

Abstract
The CNS is considered an immune privileged site because its repertoire of highly immunogenic molecules remains unseen by the immune system under normal conditions. However, the mechanism underlying the inhibition of immune reactions within the CNS environment is not known, particularly in regions containing myelin, which contains several potent proteins and lipids that are invariably recognized as foreign by immune system cells. Sulfatides constitute a major component of myelin glycolipids and are known to be capable of raising an immune response. In this study, the effect of sulfatides on mouse T cell function and differentiation was analyzed in vitro and in vivo. We found profound inhibition of sulfatide-dependent T cell proliferation which was particularly pronounced in naive T helper (Th) cells. The inhibitory effect of sulfatides on T cell function was CD1d-independent and was not related to apoptosis or necrosis but did involve the induction of anergy as confirmed by the upregulation of early growth response 2 transcription factor. A glycolipid 3-sulfate group was essential for the T cell suppression, and the T cell inhibition was galectin-4-dependent. Sulfatide stimulation in vitro led to prominent suppression of Th17 differentiation, and this was related to a decrease in susceptibility to disease in a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis. Thus, we have defined a novel mechanism of negative regulation of T cell function by endogenous brain-derived glycolipids, a family of molecules traditionally deemphasized in favor of myelin proteins in studies of CNS autoimmunity.
Keywords: autoimmune demyelination, experimental autoimmune encephalomyelitis, glycolipids, multiple sclerosis, T cells, Th17
Introduction
The CNS is an immunologically privileged site where inflammatory reactions are blocked or blunted at the blood–brain barrier to minimize collateral damage (Ransohoff and Brown, 2012). In multiple sclerosis (MS), a putative autoimmune disease of the CNS manifested by inflammation and demyelination, T helper cell (Th)–mediated autoimmunity to myelin antigens, usually proteins, has long been thought to be a major effector on its pathogenesis (Nylander and Hafler, 2012). Disturbances of various Th populations, such as Th1, Th2, and Treg, have been linked to MS susceptibility. Recently, another subtype, Th17 cells, characterized by the expression of the transcription factors retinoic acid receptor-related orphan receptor α and γt (ROR-α and ROR-γt), and the production of interleukin-17A (IL-17A), have been demonstrated as pivotal for autoimmune demyelination (Bettelli et al., 2008). However, why immune-privileged mechanisms in the CNS fail to protect the tissue from autoimmune reactions that lead to the development of MS remains an enigma.
A hallmark of MS is loss of myelin, insulating membrane laid down in segments along axons. Myelin is composed mostly of lipids assembled with proteins, including myelin/oligodendrocyte glycoprotein (MOG) (Aggarwal et al., 2011). Although myelin proteins are minor constituents of the membrane complex, historically, they have attracted the majority of attention by neuroimmunology investigators resulting in myelin lipids being less extensively studied. Sulfatides are glycolipids that form a major component of myelin (Aggarwal et al., 2011). Mice lacking sulfatides display myelin dissolution, progressive loss of nerve conduction, and early death (Bosio et al., 1996; Coetzee et al., 1996). Apart from myelin-related functions, sulfatides have been demonstrated to be endogenous lipid antigens recognized in the context of the major histocompatibility complex (MHC) class 1-like molecule, CD1d by a subset of T cells, and sulfatide-specific T cells have been implicated in the pathogenesis of experimental autoimmune encephalomyelitis (EAE), the major model for MS (Jahng et al., 2004; Zajonc et al., 2005; Kanter et al., 2006; Girardi et al., 2012). Furthermore, sulfatides have been found to influence a variety of immune cells, such as neutrophils, dendritic cells, B cells, and microglia (Konno et al., 1996; Jeon et al., 2008; Yamada et al., 2011; Kanamori et al., 2012). Thus, sulfatides may represent an important, relatively unexplored component of immune regulation in the CNS.
In MS, sulfatides have been reported to be substantially reduced both in lesions and in normal appearing adjacent white matter (Cuzner and Davison, 1973; Suzuki et al., 1973; Woelk and Borri, 1973; Yahara et al., 1982; Wilson and Tocher, 1991). To gain insight into the role of lipids during immunologic responses of relevance to MS, we have analyzed the impact of sulfatides on specific functions of Th cells, the major orchestrators of autoimmune demyelination, and have identified a potent, CD1d-independent inhibitory effect of sulfatides on the proliferation of Th cells, in particular naive Th cells. In addition, sulfatides impaired the development of Th17 cells and suppressed EAE. Thus, we draw attention to the possibility of a novel, distinct pathogenetic role for sulfatides in autoimmune demyelination.
Materials and Methods
Mice.
C57BL/6, C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J (2D2 mice), and B6.129S6-Cd1d1/Cd1d2tm1Spb/J (CD1d-deficient mice) female mice were purchased from The Jackson Laboratory and maintained for the duration of experiments. All animal protocols were approved by the Institutional Animal Care and Use Committee of the Medical University of Lodz.
Antibodies, cytokines, peptides, and other reagents for functional assays.
These consisted of the following: fluorochrome-conjugated antibodies specific for CD3, CD4, CD8, CD25, CD44, CD62L, CD69, B220, IL-17A, interferon-γ (IFN-γ), and the active form of caspase-3, which were purchased from BD Biosciences; anti-CD62L antibody (MEL-14) and control Ig from Santa Cruz Biotechnology; anti-galectin-4 polyclonal antibody and control Ig from R&D Systems; MOG35–55 peptide from Peptide 2.0; IL-2, IL-4, IL-6, and IL-12 from PeproTech; IL-1b from Sigma; TGF-β and IL-23 from R&D Systems; aryl-sulfatase from Sigma; Z-VAD-FMK from Calbiochem; and AMD 3100 from Tocris Bioscience.
Glycolipid stimulation of T cell cultures.
Brain sulfatides, brain cerebrosides, and synthetic glycolipids, N-tetracosanoyl-sulfatide, N-palmitoyl-sulfatide, N-acetyl-sulfatide and lyso-sulfatide (Matreya), were dissolved in vehicle (0.5% polysorbate 20 and 0.9% NaCl solution), at a concentration 20 mg/ml. For cell culture stimulation, single-cell suspensions of freshly isolated splenocytes or sorted T cells from C57BL/6, CD1d-deficient, or 2D2 mice were cultured for 24 h in IMDM-based cell culture medium in triplicate at a density of 2 × 105 cells per well in 200 μl U-bottomed microtiter well plates with or without the addition of glycolipids. Glycolipid stimulation caused no cell loss or death, as assessed by trypan blue (Sigma) staining. Subsequently, cells were transferred to 200 μl U-bottomed microtiter well plates with plate-bound anti-CD3 (2 μg/ml; 145–2C11), or plate-bound anti-CD3 and anti-CD28 (both at 2 μg/ml), or stimulated with concanavalin A (ConA, 5 μg/ml), and with glycolipids at the same concentration as for preincubation. CD4+ T cells, CD8+ T cells, and naive and memory CD4+ T cells were isolated, either directly from splenocytes or from cell cultures, by indirect magnetic sorting with CD4+ T Cell Isolation Kit II, CD8+ T Cell Isolation Kit II, or with CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec). To assess proliferation, after a 72 h culture, for the final 18 h of culture, 1 μCi/well 3H-labeled thymidine (PerkinElmer) was added and uptake measured using a liquid scintillation β counter (PerkinElmer). Results were expressed as counts per minute.
