

Adipose tissue dysfunction leads to increased hepatocyte HIF-1α expression, promoting first-pass incretin hormone degradation.
Abstract
The decrease in incretin effects is an important etiologic component of type 2 diabetes with unknown mechanisms. In an attempt to understand obesity-induced changes in liver oxygen homeostasis, we found that liver HIF-1α expression was increased mainly by soluble factors released from obese adipocytes, leading to decreased incretin effects. Deletion of hepatocyte HIF-1α protected obesity-induced glucose intolerance without changes in body weight, liver steatosis, or insulin resistance. In-depth mouse metabolic phenotyping revealed that obesity increased first-pass degradation of an incretin hormone GLP-1 with increased liver DPP4 expression and decreased sinusoidal blood flow rate, reducing active GLP-1 levels in peripheral circulation. Hepatocyte HIF-1α KO blocked these changes induced by obesity. Deletion of hepatocyte HIF-2α did not change liver DPP4 expression but improved hepatic steatosis. Our results identify a previously unknown pathway for obesity-induced impaired beta cell glucose response (incretin effects) and the development of glucose intolerance through inter-organ communications.
INTRODUCTION
The etiology of type 2 diabetes mellitus (T2DM) involves both insulin resistance and β cell dysfunction, and one typically needs both defects to develop the hyperglycemic diabetic state (1, 2). It has been suggested that reciprocal interaction between these two defects may be able to contribute to progressive deterioration of glucose homeostasis by increasing body insulin needs and hyperinsulinemia, exaggerating β cell stress and insulin resistance (1–3). However, whether insulin target tissues communicate with pancreatic β cells and cause β cell dysfunction is not clearly understood.
Incretins are peptide hormones produced from enteroendocrine cells in the gastrointestinal (GI) tract (4, 5). Glucose or mixed meal ingestion stimulates incretin secretion, and increased circulating incretins bind to their specific receptors on the surface of β cells, potentiating glucose-stimulated insulin secretion. In healthy humans, incretin action is responsible for approximately 60 to 70% of postprandial insulin secretion (6). However, in obesity, incretin effects are decreased even before onset of T2DM and decline further as the disease progresses with unknown mechanisms (7). Thus, in T2DM subjects, incretin effects account for only 7 to 40% of postprandial insulin secretion (8, 9).
Most incretin effects are mediated by two gut hormones: glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) (4, 5). Systemic effects of these incretin hormones are controlled mainly at the levels of secretion and inactivation. Once released, circulating incretins are rapidly inactivated by the specific peptidase enzyme dipeptidyl peptidase-4 (DPP4) (10) and are eventually cleared, mainly by the kidney (11). DPP4 is produced as a membrane-bound protein, and a soluble circulating form of DPP4 is generated through proteolytic cleavage at the base of the extracellular domain (12). DPP4 is expressed ubiquitously but most abundantly in the GI tract, kidney, liver, and endothelial cells (13).
GLP-1 is one of the most studied gut hormones fulfilling incretin criteria. GLP-1 enhances glucose-stimulated insulin secretion and suppresses glucagon secretion, whereas GIP stimulates secretion of both insulin and glucagon (14). GLP-1 is targeted by DPP4 with an in vivo half-life of 1.5 to 2 min (15). Consequently, only 10 to 15% of the secreted intact/active GLP-1 enters the systemic circulation (16). Global knockout (KO) of DPP4 causes six- to ninefold increases in plasma intact/active GLP-1 levels and improves glucose tolerance in mice (13, 17). Therefore, it is plausible that DPP4-dependent GLP-1 inactivation may be a critical regulatory step for systemic GLP-1 action. In T2DM, circulating levels of intact/active GLP-1 are decreased (5, 9, 18, 19), whereas GIP levels are increased (14). Moreover, the effect of exogenous GLP-1 to increase insulin secretion is diminished in subjects with impaired glucose tolerance (IGT) or T2DM (20) with increased plasma DPP4 activity compared to normal (21). Therefore, it appears that the decrease of intact/active GLP-1 levels is an important etiologic component of decreased incretin effects (insulin secretion) and glucose tolerance in obesity and T2DM (22). Administration of a long-acting GLP-1 receptor agonist or DPP4 inhibitor compounds improves glycemic control in type 2 diabetics. Increased adiposity and insulin resistance are associated with this defect (23); however, the mechanisms for how obesity decreases intact/active GLP-1 levels are unknown.
In the present study, we adopted interdisciplinary approaches to assess obesity-induced changes in hepatic microhemodynamics and DPP4 activity. We found that obesity increased hepatic hypoxia-inducible factor–1α (HIF-1α), leading to decreased sinusoidal blood flow rate and velocity, and increased DPP4 expression. These changes were sufficient to augment first-pass degradation of incretin hormones.
RESULTS
Obesity increases hepatic HIF-1α expression
High-fat diet (HFD) induces progressive deterioration of glucose intolerance with decreased insulin sensitivity and impaired glucose-stimulated insulin secretion in mice (24). This is similar to the clinical manifestations observed in subjects with IGT (1, 2). HIF-1α is an intracellular oxygen sensor and plays a crucial role in regulating hepatic glucose and lipid metabolism in normal and pathological conditions (25). Increased liver HIF-1α promotes liver damage, steatosis, and inflammation in a rodent model of alcoholic steatohepatitis (26). Moreover, liver HIF-1α overexpression induces lipogenic and gluconeogenic pathways (27–29). To examine the effect of obesity on liver oxygen homeostasis, we measured the expression of HIF-1α and HIF-2α in the liver of normal chow diet (NCD) and HFD mice. Protein expression of HIF-1α and mRNA levels of HIF-1α target genes such as Vegf, Pdk1, and Lox were increased in HFD mouse liver compared with NCD-fed control mice (Fig. 1, A and B). On the other hand, HIF-2α protein levels and mRNA expression of several HIF-2α–specific target genes, such as Epo, Cat, Cited2, and Irs2, were not increased in obese mouse liver (Fig. 1, A and B), suggesting that diet-induced obesity confers isoform-selective regulation of hepatic HIF-α protein expression.
Fig. 1. Increased hepatic HIF-1α expression contributes to glucose intolerance in obesity.
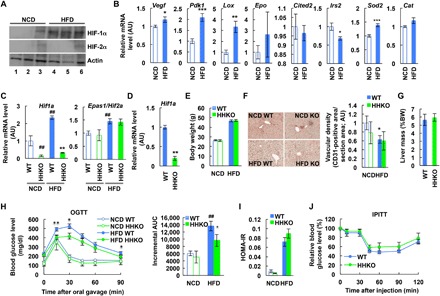
(A) Liver HIF-α expression (n = 3 mouse per group). (B) mRNA levels of HIF target genes in liver (n = 6 mice per group). Epo was barely expressed in NCD and HFD mouse liver (Ct > 30) with high variability in its expression. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Liver Hif1a and Epas1/Hif2a mRNA levels (n = 6, 4, 11, and 8 mice per group). **P < 0.01 versus lane 3, ##P < 0.01 versus lane 1. (D) mRNA levels of Hif1a in primary hepatocytes isolated from HFD mice (n = 4 mice per group). **P < 0.01. (E) Body weight (BW) (n = 6, 4, 11, and 8 mice per group). (F) Immunohistochemistry analysis of CD31-positive vascular density in liver sections (n = 4 NCD WT, 4 NCD HHKO, 11 HFD WT, and 10 HFD HHKO mice). Images were taken under ×200 magnification. *P < 0.05 versus lane 1. (G) Liver mass of HFD mice (n = 11 and 8 mice per group). (H to J) Metabolic phenotyping of HHKO mice. NCD and HFD WT and HHKO mice were subjected to oral glucose tolerance tests (OGTTs) (H) or intraperitoneal insulin tolerance tests (IPITTs) (J). HOMA-IR (I) was calculated from the data collected during OGTT. n = 6, 4, 11, and 8 mice were used per group. *P < 0.05, **P < 0.01 HFD WT versus KO; ##P < 0.01 versus lane 1. AUC, area under the curve; AU, arbitrary units. Similar results were observed in at least two separate cohort mouse studies. For statistical analysis, one-way (C; F; and H, right) or two-way (H, left) analysis of variance (ANOVA) with post hoc t tests between the individual groups or two-tailed unpaired t test (B and D) was performed. All data are presented as means ± SEM.
Deletion of hepatocyte HIF-1α improves glucose tolerance
To assess the effects of HIF-1α in obesity-associated liver dysfunction, we generated hepatocyte-specific HIF-1α KO (HHKO) mice (Hif1afl/fl:Albumin-Cre+) (Fig. 1, C and D) and measured their metabolic phenotype on NCD and HFD. Cre− Hif1a-floxed mice (Hif1afl/fl:Albumin-Cre−) did not show significant changes in liver Hif1a and Epas1/Hif2a expression, plasma insulin levels, or glucose tolerance on NCD and HFD compared with age-matched wild-type (WT) mice. Therefore, Cre− Hif1a-floxed littermates were used as controls, referred to hereafter as WT for comparison with HHKO mice. HHKO mice exhibited normal body weight, liver and adipose tissue mass, and hepatic vascular density on both NCD and HFD (Fig. 1, E to G, and fig. S1). On NCD, HHKO mice exhibited normal oral glucose tolerance (Fig. 1H). However, on HFD, HHKO mice displayed significantly improved oral glucose tolerance compared to controls (Fig. 1H).
