

Abstract
Background and Purpose
Inhibition of brain Na+-K+-Cl− cotransporter 1 (NKCC1) with bumetanide (BMT) is of interest in ischemic stroke therapy. However, its poor brain penetration limits the application. In this study, we investigated the efficacy of two novel NKCC1 inhibitors, a lipophilic BMT prodrug STS5 and a novel NKCC1 inhibitor STS66, on reducing ischemic brain injury.
Methods
Large-vessel transient ischemic stroke in normotensive C57BL/6J mice was induced with 50-min occlusion of the middle cerebral artery (t-MCAO) and reperfusion. Focal, permanent ischemic stroke in angiotensinII (AngII)-induced hypertensive C57BL/6J mice was induced by permanent occlusion of distal branches of MCA (pd-MCAO). A total of 206 mice were randomly assigned to receive vehicle DMSO, BMT, STS5 or STS66.
Results
Post-stroke BMT, STS5 or STS66 treatment significantly decreased infarct volume and cerebral swelling by ~40–50% in normotensive mice after t-MCAO but STS66-treated mice displayed better survival and sensorimotor functional recovery. STS5 treatment increased the mortality. AngII-induced hypertensive mice exhibited increased phosphorylatory activation of SPAK and NKCC1 as well as worsened infarct and neurological deficit after pd-MCAO.
Conclusions
The novel NKCC1 inhibitor STS66 is superior to BMT and STS5 in reducing ischemic infarction, swelling, and neurological deficits in large-vessel transient ischemic stroke as well as in permanent focal ischemic stroke with hypertension comorbidity.
Keywords: angiotensinII, bumetanide, bumetanide prodrug STS5, STS66, hypertension
Subject Terms: Basic Science Research, Ischemic Stroke, Hypertension
Graphical Abstract
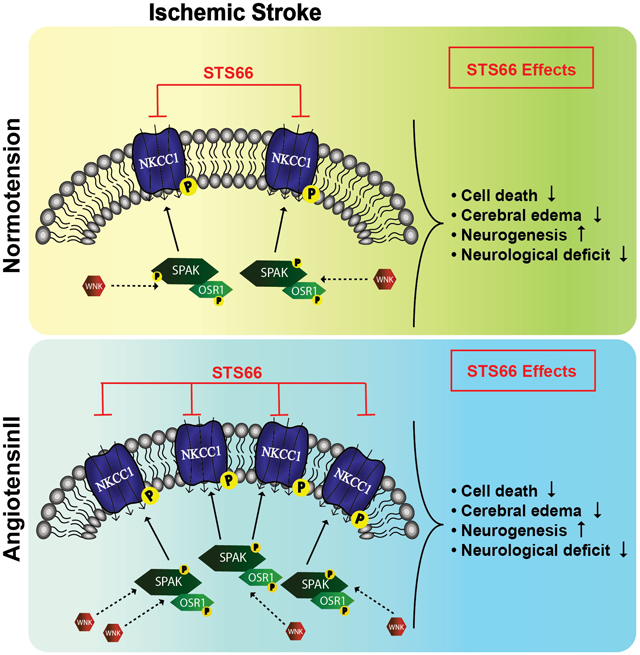
Ischemic stroke triggers moderate activation of the WNK-SPAK/OSR1 signaling pathway and expression of phosphorylated NKCC1 (pNKCCI) in normotensive brains. Stroke with hypertension comorbidity further stimulates the WNK-SPAK/OSR1-NKCC1 cascade, which contributes to worsened cerebral swelling, infarction and neurgoloifcal function deficits. Post-stroke administration of the novel NKCC1 inhibitor STS66 blocks NKCC1 protein function, reduces brain injury, and improves sensorimotor function recovery.
