Davis, C.A. , Merkel, A. , Dobin, A. , Lassmann, T. , Mortazavi, A. , Tanzer, A. , Lagarde, J. , Lin, W. , Schlesinger, F. , Xue, C. , Marinov, G.K. , Khatun, J. , Williams, B.A. , Zaleski, C. , Rozowsky, J. , Röder, M. , Kokocinski, F. , Abdelhamid, R.F. , Alioto, T. , Antoshechkin, I. , Baer, M.T. , Bar, N.S. , Batut, P. , Bell, K. , Bell, I. , Chakrabortty, S. , Chen, X. , Chrast, J. , Curado, J. , Derrien, T. , Drenkow, J. , Dumais, E. , Dumais, J. , Duttagupta, R. , Falconnet, E. , Fastuca, M. , Fejes‐Toth, K. , Ferreira, P. , Foissac, S. , Fullwood, M.J. , Gao, H. , Gonzalez, D. , Gordon, A. , Gunawardena, H. , Howald, C. , Jha, S. , Johnson, R. , Kapranov, P. , King, B. , Kingswood, C. , Luo, O.J. , Park, E. , Persaud, K. , Preall, J.B. , Ribeca, P. , Risk, B. , Robyr, D. , Sammeth, M. , Schaffer, L. , See, L.H. , Shahab, A. , Skancke, J. , Suzuki, A.M. , Takahashi, H. , Tilgner, H. , Trout, D. , Walters, N. , Wang, H. , Wrobel, J. , Yu, Y. , Ruan, X. , Hayashizaki, Y. , Harrow, J. , Gerstein, M. , Hubbard, T. , Reymond, A. , Antonarakis, S.E. , Hannon, G. , Giddings, M.C. , Ruan, Y. , Wold, B. , Carninci, P. , Guigó, R. , Gingeras, T.R. (2012) Landscape of transcription in human cells. Nature 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagano, T. , Fraser, P. (2011) No‐nonsense functions for long noncoding RNAs. Cell 145, 178–181. [DOI] [PubMed] [Google Scholar]
- 24. Guttman, M. , Donaghey, J. , Carey, B.W. , Garber, M. , Grenier, J.K. , Munson, G. , Young, G. , Lucas, A.B. , Ach, R. , Bruhn, L. , Yang, X. , Amit, I. , Meissner, A. , Regev, A. , Rinn, J.L. , Root, D.E. , Lander, E.S. (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chu, C. , Qu, K. , Zhong, F.L. , Artandi, S.E. , Chang, H.Y. (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA‐chromatin interactions. Mol. Cell 44, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cabili, M.N. , Trapnell, C. , Goff, L. , Koziol, M. , Tazon‐Vega, B. , Regev, A. , Rinn, J.L. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ørom, U.A. , Derrien, T. , Beringer, M. , Gumireddy, K. , Gardini, A. , Bussotti, G. , Lai, F. , Zytnicki, M. , Notredame, C. , Huang, Q. , Guigo, R. , Shiekhattar, R. (2010) Long noncoding RNAs with enhancer‐like function in human cells. Cell 143, 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercer, T.R. , Dinger, M.E. , Mattick, J.S. (2009) Long non‐coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. [DOI] [PubMed] [Google Scholar]
- 29. Mattick, J.S. , Amaral, P.P. , Dinger, M.E. , Mercer, T.R. , Mehler, M.F. (2009) RNA regulation of epigenetic processes. Bioessays 31, 51–59. [DOI] [PubMed] [Google Scholar]
- 30. Khalil, A.M. , Guttman, M. , Huarte, M. , Garber, M. , Raj, A. , Rivea Morales, D. , Thomas, K. , Presser, A. , Bernstein, B.E. , van Oudenaarden, A. , Regev, A. , Lander, E.S. , Rinn, J.L. (2009) Many human large intergenic noncoding RNAs associate with chromatin‐modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 106, 11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amaral, P.P. , Dinger, M.E. , Mercer, T.R. , Mattick, J.S. (2008) The eukaryotic genome as an RNA machine. Science 319, 1787–1789. [DOI] [PubMed] [Google Scholar]