Hepatocyte HIF-1α is not necessary for obesity-induced liver steatosis, inflammation, and insulin resistance
To test whether the improvement of glucose tolerance in HFD HHKO mice is associated with improved insulin sensitivity, we performed insulin tolerance tests and calculated homeostatic model assessment for insulin resistance (HOMA-IR). Unexpectedly, insulin tolerance and HOMA-IR were not changed in HFD HHKO mice compared with HFD WT mice (Fig. 1, I and J). Moreover, plasma and liver levels of triglycerides and nonesterified fatty acids, histologic features of hepatic steatosis, and mRNA expression of lipogenic Fas were all comparable in HFD WT and HHKO mice (fig. S2, A to F). Furthermore, mRNA expression of proinflammatory Tnfa and fibrogenic Lox, Acta2 [encoding alpha smooth muscle actin, (α-SMA)], and Tgfb1 was comparable in the liver of WT and HHKO mice (fig. S2G), while obesity-induced hepatic caspase-3/7 activity and serum alanine transferase activity were reduced in HHKO mice (fig. S2, H and I). Consistent with this, the ratio of Kupffer cells (KCs), recruited hepatic macrophages (RHMs), and T and B lymphocytes in overall CD45+ hepatic leukocyte population was identical in HFD WT and HHKO mice (fig. S2, J to L). Moreover, proinflammatory gene expression in each of the KC or RHM fractions was comparable between the genotypes (fig. S2M). Together, these results suggest that, unlike alcohol-induced hepatic disease (26), hepatocyte HIF-1α expression is not necessary for obesity-induced liver steatosis or inflammation.
Deletion of hepatocyte HIF-1α improves glucose tolerance by increasing GLP-1 action
Because HFD HHKO mice exhibited improved glucose tolerance without changes in insulin sensitivity, we went on to test whether insulin secretion is increased in HHKO mice. As seen in Fig. 2 (A to C), HFD HHKO mice exhibited higher HOMA-%β index with higher plasma insulin and C-peptide levels upon high glucose challenge. This occurred without changes in pancreas mass, overall pancreatic insulin content, and β cell mass (Fig. 2, D to F). Moreover, insulin–to–C-peptide ratio was comparable between HFD WT and HHKO mice (Fig. 2G). These results suggest that HHKO improved obesity-induced glucose intolerance mainly by increasing β cell insulin secretion, without changes in insulin sensitivity, β cell mass, or plasma insulin clearance.
Fig. 2. HHKO mice exhibit improved glucose tolerance by increasing plasma incretin levels and insulin secretion.
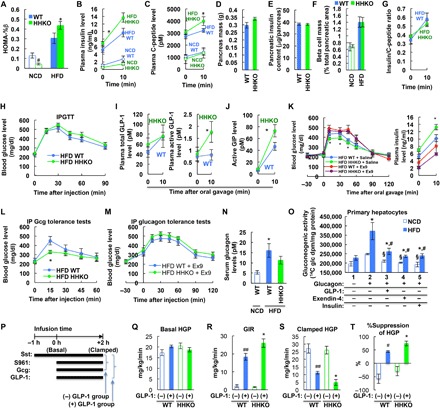
(A to C) Plasma insulin (B) and C-peptide (C) levels during OGTT in Fig. 1H mice were used for calculating HOMA-%β (A). #P < 0.05, *P < 0.05 versus WT counterparts. (D to F) Pancreas mass (D), pancreatic insulin content (E), and overall β cell mass (F) were measured in NCD and/or HFD WT and HHKO mice. n = 6 NCD WT, 4 NCD HHKO, 11 HFD WT, and 8 HFD HHKO mice. (G) Plasma insulin/C-peptide ratio in Fig. 1H mice. (H) IPGTTs. n = 11 and 10 per group. (I and J) Plasma total (I, left) and active GLP-1 (I, right) and active GIP (J) levels in 6-hour–fasted HFD WT and HHKO mice before or 10 min after glucose oral gavage (n = 5 and 7 mice per group). *P < 0.05 versus WT controls. (K) OGTTs in HFD mice conducted 30 min after saline or ex9 administration. n = 11 and 10 per group. *P < 0.05, HFD WT + saline versus HFD HHKO + saline. (L and M) Glucagon (Gcg) tolerance tests with (M) or without (L) ex9 administration 30 min before glucagon injection. n = 6 and 7 per group. *P < 0.05. (N) Fasting (6 hours) serum glucagon levels (n = 6, 10, and 8 mice per group). *P < 0.05 versus lane 1. (O) Gluconeogenesis activity assays in primary hepatocytes. n = 5 wells per group. §P < 0.05 versus NCD group in lane 2, *P < 0.05 versus HFD group in lane 1, #P < 0.05 versus HFD group in lane 2. (P) Schematic representation of the experimental procedures for hyperglycemic clamp studies in (Q) to (T). (Q to T) Hyperglycemic clamp studies. Basal HGP (Q), glucose infusion rate (GIR) (R), clamped HGP (S), and %suppression of HGP (T) were calculated as described in Materials and Methods. n = 5, 4, 9, and 7 mice per group. *P < 0.05 versus lane 2, #P < 0.05 versus lane 1, ##P < 0.01 versus lane 1. For statistical analysis, one-way (A, N, O, and R to T) or two-way (B, C, I, J, and L) ANOVA with post hoc t tests between the individual groups was performed. All data are presented as means ± SEM.
To test whether increased β cell insulin secretion in HHKO mice is associated with increased incretin action, we first measured intraperitoneal glucose tolerance (IPGTT), which does not stimulate incretin secretion. As seen in Fig. 2H, intraperitoneal glucose tolerance was comparable between HFD WT and HHKO mice. Moreover, glucose-stimulated plasma active GLP-1 and active GIP levels were significantly higher in HFD HHKO mice compared with HFD WT mice (Fig. 2, I and J). Plasma total GLP-1 levels were unchanged (Fig. 2I). To test whether improved oral glucose tolerance in HFD HHKO mice was mainly due to increased active GLP-1 levels, we injected the GLP-1 receptor antagonist exendin-9 (ex9) to HFD WT and HHKO mice and measured oral glucose tolerance. As seen in Fig. 2K, ex9 treatment impaired glucose tolerance in both HFD WT and HHKO mice. Consequently, oral glucose tolerance became comparable in HFD WT and HHKO after ex9 administration (Fig. 2K). Together, these results suggest that HHKO increased β cell insulin secretion and improved glucose tolerance by increasing circulating active GLP-1 levels.
Deletion of hepatocyte HIF-1α decreases glucagon sensitivity by increasing GLP-1 action
Increased glucagon action is an important cause of hyperglycemia, especially in insulin-resistant states such as T2DM (30). GLP-1 also contributes to glucose homeostasis by suppressing glucagon secretion and action through both insulin-dependent and insulin-independent mechanisms (31, 32). To test whether increased plasma active GLP-1 levels in HHKO mice led to a decrease in hepatic glucagon sensitivity, we performed glucagon tolerance tests with or without ex9 treatment. As seen in Fig. 2L, HFD HHKO mice exhibited decreased glucose excursion upon intraperitoneal glucagon challenge compared with HFD WT controls. Ex9 treatment increased glucagon-stimulated glucose excursion in both HFD WT and HHKO mice, and after ex9 administration, glucagon tolerance was identical in HFD WT and HHKO mice (Fig. 2M). Serum glucagon levels were slightly decreased in HHKO mice, but this did not reach statistical significance (Fig. 2N). Together, these results suggest that, in obesity, increased hepatocyte HIF-1α contributes to increased glucagon sensitivity mainly by decreasing GLP-1 action.
Beyond its insulinotropic effects, GLP-1 exerts direct effect on hepatocyte metabolism (33–35). Consistently, we observed that GLP-1 treatment decreased glucose production in cultured primary hepatocytes (Fig. 2O), with decreased glucagon-stimulated cyclic AMP (adenosine monophosphate) levels. To test whether the local hepatic GLP-1 effect is increased in HHKO mice in a quantitative fashion, we conducted hyperglycemic clamp studies (Fig. 2P). GLP-1 increases plasma insulin levels by enhancing glucose-stimulated insulin secretion, as well as by decreasing plasma GLP-1 clearance (36). Therefore, to block both insulin secretion and action and measure insulin-independent hepatic GLP-1 effects, we gave mice a constant infusion of somatostatin and S961 insulin receptor antagonist along with exogenous glucagon ± GLP-1. Basal hepatic glucose production (HGP) was comparable in HFD WT and HHKO mice (Fig. 2Q). Infusion of active GLP-1 increased the glucose infusion rate with decreased HGP in both HFD WT and HHKO mice, but these effects of GLP-1 were enhanced in HHKO mice compared with WT mice (Fig. 2, R to T).
Obesity promotes first-pass GLP-1 degradation through HIF-1α–dependent regulation of liver DPP4 expression and sinusoidal blood flow rate
The increase in plasma intact/active GLP-1 levels without changes in total GLP-1 levels in HFD HHKO mice raises a possibility that HHKO decreases GLP-1 inactivation. Because GLP-1 inactivation is mediated mainly by DPP4 (13), we assessed the effect of obesity and hepatocyte HIF-1α KO on systemic and hepatic DPP4 expression. As seen in Fig. 3A and fig. S3 (A and B), Dpp4 expression was increased selectively in liver and adipose tissue but not in the kidney or GI tract of obese mice. DPP4 protein expression and activity were also increased in both liver and serum of obese WT mice (Fig. 3, B and C, and fig. S3C). When measured separately, obesity increased DPP4 expression specifically in hepatocytes, but not in nonparenchymal hepatic cells, although Dpp4 was abundantly expressed in both hepatocytes and nonparenchymal hepatic cells (Fig. 3, D and E). In HHKO mice, serum and liver DPP4 expression or activity or liver Dpp4 mRNA levels were not increased in obesity (Fig. 3, A to F). While increased in obesity, adipose tissue Dpp4 expression was negligible compared with liver Dpp4 expression and was not affected in HHKO mice (figs. S3D and S4A).
Fig. 3. Deletion of hepatocyte HIF-1α blocks obesity-induced liver DPP4 expression and first-pass GLP-1 inactivation.
