

CATACOMB is a catalytic antagonist PRC2 subunit whose expression is induced upon DNA demethylation.
Abstract
Using biochemical characterization of fusion proteins associated with endometrial stromal sarcoma, we identified JAZF1 as a new subunit of the NuA4 acetyltransferase complex and CXORF67 as a subunit of the Polycomb Repressive Complex 2 (PRC2). Since CXORF67’s interaction with PRC2 leads to decreased PRC2-dependent H3K27me2/3 deposition, we propose a new name for this gene: CATACOMB (catalytic antagonist of Polycomb; official gene name: EZHIP). We map CATACOMB’s inhibitory function to a short highly conserved region and identify a single methionine residue essential for diminution of H3K27me2/3 levels. Remarkably, the amino acid sequence surrounding this critical methionine resembles the oncogenic histone H3 Lys27-to-methionine (H3K27M) mutation found in high-grade pediatric gliomas. As CATACOMB expression is regulated through DNA methylation/demethylation, we propose CATACOMB as the potential interlocutor between DNA methylation and PRC2 activity. We raise the possibility that similar regulatory mechanisms could exist for other methyltransferase complexes such as Trithorax/COMPASS.
INTRODUCTION
Polycomb group (PcG) proteins are highly conserved chromatin regulatory factors that play fundamental roles in several physiological processes attributed to their ability to repress transcription (1). Alterations affecting PcG proteins, including point mutations, overexpression, gene amplifications, and deletions, have been characterized as drivers of specific cancer subtypes (2). Among the catalog of PcG genetic alterations, chromosomal translocations resulting in chimeric fusion proteins are the least well understood. The first reported chromosomal translocation involving a PcG gene was a recurrent t(7;17)(p15;q21) that fuses the N-terminal region of the JAZF1 zinc finger protein to the Polycomb repressive complex 2 (PRC2) subunit SUZ12 (3). This chromosomal translocation is found specifically in endometrial stromal sarcomas (ESSs), and the resulting fusion protein retains the ability to form a complex with other core PRC2 subunits such as EZH2 and EED (4).
After identification of the JAZF1-SUZ12 chromosomal translocation, many other PcG fusions were uncovered in the ESS (5). It is now evident that most of the ESS-associated translocations involve genes belonging to the NuA4 acetyltransferase complex and PcG proteins (mainly PRC2 subunits). For instance, t(6;10)(p21;p11), t(1;6)(p34;p21), and t(5;6)(q31;p21) found in ESS specimens create the EPC1-PHF1, MEAF6-PHF1, and BRD8-PHF1 fusion proteins, respectively. EPC1, MEAF6, and BRD8 are all subunits of the NuA4 complex, whereas PHF1 is a subunit of PRC2 (2, 6). As for the aforementioned JAZF1-SUZ12 fusion, many ESS chromosomal translocations contain JAZF1 as the 5′ fusion partner of a PcG gene, such as t(6;7)(p21;p15) fusing JAZF1 and PHF1, and t(7; X)(p15; q26.1) fusing JAZF1 and BCORL1 (7). Recently, a chromosomal translocation t(X; 17)(p11.2; q21.33) between the NuA4 subunit gene MBTD1 and the uncharacterized gene CXORF67 was reported (8). In low-grade ESS, JAZF1 and CXORF67 are the only two reported genes involved in chromosomal translocations whose protein products have not been fully biochemically characterized.
While investigating the biochemical properties of the JAZF1 and CXORF67 ESS-associated fusions, we identified JAZF1 as a new subunit of the NuA4 acetyltransferase complex. In addition, we found that both CXORF67 and the MBTD1-CXORF67 fusion protein interact with PRC2. This further reinforces the notion that ESS-associated fusion proteins are characterized by the presence of an N-terminal NuA4 component and a C-terminal PcG subunit. We found that CXORF67 and the MBTD1-CXORF67 fusion protein were able to strongly decrease the catalytic products of PRC2, namely, H3K27me2/3. Because of this property, we have given this gene the name CATACOMB (catalytic antagonist of Polycomb). The CATACOMB gene is poorly conserved across metazoans; however, careful analysis of the 3′ portion contained in the MBTD1-CATACOMB fusion revealed a short stretch of amino acids that is highly conserved among eutherian mammals. This region contains a methionine flanked by amino acid sequences that resemble the H3K27M PRC2 inhibitory mutant histone, suggesting that this sequence may mediate CATACOMB suppression of PRC2 activity. Remarkably, a single amino acid substitution of this methionine in CATACOMB completely abolishes its ability to reduce H3K27me2/3 levels but does not disrupt its interaction with PRC2. We also found that CATACOMB expression is silenced through DNA methylation, and upon treatment with DNA demethylating agents, CATACOMB is expressed, binds to PRC2, and inhibits its H3K27 methyltransferase activity. Our data support an unexpected H3K27M-like activity for the inducible CATACOMB in the regulation of PRC2 catalytic activity.
RESULTS
JAZF1 is a new NuA4-interacting protein
To investigate the biochemical characteristics of the JAZF1-SUZ12 fusion protein, we isolated primary human endometrial stromal cells (hEnSCs) from a normal uterus (9). Using lentivirus-mediated transduction, we expressed a hemagglutinin (HA)–FLAG–tagged version of the JAZF1-SUZ12 fusion or JAZF1 full-length protein in primary hEnSC at low passages (fig. S1A). Mass spectrometry analysis of proteins associated with the JAZF1-SUZ12 fusion identified several known subunits of both the NuA4 and PRC2 complexes (Fig. 1A). In contrast, JAZF1 full length associates with NuA4 subunits but not PRC2, thus demonstrating that JAZF1 mediates interaction with NuA4 (Fig. 1A). As an additional control, we performed immunoprecipitation (IP) of SUZ12 full length, which does not interact with NuA4, but does, as expected, coimmunoprecipitate PRC2 subunits (Fig. 1A). Our mass spectrometry data did not detect EZH2, the main catalytic subunit of PRC2, in association with JAZF1-SUZ12; however, Western blot analyses were able to confirm this expected interaction (fig. S1B and table S1). We also validated the interaction between JAZF1 or JAZF1-SUZ12 fusion protein and several members of the NuA4 complex by Western blotting (Fig. 1B). We were able to demonstrate that JAZF1-SUZ12 mediates an interaction between the PRC2 and NuA4 complexes, as demonstrated by the interaction between the PRC2 subunit EZH2 and the NuA4 subunit TRRAP, specifically in the JAZF1-SUZ12–expressing cells (fig. S1C).
Fig. 1. JAZF1 and CATACOMB are a NuA4 and a PRC2 subunit, respectively.
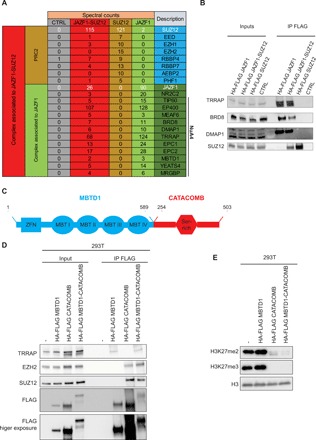
(A) Schematic representation of mass spectrometry data obtained from FLAG IPs from hEnSC wild type (WT) (CTRL), expressing the HA-FLAG versions of the JAZF1-SUZ12 fusion protein, SUZ12, or JAZF1. Numbers represent spectral counts. The far-right columns list the protein identification symbols. JAZF1 and SUZ12 (or JAZF1-SUZ12 in the fusion-expressing cells) are shown in white type face, as are the numbers for their spectral counts obtained in each experiment. The previously identified subunits for NuA4 are indicated. (B) FLAG-IP in HCT-116 cells that are either WT or express HA-FLAG versions of the JAZF1-SUZ12 fusion protein, full-length SUZ12, or full-length JAZF1. Ten percent of the input and 50% of the IP material were loaded for Western blot analysis. Antibodies used for Western blotting are indicated in the panel. (C) Schematic representation of the MBTD1-CATACOMB putative fusion protein including the domain previously reported. Numbers in black represent the amino acid residues. The MBTD1 portion is in blue, and the CATACOMB portion is in red. (D) FLAG-IP in 293T cells WT (−), expressing HA-FLAG versions of the MBTD1-CATACOMB putative fusion protein, full-length MBTD1, or full-length CATACOMB. Ten percent of the input and 50% of the IP material were loaded for Western blot analysis. Antibodies used for Western blotting are indicated in the panel. (E) Western blotting of chromatin extracts from cells described in (D). Antibodies used for Western blotting are indicated in the panel. Histone H3 (H3) served as a loading control.