Immunization and stimulation of T cell cultures with antigen.
Eight- to 12-week-old C57BL/6 mice were immunized subcutaneously over the abdominal flanks with 0.15 mg MOG35–55 peptide in 150 μl complete Freund's adjuvant (Sigma) containing 0.75 mg Mycobacterium tuberculosis (Difco Laboratories). Splenocytes in single-cell suspensions were cultured in triplicate as described above in the presence of glycolipid for 24 h followed by 72 h stimulation with MOG35–55 antigen. For 2D2 T cell responses, analogous cultures were set up from splenocytes from 8- to 12-week-old nonimmunized 2D2 mice.
CD4+ T subpopulations: in vitro differentiation.
Th1, Th2, Th17, and Treg in vitro differentiation was performed on splenic naive CD4+ T cells that had been cultured for 24 h with sulfatides and then stimulated with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (10 μg/ml), polarized for 6 d with the following: for Th1 IL-12 (10 ng/ml) in the presence of anti-IL-4 (10 μg/ml; 11B11) and anti-IFN-γ (10 μg/ml; XMG1.2); for Th2 IL-4 (40 ng/ml) in the presence of anti-IFN-γ (10 μg/ml); for Treg IL-6 (25 ng/ml), TGF-β (2 ng/ml), IL-1b (20 ng/ml), and IL-23 (20 ng/ml) in the presence of anti-IL-4 (10 μg/ml) and anti-IFN-γ (10 μg/ml) and with the addition of 40 mmNaCl for Th17; TGF-β (10 ng/ml) and IL-2 (100 U/ml) in the presence of anti-IL-4 (10 μg/ml) and anti-IFN-γ (10 μg/ml); Th0 conditions were maintained by naive CD4+ T cells cultured in the presence of anti-IL-4 (10 μg/ml) and anti-IFN-γ (10 μg/ml). After 6 d of culture in Th17 polarizing conditions, cells were assayed for cytokine production by intracellular flow cytometry.
Extraction of RNA and mRNA expression analysis.
Extraction of total RNA was performed using a mirVana kit (Ambion). For quantitative analysis of RNA expression, we performed real-time qRT-PCR with TaqMan probes (Invitrogen), using a 7500 Real Time PCR System (Applied Biosystems). mRNA expression data were normalized to that of the large RNA polymerase subunit, RPA194 (POLR1A). Expression was evaluated by the comparative cycling threshold (ΔΔCT) method.
Flow cytometry.
Four- to six-color flow cytometry analysis with LSR II (Becton Dickinson) was performed according to standard procedures. Annexin V and 7AAD staining was conducted using an Annexin V Apoptosis Detection Kit (BD Biosciences). Detection of the active form of caspase 3 was by intracellular staining using a BD Cytofix/Cytoperm Kit (BD Biosciences). For intracellular detection of cytokine production, Th cells were stimulated with 500 ng/ml phorbol dibutyrate and 500 ng/ml ionomycin in the presence of brefeldin A for 6 h. Detection of IL-17A and IFN-γ-positive CD4+ T cells was by intracellular staining using a Mouse Regulatory T cell Staining Kit (eBioscience). Flow cytometry data were analyzed with FlowJo (Tree Star).
Induction of EAE and treatment with sulfatides.
Active EAE was induced by subcutaneous immunization over the abdominal flanks of 8- to 12-week-old CD1d-deficient mice with 0.15 mg MOG35–55 peptide in 150 μl complete Freund's adjuvant containing 0.75 mg M. tuberculosis. In addition, 0.2 μg pertussis toxin (Sigma) was injected intravenously on days 0 and 2. On days 0, 2, 4, 6, and 8 after EAE induction, mice treated with sulfatides received intravenous injections of 200 μg sulfatides dissolved in 200 μl of vehicle (0.5% polysorbate 20 and 0.9% NaCl solution), to a total dose of 1 mg/sulfatides per mouse. On the same days, control mice received intravenous injections of 200 μl vehicle alone. Mice were checked daily for body weight and signs of EAE and scored on a scale of 0–5 as follows: 0, no disease; 1, weak tail or unsteady gait; 2, hind-limb paresis; 3, hind-limb paralysis; 4, hind- and fore-limb paralysis; 5, death or euthanasia.
Histopathology.
Evaluation of inflammation during EAE was performed on hematoxylin and eosin-stained paraffin sections. Spinal cords were removed from 10% phosphate-buffered formalin-perfused mice and fixed by immersion in 10% phosphate-buffered formalin. Transverse sections for light-microscope analysis were cut at 5 μm from paraffin-embedded material and stained with hematoxylin and eosin. Representative areas of lumbar spinal cord were selected for histologic evaluation.
Western blot analysis.
Cells were lysed and total lysates were resolved on SDS electrophoresis gels by standard procedures. Immunoblotting was performed with mouse primary antibodies to early growth response 2 transcription factor (EGR2) and GAPDH (Santa Cruz Biotechnology), and visualized by G:BOX Chemi (Syngene). Quantitative densitometric analysis was performed using program G:BOX Chemi-XR5 (Syngene).
Statistical analysis.
Results were compared using the program, StatGraphics Centurion XV (StatPoint Technologies). Clinical data from mice with EAE were compared using the Mann–Whitney U test. Results of proliferation and qPCR were compared using one-way ANOVA. p < 0.05 was considered statistically significant.
Results
Sulfatides inhibit T cell proliferation
Splenocytes from naive mice were preincubated for 1 d with sulfatides (0–100 μg/ml) and stimulated for proliferation either with plate-bound anti-CD3 (Fig. 1A), plate-bound anti-CD3 and anti-CD28 (Fig. 1B), or with ConA (Fig. 1C), in the presence of glycolipids. Increasing doses of sulfatides led to substantial inhibition of proliferation (Fig. 1A–C). The suppressive effect of sulfatides on T cell proliferation was accompanied by a significant decrease in expression of the myc gene family, important cell-cycle regulators in T cells (Figure 1D) (Mycko et al., 2009; Guy et al., 2013).
Figure 1.

Inhibitory effect of sulfatide on T cell proliferation. A–C, Splenocytes were stimulated with plate-bound anti-CD3 (A) with plate-bound anti-CD3 and plate-bound anti-CD28 (B), or with ConA (C) for 3 d at the sulfatide concentrations indicated, and proliferative responses were measured. Representative results (mean ± SEM) from five independent experiments are shown. D, Myc gene family expression was analyzed for 3 d in splenocytes stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides or left unstimulated. Representative results (mean ± SEM) from two independent experiments are shown. E, Splenic CD4+ and CD8+ T cells were sorted and stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides for 3 d, and proliferative responses were measured. Results represent mean ± SEM of the percentage of responses from three separate experiments compared with cultures without added sulfatides. F, Naive splenic and memory CD4+ T cells were sorted and stimulated for 3 d with plate-bound anti-CD3 plus indicated concentrations of sulfatides, and proliferative responses were measured. Results represent mean ± SEM of the percentage of the responses from three separate experiments compared with cultures without sulfatides. G, Splenocytes from mice immunized with MOG35–55 restimulated for 3 d in vitro with MOG35–55 and with indicated concentrations of sulfatides, and proliferative responses were measured. Representative results (mean ± SEM) from three independent experiments are shown. H, I, Splenocytes from 2D2 MOG35–55 TCR transgenic mice were stimulated in vitro either with MOG35–55 (H) or with plate-bound anti-CD3 or plate-bound anti-CD3 and plate-bound anti-CD28 (I) and with indicated concentrations of sulfatides for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from three independent experiments are shown. *p < 0.01 (one-way ANOVA). **p < 0.001 (one-way ANOVA).