- 32. Dinger, M.E. , Amaral, P.P. , Mercer, T.R. , Pang, K.C. , Bruce, S.J. , Gardiner, B.B. , Askarian‐Amiri, M.E. , Ru, K. , Soldà, G. , Simons, C. , Sunkin, S.M. , Crowe, M.L. , Grimmond, S.M. , Perkins, A.C. , Mattick, J.S. (2008) Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 18, 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guttman, M. , Amit, I. , Garber, M. , French, C. , Lin, M.F. , Feldser, D. , Huarte, M. , Zuk, O. , Carey, B.W. , Cassady, J.P. , Cabili, M.N. , Jaenisch, R. , Mikkelsen, T.S. , Jacks, T. , Hacohen, N. , Bernstein, B.E. , Kellis, M. , Regev, A. , Rinn, J.L. , Lander, E.S. (2009) Chromatin signature reveals over a thousand highly conserved large non‐coding RNAs in mammals. Nature 458, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao, J. , Sun, B.K. , Erwin, J.A. , Song, J.J. , Lee, J.T. (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dinger, M.E. , Amaral, P.P. , Mercer, T.R. , Mattick, J.S. (2009) Pervasive transcription of the eukaryotic genome: functional indices and conceptual implications. Brief. Funct. Genomics Proteomics 8, 407–423. [DOI] [PubMed] [Google Scholar]
- 36. Dinger, M.E. , Pang, K.C. , Mercer, T.R. , Crowe, M.L. , Grimmond, S.M. , Mattick, J.S. (2009) NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 37, D122–D126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercer, T.R. , Dinger, M.E. , Mariani, J. , Kosik, K.S. , Mehler, M.F. , Mattick, J.S. (2008) Noncoding RNAs in long‐term memory formation. Neuroscientist 14, 434–445. [DOI] [PubMed] [Google Scholar]
- 38. Mercer, T.R. , Dinger, M.E. , Sunkin, S.M. , Mehler, M.F. , Mattick, J.S. (2008) Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 105, 716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagano, T. , Mitchell, J.A. , Sanz, L.A. , Pauler, F.M. , Ferguson‐Smith, A.C. , Feil, R. , Fraser, P. (2008) The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720. [DOI] [PubMed] [Google Scholar]
- 40. Guttman, M. , Russell, P. , Ingolia, N.T. , Weissman, J.S. , Lander, E.S. (2013) Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154, 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang, L. , Park, H.J. , Dasari, S. , Wang, S. , Kocher, J.P. , Li, W. (2013) CPAT: coding‐potential assessment tool using an alignment‐free logistic regression model. Nucleic Acids Res. 41, e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin, M.F. , Jungreis, I. , Kellis, M. (2011) PhyloCSF: a comparative genomics method to distinguish protein coding and non‐coding regions. Bioinformatics 27, i275–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ingolia, N.T. , Lareau, L.F. , Weissman, J.S. (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collier, S.P. , Collins, P.L. , Williams, C.L. , Boothby, M.R. , Aune, T.M. (2012) Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J. Immunol. 189, 2084–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu, G. , Tang, Q. , Sharma, S. , Yu, F. , Escobar, T.M. , Muljo, S.A. , Zhu, J. , Zhao, K. (2013) Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 14, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spurlock, C.F. , Tossberg, J.T. , Guo, Y. , Collier, S.P. , Crooke, P.S. , Aune, T.M. (2015) Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat. Commun. 6, 6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sigova, A.A. , Mullen, A.C. , Molinie, B. , Gupta, S. , Orlando, D.A. , Guenther, M.G. , Almada, A.E. , Lin, C. , Sharp, P.A. , Giallourakis, C.C. , Young, R.A. (2013) Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 110, 2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang, H. , Nestor, C.E. , Zhao, S. , Lentini, A. , Bohle, B. , Benson, M. , Wang, H. (2013) Profiling of human CD4+ T‐cell subsets identifies the Th2‐specific noncoding RNA GATA3‐AS1. J. Allergy Clin. Immunol. 132, 1005–1008. [DOI] [PubMed] [Google Scholar]
- 49. Boehm, T. (2008) Thymus development and function. Curr. Opin. Immunol. 20, 178–184. [DOI] [PubMed] [Google Scholar]
