

Abstract
Complex karyotype (CK) with ≥3 abnormalities is detected in 10-12% of patients with acute myeloid leukemia (AML) and associated with poor prognosis. The most common unbalanced abnormalities found in CK result in loss of material from the 5q, 7q and/or 17p chromosome arms. The presence of 5q, 7q and/or 17p abnormalities denotes typical CK and their absence denotes atypical CK. Since molecular features of CK-AML are not well-characterized, we investigated mutational status of 81 leukemia/cancer-associated genes in 160 clinically well-characterized patients. They included 136 patients with ≥3 exclusively unbalanced chromosome abnormalities, 96 of whom had a typical CK and 40 atypical CK, and 24 patients with ≥1 balanced abnormality in addition to ≥2 unbalanced ones. Patients with atypical CK-AML differed from those with typical CK-AML: they carried TP53 mutations less often (P<0.001) and more often PHF6 (P=0.008), FLT3-TKD (P=0.02), MED12 (P=0.02) and NPM1 (P=0.02) mutations. They were younger (P=0.007), had higher WBC (P=0.001) and percentages of marrow (P<0.001) and blood (P=0.006) blasts, higher complete remission rates (P=0.02) and longer overall survival (P<0.001), thus indicating that atypical and typical CK-AMLs constitute distinct disease subtypes. We also identified smaller patient subsets within both typical and atypical CK-AML that differed molecularly and clinically.
Keywords: acute myeloid leukemia, complex karyotype, next-generation sequencing, gene mutations, clinical outcome
INTRODUCTION
Patients with acute myeloid leukemia (AML) presenting with a complex karyotype (CK), defined as a karyotype with ≥3 chromosome abnormalities, comprise 10-12% of all AML patients and thus constitute the second largest (following cytogenetically normal AML) cytogenetic subset of AML patients.1–4 In most studies, the definition of CK excludes karyotypes consisting of ≥3 abnormalities that include t(8;21)(q22;q22), inv(16)(p13q22)/t(16;16)(p13;q22), t(15;17)(q22;q12-21), t(9;11)(p22;q23), any balanced rearrangement involving band 11q23, or any “primary balanced abnormality”.2,3,5–10 Additionally, the 2017 European LeukemiaNet (ELN) recommendations11 also exclude from the CK category karyotypes containing such World Health Organization (WHO)-designated recurring translocations or inversions as t(6;9)(p23;q34), inv(3)(q21q26)/t(3;3)(q21;q26) and t(9;22)(q34;q11.2).
Although each chromosome in the human karyotype can participate in various structural and numerical abnormalities constituting CK in AML, the involvement of particular chromosomes or chromosome arms in these aberrations is non-random.7,12,13 Unbalanced abnormalities predominate and mostly result in loss of chromosome segments. The most commonly lost are parts of the long arm of chromosome 5 (5q), detected in ~80% of patients with CK-AML, followed by loss of material from 7q and 17p, each occurring in approximately one-half of the cases.13 Abnormalities of 5q, 7q and 17p often occur together, and ~85% of all patients with CK-AML harbor at least one of these abnormalities. Based on these data, we have divided CKs into typical and atypical categories, with the former defined as CK with ≥3 abnormalities that include 5q, 7q and/or 17p loss, and the latter as CK with ≥3 abnormalities other than the aforementioned ones.13,14
Despite being the second largest cytogenetic subset of AML, CK-AML is relatively poorly characterized at the molecular level. The high incidence of TP53 mutations and their adverse influence on CK-AML patients’ outcome are well-known,8,13,15–17 but information on the mutational involvement of other genes has only recently begun to be collected as part of studies using next-generation sequencing (NGS) in large series of AML patients.18–20 To our knowledge, no large NGS study has focused specifically on molecular characterization of CK-AML.
Therefore, we analyzed mutational status of 81 leukemia/cancer-associated genes and clinical characteristics of a clinically well-characterized cohort of 160 CK-AML patients. Our data show that patients with typical and atypical CKs differ with regard to their mutational patterns, clinical features and outcome, and thus should be considered as separate disease subtypes.