CATACOMB decreases H3K27me2/3 levels through a highly conserved sequence
We next investigated the biochemical properties of the MBTD1-CATACOMB fusion protein. It was recently reported that in glioma cells, CATACOMB interacts with PRC2, and its overexpression or deletion can decrease or increase, respectively, the H3K27me3 levels in the cells (10). The MBTD1-CATACOMB fusion protein contains the majority of the MBTD1 protein sequence (lacking only the final 39 residues) fused to the C-terminal half of CATACOMB (amino acid 254 to 503) (Fig. 1C) (8). As the JAZF1-SUZ12 fusion retains the ability to be incorporated into PRC2 through the SUZ12 moiety, we hypothesized that the putative fusion protein MBTD1-CATACOMB might have the same ability through the CATACOMB portion. Since there is no biological source available to obtain the MBTD1-CATACOMB complementary DNA (cDNA), we generated a cDNA based on the predicted fusion sequence (8). We then generated cells stably expressing an HA-FLAG–tagged version of MBTD1, CATACOMB, or the fusion MBTD1-CATACOMB. IP of these proteins shows that the MBTD1-CATACOMB fusion protein is able to interact with EZH2 (Fig. 1D). The putative fusion MBTD1-CATACOMB is also able to interact with subunits of the NuA4 complex, further suggesting the propensity of the ESS chromosomal translocation to create an aberrant NuA4-PRC2 supercomplex.
The MBTD1-CATACOMB putative fusion protein retains the ability to decrease the H3K27me2 and H3K27me3 levels similar to CATACOMB, while MBTD1 overexpression has a minor effect on the levels of these two histone marks (Fig. 1E). These results indicate that the C-terminal region of CATACOMB contained in the MBTD1-CATACOMB putative fusion is most likely responsible for binding to PRC2 and for the decrease of H3K27me2/3 levels. To investigate this hypothesis, we looked into possible protein domains that might be functionally important for CATACOMB molecular activity. The only previously described domain for CATACOMB is the serine-rich domain that is also retained in the MBTD1-CATACOMB putative fusion protein (Fig. 1C) (8). Despite minimal overall conservation of the CATACOMB gene (10), a small region that is C-terminal to the serine-rich domain is well conserved across eutherian mammals (Fig. 2A and fig. S2, A and B) (11).
Fig. 2. A conserved region in CATACOMB is responsible for H3K27me2/3 global reduction.
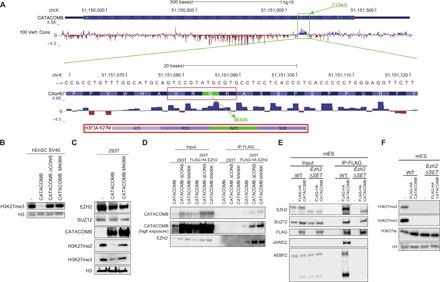
(A) Schematic representation of the CATACOMB gene locus with PhyloP-generated scores in hg19 genome assembly. Negative scores are represented by red tracks for predicted acceleration, and positive scores are represented by blue tracks, for predicted conservation in 100 vertebrates (Vert.) (species list can be found in the University of California Santa Cruz genome browser). Top: The green box indicates the highly conserved region named CONS. Bottom: A zoom-in of the CONS region at DNA and amino acid levels. The red box indicates the four amino acid residues, including M406, that resemble the sequence surrounding the H3K27M mutant histone, which is shown below for comparison. (B) Western blotting of total cell extracts from SV40 immortalized hEnSC WT (−) expressing CATACOMB, CATACOMB without the CONS domain (ΔCONS), and CATACOMB containing a methionine-to-lysine mutation in position 406 (M406K). Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control. (C) Western blotting of total cell extract from 293T cells (−) or 293T cells expressing CATACOMB or the CATACOMB M406K mutant. Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control. (D) FLAG-IP in 293T cells that are WT or express FLAG-HA EZH2. In both cells lines, we expressed untagged CATACOMB, CATACOMBΔ CONS, or CATACOMB M406K. Ten percent of the input and 50% of the IP material were loaded for Western blot analysis. Antibodies used for Western blotting are indicated in the panel. (E) FLAG-IP in mESCs that are either WT or Ezh2ΔSET. In both cells lines, we expressed FLAG-HA CATACOMB. Cells with no FLAG-HA CATACOMB expression were used as negative control (−). Five percent of the input and 30% of the IP material were loaded for Western blot analysis. Antibodies used for Western blotting are indicated in the panel. (F) Western blotting of chromatin extracts from cells described in (E). Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control.
To test whether the conserved C-terminal region mediates the effect of CATACOMB on H3K27me3 levels, we generated cells expressing a mutant lacking the conserved region from residue Pro399 to Phe418 (ΔCONS). These cells show H3K27me3 levels comparable to those observed in wild-type cells, while full-length CATACOMB-expressing cells show very low levels of H3K27me3 (Fig. 2B). These results strongly suggest that this conserved region is crucial for CATACOMB’s ability to reduce global H3K27me3. A closer examination of the amino acid sequence of the conserved region revealed a methionine residue (M406) within an amino acid sequence that resembles the residues around the Met27 in the H3K27M histone mutant (Fig. 2A). Since expression of H3K27M also globally reduces H3K27me2/3 levels (12), we hypothesized that Met406 in CATACOMB might have a similar role. Generation of cells expressing a mutant CATACOMB with M406 substituted with lysine (M406K) shows similar H3K27me2/3 levels to those observed in either wild-type cells or cells expressing CATACOMB ΔCONS (Fig. 2, B and C). Similar to the lysine-to-isoleucine substitution at the H3K27 (H3K27I) that could inhibit PRC2 activity similarly to H3K27M (13), CATCOMB M406I is able to decrease H3K27me3 levels when expressed in 293T cells (fig. S3A). However, unexpectedly, an alanine substitution at the 406 residue of CATACOMB (M406A) retains the ability to reduce H3K27me3 levels similarly to wild-type CATACOMB or CATACOMB M406I (fig. S3A). This differs from the H3K27A histone mutant, which does not reduce H3K27me2/3 levels in cells (13). These results demonstrate that the conserved M406 in the human CATACOMB protein is a key residue that mediates the global reduction of cellular H3K27me2/3 levels.