Sulfatide-induced inhibition of T cell proliferation was predominantly seen in CD4+ T cells compared with CD8+ T cells (Fig. 1E). Subsequently, we found that proliferation of sorted naive Th cells was significantly more suppressed compared with memory Th cells (Fig. 1F). Thus, naive Th cells were the most susceptible T cell population to suppression of sulfatide-induced proliferation.
To address the sulfatide effect on antigen-induced Th cell proliferation, we used either splenocytes from mice that had been immunized with MHC Class II antigen, MOG35–55 peptide (Fig. 1G) or MOG35–55 transgenic CD4+ T cells, 2D2 T cells (Fig. 1H). As expected in both settings, MOG35–55 induced vigorous proliferation of T cells. However, when T cells were stimulated with MOG35–55 in the presence of sulfatides, strong inhibition of the proliferative response was seen. Similar to results obtained from wild-type mice, 2D2 T cell proliferation induced with plate-bound anti-CD3 or with plate-bound anti-CD3 and anti-CD28 was also diminished by sulfatides (Fig. 1I). Thus, both antigenic and nonantigenic TCR-induced proliferation using either monoclonal or polyclonal TCR was strongly suppressed by sulfatides.
Sulfatide-induced suppression of T cells is independent of CD1d presentation
Sulfatides have been demonstrated to be potent inducers of immune responses via CD1d presentation (Jahng et al., 2004; Zajonc et al., 2005). To determine the role of CD1d in the process of sulfatide-induced suppression of T cell proliferation, we used CD1d-deficient mice. Intriguingly, in the presence of sulfatide, proliferation of CD1d-deficient T cells induced either with plate-bound anti-CD3 (Fig. 2A), or plate-bound anti-CD3 and anti-CD28 (Fig. 2B), was profoundly diminished from levels seen with CD1d-sufficient cells (Fig. 1A,B). Therefore, the presence of CD1d was not required for the inhibitory effects of sulfatides upon T cell proliferation.
Figure 2.

The 3-sulfate group is critical for the glycolipid inhibitory effect on T cell proliferation. A, B, Splenocytes from CD1d-deficient mice were stimulated with plate-bound anti-CD3 (A) or with plate-bound anti-CD3 and plate-bound anti-CD28 (B) and with indicated concentrations of sulfatides for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from three independent experiments are shown. C, Structure of glycolipids tested. D, Splenocytes from wild-type mice were stimulated with plate-bound anti-CD3 and with indicated concentrations of glycolipids for 3 d, and proliferative responses were measured. Results represent mean ± SEM of the percentage of the responses from three separate experiments compared with cultures without sulfatides. E, Sulfatides were preincubated for 24 h with concentrations of aryl-sulfatase at doses indicated and added to cultures of splenocytes from wild-type mice stimulated with plate-bound anti-CD3 for 3 d, and proliferative responses were measured. *p < 0.01 (one-way ANOVA). **p < 0.001 (one-way ANOVA).
Chemical structure dictates activity of glycolipids on T cell proliferation
Sulfatides are a mixture of 3-sulfate esters of cerebrosides varying with regard to the position, length, and structure of the fatty acid chain. To analyze how different moieties of sulfatides might influence T cell proliferation, we compared the effect of glycolipids that differ from sulfatides either by lacking the 3-sulfate group (cerebrosides) or by having a shortened fatty acid chain (Fig. 2C). We found that sulfatide analogs with a truncated fatty acid chain still exerted suppressive effects on T cell proliferation. In contrast, cerebrosides alone lacked any effect on T cell proliferation, even at high concentrations (Fig. 2D). This suggested that the presence of the 3-sulfate group was critical for sulfatide-induced inhibition of T cell proliferation, whereas the structure and length of fatty acid chains were not involved in this process. To confirm this finding, we preincubated sulfatides with aryl-sulfatase, an enzyme responsible for 3-sulfate ester breakdown. Indeed, sulfatide treatment with aryl-sulfatase led to the abrogation of suppression of T cell proliferation (Fig. 2E). Thus, a critical role for the 3-sulfate group for sulfatide-mediated inhibition of T cell proliferation was further confirmed.
Sulfatides do not induce apoptosis or necrosis of Th cells
To exclude the possibility that the suppressive activity of sulfatides on Th cell proliferation might be related to induction of cell death, we searched by flow cytometry for the presence of apoptotic and necrotic markers on Th cells stimulated for proliferation in the presence of sulfatides. No differences in frequency of apoptotic cells were found, characterized either by the intracellular presence of active form of caspase 3 (Fig. 3A), or by an annexin V-positive 7AAD-negative phenotype (Fig. 3B), in Th cells stimulated with or without sulfatides. In addition, the percentages of necrotic cells, characterized as annexin V-negative and 7AAD-positive cells (Fig. 3C), were no different between Th cells cultured with or without sulfatides. Furthermore, a caspase inhibitor, z-VAD-FMK, did not influence the suppressive capacity of sulfatides on Th cell proliferation (Fig. 3D). These results did not support apoptosis or necrosis as mechanisms responsible for sulfatide inhibition of Th cell proliferation.
Figure 3.
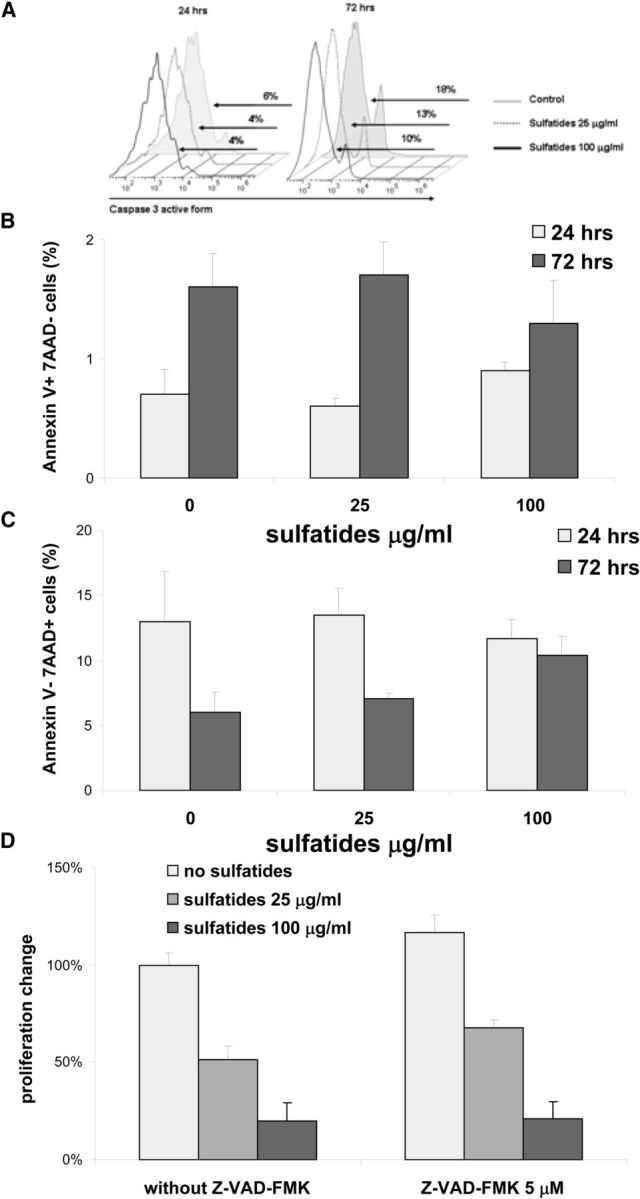
Sulfatide treatment of Th cells did not induce cell death. A–C, Splenocytes were stimulated with plate-bound anti-CD3 and with the concentrations indicated of sulfatides and percentages of intracellular active form of caspase 3 (A), annexin V-positive 7AAD-negative (B), or annexin V-negative 7AAD-positive Th cells (C) were measured by flow cytometry after a 24 or 72 h period in culture. Results are either representative (A) or mean ± SEM (B, C) from four independent experiments. Data for gated Th cells are presented. D, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides and Z-VAD-FMK for 3 d, and proliferative responses were measured. Results represent mean ± SEM of the percentage of the responses from two separate experiments compared with cultures without sulfatides and without Z-VAD-FMK.