INTRODUCTION
Plasmalemmal electroneutral NKCC are targets of the “loop” diuretic drugs furosemide (NKCC2 inhibitor) and bumetanide (NKCC1 and NKCC2 inhibitor).1 The ion transporters are involved in several brain disorders including ischemic stroke, epilepsy, traumatic brain injury, and autism.2 The underlying mechanisms are not completely understood, but are in part associated with altered cellular Na+ and Cl− homeostasis.3 Stimulation of NKCC1 activity is involved in ischemic cell damage through NKCC1-mediated Na+ and Cl− overload, cytotoxic edema, and excitotoxicity.2 Human ischemic stroke brains display upregulated total and phosphorylated NKCC1 protein expression in the ischemic penumbra, contributing to ischemic neuronal damage.4
Furosemide and bumetanide (BMT) have been explored as novel treatment options for brain disorders.2 However, BMT is heavily bound to plasma proteins (~98%) and highly ionized at physiological pH, resulting in poor penetration into the brain.3 In addition, chronic administration of BMT has potent diuretic effects.3 To overcome these problems, Erker et al. developed a lipophilic and uncharged prodrug of BMT, STS5,5 and a novel NKCC1 inhibitor STS66, which penetrate the blood-brain barrier (BBB) more easily than BMT.3 STS5 reduces seizure development in rodents via reducing intracellular Cl− concentration.5 However, it remains unknown whether STS5 and STS66 are better choices than BMT to reduce ischemic brain edema and injury after ischemic stroke.
In this study, we tested BMT, STS5, and STS66 in two stroke models, in normotensive mice with t-MCAO-mediated large vessel transient ischemic stroke and in the AngII-infused hypertensive mice with focal stroke by pd-MCAO. Hypertension is a leading risk factor for ischemic stroke, and the renin-angiotensin system plays a causal role in the pathogenesis of hypertension and stroke.6 We report here that STS66 is superior to BMT and STS5 in reducing ischemic brain injury in both models. AngII-infused mice exhibit upregulated expression of NKCC1 and its regulatory kinases after stroke.
MATERIAL AND METHODS
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion (TOP). The data that support the findings of this study are available within the article.
Animals and ischemic stroke models
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The manuscript adheres to the ARRIVE guidelines for reporting animal experiments. Transient-MCAO for 50-min was induced in C57BL/6J normotensive mice (2–3 months, male and female). Saline control or AngII-induced hypertensive C57BL/6J mice received infusion of either saline or AngII via osmotic minipumps at a rate of 1000 ng/kg/min for 14 days. Permanent focal cerebral ischemia by pd-MCAO was subsequently induced in either Saline control or AngII-infused mice as described in the Online Supplement. No experiments using 50 min of t-MCAO protocol were conducted in the saline or AngII-induced hypertensive mice because the latter group exhibited severe infarction, hemispheric swelling, and high mortality, which prevented us from assessing neurological function deficit and recovery in the t-MCAO model.
Drug development and treatment
Syntheses of STS5 and STS66 were described in the Online-only Data Supplement. DMSO (2 ml/kg body weight/day), BMT (10 mg/kg body weight/day), STS5 (13 mg/kg body weight/day) or STS66 (12 mg/kg body weight/day) were administered (i.p.). Due to the short half-life of the drugs (~ 4 hr), the initial half dose of the drugs (BMT, STS5, STS66) was given at 3 hr and the second half dose at 8 hr post MCAO, and twice daily for 1–5 days.
Neurological function tests
Neurological score, adhesive tape removal test, cylinder test, and foot-fault test were assessed in a blinded manner as described in the Online-only Data Supplement.
Immunoblotting and immunofluorescent staining
Immunoblotting for NKCC1, SPAK (Ste20-related proline alanine-rich kinase) and OSR1 (oxidative stress-responsive kinase-1) and immunofluorescent staining for MAP2 and NeuN were performed as described in the Online Supplement.
Statistics and inclusion and exclusion criteria
Animal subjects were randomly assigned into different study groups, and data analyses were performed by investigators blinded to experimental conditions. Statistical significance was determined by Student’s t-test, One- or Two-way ANOVA using the Tukey’s post-hoc test for multiple comparisons (GraphPad Prism 7.0, San Diego, CA, USA). A p-value <0.05 was considered statistically significant. The number of animals studied was 80% powered to detect 30% changes with α (2-sided) = 0.05 (Supplemental Table I). Inclusion and exclusion criteria were described in the online Supplement.