CATACOMB and its C-terminal region included in the MBTD1-CATACOMB putative fusion protein are able to interact with PRC2 (Fig. 1D). To understand whether and how this biochemical interaction could be responsible for CATACOMB’s ability to reduce the global levels of H3K27me2/3, we performed co-IP between EZH2 and the mutant CATACOMB versions described above. Both the M406K and the ΔCONS mutants were able to interact with EZH2, indicating that neither M406 nor the conserved domain is essential per se for the interaction between CATACOMB and PRC2 (Fig. 2D). Further exploring what part of CATACOMB is required for the binding to PRC2, we generated two CATACOMB mutants lacking either portion of the protein between the fusion breakpoint and the conserved domain (PRE CONS) or the part after the conserved domain (POST CONS) (fig. S3B). Both the mutants retain both EZH2-binding property and the ability to reduce H3K27me2/3 levels (fig. S3, C and D). Therefore, while the conserved domain and M406 are sufficient for reducing H3K27me2/3 levels, they are dispensable per se for CATACOMB binding to the PRC2. Therefore, our data, using different mutants, suggest multiple CATACOMB-PRC2–binding sites. To further interrogate the mechanism of CATACOMB interaction, we tested its interaction with a mutant PRC2 complex. We expressed Flag-tagged CATACOMB in wild-type mouse embryonic stem cells (mESCs) or ESCs with a homozygous deletion of the Ezh2 SET methyltransferase domain (Ezh2ΔSET) (Fig. 2, E and F). As in our previous experiments, CATACOMB expression in mESCs is associated with a marked reduction in H3K27me2/3 levels (Fig. 2F). Co-IP experiments revealed that CATACOMB is able to bind both the wild-type Ezh2 containing PRC2 and Ezh2ΔSET PRC2 (Fig. 2E). We also noticed that in these cells, CATACOMB can also interact in an EZH2 SET domain–dependent fashion with JARID2 and AEBP2, two PRC2 accessory subunits, while EZH2 binding to JARID2 is independent of its SET domain (Fig. 2E and data not shown) (2). This indicates that the CATACOMB binding to the core subunits EZH2 and SUZ12 does not require the EZH2 SET domain, which is instead required for CATACOMB binding to the PRC2 accessory subunits JARID2 and AEBP2.
The CATACOMB gene is a DNA demethylation–inducible PRC2 subunit
Since the first composition of PRC2 was identified, many subunits have been reported to be previously unidentified stoichiometric or substoichiometric components of the complex (2, 14). AU022751, the mouse homolog of CATACOMB, was initially found as an EZH1 (EZH2 ortholog)–interacting protein in mESCs, although the interaction was not further investigated (15). In that study, they used mESCs grown in serum-containing media; however, the expression of Au022751 is undetectable in mESCs grown in serum-free 2i media (fig. S4A) (16). Since mESCs grown in the presence of serum are more prone to spontaneously differentiate, to a degree that depends on several factors (17), it is plausible that AU022751 is expressed only in a subset of the spontaneously differentiated cells. This would reduce the chance of AU022751 being identified as a PRC2-interacting protein and would also explain why numerous other PRC2 biochemical studies in mESCs failed to report this interaction. PRC2 has been purified from several human cell lines, yet CATACOMB was never reported as a PRC2-interacting protein (18) until the very recent study that purified PRC2 subunits from U2OS and DAOY cancer cell lines (10). This is likely due to the lack of CATACOMB expression in most of the cell lines that were used to purify PRC2 subunits in the earlier studies. Following this hypothesis, we examined CATACOMB levels in the Cancer Cell Line Encyclopedia (CCLE) and found that the vast majority of cell lines have low or undetectable levels of CATACOMB, while U2OS and DAOY are the two cell lines with the highest expression (Fig. 3A) (19).
Fig. 3. CATACOMB expression and interaction with PRC2 complex are induced by DNA demethylation.
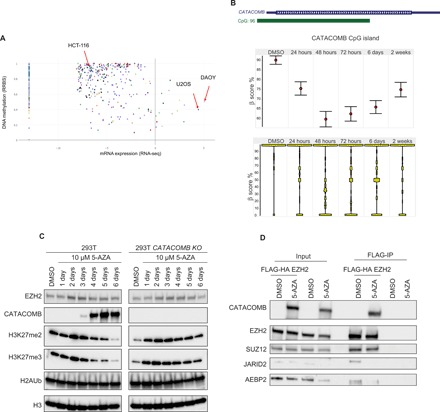
(A) CCLE-generated scatterplot to quantify CATACOMB expression and CATACOMB locus methylation levels using 826 different cell lines, each represented by a dot. The x axis represents mRNA expression levels (cells with values below 0 have no or negligible expression), and the y axis represents DNA methylation levels (0 to 1 values range from unmethylated to fully methylated). (B) Top: Schematic representation of the CATACOMB locus with the associated CpG island from the hg19 genome assembly. Middle: Star Wars plots of CG methylation status of the CATACOMB-associated CpG island during a time course with 10 μM 5-AZA treatment in 293T cells. Dimethyl sulfoxide (DMSO) (vehicle control) and the time points of treatment are indicated above. The red circles indicate the mean levels of CG methylation for each time point. Error bars represent SEs. Bottom: Same as in (B) showing total distribution of CG methylation at each time point. (C) Western blotting of total cell extracts from a time course treatment in 293T and 293T CATACOMB KO cells after a single administration of 10 μM 5-AZA. Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control. (D). FLAG-IP in 293T cells that are WT or express FLAG-HA EZH2. Both cell lines were treated with either 10 μM 5-AZA or DMSO (vehicle control) for a week. In both cells lines, we expressed FLAG-HA CATACOMB. Ten percent of the input and 50% of the IP material were loaded for Western blot analysis. Antibodies used for Western blotting are indicated in the panel.
In addition to gene expression data, the CCLE also contains information about the DNA methylation status of genes. The CATACOMB locus exhibits high levels of DNA methylation in most of the cell lines, possibly explaining why this gene is not more broadly expressed (Fig. 3A). Notably, this locus contains a large CpG island that spans more than half of the gene (Fig. 3B). In 293T cells, this CpG island is heavily methylated (β score, ~0.9), and the methylation levels can be strongly reduced by a single treatment with the demethylating agent 5-azacytidine (5-AZA) (Fig. 3B). CATACOMB CpG island methylation decreased for 2 to 3 days before beginning to recover, likely through replication-dependent dilution of the 5-AZA (Fig. 3B). The loss of CATACOMB CpG island methylation induced by the 5-AZA treatment anticorrelates with CATACOMB protein levels, suggesting that this gene might be activated by DNA demethylation (Fig. 3, B and C). Global H3K27me2/3 levels anticorrelate with CATACOMB levels during the 6-day time course following a single dose of 5-AZA treatment (Fig. 3C). Using isogenic 293T CATACOMB knockout (KO), we were able to largely rescue the decline of H3K27me2/3 levels (Fig. 3C). This indicates that the drop in the levels of these PRC2-deposited histone marks upon 5-AZA treatment is mainly due to CATACOMB induction. Global EZH2 levels did not show any major change besides a slight increase between 2 and 3 days after treatment; similarly, the PRC1 catalytic product is also largely unaffected by the treatment with 5-AZA (Fig. 3C).
To extend our observation to other cell lines with similarly high levels of DNA methylation at the CATACOMB locus and negligible or no CATACOMB expression, we treated HCT-116 colorectal cancer cells with 5-AZA (Fig. 3A). Unexpectedly, no CATACOMB protein could be detected following a single dose of 5-AZA treatment in HCT-116 cells, while the same treatment in 293T cells resulted in substantial CATACOMB protein levels (fig. S4B). However, despite a similar reduction in the DNA methylation at the CATACOMB locus in both HCT-116 and 293T cells after 24 hours of treatment with 5-AZA, the DNA methylation levels at that specific locus recover much faster in HCT-116 compared with 293T cells (Fig. 3B and fig. S4C). To understand whether failure of HCT-116 to express CATACOMB might be due to insufficient demethylation of the CATACOMB locus, we performed a 4-day treatment with 5-aza-2′-deoxycytidine (D-AZA) administered every day. This treatment regimen led to a transcriptional expression of CATACOMB after 4 days (fig. S4D). Analysis of the reads obtained in the RNA sequencing (RNA-seq) experiments revealed a frameshift mutation at the 300th amino acid residue of CATACOMB in HCT-116 cells, which is consistent with previous studies (fig. S4E) (19). Therefore, we conclude that the lack of sufficient demethylation and/or the presence of a frameshift mutation in the CATACOMB gene in HCT-116 cells might explain the absence of the CATACOMB protein upon single treatment with 5-AZA.