Sulfatide treatment of Th cells induces features of anergy
To further characterize sulfatide-mediated inhibition of T cell proliferation, we tested markers of immunologic anergy. Clonal anergy describes a phenomenon when clones of T cells can still be found alive in the circulation but are ineffective at mounting immune responses, in particular an inability to proliferate (Chappert and Schwartz, 2010). Indeed, the expression of EGR2 was significantly upregulated in Th cells stimulated with plate-bound anti-CD3 in the presence of sulfatides, at both the mRNA (Fig. 4A), and protein levels (Fig. 4B,C). Moreover, IL-2 treatment of sulfatide-stimulated cultures led to significant attenuation of the inhibitory effect on Th cell proliferation (Fig. 4D). Reversal of the inhibition of proliferation rate with IL-2 is a classical indicator of anergy (Chappert and Schwartz, 2010). Collectively, these data suggest that sulfatides are capable of inducing anergy in stimulated Th cells.
Figure 4.
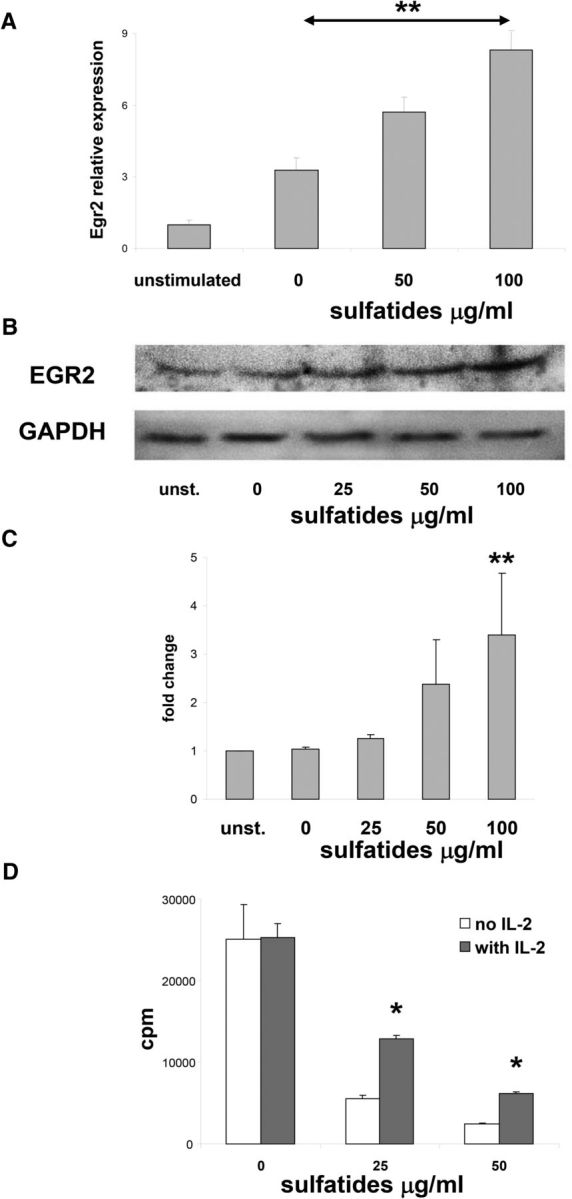
Anergy features are induced by sulfatides in Th cells. A, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides for 3 d or left untreated, and Egr2 gene expression was analyzed. Data normalized to expression levels of untreated Th cells. Representative results (mean ± SEM) from three independent experiments are shown. B, C, Western blot analysis of EGR2 and GAPDH in Th cell cultures as described in A. Representative results from three independent experiments are shown. C, EGR2 expression was quantified by densitometry, normalized to the GAPDH levels, and compared with untreated Th cells levels. D, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides and IL-2 (50 U/ml) for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from two independent experiments are shown. *p < 0.01 (one-way ANOVA). **p < 0.001 (one-way ANOVA).
Galectin-4 inhibition attenuates suppression of Th cells by sulfatides
Naive Th cells were revealed to be the most susceptible population to sulfatide-induced inhibition of proliferation. L-selectin (CD62L), a surface receptor for sulfatides (Suzuki et al., 1993; Ogawa et al., 2004), is also a marker for naive Th cells. Therefore, we decided to test whether CD62L mediated the effect of sulfatides on Th cell proliferation. Intriguingly, we found no evidence that CD62L blockage with a specific CD62L blocking antibody, MEL-14 (Gallatin et al., 1983), diminished sulfatide suppression of Th cell proliferation (Fig. 5A). Because CD62L-mediated signals specifically enhance CXCR4 expression and function of the chemokine receptor, CXCR4 (Duchesneau et al., 2007; Yamada et al., 2011), we also tested for a potential role for CXCR4 as a chemoattractant receptor in the effect of sulfatides on proliferation. Again, however, the presence of a specific CXCR4 inhibitor, AMD3100 (Bridger et al., 1995; Hatse et al., 2002), failed to influence sulfatide-induced inhibition of Th proliferation (Fig. 5B).
Figure 5.

Galectin-4 is involved in the inhibitory effect of sulfatides on Th proliferation. A, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides in the presence of anti-CD62L (3 μg/ml) or control Ig for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from two independent experiments are shown. B, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of sulfatides in the presence of AMD 3100 for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from two independent experiments are shown. C, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with indicated concentrations of the sulfatides in the presence of anti-galectin-4 (10 μg/ml) or control Ig for 3 d, and proliferative responses were measured. Representative results (mean ± SEM) from two independent experiments are shown. D, Expression of galectin-4 gene, LGALS4, in sorted ex vivo naive and memory splenic Th cells. Data normalized to expression levels of memory Th cells. Representative results (mean ± SEM) from four independent experiments are shown. *p < 0.01 (one-way ANOVA).