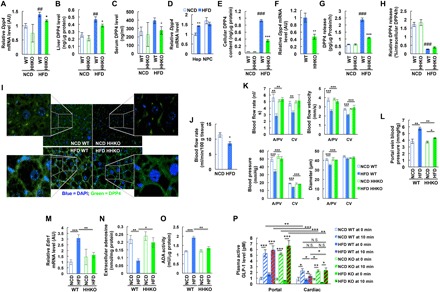
(A to C) Liver Dpp4 mRNA (A) and liver (B) and serum (C) DPP4 protein levels (n = 6, 4, 11, and 8 mice per group). *P < 0.05 versus lane 3, ##P < 0.01 versus lane 1. (D) Dpp4 mRNA expression in primary hepatocytes (Hep) and nonparenchymal cells (NPC) isolated from NCD and HFD WT mice (n = 4 mice per group). **P < 0.01 versus lane 1. (E) DPP4 protein content in primary hepatocytes isolated from NCD and HFD WT mice (n = 4 and 6 mouse per group). ###P < 0.001 versus lane 1, ***P < 0.001 versus lane 3. (F) Dpp4 mRNA expression in primary hepatocytes isolated from HFD WT and HHKO mice (n = 11 and 8 mice per group). **P < 0.01. (G) DPP4 protein release from cultured primary hepatocytes (n = 4 and 6 mouse per group). ###P < 0.001 versus lane 1, ***P < 0.001 versus lane 3. (H) Relative DPP4 release per hepatocyte DPP4 content (n = 4 and 6 mouse per group). ###P < 0.001. (I) Immunohistochemistry analysis of DPP4 in the liver of NCD and HFD WT and HHKO mice. Images were taken under ×400 magnification. (J) Blood flow rate across liver (n = 4 mice per group). *P < 0.05. DAPI, 4′,6-diamidino-2-phenylindole. (K) Liver microhemodynamics (n = 4 mice per group). **P < 0.01, ***P < 0.001. (L) Portal vein blood pressure (n = 4 mice per group). *P < 0.05, **P < 0.01. (M) Liver Edn1 mRNA expression (n = 6 NCD WT, 4 NCD KO, 11 HFD WT, and 8 HFD KO mice per group). **P < 0.01, ***P < 0.001. (N) Extracellular adenosine levels (n = 4 different mouse hepatocytes per group). *P < 0.05, **P < 0.01. (O) ADA activity in the plasma membrane fraction of hepatocytes (n = 4 different mouse hepatocytes per group). **P < 0.01, ***P < 0.001. (P) Intact/active GLP-1 levels in the portal and cardiac plasma of 6-hour–fasted mice before or 10 min after glucose oral gavage (n = 18 NCD WT, 19 HFD WT, 6 NCD HHKO, and 10 HFD HHKO mice). *P < 0.05, **P < 0.01, ***P < 0.001; N.S., not significant. For statistical analysis, one-way ANOVA (A to C, E, G, H, and K to P) with post hoc t tests between the individual groups or two-tailed unpaired t test (D, F, and J) was performed. All data are presented as means ± SEM.
HFD/obesity also increased hepatocyte DPP4 release (Fig. 3G), with increased expression of hepatic DPP4 sheddases (metalloprotease-1 and metalloprotease-14) (fig. S4B) (37). However, the increase of DPP4 expression was very large in HFD hepatocytes so that the fraction of DPP4 that is released was lesser in hepatocytes from HFD mice (Fig. 3H). HIF-1α KO did not affect the proportion of released DPP4 among the total intracellular DPP4 or the expression of DPP4 sheddases (Fig. 3H and fig. S4B). Immunohistochemistry analyses revealed that DPP4 protein was largely localized to the plasma membrane of HFD/obese hepatocytes (Fig. 3I), suggesting that obese liver is highly accumulated with membrane-bound DPP4 on the plasma membrane of hepatocytes.
Newly produced active GLP-1 is first introduced into the system through the portal circulation, and previously, it was reported that hepatic GLP-1 extraction accounts for ~44% of exogenous GLP-1 degradation (38). Therefore, we hypothesized that the HIF-1α–dependent increase in membrane-bound DPP4 expression in obese liver could enhance first-pass GLP-1 inactivation. A premise of this hypothesis is that obesity does not increase sinusoidal blood flow rate or velocity because it can decrease retention time of intact/active GLP-1 within liver before escaping to systemic blood circulation. Therefore, we assessed liver microhemodynamics in NCD and HFD WT and HHKO mice. Blood flow rate across liver was not increased but decreased in HFD/obese WT mice compared with NCD/lean WT mice (Fig. 3J). Moreover, sinusoidal blood flow rate, velocity, and pressure as well as sinusoidal diameter were substantially decreased in HFD WT mice compared with NCD WT mice (Fig. 3K). These changes were more pronounced at arteriolar/portal venular (A/PV) sinusoids than central venular (CV) sinusoids (Fig. 3K). Moreover, in HFD mice, the ratio of sinusoidal blood pressure gradient over blood flow was increased at A/PV sinusoids compared with NCD mice without changes in that at CV sinusoids. These results suggest that HFD/obesity increases sinusoidal flow resistance. Consistent with this idea, portal vein blood pressure was substantially increased in HFD mice compared with NCD mice (Fig. 3L). These changes in hepatic microcirculation were associated with increased liver Edn1 (encoding endothelin-1, a vasoconstrictor) expression (Fig. 3M). Moreover, the levels of extracellular adenosine, a purine metabolite producing vasodilation, were decreased in cultured HFD WT mouse hepatocytes, with increased adenosine deaminase (ADA) activity in the plasma membrane fraction of the cells compared with NCD WT mouse hepatocytes (Fig. 3, N and O). Consistently, mRNA expression of Ada was increased by HFD (fig. S4C).
In HHKO mice, sinusoidal blood flow rate, velocity, and pressure as well as sinusoidal diameter were not decreased upon HFD and were higher than those in HFD WT mice (Fig. 3K). These changes were associated with lower Edn1 and Ada expression in HFD HHKO mice compared with HFD WT mice (Fig. 3M and fig. S4C). Moreover, extracellular adenosine levels were higher in the conditioned media of HFD HHKO mouse hepatocytes compared with those of HFD WT mouse hepatocytes, along with decreased ADA activity in the plasma membrane fraction (Fig. 3, N and O). Consistently, treatment with a HIF-1α stabilizing agent, CoCl2, increased Edn1, Ada, and Dpp4 mRNA expression in WT hepatocytes but not in HIF-1α KO hepatocytes (fig. S4D). On the other hand, expression of Entpd1 and Nt5e encoding the enzymes important for extracellular adenosine generation (CD39 and CD73) was unchanged in HFD HHKO mice compared with HFD WT mice (fig. S4C). These results suggest that obesity-induced hepatocyte HIF-1α can promote sinusoidal vasoconstriction and flow resistance by increasing endothelin-1 expression and decreasing extracellular adenosine levels, causing decreased sinusoidal blood flow rate.
Last, we tested whether these HIF-1α–dependent changes in sinusoidal blood flow and liver DPP4 expression in obesity led to increased first-pass GLP-1 degradation. To this end, we measured active GLP-1 levels in the portal and peripheral (cardiac) plasma of 6-hour–fasted NCD and HFD mice before and 10 min after high glucose challenge. As expected, upon fasting, intact/active GLP-1 levels were comparable in the portal and peripheral plasma (Fig. 3P). Upon glucose challenge, plasma active GLP-1 levels were increased in both portal and cardiac plasma in all mice (Fig. 3P). Moreover, intact/active GLP-1 levels were substantially higher in the portal plasma compared with peripheral plasma, consistent with the view that hepatic GLP-1 elimination is induced upon high glucose challenge. In NCD WT mice, glucose-stimulated plasma active GLP-1 levels were ~56% lower in peripheral plasma compared with portal plasma (Fig. 3P, lane 2 versus lane 10). In HFD/obese mice, this difference was increased to ~76% (Fig. 3P, lane 4 versus lane 12), suggesting that obesity causes ~35% increase in first-pass GLP-1 inactivation.
On NCD, intact/active GLP-1 levels were comparable in WT and HHKO mice in both portal and peripheral plasma (Fig. 3P, lane 10 versus lane 14). However, on HFD, HHKO mice exhibited significantly increased glucose-stimulated peripheral plasma intact/active GLP-1 levels (Fig. 3P, lane 12 versus lane 16) without changes in the portal plasma intact/active GLP-1 levels (Fig. 3P, lane 4 versus lane 8). This led to ~67% increase in peripheral active GLP-1 levels in HFD HHKO mice compared with HFD WT mice. These results suggest that deletion of hepatocyte HIF-1α led to a decrease in glucose-stimulated first-pass intact/active GLP-1 degradation by preventing obesity-induced liver DPP4 expression.
Hepatocyte HIF-2α does not regulate liver DPP4 expression
Both HIF-1α and HIF-2α can be induced in hypoxic conditions and mediate hypoxic responses with many common target genes. However, distinct or opposite physiological roles of HIF-1α and HIF-2α have also been reported in certain (patho-)physiological contexts including obese adipocytes (25, 39–41). In liver, it was reported that HIF-2α overexpression improves glucose and glucagon tolerance and hepatic insulin sensitivity (40, 42). Therefore, we hypothesized that hepatic HIF-2α can counteract HIF-1α, at least functionally, like in adipocytes (39). To address this question in the context of obesity, we generated hepatocyte-specific HIF-2α KO (H2HKO) mice (Hif2afl/fl:Albumin-Cre+) (Fig. 4A) and measured their metabolic phenotypes. Cre− Hif2a-floxed littermates (Hif2afl/fl:Albumin-Cre−) were used as WT controls for H2HKO mice. Unexpectedly, H2HKO mice exhibited normal body weight gain (Fig. 4B) and comparable glucose and insulin tolerance compared with WT mice on both NCD and HFD (Fig. 4, C to E). Moreover, glucagon tolerance was also unchanged in HFD H2HKO mice compared with HFD WT controls (Fig. 4F). Consistent with this, plasma and hepatic DPP4 protein levels and mRNA expression of hepatic Dpp4 and Irs2 were also unchanged in H2HKO mice (Fig. 4, G to I). These results suggest that hepatocyte HIF-2α is dispensable for the maintenance of normal glucose and insulin tolerance and for the development of glucose, glucagon, and insulin intolerance in obesity. On the other hand, HFD H2HKO mice exhibited lower levels of hepatic triglyceride and nonesterified fatty acid content with decreased expression of lipogenic Fas compared with HFD WT controls (Fig. 4, I to K), suggesting that hepatocyte HIF-2α expression contributes to obesity-induced hepatic steatosis. These results are consistent with a previous report that deletion of Hif2a/Epas1 improves liver steatosis in Von Hippel-Lindau (VHL) KO background in mice (43).