To confirm that the 5-AZA inducible CATACOMB can be incorporated into PRC2, IP with FLAG-EZH2 was performed in 293T cells treated with 5-AZA. We found that the endogenous inducible CATACOMB is efficiently incorporated in PRC2 (Fig. 3D). The interaction between EZH2 and the accessory subunits AEBP2 and JARID2 is strongly inhibited by the 5-AZA treatment. Together, these data indicate that CATACOMB is a DNA demethylation–inducible subunit of PRC2.
CATACOMB and PRC2 catalytic activity are required for cellular proliferation
To further investigate the role of CATACOMB in cells in which it is normally expressed, we turned to U2OS cells. The endogenous binding between CATACOMB and PRC2 subunits in U2OS was reported in a prior study (10). To estimate the fraction of PRC2 interacting with CATACOMB, we performed size exclusion chromatography with U2OS nuclear extracts. We found very similar elution profiles of the core PRC2 subunits and CATACOMB, suggesting that a substantial fraction of CATACOMB is incorporated into PRC2 (fig. S5A). Since we were unable to detect any H3K27me3 as determined by Western blotting (Fig. 4A) (20), we therefore asked whether KO of CATACOMB in U2OS cells could lead to increased H3K27me3 levels or whether it would cause any phenotypic change in the cells. Using CRISPR-Cas9 technology, we generated two CATACOMB KO U2OS clones that show statistically significant difference in the proliferation rate compared with the wild-type U2OS parental cell line (Fig. 4A). The H3K27me2/3 levels that are undetectable in wild-type U2OS can be detected in the CATACOMB KO U2OS clones (Fig. 4A), further supporting that CATACOMB functions in reducing H3K27me2/3 levels. It is also worth mentioning that while the proliferation rate difference is quite modest for clone #1 compared with the parental cells, this difference is more pronounced for clone #2, which shows even higher H3K27me3 and lower H3K27me2 levels than those in clone #1 (Fig. 4A).
Fig. 4. CATACOMB and PRC2 catalytic activity are required for U2OS proliferation.
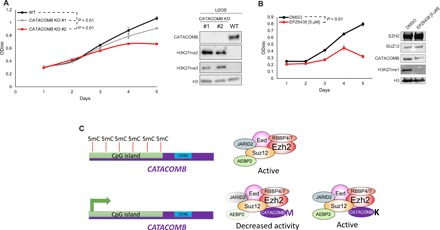
(A) Left: Growth curve comparison between U2OS WT parental cells and U2OS CATACOMB KO clones. Absorbance of crystal violet in 10% acetic acid was used to represent the cell number and plotted as optical density (OD) (λ = 590). n = 3. Error bars represent SDs. Right: Western blotting of total cell extracts from cells described in the left part of the panel. Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control. P values were calculated using the areas under the curves. (B) Left: Growth curve comparison between U2OS cells treated with 5 μM EPZ6438 (EZH2 inhibitor) or DMSO (vehicle control). Cells were pretreated for 6 days, every other day for 6 days, and kept on the same regimen during the 5 days that the proliferation was assessed. Absorbance of crystal violet in 10% acetic acid was used to represent the cell number and plotted as OD (λ = 590). n = 3. Error bars represent SDs. Right: Western blotting of total cell extracts from cells shown in the left panel. Antibodies used for Western blotting are indicated in the panel. H3 served as a loading control. (C) Working model proposed by our study. CATACOMB is kept silenced by DNA methylation; once demethylation occurs, the gene is activated, the protein product binds to the PRC2, and through the conserved M406 residue decreases PRC2 activity. Single methionine-to-lysine mutation (M406K) abolishes CATACOMB inhibitory activity toward PRC2 without disrupting the binding between CATACOMB and PRC2.
Although H3K27me3 is undetectable in wild-type U2OS cells by Western blotting, H3K27me3 can be detected by chromatin IP followed by sequencing (ChIP-seq) (21). This closely resembles the molecular features of the H3K27M-positive midline glioma cells that have undetectable levels of H3K27me3 as determined by Western blotting or immunohistochemistry, but appreciable H3K27me3 can be identified by ChIP-seq (22–24). In addition, depleting CATACOMB from U2OS or depleting H3K27M from midline glioma cells is correlated with increased H3K27me2/3 levels (Fig. 4A and fig. S4B) (25). Although this might be interpreted as a tumor-suppressive role for PRC2 catalytic activity in U2OS cells, just as in midline glioma cells, treatment with an EZH2 catalytic inhibitor has a significant negative effect on cell proliferation (Fig. 4B). Other studies using an RNA interference approach against PRC2 subunits in U2OS cells also found that PRC2 was required for cell proliferation in U2OS cells (26, 27). To demonstrate effective target inhibition of PRC2 by EPZ6438 in U2OS cells, we probed H3K27me1 levels, the only PRC2-implemented histone modification we were able to detect by Western blot analysis in U2OS wild-type cells (28). Treatment with the EZH2 catalytic inhibitor led to a strong decrease in H3K27me1 levels in U2OS cells compared with those in DMSO-treated cells (Fig. 4B).
It is important to underline that the proliferation defect induced by EPZ6438 is independent of the transcriptional activation of the CDKN2A locus, a well-known PRC2 target in several cell types (fig. S5C). This is in complete agreement with previous studies, and it is consistent with the fact that the CDKN2A locus is methylated at the DNA level in U2OS cells (29). This further supports the idea that PRC2-mediated proliferation control is largely CDKN2A independent (22, 30). Together, these results point toward oncogenic and tumor-suppressive functions for PRC2 catalytic activity in CATACOMB-expressing cells. These dual functions might be explained by aberrant genomic distribution of PRC2 and its activity that, at the same time, restrains transcription of tumor suppressor genes (e.g., pro-differentiation genes, cell cycle inhibitors) and releases repression of proto-oncogenes (e.g., stem cell–specific transcription factors).
DISCUSSION
Increasing evidence points toward chromatin dysfunction as a hallmark of cancer (31) with a large number of chromatin modifier genes mutated across multiple cancer subtypes (32). ESSs have a particular recurrent genetic alteration that involves chromosomal translocations fusing several genes encoding subunits of the NuA4 acetyltransferase complex to the PRC2 methyltransferase complex. The most common translocation found in ESSs is the JAZF1-SUZ12 fusion (5). Despite its frequent translocation in ESS, the molecular function of JAZF1 was unknown prior to this study. We identify JAZF1 as a bona fide NuA4-interacting protein and also shows that the JAZF1-SUZ12 fusion protein retains the ability to interact with both NuA4 and PRC2 acting de facto as a molecular bridge between the two complexes (Fig. 1, A and B, and fig. S1C).
In this study, we also identified interesting biochemical properties of CATACOMB, the C-terminal partner of the putative fusion protein MBTD1-CATACOMB (8). While MBTD1 is a known subunit of NuA4, CATACOMB was only recently identified as a PRC2-interacting protein (10, 33). We demonstrate that the putative MBTD1-CATACOMB binds both the TRRAP (NuA4 subunit) and PRC2 core subunits (EZH2 and SUZ12) and that the CATACOMB moiety, but not MBTD1, is required for binding to PRC2. Furthermore, we prove that the MBTD1-CATACOMB putative fusion is able to induce global reduction of the H3K27me2/3 levels in cells (Fig. 1E). Further investigation into the 3′ half of CATACOMB contained in the chromosomal translocation unexpectedly revealed a conserved sequence that is essential for the ability of CATACOMB to suppress cellular H3K27me2/3 levels (Fig. 2, A and B, and fig. S2, A and B). A methionine residue (M406) contained in this conserved region is crucial for this activity (Fig. 2C). Notably, the surrounding region of the M406 is similar to the sequence surrounding M27 in the H3K27M histone mutant that can also cause global reduction of the global H3K27me2/3 levels (Fig. 2A) (34). It is although worth recognizing that substitution of the CATACOMB M406 with other hydrophobic amino acids such as isoleucine or alanine does not alter CATACOMB’s ability to reduce H3K27me2/3 levels (fig. S3A). However, only the lysine-to-methionine or lysine-to-isoleucine H3K27 histone mutants, but not any other substitution to other hydrophobic amino acids (such as alanine), are able to reduce H3K27me2/3 levels (13). This suggests structural differences in the inhibitory regions of these two proteins.