Sulfatides have been identified as high-affinity ligands for galectin-4 in enterocytes and neurons (Delacour et al., 2005; Stancic et al., 2012). Thus, we decided to test for a role for galectin-4 in sulfatide inhibition of Th cell proliferation. We found that galectin-4 blockage with a specific antibody led to significant attenuation of sulfatide suppression of proliferation (Fig. 5C). Expression of the galectin-4 gene, LGALS4, appeared to be significantly higher in naive Th compared with memory Th cells (Fig. 5D). These data suggested that interaction between galectin-4 and sulfatides played a functional role in sulfatide-mediated inhibition of naive Th cells.
Sulfatides inhibit Th17 cell differentiation
Using flow cytometry, the effect of sulfatides was examined on the expression of early Th cell activation markers (Fig. 6A). No difference was found in the frequency of CD69+ or CD25+ expressing Th cells stimulated with plate-bound anti-CD3 with or without added sulfatides. These data suggested that early TCR signaling in Th cells was not blocked by sulfatides. To assess the later effects of sulfatides on TCR stimulation, we analyzed the gene expression pattern of naive Th cells stimulated with plate-bound anti-CD3 for 3 d (Fig. 6B). No change in expression of Gata3 (Th2 marker), Tbx21, Ifng (both Th1 markers), or Foxp3 (Treg marker) was seen between cells stimulated with or without sulfatides, suggesting no changes in Th1, Th2, or Treg development. In contrast, levels of IL17a and Rora, markers for Th17, were clearly downregulated in cultures treated with sulfatides, proportional to glycolipid concentration (Fig. 6B). When naive Th cells were actively polarized for 6 d with a cytokine mixture favoring Th1, Th2, Th17, and Treg, only Th17 cultures with sulfatides showed a decrease of marker gene expression (Fig. 6C). This finding has been corroborated by an analysis that revealed a gradual decrease of IL-17A-positive Th cells in Th17-polarized cultures with increasing concentrations of sulfatides (Fig. 6D). These findings demonstrated that sulfatide treatment of Th cells in vitro strongly inhibited Th17 differentiation.
Figure 6.

Sulfatide treatment of Th cells inhibits Th17 development. A, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 for 24 h and without or with the sulfatides (50 μg/ml) or left untreated, and the percentage of CD69- or CD25-expressing cells measured by flow cytometry. Representative results (mean ± SEM) from four independent experiments are shown. B, Sorted splenic Th cells were stimulated with plate-bound anti-CD3 and with the concentrations indicated of the sulfatides or left untreated for 3 d, and levels of expression of Gata3 (Th2 marker), Tbx21, Ifng (both Th1 markers), Foxp3 (Treg marker), and Rora, Il17a (both Th17 markers) measured. Data normalized to expression levels in untreated Th cell cultures. Representative results (mean ± SEM) from four independent experiments are shown. C, Splenic naive Th cells were polarized toward Th0, Th1, Th2, Th17, and Treg for 6 d in the presence of indicated concentrations of sulfatides. Data represent expression levels of indicated marker genes, normalized to expression levels of Th0 cultures without sulfatide stimulation. Representative results (mean ± SEM) from three independent experiments are shown. D, Naive splenic Th cells were polarized toward Th17 cells in the presence of indicated concentrations of sulfatides and were assayed for the secretion of IL-17 and IFN-γ by intracellular flow cytometry. Representative results from four independent experiments are shown. *p < 0.03 (ANOVA). **p < 0.001 (ANOVA).
Sulfatide administration in vivo abrogates EAE
To translate the sulfatide effect on T cells in vitro into an in vivo paradigm, sulfatides were administrated to mice that had been sensitized for EAE. CD1d-deficient mice on a C57BL/6 background are as susceptible to EAE as wild-type animals (Jahng et al., 2001; Singh et al., 2001), and these mice were used to avoid the influence of CD1d-mediated sulfatide presentation to NKT cells during EAE. Vehicle-treated CD1d-deficient mice were found to develop typical clinical EAE beginning day 10, with a remission phase beginning day 40 after immunization (Fig. 7A). In contrast, CD1d-deficient mice treated with sulfatides showed minimal and delayed signs that cleared by day 30. Sulfatide-treated mice also lacked weight loss, in contrast to the vehicle-treated group (Fig. 7B). Pathology of the CNS of vehicle treated animals showed typical infiltrates of mononuclear cells in the spinal cord and very few inflammatory cells in sulfatide-treated mice (Fig. 7C). Thus, in vivo data corroborated the immunosuppressive properties of sulfatides observed in vitro and demonstrated their involvement in CD1d-independent control mechanisms.
Figure 7.
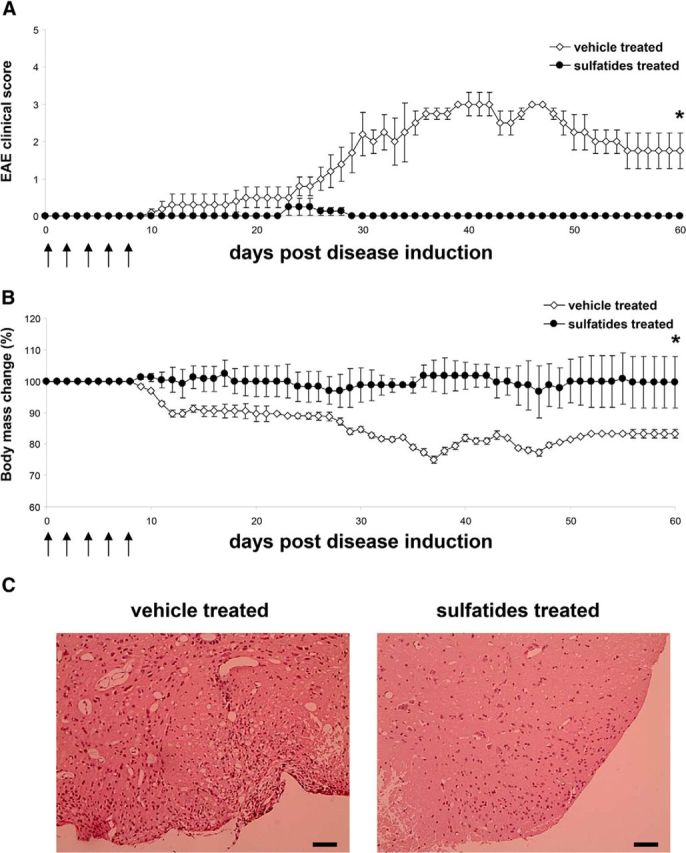
Sulfatide treatment suppresses EAE. A, B, CD1d-deficient mice sensitized for EAE with MOG35–55 were treated with five intravenous injections of sulfatides or vehicle. Arrows indicate days of sulfatide or vehicle treatment. A, Clinical scores presented as mean ± SEM. B, Body weight changes of mice during sulfatide or vehicle-treated EAE presented as mean percentage ± SEM, compared with weight on day 0. Five mice per group used. *p < 0.01 (Mann–Whitney U test). C, Histopathology of CNS lesions in vehicle- and sulfatide-treated CD1d-deficient mice sensitized for EAE with MOG35–55, as described in A. B, Photomicrographs taken from 5 μm sections of paraffin-embedded materials of lumbar spinal cord sampled during acute EAE (day 30) stained with hematoxylin and eosin. In vehicle-treated mice, inflammatory infiltrates typical of EAE were seen within the meninges. The spinal cord surface is gliotic and demyelinated. In sulfatide-treated mice, inflammatory cells are scarce. Only a small number are seen within the meninges below, and there is some fragmentation of myelin. Scale bars, 100 μm.