RESULTS
Following the t-MCAO with large-vessel ischemic stroke, the Veh-control mice exhibited large infarction and hemispheric edema (Figure 1A-B). BMT, STS5, and STS66 (given at equimolar dose of 10 mg/kg BMT) were equally effective in reducing infarction (p < 0.05, Figure 1B). At a reduced dose (equimolar dose of 2 mg/kg BMT), BMT or STS5 administration showed no effects (Figure 1C). But, STS66-treated mice exhibited significantly reduced infarct volume and hemispheric swelling. In assessing their impact on post-stroke recovery, all drug-treated mice showed similar body weight recovery (Figure 2A). About 30% of the Veh-control animals died, compared to low mortality in the BMT-treated group (~16.7%), and high mortality in the STS5-treated group (~ 66.7%). But, zero mortality was detected in the STS66-treated group (Figure 2A). No animals died in the reduced drug dose treatments within 24 hr after t-MCAO. The better outcome of the STS66-treated mice was further illustrated by significant improvement in the neurological scoring test, the foot-fault test, and the adhesive contact test (Figure 2B-C). The Veh-control mice did not show post-stroke neurological function recovery. Sham-operated C57BL/6j mice exhibited no mortality and minimum neurological deficit, or body weight loss.
Figure 1. Efficacy of novel NKCC1 inhibitors in reducing ischemic infarction and cerebral edema in mice after t-MCAO.

(A) Experimental protocol in normotensive mice and representative TTC staining images. (B) Infarct and swelling. Data are mean ± SD, n = 6-15 (male, n = 3-9; female, n = 3-6), * p < 0.05. (C) Mice treated with drugs at an equimolar dose of 2 mg/kg BMT. Data are mean ± SD, n = 5-9 (male, n = 3-5; female, n = 2-4).
Figure 2. Efficacy of novel NKCC1 inhibitors on post-stroke survival and neurological function in mice after t-MCAO.

(A) Changes of body weight and survival. (B) Neurological score, corner test, foot-fault test. (C) Adhesive removal and contact tests. Data are mean ± SD, n = 5-9 (male, n = 3-6; female, n = 2-3).
The AngII-infused mice (Supplemental Figure I) exhibited focal infarct and swelling at 24 hr after pd-MCAO (Figure 3A). The BMT treatment did not reduce infarction or swelling. But the STS66-treated mice showed significantly smaller infarct and less brain edema (Figure 3A). Pd-MCAO triggered nearly zero mortality in the Veh-, BMT- or STS66-treated group (Supplemental Figure IIC). However, STS5-treated mice exhibited ~20% mortality and severe body weight loss, likely due to non-neural factors (Supplemental Figure IIC). Moreover, the STS66-treated mice displayed significantly better neurological function than the other three groups (Supplemental Figure IID). Assessing the peri-lesion areas (penumbra) in the Veh-control mice revealed significantly decreased MAP2 intensity (Figure 3B) than in the BMT- or STS66-treated mice. The STS66-treated mice displayed ~ 40% higher number of NeuN+ cells than the BMT-treated mice (Figure 3B, and Supplemental Figure III C-D).
Figure 3. Efficacy of novel NKCC1 inhibitors against permanent focal ischemic brain injury with hypertension comorbidity.

(A) Infarct and swelling. Data are mean ± SD. n = 4-8. (B) Representative NeuN, MAP2, and To-pro-3 staining in cortical peri-lesion areas from AngII Veh-, AngII BMT-, and AngII STS66-treated mice after 24 hr pd-MCAO. (C) Representative immunoblots and quantification. Data are mean ± SEM, n = 6.
AngII-infused mouse brains showed higher level of pNKCC1 (pThr206) in the non-ischemic hemisphere tissues (Figure 3C). Ischemic stroke further increased pSPAK (pSer383) and pOSR1 (pSer325), tSPAK in the Veh-, BMT-, and STS66-treated brains (Figure 3C). STS66 treatment completely blocked the ischemia-induced elevation of pNKCC1 (Figure 3C).