We demonstrated that CATACOMB binding to PRC2 is not mediated by the inhibitory M406 residue or the immediately surrounding conserved region. It is also important to reiterate that the SET domain of EZH2, essential for H3K27me3 catalysis, is not essential for the binding to CATACOMB (Fig. 2, D to F). Similarly, PRC2 binding to H3K27M-containing nucleosomes is not dominated by the M27 residue but rather is mediated by the DNA, although, as in CATACOMB, the methionine residue is definitely required for the inhibitory effect on the PRC2 (34, 35).
Histone H3K27M and CATACOMB expression are mutually exclusive in a subgroup of posterior fossa ependymomas, and a subset of ependymomas were previously reported to be characterized by very low H3K27me3 levels (10, 36). This reinforces the idea that H3K27M and CATACOMB have similar molecular and pathological functions. However, while the H3K27M presence correlates with a poorer prognosis in midline gliomas (37), there is currently no evidence that CATACOMB expression is a negative prognostic factor in ependymomas. Nonetheless, previous studies implicated a worse overall survival in patients with ependymoma bearing tumors with low H3K27me3 levels and also in patients belonging to the molecular group A (high levels of CATACOMB) compared with patients belonging to the group B (low or absent levels of CATACOMB) (10, 36, 38). Given the high expression levels of CATACOMB in group A posterior ependymoma fossa and the ability to reduce global H3K27me3 levels, it is reasonable to anticipate its potential role as a negative prognostic factor in these tumors. The two known ESSs bearing MBTD1-CATACOMB chromosomal translocation were classified as FIGO (International Federation of Gynecology and Obstetrics) stage III (10). This is an advanced stage for low-grade ESS (7), hypothetically supporting a correlation between advanced-stage low-grade ESS and the presence of this specific chromosomal translocation; however, the very small number of cases precludes any solid conclusion.
The architecture of the CATACOMB gene locus is quite remarkable. A large CpG island represents more than half of the gene and is generally methylated in cell lines (Fig. 3A). We show that treatment with a demethylating agent can activate the expression of this gene and, at least in one cell line, lead to protein accumulation (Fig. 3, B and C). The reduction of the H3K27me2/3 levels upon 5-AZA treatment is largely dependent on CATACOMB induction, which is efficiently incorporated into PRC2. CATACOMB, to the best of our knowledge, is the only DNA demethylation–inducible subunit incorporated into a chromatin modifier multiprotein complex (Figs. 3D and 4C).
The inducibility of CATACOMB might have important functions during development and implications for oncogenesis. An interesting but largely speculative possibility at the moment could be that the large CpG island in the CATACOMB locus works as a sensor of global CpG methylation levels, whereby global reductions in DNA methylation lead to induction of CATACOMB transcription, with the resulting protein attenuating PRC2 activity to prevent aberrant H3K27me2/3 deposition at newly DNA-demethylated CpG islands, the classic targeting sequence for PRC2 (39). Several lines of evidence already support this idea, including the expression of CATACOMB in globally DNA-hypomethylated tumors (36) and the mislocalization of PRC2 and H3K27me3 in DNA methylation–deficient cells (40). It is also worth noting that H3K27M histone mutant–positive high-grade gliomas are also characterized by global DNA hypomethylation (41). Therefore, it would not be unexpected if H3K27M and CATACOMB play the same pathological role in hypomethylated tumors.
In accordance with the same line of thought, it is worth noticing that CATACOMB is also highly expressed in placental trophoblasts (42). This is not completely unexpected since CATACOMB is a eutherian-specific gene; therefore, it seems plausible that it might play a role during placenta development. While after the first wave of global demethylation the inner cell mass cells establish de novo methylation, the DNA in the trophectoderm-derived trophoblasts remain hypomethylated, potentially explaining the high expression of CATACOMB in these specific cells (43).
While our manuscript was under review, another study reached similar conclusions about the role of CXORF67 (CATACOMB) as an endogenous analog of the H3K27M histone mutant and named the gene EZHIP, which is now the official gene symbol (44). In this study, the authors also showed the direct PRC2 inhibitory ability of CATACOMB by performing a PRC2 methyltransferase in vitro assay in the presence of a peptide generated from the conserved region of CATACOMB.
In conclusion, it is important to underline the contribution of chromatin pathway deregulation in cancer and how much can be learned from seemingly peculiar alterations that occur in very rare tumors such as ESS. These results can be applied to the biology of other tumors or even to normal biological processes and can help move forward our understanding of how these large multiprotein chromatin modifier complexes work in cellular and developmental contexts. Our findings in this study demonstrate that an endogenous inducible gene, CATACOMB, once expressed, can regulate the catalytic activity of PRC2 and opens the possibility that this mechanism of regulation of histone modifications through the expression of antagonistic subunits may also exist for Trithorax and other histone-modifying enzyme complexes.
MATERIALS AND METHODS
Cell lines, transfection, and viral transduction
293T, HCT-116, and U2OS were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Gaithersburg, MD) containing 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Gibco) in a CO2 incubator (5% CO2) with normal atmospheric oxygen tension (~21% O2). mESCs were grown in 2i medium as described in a previous study (16). Human primary endometrial stromal cells (hEnSCs) were derived and maintained in culture as previously described (9). Low-passage (up to 12) hEnSCs were used to perform the experiments described in this study. Transfections and viral transduction were performed using previously described methods (30). Specifically for hEnSC viral transduction, we used concentrated virus stocks. These were generated by ultracentrifugation at 25,000 rpm for 2.5 hours of supernatant containing virus using 32.4-ml OptiSeal tubes (Beckman Coulter) with the SW 32 Ti rotor (Beckman Coulter) and the Optima XPN-100 ultracentrifuge (Beckman Coulter). In this case, 2 × 106 hEnSCs were infected by adding lentivirus particles coming from the equivalent of two 10-cm dish (approximately 20 ml) supernatants resuspended in 10 to 20 μl of DMEM in hEnSC-specific medium (9). Polybrene was added as previously described (30). The multiplicity of infection for hEnSC used was ~1. All the cells transduced with lentivirus were positively selected using the appropriate antibiotic according to the resistance expressed by the lentiviral vector [puromycin (2 μg/ml), hygromycin (300 to 500 μg/ml), and neomycin (500 to 900 μg/ml)] until untransduced control cells treated with antibiotic were cleared from the plate. All the cells presented expressing exogenous proteins in this study were stably transduced unless indicated in the figure legend.