Discussion
Neuroimmunologic studies on CNS autoimmune demyelinating disease models have traditionally emphasized pathogenetic roles for myelin protein-related molecules presented in the context of Class II MHC molecules. In the present study, using similar paradigms, we have focused on myelin-derived glycolipids and MHC Class I molecules and have revealed a significant effect of sulfatides on T cell proliferation and differentiation. Also, dose-dependent suppression of T cell proliferation by these glycolipids was shown for both antigen-specific and nonantigen-specific TCR stimulation. Sulfatides failed to induce apoptosis or necrosis in vitro. Instead, features of anergy after sulfatide stimulation of Th cells were evident. Sulfatide-induced inhibition of T cell proliferation was most pronounced in naive Th cells and was mediated by galectin-4. In addition, sulfatides inhibited Th17 differentiation and suppressed the development of EAE. Thus, sulfatides in several ways similar to protein antigens, can function as strong modulators of Th cell functions relevant to autoimmune demyelination and CNS immune-protection mechanisms.
In contrast to myelin protein antigens, which are largely presented by MHC Class II molecules, lipid antigens have been shown to be presented to T cells by MHC Class I CD1 molecules with a model antigen, sponge-derived α-galactosylceramide that activates an invariant subset of T cells, Type I NKT cell (Adams and Luoma, 2013). Sulfatides have been reported to be self-antigens, recognized by another T cell subset, Type II NKT cells, in the context of CD1d (Girardi et al., 2012; Patel et al., 2012). These sulfatide-specific T cells have been implicated in the pathogenesis of EAE, and sulfatide immunization in CD1d-sufficient mice was found to be protective for development of EAE by direct activation of Type II NKT cells (Jahng et al., 2004). Another study reported that coimmunization of SJL/J mice with sulfatides plus myelin peptide resulted in a more severe EAE via CD1d-mediated induction of anti-sulfatide antibodies (Kanter et al., 2006). Interestingly, when we tested CD1d-deficient animals, CD1d was found to be dispensable for the suppression of T cell proliferation by sulfatides. These data indicate that, depending on CD1d stimulation, sulfatides might have an opposite effect on EAE. Interestingly, lyso-sulfatide, which lacks an acyl chain and binds weakly to mouse CD1d (Roy et al., 2008), was still a potent suppressor of Th cell proliferation. To further support the CD1d-independent activity of sulfatides, we sensitized CD1d-deficient animals for EAE and treated them with these glycolipids. Sulfatide treatment led to almost complete protection from disease, both clinically and histologically. Interestingly, a recent study has reported a similar, CD1d-independent phospholipid inhibitory effect on T cell responses and the course of EAE (Ho et al., 2012). Thus, a spectrum of brain lipids may exert previously unappreciated CD1d-independent actions on T cell function.
In a search for molecules other than CD1d that might mediate the suppressive effect of sulfatide on Th cell proliferation, CD62L expression was examined. CD62L has been reported to be an independent surface receptor for sulfatides (Suzuki et al., 1993; Laudanna et al., 1994; Ogawa et al., 2004; Delacour et al., 2005; Merten et al., 2005). Sulfatide-induced activation of CD62L has been linked to activation of the NFkB pathway in glial cells (Jeon et al., 2008), and neutrophils (Turutin et al., 2003), in the inhibition of 5-lipoxygenase in polymorphonuclear granulocytes (Sud'ina et al., 2001), and in the inhibition of Ig production by B cells (Konno et al., 1996). Antibody blockage of CD62L failed to modify sulfatide suppression of Th cell proliferation. These data point to a CD62L-independent mode of action of sulfatides in Th cell signaling (Ding et al., 1997, 2000). Because CD62L-mediated signals enhance CXCR4 expression and function (Ding et al., 2003; Duchesneau et al., 2007), Th cell proliferation was tested with the specific inhibition of CXCR4. Again, blocking of this chemoattractant receptor CXCR4 did not rescue proliferation of Th cells upon stimulation with sulfatides. Finally, blocking of another sulfatide receptor, galectin-4 (Delacour et al., 2005; Ideo et al., 2007), resulted in a diminished inhibitory effect of sulfatides on Th cell proliferation. Galectin-4 belongs to a family of carbohydrate-binding proteins that are able to target branch-end epitopes of glycan chains and regulate diverse processes, such as cell adhesion, differentiation, growth, and migration (Rabinovich and Croci, 2012). Thus, galectin-4 has been found to be highly expressed in the mammalian gastrointestinal tract where its main function involves binding to clustered N-glycans of distinct glycoproteins enabling maintenance of epithelial polarity (Delacour et al., 2005; Stechly et al., 2009). More recently, galectin-4 has been found to be a critical component in the membrane of neurons (Storan et al., 2004; Velasco et al., 2013). In particular, galectin-4 was found to regulate neuronal control over oligodendrocyte maturation and myelination (Wei et al., 2007; Stancic et al., 2012). Galectin-4 expression on Th cells has not been reported. However, in this study, we found that naive Th cells expressed significantly more mRNA for galectin-4 than memory Th cells. Thus, interaction between sulfatides and galectin-4 may explain the particular susceptibility of naive Th cells to sulfatide-induced inhibition of proliferation. Further studies are warranted to understand more the function of galectin-4 in Th cell activity.
Regarding the effect of sulfatides on TCR signaling in Th cells, we showed that early TCR signaling was unaffected, as demonstrated by the normal upregulation of surface activation markers, CD69 and CD25. However, upregulation of myc gene expression, resulting from full activation of the TCR immunoreceptor tyrosine-based activation motifs (Guy et al., 2013), was inhibited by sulfatide stimulation. These data suggest selective modulation of TCR activation as a mechanism of sulfatide action. Interestingly, Th17 differentiation was particularly affected after sulfatide stimulation, as seen by downregulation of the Th17 marker genes, Rora and Il17a, and by inhibition in vitro of the development of IL-17A-producing Th cells. Furthermore, sulfatides induced features of Th cell anergy. These two effects of sulfatides might be linked because induction of Egr-2, a transcription factor essential for the induction of T cell anergy, has also been reported to regulate Th17 development negatively (Iwasaki et al., 2013; Miao et al., 2013). Because CD1d knock-out mice showed an identical course of EAE after MOG35–55 immunization to wild-type mice (Jahng et al., 2001; Singh et al., 2001; Mars et al., 2008) and EAE is primarily driven by Th17 (Bettelli et al., 2008; Ghoreschi et al., 2011), our results showing EAE protection with sulfatides further supports our data in vitro on the impact of sulfatides on Th cell differentiation. However, as we used T cells preincubated with sulfatides before further EAE experimentations, we are unable to conclude how sulfatides might affect fully differentiated encephalitogenic effector cells operating during the peak of the disease.
Sulfatides constitute a major component of myelin lipids within the CNS. Based on our results, we propose that, under normal conditions, sulfatides present on the surface of the intact myelin sheath serve as inhibitory signals for T cell activation and thus prevent the potential induction of autoimmunity within the CNS. In this way, sulfatides might contribute to the phenomenon of immunologic privilege in the brain. However, under conditions when CNS blood–brain damage occurs resulting in myelin destruction and loss of sulfatide, the above mechanisms might become impaired and significant and may lead or contribute to the spectrum of neuroimmunologic events traditionally considered to be related to myelin protein antigens. These findings have potential novel implications of therapeutic relevance to MS.