DISCUSSION
We report here that the novel NKCC1 inhibitor STS66 is superior to BMT in reducing ischemic infarction, neuronal death, cerebral swelling, and neurological deficits in large vessel transient ischemic stroke as well as in permanent focal ischemic stroke with hypertension comorbidity. Most importantly, AngII infusion stimulated the brain WNK-SPAK-NKCC1 signaling pathway and exacerbated somatosensory and motor cortex infarction and sensory-motor deficits after pd-MCAO. AngII infusion has been reported to increase BP via phosphorylating SPAK and NKCC1 in the mouse aorta,7 but, our study provides the first evidence that the WNK-SPAK-NKCC1 cascade is stimulated in ischemic brains in the AngII-induced hypertensive mice. Future experiments validating a role of brain WNK-SPAK-NKCC1 cascade in the AngII-mediated exacerbation of ischemic brain damage using the large vessel t-MCAO model would further strengthen our view that the brain WNK-SPAK-NKCC1 complex presents a therapeutic target for focal ischemic stroke with comorbid hypertension.
Supplementary Material
Acknowledgments
Funding: This study was supported by NIH Grant R01 NS38118 (DS), NIH R01 NS043277 (DS, BJM), Biogen, supporting patient enrollment for CHARM trial (BJM), and Ministry of Science and Technology (MOST:106–2314-B-016–043-MY3), Taiwan (SSY).
Footnotes
U.S. Provisional Application No. 62/653,708, entitled “Bumetanide derivatives for the therapy of stroke and other neurological diseases/disorders involving NKCCs”, filed April 6, 2018
Disclosures: Dr. Molyneaux has received research support and consulting honoraria from Biogen and is a member of the CHARM trial scientific advisory board for Biogen. The other authors report no conflicts.
Contributor Information
Huachen Huang, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA; Department of Neurology, First affiliate Hospital, Harbin Medical University, Harbin, Heilongjiang, China.
Mohammad Iqbal H. Bhuiyan, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Tong Jiang, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Shanshan Song, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Sandhya Shankar, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Taraneh Taheri, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Eric Li, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Philipp Schreppel, Department of Pharmaceutical Chemistry, University of Vienna, Vienna, Austria.
Michael Hintersteininger, Department of Pharmaceutical Chemistry, University of Vienna, Vienna, Austria.
Sung-Sen Yang, Division of Nephrology, Department of Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan.
Shih-Hua Lin, Division of Nephrology, Department of Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan.
Bradley J. Molyneaux, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
Zhongling Zhang, Department of Neurology, First affiliate Hospital, Harbin Medical University, Harbin, Heilongjiang, China.
Thomas Erker, Department of Pharmaceutical Chemistry, University of Vienna, Vienna, Austria.
Dandan Sun, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA; Pittsburgh Institute for Neurodegenerative Disorders, University of Pittsburgh, Pittsburgh, PA, USA.
REFERENCES
- 1.Markadieu N, Delpire E. Physiology and pathophysiology of slc12a1/2 transporters. Pflugers Archiv : European journal of physiology. 2014;466:91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang H, Song S, Banerjee S, Jiang T, Zhang J, Kahle KT, et al. The wnk-spak/osr1 kinases and the cation-chloride cotransporters as therapeutic targets for neurological diseases. Aging and disease. 2019;10:00–00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tollner K, Brandt C, Topfer M, Brunhofer G, Erker T, Gabriel M, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014;75:550–562 [DOI] [PubMed] [Google Scholar]
- 4.Bhuiyan MIH, Song S, Yuan H, Begum G, Kofler J, Kahle KT, et al. Wnk-cab39-nkcc1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. J Cereb Blood Flow Metab. 2017;37:2780–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erker T, Brandt C, Tollner K, Schreppel P, Twele F, Schidlitzki A, et al. The bumetanide prodrug bum5, but not bumetanide, potentiates the antiseizure effect of phenobarbital in adult epileptic mice. Epilepsia. 2016;57:698–705 [DOI] [PubMed] [Google Scholar]
- 6.Unger T The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol. 2002;89:3A–9A [DOI] [PubMed] [Google Scholar]
- 7.Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, et al. Dietary salt intake regulates wnk3-spak-nkcc1 phosphorylation cascade in mouse aorta through angiotensin ii. Hypertension. 2013;62:872–878 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.