- 50. Takahama, Y. (2006) Journey through the thymus: stromal guides for T‐cell development and selection. Nat. Rev. Immunol. 6, 127–135. [DOI] [PubMed] [Google Scholar]
- 51. Kurobe, H. , Liu, C. , Ueno, T. , Saito, F. , Ohigashi, I. , Seach, N. , Arakaki, R. , Hayashi, Y. , Kitagawa, T. , Lipp, M. , Boyd, R.L. , Takahama, Y. (2006) CCR7‐dependent cortex‐to‐medulla migration of positively selected thymocytes is essential for establishing central tolerance. Immunity 24, 165–177. [DOI] [PubMed] [Google Scholar]
- 52. Holländer, G. , Gill, J. , Zuklys, S. , Iwanami, N. , Liu, C. , Takahama, Y. (2006) Cellular and molecular events during early thymus development. Immunol. Rev. 209, 28–46. [DOI] [PubMed] [Google Scholar]
- 53. Farber, D.L. , Yudanin, N.A. , Restifo, N.P. (2014) Human memory T cells: generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 14, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Youngblood, B. , Hale, J.S. , Ahmed, R. (2013) T‐cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 139, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pepper, M. , Jenkins, M.K. (2011) Origins of CD4(+) effector and central memory T cells. Nat. Immunol. 12, 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shih, H.Y. , Sciumè, G. , Poholek, A.C. , Vahedi, G. , Hirahara, K. , Villarino, A.V. , Bonelli, M. , Bosselut, R. , Kanno, Y. , Muljo, S.A. , O'Shea, J.J. (2014) Transcriptional and epigenetic networks of helper T and innate lymphoid cells. Immunol. Rev. 261, 23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKenzie, A.N. , Spits, H. , Eberl, G. (2014) Innate lymphoid cells in inflammation and immunity. Immunity 41, 366–374. [DOI] [PubMed] [Google Scholar]
- 58. Diefenbach, A. , Colonna, M. , Koyasu, S. (2014) Development, differentiation, and diversity of innate lymphoid cells. Immunity 41, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pang, K.C. , Dinger, M.E. , Mercer, T.R. , Malquori, L. , Grimmond, S.M. , Chen, W. , Mattick, J.S. (2009) Genome‐wide identification of long noncoding RNAs in CD8+ T cells. J. Immunol. 182, 7738–7748. [DOI] [PubMed] [Google Scholar]
- 60. Ranzani, V. , Rossetti, G. , Panzeri, I. , Arrigoni, A. , Bonnal, R.J. , Curti, S. , Gruarin, P. , Provasi, E. , Sugliano, E. , Marconi, M. , De Francesco, R. , Geginat, J. , Bodega, B. , Abrignani, S. , Pagani, M. (2015) The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc‐MAF‐4. Nat. Immunol. 16, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Collier, S.P. , Henderson, M.A. , Tossberg, J.T. , Aune, T.M. (2014) Regulation of the Th1 genomic locus from Ifng through Tmevpg1 by T‐bet. J. Immunol. 193, 3959–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang, F. , Zhang, H. , Mei, Y. , Wu, M. (2014) Reciprocal regulation of HIF‐1α and lincRNA‐p21 modulates the Warburg effect. Mol. Cell 53, 88–100. [DOI] [PubMed] [Google Scholar]
- 63. Wu, G. , Cai, J. , Han, Y. , Chen, J. , Huang, Z.P. , Chen, C. , Cai, Y. , Huang, H. , Yang, Y. , Liu, Y. , Xu, Z. , He, D. , Zhang, X. , Hu, X. , Pinello, L. , Zhong, D. , He, F. , Yuan, G.C. , Wang, D.Z. , Zeng, C. (2014) LincRNA‐p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130, 1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spurlock, C.F. , Tossberg, J.T. , Matlock, B.K. , Olsen, N.J. , Aune, T.M. (2014) Methotrexate inhibits NF‐κB activity via long intergenic (noncoding) RNA‐p21 induction. Arthritis Rheumatol. 