Footnotes
This work was supported in part by National Science Centre Poland Grant PRELUDIUM 2011/01/N/NZ4/04962 to B.S., Polish-Swiss Research Programme Grant PSPB-007/2010 to M.P.M., National Center for Research and Development Grant ERA.NET-RUS/04/2012 to M.P.M., National Science Centre Poland Grant MAESTRO to K.W.S., National Science Centre Poland Grant PRELUDIUM to M.C., and National Multiple Sclerosis Society Grant RG 1001-K-11 to C.S.R. We thank Patricia Cobban-Bond for preparation of the manuscript.
The authors declare no competing financial interests.
References
- Adams EJ, Luoma AM. The adaptable major histocompatibility complex (MHC) fold: structure and function of nonclassical and MHC class I-like molecules. Annu Rev Immunol. 2013;31:529–561. doi: 10.1146/annurev-immunol-032712-095912. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Yurlova L, Simons M. Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol. 2011;21:585–593. doi: 10.1016/j.tcb.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio A, Binczek E, Stoffel W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc Natl Acad Sci U S A. 1996;93:13280–13285. doi: 10.1073/pnas.93.23.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridger GJ, Skerlj RT, Thornton D, Padmanabhan S, Martellucci SA, Henson GW, Abrams MJ, Yamamoto N, De Vreese K, Pauwels R. Synthesis and structure-activity relationships of phenylenebis(methylene)-linked bis-tetraazamacrocycles that inhibit HIV replication: effects of macrocyclic ring size and substituents on the aromatic linker. J Med Chem. 1995;38:366–378. doi: 10.1021/jm00002a019. [DOI] [PubMed] [Google Scholar]
- Chappert P, Schwartz RH. Induction of T cell anergy: integration of environmental cues and infectious tolerance. Curr Opin Immunol. 2010;22:552–559. doi: 10.1016/j.coi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/S0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Cuzner ML, Davison AN. Changes in cerebral lysosomal enzyme activity and lipids in multiple sclerosis. J Neurol Sci. 1973;19:29–36. doi: 10.1016/0022-510X(73)90053-1. [DOI] [PubMed] [Google Scholar]
- Delacour D, Gouyer V, Zanetta JP, Drobecq H, Leteurtre E, Grard G, Moreau-Hannedouche O, Maes E, Pons A, André S, Le Bivic A, Gabius HJ, Manninen A, Simons K, Huet G. Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. J Cell Biol. 2005;169:491–501. doi: 10.1083/jcb.200407073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Kawashima H, Suzuki Y, Suzuki T, Ward PA, Miyasaka M. A sulfatide receptor distinct from L-selectin is involved in lymphocyte activation. FEBS Lett. 1997;418:310–314. doi: 10.1016/S0014-5793(97)01400-2. [DOI] [PubMed] [Google Scholar]
- Ding Z, Kawashima H, Miyasaka M. Sulfatide binding and activation of leukocytes through an L-selectin-independent pathway. J Leukoc Biol. 2000;68:65–72. doi: 10.1189/jlb.1938-3673. [DOI] [PubMed] [Google Scholar]
- Ding Z, Issekutz TB, Downey GP, Waddell TK. L-selectin stimulation enhances functional expression of surface CXCR4 in lymphocytes: implications for cellular activation during adhesion and migration. Blood. 2003;101:4245–4252. doi: 10.1182/blood-2002-06-1782. [DOI] [PubMed] [Google Scholar]
- Duchesneau P, Gallagher E, Walcheck B, Waddell TK. Up-regulation of leukocyte CXCR4 expression by sulfatide: an L-selectin-dependent pathway on CD4+ T cells. Eur J Immunol. 2007;37:2949–2960. doi: 10.1002/eji.200737118. [DOI] [PubMed] [Google Scholar]
- Gallatin WM, Weissman IL, Butcher EC. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature. 1983;304:30–34. doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Hirahara K, O'Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011;32:395–401. doi: 10.1016/j.it.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi E, Maricic I, Wang J, Mac TT, Iyer P, Kumar V, Zajonc DM. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat Immunol. 2012;13:851–856. doi: 10.1038/ni.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, Xie J, Dempsey PJ, Crawford HC, Aifantis I, Davis MM, Vignali DA. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. 2013;14:262–270. doi: 10.1038/ni.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–262. doi: 10.1016/S0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- Ho PP, et al. Identification of naturally occurring fatty acids of the myelin sheath that resolve neuroinflammation. Sci Transl Med. 2012;4:137ra173. doi: 10.1126/scitranslmed.3003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideo H, Seko A, Yamashita K. Recognition mechanism of galectin-4 for cholesterol 3-sulfate. J Biol Chem. 2007;282:21081–21089. doi: 10.1074/jbc.M703770200. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Fujio K, Okamura T, Yanai A, Sumitomo S, Shoda H, Tamura T, Yoshida H, Charnay P, Yamamoto K. Egr-2 transcription factor is required for Blimp-1-mediated IL-10 production in IL-27-stimulated CD4(+) T cells. Eur J Immunol. 2013;43:1063–1073. doi: 10.1002/eji.201242942. [DOI] [PubMed] [Google Scholar]
- Jahng AW, Maricic I, Pedersen B, Burdin N, Naidenko O, Kronenberg M, Koezuka Y, Kumar V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1789–1799. doi: 10.1084/jem.194.12.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. 2004;199:947–957. doi: 10.1084/jem.20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol. 2008;181:8077–8087. doi: 10.4049/jimmunol.181.11.8077. [DOI] [PubMed] [Google Scholar]
- Kanamori M, Tasumi Y, Iyoda T, Ushida M, Inaba K. Sulfatide inhibits alpha-galactosylceramide presentation by dendritic cells. Int Immunol. 2012;24:129–136. doi: 10.1093/intimm/dxr108. [DOI] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Konno A, Nunogami K, Wada T, Yachie A, Suzuki Y, Takahashi N, Suzuki T, Miyamoto D, Kiso M, Hasegawa A, Miyawaki T. Inhibitory action of sulfatide, a putative ligand for L-selectin, on B cell proliferation and Ig production. Int Immunol. 1996;8:1905–1913. doi: 10.1093/intimm/8.12.1905. [DOI] [PubMed] [Google Scholar]
- Laudanna C, Constantin G, Baron P, Scarpini E, Scarlato G, Cabrini G, Dechecchi C, Rossi F, Cassatella MA, Berton G. Sulfatides trigger increase of cytosolic free calcium and enhanced expression of tumor necrosis factor-alpha and interleukin-8 mRNA in human neutrophils: evidence for a role of L-selectin as a signaling molecule. J Biol Chem. 1994;269:4021–4026. [PubMed] [Google Scholar]