Fig. 4. Deletion of hepatocyte HIF-2α does not affect obesity-induced glucose, glucagon, and insulin intolerance.
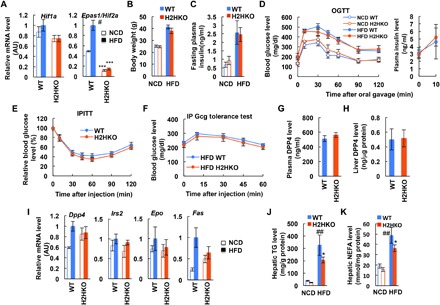
(A) Liver Hif1a and Epas1/Hif2a mRNA levels (n = 4, 4, 6, and 9 mice per group). ***P < 0.0001 versus WT counterparts, #P < 0.05 versus NCD WT. (B) Body weight (n = 4, 5, 6, and 9 mice per group). (C) Fasting plasma insulin levels (n = 4, 5, 6, and 9 mice per group). (D) OGTTs and plasma insulin levels during OGTT (n = 4, 5, 6, and 9 mice per group). (E) IPITTs in HFD mice (n = 6 WT and 9 KO mice). (F) Glucagon tolerance tests in HFD mice (n = 11 WT and 8 KO mice). (G) Plasma DPP4 levels in HFD mice (n = 6 WT and 9 KO mice). (H) Liver DPP4 expression in HFD mice (n = 6 WT and 9 KO mice). (I) mRNA levels in liver (n = 4, 4, 6, and 9 mice per group). (J and K) Liver triglyceride (TG) (J) or nonesterified fatty acid (NEFA) (K) content (n = 6 WT and 9 KO mice). *P < 0.05 versus lane 3, ##P < 0.01 versus lane 1. For statistical analysis, one-way ANOVA with post hoc t tests between the individual groups was performed. All data are presented as means ± SEM.
Adipose tissue dysfunction induces liver HIF-1α and Dpp4 expression
Next, we sought to identify the mechanism for how obesity confers increased liver HIF-1α expression. Previously, we and others reported that HFD/obesity decreases oxygen tension in adipose tissue (44, 45), and this leads to increased adipocyte HIF-1α expression, triggering adipose tissue inflammation and dysfunction (3, 39, 46, 47). Consistent with previous reports, adipose tissue oxygen tension was decreased in HFD mice (fig. S5A). Interstitial oxygen tension was also moderately reduced in the liver but not in the skeletal muscle and kidney of HFD WT mice compared with NCD controls (Fig. 5A and fig. S5, A and B). To test whether this moderate decrease in hepatic interstitial oxygen tension is sufficient to induce hepatocyte HIF-1α expression, we incubated primary mouse hepatocytes at various oxygen conditions, including 2 and 3% oxygen equivalent to the interstitial oxygen tension in the liver of lean and obese mice, respectively (Fig. 5A). As seen in Fig. 5B and fig. S5 (C and D), Hif1a mRNA and protein levels were not different at 2 and 3% oxygen conditions. These results suggest that the obesity-induced decrease in hepatic interstitial oxygen tension is not severe enough to trigger increased hepatocyte HIF-1α expression. In contrast, HIF-2α levels were slightly lower at 2% compared with 3% oxygen condition (Fig. 5B).
Fig. 5. Adipose tissue dysfunction induces increased liver HIF-1α expression.
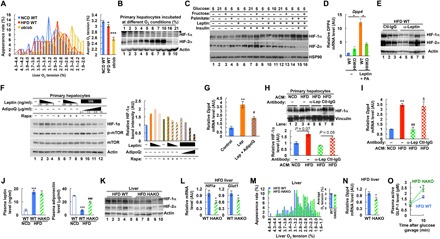
(A) Liver interstitial oxygen tension (n = 4 mice per group). *P < 0.05, ***P < 0.001 versus lane 1. (B) HIF-α expression in primary hepatocytes incubated at different oxygen conditions for 3 hours. Arrowhead indicates nonspecific bands. (C) HIF-α expression in primary hepatocytes incubated with different combinations of leptin (100 ng/ml), insulin (100 nM), palmitic acid (400 μM), and fructose (5 mM) at three different concentrations of glucose (5, 10, and 21 mM) for 4 hours. (D) Dpp4 mRNA expression in WT and HIF-1α KO (HHKO) hepatocytes treated with leptin (100 ng/ml) + palmitic acid (PA) (400 μM) for 24 hours. *P < 0.05. (E) HIF-α expression in the liver of HFD WT mice treated with control immunoglobulin G (Ctl-IgG) or anti-leptin antibodies for 1 hour (n = 3 and 5 mice, respectively). (F) Western blot analysis of HIF-α and phosphorylated and total mTOR levels in primary hepatocytes incubated with different concentrations of leptin and/or adiponectin (AdipoQ) in the presence or absence of rapamycin (Rapa; 1 μM) for 3 hours. (G) Dpp4 mRNA expression in primary hepatocytes incubated with leptin (Lep) ± adiponectin for 16 hours. **P < 0.01 versus lane 1, #P < 0.01 versus lane 2. (H and I) HIF-1α protein expression (n = 5, 2, 2, and 3) (H) and Dpp4 mRNA levels (n = 3 samples per group) (I) in primary hepatocytes incubated with ACM of NCD or HFD WT mice for 3 hours (H) or 18 hours (I). For leptin neutralization, HFD ACM was incubated with control IgG or anti-leptin antibodies for 1 hour at 4°C before incubation with hepatocytes. **P < 0.05 versus lane 1, #P < 0.01 versus lane 2, $P < 0.05 versus lane 3. (J) Plasma leptin and adiponectin levels (n = 2, 9, and 9 mice per group). ***P < 0.001 versus lane 1, ###P < 0.001 versus lane 2. (K) Liver HIF-α expression. (L) Liver Hif1a and Glut1 mRNA expression (n = 10 mice per group). *P < 0.05. (M) Interstitial oxygen tension in the liver of HFD mice (n = 4 per group). (N) mRNA expression of Dpp4 in the liver of HFD mice (n = 10 mice per group). *P < 0.05. (O) Plasma intact/active GLP-1 levels in 6-hour–fasted HFD mice before or 10 min after glucose oral gavage (n = 7 mice per group). *P < 0.05. For statistical analysis, one-way (A, D, and G to J) or two-way (O) ANOVA with post hoc t tests between the individual groups or two-tailed unpaired t test (L and N) was performed. All data are presented as means ± SEM.
Next, we turned our attention toward signaling pathways that can stimulate HIF-1α expression in hepatocytes. Incubation of primary hepatocytes with different combinations of hormones and nutrients that are commonly increased in obesity revealed that leptin, insulin, fructose, and high levels of palmitic acid and glucose can interactively increase hepatocyte HIF-1α expression (Fig. 5C and fig. S5E). Among these, the combination of leptin + palmitic acid was sufficient to induce maximal HIF-1α expression. Moreover, leptin + palmitic acid treatment stimulated Dpp4, Edn1, and Ada expression in culture hepatocytes, which was blunted in HIF-1α KO hepatocytes (Fig. 5D and fig. S5F). In sharp contrast, HIF-2α expression was decreased by the combination of leptin + palmitic acid + insulin (Fig. 5C). To test whether increased plasma leptin concentrations are critical for obesity-induced hepatic HIF-1α expression, we injected anti-leptin–neutralizing antibodies into HFD/obese mice. As seen in Fig. 5E, acute neutralization of circulating leptin markedly decreased hepatic HIF-1α expression.
Plasma levels of an insulin-sensitizing hormone, adiponectin, are decreased in obesity, and adiponectin counteracts leptin in certain physiological contexts (48). Therefore, we tested whether decreased plasma adiponectin levels can also contribute to the increase in liver HIF-1α expression in obesity. As seen in Fig. 5F (lane 2 versus lanes 9 to 12), increasing adiponectin concentrations markedly reduced leptin-stimulated hepatocyte HIF-1α expression. Moreover, adiponectin treatment decreased leptin-induced Dpp4 expression in hepatocytes (Fig. 5G). Further mechanistic studies revealed that leptin stimulated mammalian target of rapamycin (mTOR) phosphorylation in hepatocytes, and inhibition of mTOR activation by pretreatment with rapamycin blocked leptin-stimulated HIF-1α expression (Fig. 5F). Moreover, adiponectin suppressed leptin-stimulated phosphorylated mTOR levels (Fig. 5F). These results suggest that leptin stimulates HIF-1α expression through an mTOR-dependent signaling pathway, and adiponectin counteracts it. Together, these results suggest that increased plasma leptin and free fatty acids (FFAs) and decreased adiponectin levels are associated with increased liver HIF-1α expression.
Because leptin and adiponectin are adipocyte-specific hormones and the increase in plasma FFA levels in obesity is mainly due to increased lipolysis in adipose tissue, these results suggest that adipose tissue dysfunction contributes to the increase in hepatic HIF-1α expression. To directly test whether obese adipocytes can signal to hepatocytes to increase HIF-1α expression, we incubated primary mouse hepatocytes with adipocyte-conditioned medium (ACM) collected from NCD/lean or HFD/obese WT adipocytes. As seen in Fig. 5H, hepatocytes incubated with ACM of HFD WT mice expressed higher levels of HIF-1α compared with hepatocytes incubated with ACM of NCD WT mice. Moreover, preincubation of HFD WT ACM with leptin-neutralizing antibodies blunted the effects to increase hepatocyte HIF-1α expression (Fig. 5H). Consistent with this, Dpp4 expression was higher in hepatocytes incubated with HFD-ACM compared with NCD-ACM, and these effects were blocked by anti-leptin–neutralizing antibody treatment (Fig. 5I).
Because Hif1a mRNA expression and protein levels were increased in the liver of HFD mice compared with NCD mice, we tested whether obese ACM or leptin can also increase Hif1a mRNA expression. Incubation of hepatocytes with obese ACM or leptin for 3 hours did not increase Hif1a mRNA expression (fig. S5G), suggesting that obese ACM or leptin can increase HIF-1α protein levels mainly at the posttranscriptional level. However, 24-hour incubation with obese ACM increased Hif1a mRNA expression, and this effect was sensitive to the nuclear factor κB (NF-κB) inhibitor Bay11-7082 but not to leptin-neutralizing antibody (fig. S5H). These results are consistent with previous reports that NF-κB can transactivate Hif1a (49) and obese adipocytes release inflammatory cytokines stimulating NF-κB (3). Together, these results suggest that obese adipocytes can signal to hepatocytes to increase Hif1a mRNA and protein expression, but only leptin mediates the latter.