Vectors and cloning
JAZF1, JAZF1-SUZ12, and MBTD1 cDNA used in this study were cloned as gBlocks (IDT) using Gibson Assembly (E5510, New England BioLabs) into kanamycin-resistant pUC57 containing AttL sites for Gateway recombination. All the reactions were performed according to the manufacturer’s instructions. We used EF1α HA-FLAG PURO DEST (gift from D. Pasini) and pLenti CMV Hygro DEST (a gift from E. Campeau and P. Kaufman; #17454, Addgene) destination vectors. CATACOMB cDNA was generated as a polymerase chain reaction (PCR) product from pCMV SPORT6 CATACOMB (MHS6278-202759346, Dharmacon) and cloned into kanamycin-resistant pUC57 by Gibson Assembly. All mutations reported in this study were generated by PCR in the pUC57 CATACOMB plasmid. CATACOMB cDNA was then shuffled into destination vectors by Gateway recombination. The MBTD1-CATACOMB fusion gene was generated by digesting pUC57 CATACOMB plasmid with BmgBI restriction enzyme and inserting a MBTD1 cDNA gBlock (IDT) lacking the last 139 nucleotides by Gibson Assembly. We then removed unnecessary DNA sequences by PCR and obtained a MBTD1-CATACOMB cDNA as identified in two ESS samples (8). The resulting cDNA was shuffled in EF1α HA-FLAG PURO DEST by Gibson Assembly. All the vectors used in this study were Sanger sequence verified.
IP and mass spectrometry
Total cell pellets were incubated with nuclear prep [10 mM tris-HCl (pH 7.6), 100 mM NaCl, 2 mM MgCl2, 0.3 M sucrose, 0.25% NP-40] supplemented with protein inhibitor cocktail (Roche) for 10 min at 4°C. Nuclei were collected after brief centrifugation and lysed in S300 buffer [25 mM tris-HCl (pH 7.6), 300 mM NaCl, 10% glycerol, 0.25% NP-40] supplemented with a protease inhibitor cocktail (Roche). Samples were cleared from insoluble material by centrifugation at 20,000g for 15 min at 4°C. IP was performed using anti-FLAG agarose beads (M2, Sigma-Aldrich) with overnight rocking at 4°C.Beads were then washed five times using S300 buffer. Elution was performed using 3X FLAG peptide (200 μg/ml) (APExBIO) added to the elution buffer (tris-buffered saline 1X, 50 mM NaCl) for mass spectrometry or S300 for Western blot and incubated with rocking at 4°C for 30 min. Eluted proteins were mixed 1:1 with 2X Laemmli buffer for Western blot. Input (15%) was saved before FLAG-IP and mixed 1:1 with 2X Laemmli buffer for Western blot. For mass spectrometry, eluted proteins were precipitated O/N on ice with 60 mM tris-HCl (pH 8.5) and 20% trichloroacetic acid. Tubes containing precipitated proteins were centrifuged at 20,000g for 1 hour at 4°C.The supernatant was gently discarded, and the pellet was washed twice with ice-cold acetone. Any residual acetone was carefully removed, and the protein pellet was processed for mass spectrometry. Mass spectrometry analysis was performed as described in a previous study (45).
Western blot and antibodies
For total cell extracts, cell pellets were lysed using S300 buffer and incubated on ice for 5 min. After incubation, samples were sonicated for 5 min (30 s on +30 s off) using Bioruptor Pico (Diagenode) at 4°C. Total protein concentration of each sample was determined using Bradford reagent (Sigma-Aldrich) and then normalized across samples. Samples were then mixed 1:4 with 4X Laemmli and boiled for 5 min. For chromatin extract, cell pellets were incubated with S300 buffer for 5 min on ice and then centrifuged at 20,000g for 15 min at 4°C. The pellet obtained was washed once with buffer S300 and centrifuged at 20,000g for 15 min at 4°C. The pellet was then resuspended in 2X Laemmli, boiled for 10 min, then sonicated for 5 min (30 s on +30 s off) using Bioruptor Pico (Diagenode) at room temperature. The sample was then boiled again for 5 min. SDS–polyacrylamide gel electrophoresis and Western blotting were performed as previously described (30). Primary antibodies used were the as follows: 1:1000 anti-TRRAP antibody (EPR14952) (#ab183517, Abcam), 1:1000 anti-BRD8 antibody (#A300-219A-M, Bethyl), 1:1000 anti-DMAP1 (#PA1-886, Thermo Fisher Scientific), 1:1000 anti-SUZ12 (#sc-46264, Santa Cruz), 1:1000 anti-EZH2 (#5246, Cell Signaling Technology), 1:1000 anti-FLAG (#F3165, Sigma-Aldrich), 1:1000 anti-HA (HA-Tag #3724, Cell Signaling Technology), 1:1000 anti-H3K27me2 (#9728, Cell Signaling Technology), 1:1000 anti-H3K27me3 (#9733, Cell Signaling Technology), 1:1000 H3K27ac (#8173, Cell Signaling Technology), 1:1000 anti-CXORF67 (#HPA004003, Sigma-Aldrich), or affinity-purified anti-CATACOMB (1 μg/ml) (homemade rabbit polyclonal raised against a C-terminal region of the protein), 1:1000 anti-JARID2 (#13594, Cell Signaling Technology), 1:1000 anti-AEBP2 (#14129, Cell Signaling Technology), 1:1000 anti–β-tubulin (#sc-9104, Santa Cruz), 1:1000 H3K27me1 (#07-448, Sigma-Aldrich), and 1:1000 anti-H3 (#14269S, Cell Signaling Technology). Appropriate secondary antibodies according to primary hosts were used as previously described (30).
DNA methylation analysis
Modified reduced representation bisulfite sequencing (RRBS) was performed as previously described (46–48).
CRISPR-mediated gene editing
To generate U2OS and 293T CATACOMB KO cells, we used two guides (sequence 1: 5′-CACCGCAACCAGGGTCGACCCCAG-3′; sequence 2: 5′-CACCGACAGCAGGTTAAGTGCTAGG-3′); each of them was cloned in pX459 V2.0 (a gift from F. Zhang, Addgene plasmid #62988). Both plasmids were cotransfected in U2OS using Lipofectamine 3000 (Thermo Fisher Scientific) or using calcium phosphate in 293T. Cells were selected with puromycin (2 μg/ml) for 2 days and then switched to medium without antibiotic to avoid plasmid integration. Clones were then obtained by limited dilution single cell cloning. Clones were screened by PCR for CATACOMB locus deletion and then verified by Western blot analysis. To generate the mESC Ezh2ΔSET, we used two guides (sequence 1: 5′-CACCGGGGTCTAGTGATTCCCCTC-3′; sequence 2: 5′-CACCGTTTCACGTAAGTACTCCAG-3′); each of them was cloned in pX459 V2.0. Both plasmids were electoporated in mESCs. Cells were selected with puromycin (2 μg/ml) for 2 days and then switched to medium without antibiotic to avoid plasmid integration. Cells were then plated at low density, and single colonies were picked. Clones were screened by PCR for the Ezh2 SET domain deletion and then verified by Western blot.
Cell growth and treatments
293T and HCT-116 cells were treated with 10 μM 5-AZA (Sigma-Aldrich) dissolved in DMSO. Treatment was administered only once for each time course experiment performed in this study. HCT-116 was also treated for 4 days with daily administration of 10 μM 5-aza-2′-deoxycytidine. DMSO was used as vehicle control. U2OS cells were treated with 5 μM EZH2 inhibitor EPZ6438 (Selleckchem) dissolved in DMSO. EPZ6438 treatment was administered every other day for the entire duration of the experiment (6 days pretreatment +5 days growth curve). DMSO was used as vehicle control. Growth curves were performed as previously described (30).
Size exclusion chromatography
U2OS nuclear extracts were prepared as previously described (IP and mass spectrometry section). One hundred microliters of extract with a concentration of 10 μg/μl was fractioned by size exclusion chromatography over a Superose 6 PC 3.2/30. Fractions were then analyzed by Western blot.
Fluorescence-activated cell sorting analysis
U2OS nuclei were stained for 20 min using 1:100 Alexa Fluor 647–conjugated anti-H3K27me3 antibody in phosphate-buffered saline 0.1% bovine serum albumin and then washed twice with the same buffer before fluorescence-activated cell sorting (FACS) analysis. Unstained cells were used as negative control. Cells were analyzed by FACS as previously described (30).