66, 2947–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yoon, J.H. , Abdelmohsen, K. , Srikantan, S. , Yang, X. , Martindale, J.L. , De, S. , Huarte, M. , Zhan, M. , Becker, K.G. , Gorospe, M. (2012) LincRNA‐p21 suppresses target mRNA translation. Mol. Cell 47, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huarte, M. , Guttman, M. , Feldser, D. , Garber, M. , Koziol, M.J. , Kenzelmann‐Broz, D. , Khalil, A.M. , Zuk, O. , Amit, I. , Rabani, M. , Attardi, L.D. , Regev, A. , Lander, E.S. , Jacks, T. , Rinn, J.L. (2010) A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sánchez, Y. , Segura, V. , Marín‐Béjar, O. , Athie, A. , Marchese, F.P. , González, J. , Bujanda, L. , Guo, S. , Matheu, A. , Huarte, M. (2014) Genome‐wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 5, 5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Warburg, O. (1956) On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- 69. Iyer, N.V. , Kotch, L.E. , Agani, F. , Leung, S.W. , Laughner, E. , Wenger, R.H. , Gassmann, M. , Gearhart, J.D. , Lawler, A.M. , Yu, A.Y. , Semenza, G.L. (1998) Cellular and developmental control of O2 homeostasis by hypoxia‐inducible factor 1 alpha. Genes Dev. 12, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maxwell, P.H. , Wiesener, M.S. , Chang, G.W. , Clifford, S.C. , Vaux, E.C. , Cockman, M.E. , Wykoff, C.C. , Pugh, C.W. , Maher, E.R. , Ratcliffe, P.J. (1999) The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399, 271–275. [DOI] [PubMed] [Google Scholar]
- 71. Jaakkola, P. , Mole, D.R. , Tian, Y.M. , Wilson, M.I. , Gielbert, J. , Gaskell, S.J. , von Kriegsheim, A. , Hebestreit, H.F. , Mukherji, M. , Schofield, C.J. , Maxwell, P.H. , Pugh, C.W. , Ratcliffe, P.J. (2001) Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- 72. Willingham, A.T. , Orth, A.P. , Batalov, S. , Peters, E.C. , Wen, B.G. , Aza‐Blanc, P. , Hogenesch, J.B. , Schultz, P.G. (2005) A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309, 1570–1573. [DOI] [PubMed] [Google Scholar]
- 73. Sharma, S. , Findlay, G.M. , Bandukwala, H.S. , Oberdoerffer, S. , Baust, B. , Li, Z. , Schmidt, V. , Hogan, P.G. , Sacks, D.B. , Rao, A. (2011) Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA‐protein scaffold complex. Proc. Natl. Acad. Sci. USA 108, 11381–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tsao, H.W. , Tai, T.S. , Tseng, W. , Chang, H.H. , Grenningloh, R. , Miaw, S.C. , Ho, I.C. (2013) Ets‐1 facilitates nuclear entry of NFAT proteins and their recruitment to the IL‐2 promoter. Proc. Natl. Acad. Sci. USA 110, 15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vigneau, S. , Rohrlich, P.S. , Brahic, M. , Bureau, J.F. (2003) Tmevpg1, a candidate gene for the control of Theiler's virus persistence, could be implicated in the regulation of gamma interferon. J. Virol. 77, 5632–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gomez, J.A. , Wapinski, O.L. , Yang, Y.W. , Bureau, J.F. , Gopinath, S. , Monack, D.M. , Chang, H.Y. , Brahic, M. , Kirkegaard, K. (2013) The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon‐γ locus. Cell 152, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aune, T.M. , Spurlock, C.F. (2015) Long non‐coding RNAs in innate and adaptive immunity. Virus Res. doi: 10.1016/j.virusres.2015.07.003 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 78. Lee, G.R. , Spilianakis, C.G. , Flavell, R.A. (2005) Hypersensitive site 7 of the Th2 locus control region is essential for expressing Th2 cytokine genes and for long‐range intrachromosomal interactions. Nat. Immunol. 6, 42–48. [DOI] [PubMed] [Google Scholar]