Adipose tissue hypoxia leads to increased liver HIF-1α and Dpp4 expression in obesity
Our results suggest that in obesity, dysfunctional adipocytes promote hepatic HIF-1α expression, causing increased hepatic DPP4 expression and first-pass GLP-1 inactivation. To test this concept of a multistep inter-organ communication (fig. S6) at the organismic level, we used HIF-1α adipocyte KO (HAKO) mice. Previously, we and others reported that HAKO mice are protected from obesity-induced adipose tissue inflammation and dysfunction (3, 39, 44, 46, 47). Moreover, HFD HAKO mice exhibit several features of metabolically normal obese (MNO) subjects, distinct from metabolically abnormal (MAO) subjects, including decreased plasma leptin–to–adiponectin ratio (Fig. 5J) and plasma FFA levels upon insulin stimulation (39). Western blot analyses revealed that liver HIF-1α expression was decreased in HFD HAKO compared with HFD WT mice (Fig. 5K), with decreased Hif1a and Glut1 mRNA levels (Fig. 5L). To test whether the decrease in Hif1a mRNA expression is associated with decreased NF-κB activity, we measured phosphorylated p65 NF-κB levels in the liver of HFD WT and HAKO mice. As seen in fig. S7, phosphorylated p65 NF-κB levels were reduced in HFD HAKO mouse liver compared with HFD WT controls. On the other hand, the decrease in liver HIF-1α expression was not associated with increased hepatic oxygen tension (Fig. 5M), consistent with the idea that obesity-induced decreased liver interstitial oxygen tension is not low enough to increase hepatocyte HIF-1α expression. HFD HAKO mice expressed decreased levels of hepatic Dpp4 compared with HFD WT controls (Fig. 5N). Consistent with these results, plasma intact/active GLP-1 levels were significantly increased in HFD HAKO mice compared with HFD WT controls (Fig. 5O). These changes were observed without a decrease of adipose tissue Dpp4 expression (fig. S8). Together, these results suggest that adipocyte HIF-1α does not regulate adipocyte Dpp4 but can contribute to decreased incretin effects by regulating liver HIF-1α and Dpp4 levels.
DISCUSSION
Decreased circulating intact/active GLP-1 levels and incretin effects are important etiologic components of T2DM (22). Increased adiposity and insulin resistance are associated with this defect (23), but the underlying mechanisms are poorly understood. Here, we demonstrate a previously unknown mechanism for how obesity decreases intact/active GLP-1 levels by triggering increased hepatic HIF-1α expression and enhancing first-pass GLP-1 inactivation. We observed that the obesity-induced changes in circulating adipokines (i.e., increased leptin/adiponectin ratio) and FFA levels stimulate an isoform-selective increase in hepatic HIF-1α expression. This led to increased hepatic DPP4 expression and sinusoidal flow resistance (with increased Edn1 expression and decreased extracellular adenosine levels). Deletion of hepatocyte HIF-1α blocked obesity-induced hepatic DPP4 expression and sinusoidal flow resistance, decreasing first-pass GLP-1 degradation and increasing plasma active GLP-1 and insulin levels upon glucose challenge. These changes were sufficient to cause improved glucose and glucagon tolerance in HFD/obese glucose-intolerant mice. Administration of a GLP-1 receptor antagonist erased all of these beneficial effects on glycemic control, indicating that the effects of hepatocyte HIF-1α KO are mainly mediated by increased GLP-1 activity. Hepatocyte HIF-1α KO did not change insulin sensitivity, hepatic steatosis, or inflammation. Together, our results suggest that hepatocyte HIF-1α mediates obesity-induced multi-organ communication triggered by adipokine dysregulation, leading to impaired GLP-1 effects and glucose intolerance through increased first-pass GLP-1 inactivation.
While Dpp4 is ubiquitously expressed, several lines of evidence indicate that plasma intact/active GLP-1 levels are determined by specific spatiotemporal regulation of DPP4. For example, intact/active GLP-1 levels and incretin effects are decreased with increased plasma DPP4 activity in T2DM patients (21), whereas the rate of intravenously injected exogenous GLP-1 elimination is comparable in normal and T2DM subjects in the fasted state (15). Oral administration of a low-dose DPP4 inhibitor, which is effective only in the intestinal area (but in peripheral blood), exerts full glucose lowering effects with a partial increase in intact GLP-1 levels (50). Recently, Mulvihill et al. (13) reported that, although DPP4 is highly expressed in both endothelial cells and enterocytes, deletion of enterocyte DPP4 does not change plasma active GLP-1 levels or glucose tolerance. In contrast, deletion of endothelial cell DPP4 increases plasma intact/active GLP-1 levels and improves oral glucose tolerance. Moreover, previously, it was reported that hepatic GLP-1 extraction accounts for ~44% of intact GLP-1 inactivation in pigs (38). These results raise the possibility that post-enteric (e.g., the portohepatic) DPP4 expression/activity is a critical determinant of circulating intact/active GLP-1 levels. However, it is not known whether this hepatic GLP-1 inactivation is increased in obesity.
In the present study, we found that glucose-stimulated intact/active GLP-1 levels were ~56% lower in peripheral plasma compared with portal plasma in normal lean mice. This difference was increased to ~76% in HFD/obese mice, suggesting that obesity increases hepatic active GLP-1 elimination. In obese mice, Dpp4 expression was increased selectively in liver to hepatocytes and was not observed in nonparenchymal cells. Moreover, a substantial amount of hepatocyte-produced DPP4 was accumulated on the plasma membrane in obesity. Given that newly produced GLP-1 is first introduced into the portal circulation and that hepatic sinusoidal system represents a fenestrated capillary structure, it is plausible that increased liver DPP4 expression potentiates first-pass incretin hormone degradation in obesity. The increase in liver DPP4 expression was accompanied by decreased sinusoidal blood flow rate and velocity, which would provide more time for the increased liver DPP4 to mediate intact/active GLP-1 degradation in obesity. In HHKO mice, the obesity-induced decrease in peripheral-to-portal active GLP-1 ratio was blocked with normalization of the hepatic and plasma DPP4 levels and sinusoidal blood flow rate and velocity, suggesting that hepatocyte HIF-1α mediates those obesity-induced changes to increase first-pass active GLP-1 degradation. How hepatic HIF-1α and DPP4 confers selectivity to postprandial GLP-1 regulation remains an open question.
In regard to the mechanism by which hepatocyte HIF-1α increases sinusoidal flow resistance, we observed that deletion of HIF-1α blocks obesity-induced increased Edn1 expression and decreases extracellular adenosine levels. This is consistent with previous reports that show HIF-1α–mediated induction of Edn1 and ADA expression/activity (51, 52). ADA is a soluble protein readily detectable in blood circulation. Membrane-bound DPP4 binds to the soluble ADA, which facilitates its accumulation in the extracellular space where it acts to reduce local adenosine signaling (52). Therefore, obesity-induced increased liver membrane-bound DPP4 levels can also contribute to sinusoidal vasoconstriction by decreasing the local extracellular levels of adenosine.
Active GLP-1 levels were markedly higher in portal plasma compared with peripheral plasma, emphasizing potential local hepatic GLP-1 effects. Despite the ongoing controversy about the presence of GLP-1 receptor in hepatocytes (53–55), it has been shown that GLP-1 receptor agonist treatment directly regulates hepatocyte metabolism independent of its insulinotropic action (31–35, 56). Consistent with these reports, we observed that GLP-1 treatment decreases HGP in cultured hepatocytes. Moreover, hyperglycemic clamp studies demonstrated that GLP-1 suppresses hepatic glucagon action in the presence of S961 and somatostatin. These results suggest that GLP-1 can also suppress HGP independent of its insulinotropic action, which may be more important, particularly in insulin-resistant states. In obese HHKO mice, the decrease in hepatic DPP4 expression was associated with increased GLP-1–dependent HGP suppression. These results emphasize the importance of hepatic HIF-1α–dependent DPP4 regulation in determining local and systemic GLP-1 effects and glucose metabolism. Further studies are required to determine the signaling pathway of how GLP-1 suppresses HGP in an insulin-independent fashion.
In HFD HAKO mice, increased hepatic HIF-1α expression and decreased interstitial oxygen tension were dissociated without changes in body weight. Moreover, incubation of hepatocytes at 2% oxygen (equivalent to oxygen tension in obese liver) did not increase HIF-1α expression compared with 3% oxygen (equivalent to oxygen tension in lean liver). These results suggest that obesity-induced hepatic HIF-1α expression was not primarily due to decreased interstitial oxygen tension. Rather, treatment with hormones and nutrients that are increased in the blood circulation of obese mice interactively increased hepatocyte HIF-1α expression. The leptin + palmitic acid combination synergistically increased HIF-1α expression to a maximal level compared to all other factors and combinations except adiponectin, which suppressed leptin-stimulated HIF-1α stabilization. These results suggest that the combinatorial changes in plasma leptin, adiponectin, and FFA levels are key to the increase in hepatic HIF-1α expression in obesity. To test whether inhibition of leptin is sufficient to rebalance the effect of these combinatorial changes to increase liver HIF-1α expression, we used leptin-neutralizing antibodies. Administration of leptin-neutralizing antibodies markedly decreased obesity-induced hepatic HIF-1α expression. Moreover, increased adiponectin and decreased FFA levels in the plasma of HAKO mice were associated with decreased liver HIF-1α expression, even without changes in plasma leptin levels. Together, these results suggest that the combinatorial changes in plasma leptin, adiponectin, and FFA levels, but not the changes in single-factor (e.g., leptin) levels, are necessary for the full increase in liver HIF-1α expression in obesity.