- Mars LT, Gautron AS, Novak J, Beaudoin L, Diana J, Liblau RS, Lehuen A. Invariant NKT cells regulate experimental autoimmune encephalomyelitis and infiltrate the central nervous system in a CD1d-independent manner. J Immunol. 2008;181:2321–2329. doi: 10.4049/jimmunol.181.4.2321. [DOI] [PubMed] [Google Scholar]
- Merten M, Beythien C, Gutensohn K, Kühnl P, Meinertz T, Thiagarajan P. Sulfatides activate platelets through P-selectin and enhance platelet and platelet-leukocyte aggregation. Arterioscler Thromb Vasc Biol. 2005;25:258–263. doi: 10.1161/01.ATV.0000149675.83552.83. [DOI] [PubMed] [Google Scholar]
- Miao T, Raymond M, Bhullar P, Ghaffari E, Symonds AL, Meier UC, Giovannoni G, Li S, Wang P. Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J Immunol. 2013;190:58–65. doi: 10.4049/jimmunol.1200868. [DOI] [PubMed] [Google Scholar]
- Mycko MP, Ferrero I, Wilson A, Jiang W, Bianchi T, Trumpp A, MacDonald HR. Selective requirement for c-Myc at an early stage of V(alpha)14i NKT cell development. J Immunol. 2009;182:4641–4648. doi: 10.4049/jimmunol.0803394. [DOI] [PubMed] [Google Scholar]
- Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122:1180–1188. doi: 10.1172/JCI58649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa D, Shikata K, Honke K, Sato S, Matsuda M, Nagase R, Tone A, Okada S, Usui H, Wada J, Miyasaka M, Kawashima H, Suzuki Y, Suzuki T, Taniguchi N, Hirahara Y, Tadano-Aritomi K, Ishizuka I, Tedder TF, Makino H. Cerebroside sulfotransferase deficiency ameliorates L-selectin-dependent monocyte infiltration in the kidney after ureteral obstruction. J Biol Chem. 2004;279:2085–2090. doi: 10.1074/jbc.M305809200. [DOI] [PubMed] [Google Scholar]
- Patel O, Pellicci DG, Gras S, Sandoval-Romero ML, Uldrich AP, Mallevaey T, Clarke AJ, Le Nours J, Theodossis A, Cardell SL, Gapin L, Godfrey DI, Rossjohn J. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat Immunol. 2012;13:857–863. doi: 10.1038/ni.2372. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity. 2012;36:322–335. doi: 10.1016/j.immuni.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy KC, Maricic I, Khurana A, Smith TR, Halder RC, Kumar V. Involvement of secretory and endosomal compartments in presentation of an exogenous self-glycolipid to type II NKT cells. J Immunol. 2008;180:2942–2950. doi: 10.4049/jimmunol.180.5.2942. [DOI] [PubMed] [Google Scholar]
- Singh AK, Wilson MT, Hong S, Olivares-Villagómez D, Du C, Stanic AK, Joyce S, Sriram S, Koezuka Y, Van Kaer L. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801–1811. doi: 10.1084/jem.194.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancic M, Slijepcevic D, Nomden A, Vos MJ, de Jonge JC, Sikkema AH, Gabius HJ, Hoekstra D, Baron W. Galectin-4, a novel neuronal regulator of myelination. Glia. 2012;60:919–935. doi: 10.1002/glia.22324. [DOI] [PubMed] [Google Scholar]
- Stechly L, Morelle W, Dessein AF, André S, Grard G, Trinel D, Dejonghe MJ, Leteurtre E, Drobecq H, Trugnan G, Gabius HJ, Huet G. Galectin-4-regulated delivery of glycoproteins to the brush border membrane of enterocyte-like cells. Traffic. 2009;10:438–450. doi: 10.1111/j.1600-0854.2009.00882.x. [DOI] [PubMed] [Google Scholar]
- Storan MJ, Magnaldo T, Biol-N′Garagba MC, Zick Y, Key B. Expression and putative role of lactoseries carbohydrates present on NCAM in the rat primary olfactory pathway. J Comp Neurol. 2004;475:289–302. doi: 10.1002/cne.20167. [DOI] [PubMed] [Google Scholar]
- Sud'ina GF, Brock TG, Pushkareva MA, Galkina SI, Turutin DV, Peters-Golden M, Ullrich V. Sulphatides trigger polymorphonuclear granulocyte spreading on collagen-coated surfaces and inhibit subsequent activation of 5-lipoxygenase. Biochem J. 2001;359:621–629. doi: 10.1042/0264-6021:3590621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kamoshita S, Eto Y, Tourtellotte WW, Gonatas JO. Myelin in multiple sclerosis: composition of myelin from normal-appearing white matter. Arch Neurol. 1973;28:293–297. doi: 10.1001/archneur.1973.00490230029002. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Toda Y, Tamatani T, Watanabe T, Suzuki T, Nakao T, Murase K, Kiso M, Hasegawa A, Tadano-Aritomi K. Sulfated glycolipids are ligands for a lymphocyte homing receptor, L-selectin (LECAM-1), binding epitope in sulfated sugar chain. Biochem Biophys Res Commun. 1993;190:426–434. doi: 10.1006/bbrc.1993.1065. [DOI] [PubMed] [Google Scholar]
- Turutin DV, Kubareva EA, Pushkareva MA, Ullrich V, Sud'ina GF. Activation of NF-kappa B transcription factor in human neutrophils by sulphatides and L-selectin cross-linking. FEBS Lett. 2003;536:241–245. doi: 10.1016/S0014-5793(03)00061-9. [DOI] [PubMed] [Google Scholar]
- Velasco S, Díez-Revuelta N, Hernández-Iglesias T, Kaltner H, André S, Gabius HJ, Abad-Rodríguez J. Neuronal Galectin-4 is required for axon growth and for the organization of axonal membrane L1 delivery and clustering. J Neurochem. 2013;125:49–62. doi: 10.1111/jnc.12148. [DOI] [PubMed] [Google Scholar]
- Wei Q, Eviatar-Ribak T, Miskimins WK, Miskimins R. Galectin-4 is involved in p27-mediated activation of the myelin basic protein promoter. J Neurochem. 2007;101:1214–1223. doi: 10.1111/j.1471-4159.2007.04488.x. [DOI] [PubMed] [Google Scholar]
- Wilson R, Tocher DR. Lipid and fatty acid composition is altered in plaque tissue from multiple sclerosis brain compared with normal brain white matter. Lipids. 1991;26:9–15. doi: 10.1007/BF02544017. [DOI] [PubMed] [Google Scholar]
- Woelk H, Borri P. Lipid and fatty acid composition of myelin purified from normal and MS brains. Eur Neurol. 1973;10:250–260. doi: 10.1159/000114281. [DOI] [PubMed] [Google Scholar]
- Yahara S, Kawamura N, Kishimoto Y, Saida T, Tourtellotte WW. A change in the cerebrosides and sulfatides in a demyelinating nervous system: development of the methodology and study of multiple sclerosis and Wallerian degeneration. J Neurol Sci. 1982;54:303–315. doi: 10.1016/0022-510X(82)90191-5. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kubo H, Kobayashi S, Ishizawa K, He M, Suzuki T, Fujino N, Kunishima H, Hatta M, Nishimaki K, Aoyagi T, Tokuda K, Kitagawa M, Yano H, Tamamura H, Fujii N, Kaku M. The increase in surface CXCR4 expression on lung extravascular neutrophils and its effects on neutrophils during endotoxin-induced lung injury. Cell Mol Immunol. 2011;8:305–314. doi: 10.1038/cmi.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajonc DM, Maricic I, Wu D, Halder R, Roy K, Wong CH, Kumar V, Wilson IA. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J Exp Med. 2005;202:1517–1526. doi: 10.1084/jem.20051625. [DOI] [PMC free article] [PubMed] [Google Scholar]