Protein sequence analysis
Database (refseq_protein, 30 July 2018) searches were performed with PSI-BLAST (49). Representative sequences were aligned with MUSCLE (50) and displayed with JALVIEW (51). The phylogenetic relationship of species with CATACOMB homologs was estimated by TimeTree (52), with Tupaia glis representing the Tupaia genus in TimeTree. The tree was visualized at the iTOL server itol.embl.de (53). Marsupial and Monotreme genomes were also searched with tblastn using CATACOMB sequences from various eutherian clades to exclude the possibility that the failure to identify CATACOMB homologs outside eutheria with PSI-BLAST was due to lack of proper gene prediction.
RNA extraction and RNA-seq
RNA extraction and RNA-seq were performed as described in our previous study (22).
Supplementary Material
Acknowledgments
We thank all the members of Shilatifard’s laboratory for the very fruitful discussions, important inputs, and comments. We thank M. Dyson for crucial advice on primary endometrial stromal cell derivation and growth, S. Y. Kim for technical support, and B. Cenik for suggesting the name CATACOMB. We thank D. Pasini for sharing several plasmids. Funding: This work was supported by grants from the NIH, R00DC013805 to J.N.S., K08HL128867 to B.D.S., R50CA211428 to E.R.S., R37-HD38691 to S.E.B., and R35CA197569 to A.S. Ethics statement: Primary cells used in this study were part of a protocol approved by the Northwestern Institutional Review Board for Human Research (1375-005). Author contributions: A.P. designed the study, performed most of the experiments, and wrote the manuscript under the supervision of A.S. E.R.S. performed the mammalian conservation analysis and edited the manuscript. M.A.J.M. generated the mESC Ezh2ΔSET, performed the demethylation experiment with 5-aza-2′-deoxycytidine, and edited the manuscript. M.U. provided technical support with cell culture and CRISPR-Cas9 experiments. N.K. and J.N.S. performed and analyzed the mass spectrometry experiment. K.A.H. and B.D.S. performed and analyzed the DNA methylation assays. C.A.R. provided technical help with the FACS experiment. D.C.M., B.D.Y., and S.E.B. provided the human endometrial cells and technical help to grow them. R.A.R. provided crucial reagents for experiments and important discussions that led to the identification of the M406 in CATACOMB. E.J.R. provided technical help with the next-generation sequencing (NGS) experiments. All the authors commented on the manuscript and approve its content. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All the raw and processed data generated from NGS experiments are deposited in the National Center for Biotechnology Information GEO database under accession number GSE131626.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaax2887/DC1
Fig. S1. JAZF1-SUZ12 is a molecular bridge between NuA4 and PRC2.
Fig. S2. CATACOMB is a eutherian-specific gene.
Fig. S3. CATACOMB M406I/A and two deletion mutants retain the ability to reduce H3K27me2/3 levels in the cells.
Fig. S4. CATACOMB expression in different cells is achieved by different demethylating agent treatment regimen.
Fig. S5. H3K27me3 is present in U2OS and increases upon CATACOMB KO.
Table S1. FLAG-IP mass spectrometry results from hEnSC, expressing JAZF1-SUZ12 fusion, SUZ12 full length, or JAZF1 full length.
REFERENCES AND NOTES
- 1.Piunti A., Pasini D., Epigenetic factors in cancer development: Polycomb group proteins. Future Oncol. 7, 57–75 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Piunti A., Shilatifard A., Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Koontz J. I., Soreng A. L., Nucci M., Kuo F. C., Pauwels P., van den Berghe H., Cin P. D., Fletcher J. A., Sklar J., Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 6348–6353 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G., Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Hrzenjak A., JAZF1/SUZ12 gene fusion in endometrial stromal sarcomas. Orphanet J. Rare Dis. 11, 15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyon Y., Côté J., The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14, 147–154 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Tuyaerts S., Amant F., Endometrial stromal sarcomas: A revision of their potential as targets for immunotherapy. Vaccines 6, E56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewaele B., Przybyl J., Quattrone A., Finalet Ferreiro J., Vanspauwen V., Geerdens E., Gianfelici V., Kalender Z., Wozniak A., Moerman P., Sciot R., Croce S., Amant F., Vandenberghe P., Cools J., Debiec-Rychter M., Identification of a novel, recurrent MBTD1-CXorf67 fusion in low-grade endometrial stromal sarcoma. Int. J. Cancer 134, 1112–1122 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Dyson M. T., Roqueiro D., Monsivais D., Ercan C. M., Pavone M. E., Brooks D. C., Kakinuma T., Ono M., Jafari N., Dai Y., Bulun S. E., Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLOS Genet. 10, e1004158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pajtler K. W., Wen J., Sill M., Lin T., Orisme W., Tang B., Hübner J. M., Ramaswamy V., Jia S., Dalton J. D., Haupfear K., Rogers H. A., Punchihewa C., Lee R., Easton J., Wu G., Ritzmann T. A., Chapman R., Chavez L., Boop F. A., Klimo P., Sabin N. D., Ogg R., Mack S. C., Freibaum B. D., Kim H. J., Witt H., Jones D. T. W., Vo B., Gajjar A., Pounds S., Onar-Thomas A., Roussel M. F., Zhang J., Taylor J. P., Merchant T. E., Grundy R., Tatevossian R. G., Taylor M. D., Pfister S. M., Korshunov A., Kool M., Ellison D. W., Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollard K. S., Hubisz M. J., Rosenbloom K. R., Siepel A., Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammad F., Helin K., Oncohistones: Drivers of pediatric cancers. Genes Dev. 31, 2313–2324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D., Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuettengruber B., Bourbon H. M., Di Croce L., Cavalli G., Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Maier V. K., Feeney C. M., Taylor J. E., Creech A. L., Qiao J. W., Szanto A., Das P. P., Chevrier N., Cifuentes-Rojas C., Orkin S. H., Carr S. A., Jaffe J. D., Mertins P., Lee J. T., Functional proteomic analysis of repressive histone Methyltransferase complexes reveals ZNF518B as a G9A regulator. Mol. Cell. Proteomics 14, 1435–1446 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao K., Collings C. K., Morgan M. A., Marshall S. A., Rendleman E. J., Ozark P. A., Smith E. R., Shilatifard A., An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci. Adv. 4, eaap8747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamm C., Pijuan Galitó S., Annerén C., A comparative study of protocols for mouse embryonic stem cell culturing. PLOS ONE 8, e81156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauri S., Comoglio F., Seimiya M., Gerstung M., Glatter T., Hansen K., Aebersold R., Paro R., Gstaiger M., Beisel C., A high-density map for navigating the human Polycomb complexome. Cell Rep. 17, 583–595 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A., The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng H., Tillman H., Wu G., Davidoff A. M., Yang J., Frequent epigenetic alterations in polycomb repressive complex 2 in osteosarcoma cell lines. Oncotarget 9, 27087–27091 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easwaran H., Johnstone S. E., van Neste L., Ohm J., Mosbruger T., Wang Q., Aryee M. J., Joyce P., Ahuja N., Weisenberger D., Collisson E., Zhu J., Yegnasubramanian S., Matsui W., Baylin S. B., A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 22, 837–849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piunti A., Hashizume R., Morgan M. A., Bartom E. T., Horbinski C. M., Marshall S. A., Rendleman E. J., Ma Q., Takahashi Y.-h., Woodfin A. R., Misharin A. V., Abshiru N. A., Lulla R. R., Saratsis A. M., Kelleher N. L., James C. D., Shilatifard A., Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castel D., Philippe C., Calmon R., le Dret L., Truffaux N., Boddaert N., Pagès M., Taylor K. R., Saulnier P., Lacroix L., Mackay A., Jones C., Sainte-Rose C., Blauwblomme T., Andreiuolo F., Puget S., Grill J., Varlet P., Debily M.