Because leptin and adiponectin are adipocyte-specific hormones, and increased plasma FFA levels in obesity are mainly due to increased lipolysis in adipose tissue, it may be reasonable to conclude that adipose tissue dysfunction leads to increased hepatocyte HIF-1α expression and subsequent decrease in incretin effects. These results are consistent with a previous report that adipocyte HIF-1α KO mice exhibit increased plasma active GLP-1 levels in obesity (57). It was also reported that obesity-induced hepatic DPP4 production can stimulate adipose tissue inflammation and insulin resistance through an endocrine mechanism (58, 59). Therefore, it is likely that adipose tissue inflammation/dysfunction and hepatic HIF-1α–mediated DPP4 expression can constitute a feed-forward mechanism to exaggerate the disease pathology.
Here, we report a previously unknown pathway that obesity enhances plasma GLP-1 inactivation and impairs glucose tolerance by inducing HIF-1α–dependent liver DPP4 expression and sinusoidal flow resistance. Because hepatocyte-specific deletion of HIF-1α exerted beneficial effects to improve glucose tolerance without changing body weight in mice, it is reasonable to question whether the hepatic HIF-1α–DPP4 pathway is associated with metabolic health in obese humans (e.g., MAO versus MNO). In addition, it is possible that liver-specific inhibition of either or both HIF-α isoforms may be sufficient to improve T2DM and nonalcoholic fatty liver diseases in humans. While strongly supported by mouse genetics, all of these hypotheses remain to be tested in humans in the future.
MATERIALS AND METHODS
Study design
The current study was initially designed to (i) determine the effect of obesity on liver oxygen tension and HIF-1α and HIF-2α expression and (ii) define what role hepatocyte HIF-1α and HIF-2α play in obesity-associated changes in liver inflammation, steatosis, insulin resistance, and glucose intolerance. Initial results showed that, in HFD/obese states without massive liver damage and fibrosis, hepatocyte HIF-1α is not necessary for liver inflammation and insulin resistance but can reduce glucose-stimulated insulin secretion with decreased plasma active GLP-1 levels. The following studies were designed to (i) define what role hepatocytes HIF-1α and HIF-2α play in obesity-induced decreased incretin effects (glucose-stimulated insulin secretion) and plasma active GLP-1 degradation, (ii) define what role hepatocyte HIF-1α plays in obesity-induced liver microhemodynamics and portal blood pressure, and (iii) determine how obesity confers an isoform-selective induction of liver HIF-1α expression. For in vivo studies, age-matched Cre− Hif1afl/fl or Hif2afl/fl littermates were caged together with Cre+ KO mice and were used as WT controls. Animal numbers for each study type were determined by the investigators on the basis of data from previous similar experiments or from pilot studies. Results were confirmed in at least two separate cohort mouse experiments. For in vitro studies, at least three biological replicates were used. For in vivo experiments and biochemical assays, mouse or sample identities were not blinded, but in most cases, the sequence of analyzed tubes and mice was randomized.
Animals
To generate adipocyte- or hepatocyte-specific HIF-1α or HIF-2α KO mice, Hif1afl/fl or Hif2afl/fl mice were crossed to mice expressing Cre recombinase under the control of the Adiponectin or Albumin promoter, respectively. To obtain relatively equal numbers of KO and WT littermates, breeding pairs were set up by mating Hif1afl/fl(or Hif2afl/fl):Cre+ and Hif1afl/fl(or Hif2afl/fl):Cre− mice. All mice were on the C57BL6 background and housed in colony cages in 12-hour light/12-hour dark cycles. For HFD studies, 7- to 8-week-old male mice were subjected to 60% HFD for 10 weeks or the indicated time periods (Research Diets Inc., catalog no. D12492). Glucose and insulin tolerance tests were performed as described previously (24, 39, 60). For glucagon tolerance, mice were fasted for 6 hours and intraperitoneally injected with glucagon (15 μg/kg), and blood glucose levels were measured at indicated time points. For the glucose or glucagon tolerance tests with ex9 pretreatment, mice were fasted for 5.5 hours. After measuring blood glucose levels at time −30 min, mice were intraperitoneally injected with 5 μg of ex9. Thirty minutes later (time 0), blood glucose levels were measured at indicated time points. Hyperglycemic clamp experiments were designed and optimized for measuring liver-specific GLP-1 effects and sensitivity. For these studies, mice were surgerized for jugular vein cannulation. After 5 days of recovery, mice were fasted for 6 hours and infused with d-[3H]glucose (PerkinElmer) plus somatostatin (6 μg/kg per min) for 60 min. After tracer equilibration, blood samples were collected at −10 and 0 min (basal). Glucose (50% dextrose) and tracer (5 μCi/hour) plus somatostatin (6 μg/kg per min), glucagon (50 ng/kg per min), S961 (5 nmol/kg per min), and active GLP-1 (20 ng/kg per min) were then infused into the jugular vein. GLP-1 stimulates β cell insulin secretion and suppresses plasma insulin clearance and alpha cell glucagon secretion; however, we wanted to measure liver-specific GLP-1 effects independent of insulinotropic action. Even with a relatively high-dose somatostatin infusion, plasma insulin levels were significantly increased by GLP-1 infusion to supraphysiological levels. Therefore, we also infused S961 insulin receptor antagonist along with somatostatin and glucagon during the glucose clamp studies. Because S961 is a competitive inhibitor of insulin receptor, the dose of S961 was determined experimentally as a minimal dose that blocks insulin effects at the experimental condition. Since exogenous glucagon infusion in the presence of S961 and somatostatin caused hyperglycemia, to obtain proper window for HGP suppression by GLP-1, glucose levels were clamped at 600 mg/dl. Blood glucose levels were monitored every 10 min, and glucose infusion rate was adjusted as necessary. Steady-state blood glucose levels were maintained at 600 ± 10 mg/dl for the last 20 min or longer, without changing glucose infusion rate, and blood samples were collected at 110 and 120 min (clamped). Specific activity and insulin levels were measured from the basal and clamped plasma samples. All animal procedures were performed in accordance with an Institutional Animal Care and Use Committee–approved protocol and the research guidelines for the use of laboratory animals of the University of California San Diego or the Korea Advanced Institute of Science and Technology.
Tissue interstitial oxygen tension
The oxygen partial pressure was measured using carbon fiber electrodes (Carbostar-1, Kation Scientific, Minneapolis, MN). The tip of the electrode was coated with 5% Nafion (Sigma-Aldrich, St. Louis, MO) to increase oxygen specificity. The process consisted of three individual Nafion coats. The microelectrodes were polarized at −0.8 V relative to a silver-silver chloride reference electrode (Cypress Systems, Lawrence, KS). Oxygen measurements were performed after 3-hour fasting using the two-electrode system (working and reference electrode), and the current generated was measured with a potentiostat and electrometer (Keithley model 610C; Cleveland, OH). The microelectrodes are calibrated at 37°C with 0, 5, 10, and 21% O2 gases (Airgas, Los Angeles, CA), and tissues were superfused (0.1 ml/min) with physiological Krebs salt solution. The tissue was maintained at 35° to 37°C by the heated Krebs solution. The solution was spread on the tissue as a thin film, drained into a platter, and drawn off by suction. The solution was equilibrated with 95% N2 and 5% carbon dioxide, which maintained the superfusate at a pH of 7.4 and minimized oxygen delivery to the tissue from the atmosphere. Oxygen measurements were made by penetrating the tissue with the microelectrode tip. The reference electrode was placed in the bath, and the microelectrode was placed in a shielded holder and advanced toward the measurement site with a micromanipulator. A long-working distance ×10 Leitz objective was used to direct the electrode to the measurement site. Before measurements, the electrode tip was immersed in the supernatant suffusion solution and the current was registered. The supernatant suffusion solution was set as 0 mmHg reference point. Upon introduction into the tissue, the microelectrodes responded with a time constant that was estimated to be of the order of 10 s. A stable reading was obtained within 30 s, and upon reaching the current plateau value, the electrode was extracted from the tissue and the tip was maintained within the suffusing saline solution. The unit of tissue O2 tension was converted from mmHg to % later. Oxygen tension was measured at >30 different regions of each mouse liver. Appearance rate denotes chances for observing indicated O2 tension in a random spot of the mouse liver.
Microhemodynamics
A video image–shearing method was used to measure vessel diameter (D). Changes in arteriolar and venular diameter from baseline were used as indicators of a change in vascular tone. Arteriolar and venular centerline velocities were measured on-line using the photodiode cross-correlation method (Photo Diode/Velocity Tracker Model 102B, Vista Electronics, San Diego, CA). The measured centerline velocity (V) was corrected according to vessel size to obtain the mean red blood cell velocity. Blood flow (Q) was calculated from the measured values as Q = π × V (D/2)2.
Histology
Immunohistochemistry analyses and β cell mass were performed as described previously (60). Images were captured using a NanoZoomer slide scanner system with NanoZoomer Digital Pathology software (Hamamatsu) or confocal fluorescence microscopy. Microscopic images were analyzed using ImageJ software.
Plasma/serum and tissue hormone concentration and content
Plasma insulin (ALPCO), C-peptide (ALPCO), DPP4 (Thermo Fisher Scientific), active and total GLP-1 (ALPCO), GIP (Crystal Chem), and glucagon (Mercodia) levels were measured by enzyme-linked immunosorbent assay (ELISA) in accordance with the manufacturers’ instruction. To prevent degradation of active GLP-1 or active GIP by DPP4 during or after sample collection, DPP4 inhibitor (Millipore, catalog no. DPP4-010) was briefly added to each tube after sample collection. Plasma and tissue DPP4 activity was measured with a fluorometric assay kit (BioVision). Pancreatic insulin content was determined as described previously (60). Briefly, whole pancreas from each mouse was ground in 10 ml of acidic ethanol using a Polytron tissue homogenizer (Kinematica), and insulin levels were measured by ELISA in the tissue lysates. Total insulin content was calculated by multiplying the lysate insulin concentration, dilution factor, and total volume of the tissue lysate.