- 79. Kim, A. , Dean, A. (2012) Chromatin loop formation in the β‐globin locus and its role in globin gene transcription. Mol. Cells 34, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Walsh, K.M. , de Smith, A.J. , Chokkalingam, A.P. , Metayer, C. , Roberts, W. , Barcellos, L.F. , Wiemels, J.L. , Buffler, P.A. (2013) GATA3 risk alleles are associated with ancestral components in Hispanic children with ALL. Blood 122, 3385–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang, Y. , Zhong, H. , Xie, X. , Chen, C.Y. , Huang, D. , Shen, L. , Zhang, H. , Chen, Z.W. , Zeng, G. (2015) Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T‐cell immune responses in tuberculosis infection. Proc. Natl. Acad. Sci. USA 112, E3883–E3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sehgal, L. , Mathur, R. , Braun, F.K. , Wise, J.F. , Berkova, Z. , Neelapu, S. , Kwak, L.W. , Samaniego, F. (2014) FAS‐antisense 1 lncRNA and production of soluble versus membrane Fas in B‐cell lymphoma. Leukemia 28, 2376–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kabacik, S. , Manning, G. , Raffy, C. , Bouffler, S. , Badie, C. (2015) Time, dose and ataxia telangiectasia mutated (ATM) status dependency of coding and noncoding RNA expression after ionizing radiation exposure. Radiat. Res. 183, 325–337. [DOI] [PubMed] [Google Scholar]
- 84. Schönland, S.O. , Lopez, C. , Widmann, T. , Zimmer, J. , Bryl, E. , Goronzy, J.J. , Weyand, C.M. (2003) Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc. Natl. Acad. Sci. USA 100, 13471–13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gazzaniga, F.S. , Blackburn, E.H. (2014) An antiapoptotic role for telomerase RNA in human immune cells independent of telomere integrity or telomerase enzymatic activity. Blood 124, 3675–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Farh, K.K. , Marson, A. , Zhu, J. , Kleinewietfeld, M. , Housley, W.J. , Beik, S. , Shoresh, N. , Whitton, H. , Ryan, R.J. , Shishkin, A.A. , Hatan, M. , Carrasco‐Alfonso, M.J. , Mayer, D. , Luckey, C.J. , Patsopoulos, N.A. , De Jager, P.L. , Kuchroo, V.K. , Epstein, C.B. , Daly, M.J. , Hafler, D.A. , Bernstein, B.E. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hrdlickova, B. , Kumar, V. , Kanduri, K. , Zhernakova, D.V. , Tripathi, S. , Karjalainen, J. , Lund, R.J. , Li, Y. , Ullah, U. , Modderman, R. , Abdulahad, W. , Lähdesmäki, H. , Franke, L. , Lahesmaa, R. , Wijmenga, C. , Withoff, S. (2014) Expression profiles of long non‐coding RNAs located in autoimmune disease‐associated regions reveal immune cell‐type specificity. Genome Med. 6, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kumar, V. , Westra, H.J. , Karjalainen, J. , Zhernakova, D.V. , Esko, T. , Hrdlickova, B. , Almeida, R. , Zhernakova, A. , Reinmaa, E. , Võsa, U. , Hofker, M.H. , Fehrmann, R.S. , Fu, J. , Withoff, S. , Metspalu, A. , Franke, L. , Wijmenga, C. (2013) Human disease‐associated genetic variation impacts large intergenic non‐coding RNA expression. PLoS Genet. 9, e1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Leggett, R.M. , Ramirez‐Gonzalez, R.H. , Clavijo, B.J. , Waite, D. , Davey, R.P. (2013) Sequencing quality assessment tools to enable data‐driven informatics for high throughput genomics. Front. Genet. 4, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liao, Y. , Smyth, G.K. , Shi, W. (2013) The Subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. , Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Trapnell, C. , Hendrickson, D.G. , Sauvageau, M. , Goff, L. , Rinn, J.L. , Pachter, L. (2013) Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nat. Biotechnol. 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. , Pimentel, H. , Salzberg, S.L. , Rinn, J.L. , Pachter, L. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Robinson, M.D. , McCarthy, D.J. , Smyth, G.K. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Love, M.I. , Huber, W. , Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Leng, N. , Dawson, J.A. , Thomson, J.A. , Ruotti, V. , Rissman, A.I. , Smits, B.M. , Haag, J.D. , Gould, M.N. , Stewart, R.M. , Kendziorski, C. (2013) EBSeq: an empirical Bayes hierarchical model for inference in RNA‐seq experiments. Bioinformatics 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hardcastle, T.J. , Kelly, K.A. (2010) baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Anders, S. , McCarthy, D.J. , Chen, Y. , Okoniewski, M. , Smyth, G.K. , Huber, W. , Robinson, M.D. (2013) Count‐based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- 99. Chen, H.I. , Liu, Y. , Zou, Y. , Lai, Z. , Sarkar, D. , Huang, Y. , Chen, Y. (2015) Differential expression analysis of RNA sequencing data by incorporating non‐exonic mapped reads. BMC Genomics 16 (Suppl 7), S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ye, J. , McGinnis, S. , Madden, T.L. (2006) BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Finn, R.D. , Clements, J. , Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Thorvaldsdóttir, H. , Robinson, J.T. , Mesirov, J.P. (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. , Mesirov, J.P. (2011) Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Herrin, B.R. , Cooper, M.D. (2010) Alternative adaptive immunity in jawless vertebrates. J. Immunol. 185, 1367–1374. [DOI] [PubMed] [Google Scholar]
- 105. Eivazova, E.R. , Aune, T.M. (2004) Dynamic alterations in the conformation of the Ifng gene region during T helper cell differentiation. Proc. Natl. Acad. Sci. USA 101, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dekker, J. , Rippe, K. , Dekker, M. , Kleckner, N. (2002) Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- 107. Splinter, E. , de Wit, E. , Nora, E.P. , Klous, P. , van de Werken, H.J. G. , Zhu, Y. , Kaaij, L.J. T. , van Ijcken, W. , Gribnau, J. , Heard, E. , de Laat, W. (2011) The inactive X chromosome adopts a unique three‐dimensional conformation that is dependent on Xist RNA. Genes Dev. 25, 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Collins, P.L. , Chang, S. , Henderson, M. , Soutto, M. , Davis, G.M. , McLoed, A.G. , Townsend, M.J. , Glimcher, L.H. , Mortlock, D.P. , Aune, T.M. (2010) Distal regions of the human IFNG locus direct cell type‐specific expression. J. Immunol. 185, 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Collins, P.L. , Henderson, M.A. , Aune, T.M. (2012) Diverse functions of distal regulatory elements at the IFNG locus. J. Immunol. 188, 1726–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chang, S. , Aune, T.M. (2007) Dynamic changes in histone‐methylation “marks” across the locus encoding interferon‐gamma during the differentiation of T helper type 2 cells. Nat. Immunol. 8, 723–731. [DOI] [PubMed] [Google Scholar]
- 111. Euskirchen, G.M. , Rozowsky, J.S. , Wei, C.L. , Lee, W.H. , Zhang, Z.D. , Hartman, S. , Emanuelsson, O. , Stolc, V. , Weissman, S. , Gerstein, M.B. , Ruan, Y. , Snyder, M. (2007) Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array‐ and sequencing‐based technologies. Genome Res. 17, 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gupta, R.A. , Shah, N. , Wang, K.C. , Kim, J. , Horlings, H.M. , Wong, D.J. , Tsai, M.C. , Hung, T. , Argani, P. , Rinn, J.L. , Wang, Y. , Brzoska, P. , Kong, B. , Li, R. , West, R.B. , van de Vijver, M.J. , Sukumar, S. , Chang, H.Y. (2010) Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]