-A., Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130, 815–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashizume R., Andor N., Ihara Y., Lerner R., Gan H., Chen X., Fang D., Huang X., Tom M. W., Ngo V., Solomon D., Mueller S., Paris P. L., Zhang Z., Petritsch C., Gupta N., Waldman T. A., James C. D., Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 20, 1394–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silveira A. B., Kasper L. H., Fan Y., Jin H., Wu G., Shaw T. I., Zhu X., Larson J. D., Easton J., Shao Y., Yergeau D. A., Rosencrance C., Boggs K., Rusch M. C., Ding L., Zhang J., Finkelstein D., Noyes R. M., Russell B. L., Xu B., Broniscer A., Wetmore C., Pounds S. B., Ellison D. W., Zhang J., Baker S. J., H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 137, 637–655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun R., Shen J., Gao Y., Zhou Y., Yu Z., Hornicek F., Kan Q., Duan Z., Overexpression of EZH2 is associated with the poor prognosis in osteosarcoma and function analysis indicates a therapeutic potential. Oncotarget 7, 38333–38346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasini D., Bracken A. P., Jensen M. R., Lazzerini Denchi E., Helin K., Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23, 4061–4071 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrari K. J., Scelfo A., Jammula S. G., Cuomo A., Barozzi I., Stützer A., Fischle W., Bonaldi T., Pasini D., Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 29.McGarvey K. M., Greene E., Fahrner J. A., Jenuwein T., Baylin S. B., DNA methylation and complete transcriptional silencing of cancer genes persist after depletion of EZH2. Cancer Res. 67, 5097–5102 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Piunti A., Rossi A., Cerutti A., Albert M., Jammula S., Scelfo A., Cedrone L., Fragola G., Olsson L., Koseki H., Testa G., Casola S., Helin K., d’Adda di Fagagna F., Pasini D., Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 5, 3649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan M. A., Shilatifard A., Chromatin signatures of cancer. Genes Dev. 29, 238–249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feinberg A. P., Koldobskiy M. A., Göndör A., Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 17, 284–299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquet K., Fradet-Turcotte A., Avvakumov N., Lambert J.-P., Roques C., Pandita R. K., Paquet E., Herst P., Gingras A. C., Pandita T. K., Legube G., Doyon Y., Durocher D., Côté J., The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol. Cell 62, 409–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herz H.-M., Morgan M., Gao X., Jackson J., Rickels R., Swanson S. K., Florens L., Washburn M. P., Eissenberg J. C., Shilatifard A., Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X., Paucek R. D., Gooding A. R., Brown Z. Z., Ge E. J., Muir T. W., Cech T. R., Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 24, 1028–1038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayliss J., Mukherjee P., Lu C., Jain S. U., Chung C., Martinez D., Sabari B., Margol A. S., Panwalkar P., Parolia A., Pekmezci M., McEachin R. C., Cieslik M., Tamrazi B., Garcia B. A., la Rocca G., Santi M., Lewis P. W., Hawkins C., Melnick A., David Allis C., Thompson C. B., Chinnaiyan A. M., Judkins A. R., Venneti S., Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med. 8, 366ra161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu V. M., Alvi M. A., McDonald K. L., Daniels D. J., Impact of the H3K27M mutation on survival in pediatric high-grade glioma: A systematic review and meta-analysis. J. Neurosurg. Pediatr. 23, 308–316 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Pajtler K. W., Witt H., Sill M., Jones D. T. W., Hovestadt V., Kratochwil F., Wani K., Tatevossian R., Punchihewa C., Johann P., Reimand J., Warnatz H. J., Ryzhova M., Mack S., Ramaswamy V., Capper D., Schweizer L., Sieber L., Wittmann A., Huang Z., van Sluis P., Volckmann R., Koster J., Versteeg R., Fults D., Toledano H., Avigad S., Hoffman L. M., Donson A. M., Foreman N., Hewer E., Zitterbart K., Gilbert M., Armstrong T. S., Gupta N., Allen J. C., Karajannis M. A., Zagzag D., Hasselblatt M., Kulozik A. E., Witt O., Collins V. P., von Hoff K., Rutkowski S., Pietsch T., Bader G., Yaspo M. L., von Deimling A., Lichter P., Taylor M. D., Gilbertson R., Ellison D. W., Aldape K., Korshunov A., Kool M., Pfister S. M., Molecular classification of ependymal tumors across All CNS compartments, histopathological grades, and age groups. Cancer Cell 27, 728–743 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riising E. M., Comet I., Leblanc B., Wu X., Johansen J. V., Helin K., Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 55, 347–360 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Cooper S., Dienstbier M., Hassan R., Schermelleh L., Sharif J., Blackledge N. P., de Marco V., Elderkin S., Koseki H., Klose R., Heger A., Brockdorff N., Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 7, 1456–1470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bender S., Tang Y., Lindroth A. M., Hovestadt V., Jones D. T. W., Kool M., Zapatka M., Northcott P. A., Sturm D., Wang W., Radlwimmer B., Højfeldt J. W., Truffaux N., Castel D., Schubert S., Ryzhova M., Şeker-Cin H., Gronych J., Johann P. D., Stark S., Meyer J., Milde T., Schuhmann M., Ebinger M., Monoranu C. M., Ponnuswami A., Chen S., Jones C., Witt O., Collins V. P., von Deimling A., Jabado N., Puget S., Grill J., Helin K., Korshunov A., Lichter P., Monje M., Plass C., Cho Y. J., Pfister S. M., Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A. K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Robinson W. P., Price E. M., The human placental methylome. Cold Spring Harb. Perspect. Med. 5, a023044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hübner J.-M., Müller T., Papageorgiou D. N., Mauermann M., Krijgsveld J., Russell R. B., Ellison D. W., Pfister S. M., Pajtler K. W., Kool M., EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019, noz058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hickox A. E., Wong A. C. Y., Pak K., Strojny C., Ramirez M., Yates J. R. III, Ryan A. F., Savas J. N., Global analysis of protein expression of inner ear hair cells. J. Neurosci. 37, 1320–1339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L., Ozark P. A., Smith E. R., Zhao Z., Marshall S. A., Rendleman E. J., Piunti A., Ryan C., Whelan A. L., Helmin K. A., Morgan M. A., Zou L., Singer B. D., Shilatifard A., TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 4, eaau6986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGrath-Morrow S. A., Ndeh R., Helmin K. A., Chen S. Y., Anekalla K. R., Abdala-Valencia H., D’Alessio F. R., Collaco J. M., Singer B. D., DNA methylation regulates the neonatal CD4+ T-cell response to pneumonia in mice. J. Biol. Chem. 293, 11772–11783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walter J. M., Helmin K. A., Abdala-Valencia H., Wunderink R. G., Singer B. D., Multidimensional assessment of alveolar T cells in critically ill patients. JCI Insight 3, 123287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J., Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar S., Stecher G., Suleski M., Hedges S. B., TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Letunic I., Bork P., Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaax2887/DC1
Fig. S1. JAZF1-SUZ12 is a molecular bridge between NuA4 and PRC2.
Fig. S2. CATACOMB is a eutherian-specific gene.
Fig. S3. CATACOMB M406I/A and two deletion mutants retain the ability to reduce H3K27me2/3 levels in the cells.
Fig. S4. CATACOMB expression in different cells is achieved by different demethylating agent treatment regimen.
Fig. S5. H3K27me3 is present in U2OS and increases upon CATACOMB KO.
Table S1. FLAG-IP mass spectrometry results from hEnSC, expressing JAZF1-SUZ12 fusion, SUZ12 full length, or JAZF1 full length.