Gluconeogenic activity in primary hepatocytes
To isolate primary hepatocytes, mice were infused through the inferior vena cava with a calcium-free Hepes-phosphate buffer (pH 7.4) for 10 min followed by a collagenase solution (Liberase TM, Roche) for 10 min. The digested livers were excised, and hepatocytes were collected and washed five times in buffer by centrifuging at 70g for 5 min. Cells were further purified by centrifugation (2400g for 10 min) over a Percoll density gradient (1.06 g/ml). Primary mouse hepatocytes were allowed to attach for 6 hours on collagen-coated plates in Williams’ Medium E (Life Technologies, catalog no. 12551-032) fortified with nonessential amino acids, GlutaMAX (Life Technologies, catalog no. 35050-061), antibiotics, 10% fetal bovine serum, and dexamethasone (10 nM) and cultured overnight in the same medium without serum. Cultures were then washed in Hepes phosphate-salt-bicarbonate (HPSB) buffer (10 mM Hepes, 4 mM KCl, 125 mM NaCl, 0.85 mM KH2PO4, 1.25 mM Na2HPO4, 1 mM MgCl2, 1 mM CaCl2, and 15 mM NaHCO3) containing 0.2% FFA-free bovine serum albumin (BSA) and incubated in the same buffer containing GLP-1 (100 nM), Exendin-4 (30 nM), insulin (10 nM), and/or glucagon (10 ng/ml) and substrates for 3 hours in a 5% CO2 incubator. 14C-pyruvate (2 mM, 0.5 μCi pyruvate per incubation) was used as substrate. Incubations were carried out in 0.5-ml buffer in 24-well plates containing 0.25 million cells per well. At the end of incubation, the buffer solutions were transferred to 1.7-ml microcentrifuge tubes and added with 0.25 ml of 5% ZnSO4 and 0.25 ml of 0.3 N Ba(OH)2 suspensions to each tube, followed by addition of 0.5 ml of water. After centrifugation, supernatants were transferred to a fresh set of tubes and assayed for radiolabeled glucose released into the medium by separation of radiolabeled glucose by mixed-bed ion exchange resins, AG 501-X8 resins (Bio-Rad). Two hundred milligrams of resins was added to each tube, vortexed intermittently for 15 min, and centrifuged, and the supernatants were transferred to scintillation vials for counting radioactivity. Cells on the plates were dissolved in 1 N NaOH for protein estimation.
ADA activity and extracellular adenosine level measurements
Primary mouse hepatocytes cultured in six-well plates were washed with HPSB buffer containing 0.2% FFA-free BSA, 2 mM glucose, and 2 mM pyruvate and then incubated in the same buffer for 3 hours in a 5% CO2 incubator. Supernatant and cell pellets were collected and saved for further assays. Extracellular adenosine levels were determined in the hepatocyte-conditioned media using a colorimetric assay kit (BioVision, catalog no. K327). Plasma membrane fraction of the cell pellets (Abcam, catalog no. ab65400) was subjected to ADA activity assays using a colorimetric assay kit (BioVision, catalog no. K321).
Western blot analysis
Tissues and cells were lysed in lysis buffer [20 mM tris-HCl (pH 7.4), 100 mM NaCl, 1.5 mM MgCl2, and 0.1% (v/v) NP-40] containing a protease and phosphatase inhibitor cocktail (Roche Diagnostics) and then centrifuged at 13,000 rpm for 15 min at 4°C. The supernatants were separated in SDS–polyacrylamide gel electrophoresis gels (Bio-Rad) and electrotransferred to polyvinylidene difluoride membranes. The membranes were blocked for 2 hours in tris-buffered saline with Tween 20 (TBST) [10 mM tris-HCl and 0.1% Triton X-100 (pH 7.4)] containing 5% BSA and then incubated with specific antibodies at 4°C overnight: HIF-1α (Abcam, catalog no. ab-2185), HIF-2α (Novus Biologicals, catalog no. NB100-122), heat shock protein 90 (HSP90) (Santa Cruz Biotechnology, catalog no. SC-13119), actin (Sigma-Aldrich, catalog no. A2228), and vinculin (Cell Signaling Technology, catalog no. 4650). After washing with fresh TBST, the membrane was incubated with secondary antibody conjugated with horseradish peroxidase specific to rabbit or mouse immunoglobulin G (1:5000 dilution; Jackson ImmunoResearch Laboratories) and visualized using the enhanced chemiluminescence system (Millipore, catalog no. WBKLS0050) followed by autoradiography or Bio-Rad ChemiDoc XRS+ imaging system. The intensity of the bands in the autoradiograms was measured using ImageJ software.
Quantitative real-time polymerase chain reaction
Total RNA was extracted with TRIzol reagent (Invitrogen) or RNeasy Mini Kit (Qiagen, Hilden, Germany). Synthesis of complementary DNA (cDNA) was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time polymerase chain reaction (PCR) was performed using Power SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences are available upon request.
Liver cell sorting
To isolate primary hepatocytes and nonparenchymal cells, livers were perfused and digested using pronase/collagenase method. Single-cell suspensions were centrifuged at 50g for 5 min to pellet the hepatocyte fraction. The remaining nonparenchymal cell fraction was collected and used for immediate analysis or subjected to further fractionation. After gradient centrifugation (15% Nycodenz), hepatic immune cells were sorted (FACSAria, BD Biosciences) or analyzed by flow cytometry (FACSCanto, BD Biosciences; FlowJo software, Treestar). For flow cytometry analysis, cells were stained with LIVE/DEAD Aqua to exclude dead cells and blocked with CD16/32 monoclonal antibody (Thermo Fisher Scientific, catalog no. 14-0161-81). The fluorescence-labeled antibodies against CD45 (eBioscience, catalog no. 83-0451042) for leukocytes; CD11b (eBioscience, catalog no. 11-012-82), F4/80 (eBioscience, catalog no. 25-4801-82), and Ly6C (Thermo Fisher Scientific, catalog no. 12-5932-80) for hepatic macrophages; or CD3 (eBioscience, catalog no. 56-0032-82), CD4 (eBioscience, catalog no. 17-0041-82), and CD8 (BioLegend, catalog no. 100722) for lymphocytes were used.
Statistical analysis
The results are shown as means ± SEM. Statistical analyses were performed by Student’s t test (for comparison between two groups) or one- or two-way analysis of variance (ANOVA) (for comparison between multiple groups or in two groups at multiple time points, respectively, with post hoc t tests between the individual groups); P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank J. Olefsky and D. Brenner for critical comments and discussion on this paper. We thank C. Walser, J. Pimentel, J. B. Seo, and W. K. Choi for supporting surgical instrumentation of animals for microhemodynamics studies, mammalian cell culture, and/or biochemical assays. Funding: This study was supported by UCSD/UCLA Diabetes Research Center P&F grant DK063491 and National Heart, Lung, and Blood Institute grants R01 HL126945 and R01 HL138116. Author contributions: Y.S.L. conceived and supervised the project, acquired funding, performed and designed the majority of experiments, and wrote the manuscript. M.R. performed glucose clamp experiments and supported the biochemical assays. P.C. measured hepatic interstitial O2 tension and hemodynamics. G.K.B. performed in vitro hepatocyte glucose production assays and supported liver cell fractionation. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw4176/DC1
Fig. S1. Epididymal white adipose tissue mass in HFD WT and HHKO mice.
Fig. S2. Hepatocyte HIF-1α is not necessary for obesity-induced hepatic steatosis and inflammation.
Fig. S3. Dpp4 expression in different tissues from lean and obese mice.
Fig. S4. mRNA expression analysis in WT and HHKO mouse adipose tissue, liver, and primary hepatocytes.
Fig. S5. HIF-1α and its target gene expression in hepatocytes at different oxygen conditions or upon ACM or leptin + palmitic acid treatment.
Fig. S6. Schematic representation of a novel model of how obesity enhances GLP-1 inactivation and decreases incretin effects.
Fig. S7. Western blot analysis of phosphorylated p65 NF-κB expression in the liver of HFD WT and HAKO mice.
Fig. S8. Epididymal white adipose tissue expression of Hif1a, Epas1/Hif2a, and Dpp4 mRNAs.
REFERENCES AND NOTES
- 1.Olefsky J. M., Glass C. K., Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Halban P. A., Polonsky K. S., Bowden D. W., Hawkins M. A., Ling C., Mather K. J., Powers A. C., Rhodes C. J., Sussel L., Weir G. C., beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37, 1751–1758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y. S., Wollam J., Olefsky J. M., An integrated view of immunometabolism. Cell 172, 22–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drucker D. J., Habener J. F., Holst J. J., Discovery, characterization, and clinical development of the glucagon-like peptides. J. Clin. Invest. 127, 4217–4227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holst J. J., Knop F. K., Vilsboll T., Krarup T., Madsbad S., Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 34 (suppl. 2), S251–S257 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nauck M., Stöckmann F., Ebert R., Creutzfeldt W., Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29, 46–52 (1986). [DOI] [PubMed] [Google Scholar]
- 7.Knop F. K., Aaboe K., Vilsbøll T., Vølund A., Holst J. J., Krarup T., Madsbad S., Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes. Metab. 14, 500–510 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Faerch K., Torekov S. S., Vistisen D., Johansen N. B., Witte D. R., Jonsson A., Pedersen O., Hansen T., Lauritzen T., Sandbæk A., Holst J. J., Jørgensen M. E., GLP-1 response to oral glucose is reduced in prediabetes, screen-detected type 2 diabetes, and obesity and influenced by sex: The ADDITION-PRO study. Diabetes 64, 2513–2525 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Vilsbøll T., Krarup T., Deacon C. F., Madsbad S., Holst J. J., Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 50, 609–613 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Deacon C. F., Johnsen A. H., Holst J. J., Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J. Clin. Endocrinol. Metab. 80, 952–957 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Meier J. J., Nauck M. A., Kranz D., Holst J. J., Deacon C. F., Gaeckler D., Schmidt W. E., Gallwitz B., Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 53, 654–662 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Mulvihill E. E., Drucker D. J., Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 35, 992–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulvihill E. E., Varin E. M., Gladanac B., Campbell J. E., Ussher J. R., Baggio L. L., Yusta B., Ayala J., Burmeister M. A., Matthews D., Bang K. W. A., Ayala J. E., Drucker D. J., Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metab. 25, 152–165 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Seino Y., Fukushima M., Yabe D., GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 1, 8–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vilsboll T., Agersø H., Krarup T., Holst J. J., Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J. Clin. Endocrinol. Metab. 88, 220–224 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Holst J. J., The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Conarello S. L., Li Z., Ronan J., Roy R. S., Zhu L., Jiang G., Liu F., Woods J., Zycband E., Moller D. E., Thornberry N. A., Zhang B. B., Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 100, 6825–6830 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pala L., Ciani S., Dicembrini I., Bardini G., Cresci B., Pezzatini A., Giannini S., Mannucci E., Rotella C. M., Relationship between GLP-1 levels and dipeptidyl peptidase-4 activity in different glucose tolerance conditions. Diabet. Med. 27, 691–695 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Manell H., Staaf J., Manukyan L., Kristinsson H., Cen J., Stenlid R., Ciba I., Forslund A., Bergsten P., Altered plasma levels of glucagon, GLP-1 and glicentin during OGTT in adolescents with obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 1181–1189 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Kjems L. L., Holst J. J., Vølund A., Madsbad S., The influence of GLP-1 on glucose-stimulated insulin secretion: Effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 52, 380–386 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Lamers D., Famulla S., Wronkowitz N., Hartwig S., Lehr S., Ouwens D. M., Eckardt K., Kaufman J. M., Ryden M., Müller S., Hanisch F.-G., Ruige J., Arner P., Sell H., Eckel J., Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 60, 1917–1925 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iepsen E. W., Torekov S. S., Holst J. J., Therapies for inter-relating diabetes and obesity - GLP-1 and obesity. Expert. Opin. Pharmacother. 15, 2487–2500 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Matikainen N., Bogl L. H., Hakkarainen A., Lundbom J., Lundbom N., Kaprio J., Rissanen A., Holst J. J., Pietiläinen K. H., GLP-1 responses are heritable and blunted in acquired obesity with high liver fat and insulin resistance. Diabetes Care 37, 242–251 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Lee Y. S., Li P., Huh J. Y., Hwang I. J., Lu M., Kim J. I., Ham M., Talukdar S., Chen A., Lu W. J., Bandyopadhyay G. K., Schwendener R., Olefsky J., Kim J. B., Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60, 2474–2483 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keith B., Johnson R. S., Simon M. C., HIF1alpha and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nath B., Levin I., Csak T., Petrasek J., Mueller C., Kodys K., Catalano D., Mandrekar P., Szabo G., Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 53, 1526–1537 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim W. Y., Safran M., Buckley M. R. M., Ebert B. L., Glickman J., Bosenberg M., Regan M., Kaelin W. G., Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 25, 4650–4662 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tajima T., Goda N., Fujiki N., Hishiki T., Nishiyama Y., Senoo-Matsuda N., Shimazu M., Soga T., Yoshimura Y., Johnson R. S., Suematsu M., HIF-1α is necessary to support gluconeogenesis during liver regeneration. Biochem. Biophys. Res. Commun. 387, 789–794 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Choi J. H., Park M. J., Kim K. W., Choi Y. H., Park S. H., An W. G., Yang U. S., Cheong J., Molecular mechanism of hypoxia-mediated hepatic gluconeogenesis by transcriptional regulation. FEBS Lett. 579, 2795–2801 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Lee Y. H., Wang M.-Y., Yu X.-X., Unger R. H., Glucagon is the key factor in the development of diabetes. Diabetologia 59, 1372–1375 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Dardevet D., Moore M. C., Neal D., DiCostanzo C. A., Snead W., Cherrington A. D., Insulin-independent effects of GLP-1 on canine liver glucose metabolism: Duration of infusion and involvement of hepatoportal region. Am. J. Physiol. Endocrinol. Metab. 287, E75–E81 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Drucker D. J., Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Ding X., Saxena N. K., Lin S., Gupta N. A., Anania F. A., Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 43, 173–181 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahman K., Liu Y., Kumar P., Smith T., Thorn N. E., Farris A. B., Anania F. A., C/EBP homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab. Invest. 96, 895–908 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma S., Mells J. E., Fu P. P., Saxena N. K., Anania F. A., GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLOS ONE 6, e25269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahrén B., Thomaseth K., Pacini G., Reduced insulin clearance contributes to the increased insulin levels after administration of glucagon-like peptide 1 in mice. Diabetologia 48, 2140–2146 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Röhrborn D., Eckel J., Sell H., Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett. 588, 3870–3877 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Deacon C. F., Pridal L., Klarskov L., Olesen M., Holst J. J., Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am. J. Phys. 271, E458–E464 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Lee Y. S., Kim J.-w., Osborne O., Oh D. Y., Sasik R., Schenk S., Chen A., Chung H., Murphy A., Watkins S. M., Quehenberger O., Johnson R. S., Olefsky J. M., Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taniguchi C. M., Finger E. C., Krieg A. J., Wu C., Diep A. N., LaGory E. L., Wei K., McGinnis L. M., Yuan J., Kuo C. J., Giaccia A. J., Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 19, 1325–1330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moon J.-O., Welch T. P., Gonzalez F. J., Copple B. L., Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G582–G592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramakrishnan S. K., Ramakrishnan S. K., Zhang H., Takahashi S., Centofanti B., Periyasamy S., Weisz K., Chen Z., Uhler M. D., Rui L., Gonzalez F. J., Shah Y. M., HIF2α is an essential molecular brake for postprandial hepatic glucagon response independent of insulin signaling. Cell Metab. 23, 505–516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rankin E. B., Rha J., Selak M. A., Unger T. L., Keith B., Liu Q., Haase V. H., Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell. Biol. 29, 4527–4538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun K., Halberg N., Khan M., Magalang U. J., Scherer P. E., Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 33, 904–917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seo J. B., Riopel M., Cabrales P., Huh J. Y., Bandyopadhyay G. K., Andreyev A. Y., Murphy A. N., Beeman S. C., Smith G. I., Klein S., Lee Y. S., Olefsky J. M., Knockdown of ANT2 reduces adipocyte hypoxia and improves insulin resistance in obesity. Nat. Metab. 1, 86–97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang C., Qu A., Matsubara T., Chanturiya T., Jou W., Gavrilova O., Shah Y. M., Gonzalez F. J., Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K. Y., Gesta S., Boucher J., Wang X. L., Kahn C. R., The differential role of Hif1beta/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 14, 491–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stern J. H., Rutkowski J. M., Scherer P. E., Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 23, 770–784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frede S., Stockmann C., Freitag P., Fandrey J., Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem. J. 396, 517–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waget A., Cabou C., Masseboeuf M., Cattan P., Armanet M., Karacav M., Castel J., Garret C., Payros G., Maida A., Sulpice T., Holst J. J., Drucker D. J., Magnan C., Burcelin R., Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 152, 3018–3029 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Hu J., Discher D. J., Bishopric N. H., Webster K. A., Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem. Biophys. Res. Commun. 245, 894–899 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Eltzschig H. K., Faigle M., Knapp S., Karhausen J., Ibla J., Rosenberger P., Odegard K. C., Laussen P. C., Thompson L. F., Colgan S. P., Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26. Blood 108, 1602–1610 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta N. A., Mells J., Dunham R. M., Grakoui A., Handy J., Saxena N. K., Anania F. A., Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 51, 1584–1592 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svegliati-Baroni G., Saccomanno S., Rychlicki C., Agostinelli L., De Minicis S., Candelaresi C., Faraci G., Pacetti D., Vivarelli M., Nicolini D., Garelli P., Casini A., Manco M., Mingrone G., Risaliti A., Frega G. N., Benedetti A., Gastaldelli A., Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 31, 1285–1297 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Panjwani N., Mulvihill E. E., Longuet C., Yusta B., Campbell J. E., Brown T. J., Streutker C., Holland D., Cao X., Baggio L. L., Drucker D. J., GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE−/− mice. Endocrinology 154, 127–139 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Nishizawa M., Moore M. C., Shiota M., Gustaversuson S. M., Snead W. L., Neal D. W., Cherrington A. D., Effect of intraportal glucagon-like peptide-1 on glucose metabolism in conscious dogs. Am. J. Physiol. Endocrinol. Metab. 284, E1027–E1036 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Kihira Y., Miyake M., Hirata M., Hoshina Y., Kato K., Shirakawa H., Sakaue H., Yamano N., Izawa-Ishizawa Y., Ishizawa K., Ikeda Y., Tsuchiya K., Tamaki T., Tomita S., Deletion of hypoxia-inducible factor-1alpha in adipocytes enhances glucagon-like peptide-1 secretion and reduces adipose tissue inflammation. PLOS ONE 9, e93856 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghorpade D. S., Ozcan L., Zheng Z., Nicoloro S. M., Shen Y., Chen E., Blüher M., Czech M. P., Tabas I., Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baumeier C., Schlüter L., Saussenthaler S., Laeger T., Rödiger M., Alaze S. A., Fritsche L., Häring H.-U., Stefan N., Fritsche A., Schwenk R. W., Schürmann A., Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol. Metab. 6, 1254–1263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee Y. S., Morinaga H., Kim J. J., Lagakos W., Taylor S., Keshwani M., Perkins G., Dong H., Kayali A. G., Sweet I. R., Olefsky J., The fractalkine/CX3CR1 system regulates beta cell function and insulin secretion. Cell 153, 413–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw4176/DC1
Fig. S1. Epididymal white adipose tissue mass in HFD WT and HHKO mice.
Fig. S2. Hepatocyte HIF-1α is not necessary for obesity-induced hepatic steatosis and inflammation.
Fig. S3. Dpp4 expression in different tissues from lean and obese mice.
Fig. S4. mRNA expression analysis in WT and HHKO mouse adipose tissue, liver, and primary hepatocytes.
Fig. S5. HIF-1α and its target gene expression in hepatocytes at different oxygen conditions or upon ACM or leptin + palmitic acid treatment.
Fig. S6. Schematic representation of a novel model of how obesity enhances GLP-1 inactivation and decreases incretin effects.
Fig. S7. Western blot analysis of phosphorylated p65 NF-κB expression in the liver of HFD WT and HAKO mice.
Fig. S8. Epididymal white adipose tissue expression of Hif1a, Epas1/Hif2a, and Dpp4 mRNAs.