METHODS
Patients, treatment, and cytogenetic studies
Among 1602 adults diagnosed with de novo AML (other than acute promyelocytic leukemia) whose pretreatment bone marrow (BM) or blood samples were subjected to NGS analysis,14 we identified 208 patients with ≥3 chromosome abnormalities. However, based on the 2017 ELN recommendations,11 we excluded 48 patients with ≥3 abnormalities that included the WHO-designated recurring balanced abnormalities, thus leaving 160 patients with CK-AML who are the subject of this study (Figure 1).
Figure 1.
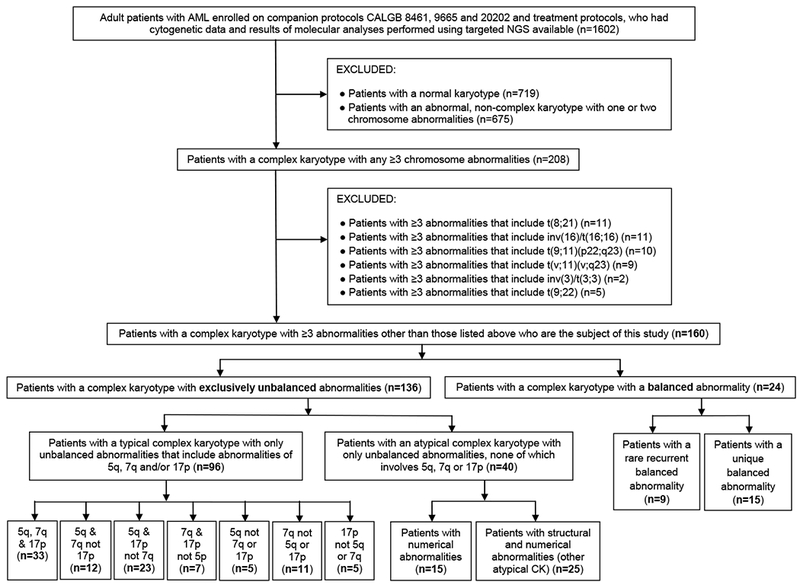
Overview of the study design. AML denotes acute myeloid leukemia; CALGB, Cancer and Leukemia Group B; CK, complex karyotype; NGS, next-generation sequencing.
All 160 patients were enrolled onto Cancer and Leukemia Group B (CALGB) front-line treatment protocols,21–34 and all patients, except nine,34 received intensive induction with cytarabine and an anthracycline (for details see Supplementary Information). CALGB is now part of the Alliance for Clinical Trials in Oncology (Alliance). The median follow-up time for living patients was 6.8 years (range, 3.2-10.2). All protocols were approved by the institutional review board of each participating institution, and written informed consent for the research use of their specimens was obtained from all patients before enrollment in accordance with the Declaration of Helsinki.
Cytogenetic analyses were performed by the CALGB/Alliance-approved institutional laboratories. Pretreatment BM and/or blood samples were subjected to short-term (24- to 48-hour) unstimulated cultures, and karyotypes were interpreted according to the International System for Human Cytogenetic Nomenclature.35 All results were reviewed centrally.36
Subsets of complex karyotype
Based on the presence or absence of specific chromosome abnormalities, we discerned subsets of CK (Figure 1). First, we separated CKs that included ≥1 non-WHO-designated balanced abnormalities (e.g., reciprocal translocations, inversions; n=24), either known to be recurrent37 (n=9) or hitherto not reported in the literature37 (n=15; thereafter referred to as “unique”), from CKs containing exclusively unbalanced abnormalities (n=136). We did this because balanced chromosome abnormalities are considered to represent primary chromosome abnormalities and almost always create gene fusions that play a pivotal role in leukemogenesis.
Among patients with CK with only unbalanced chromosome abnormalities (n=136), we identified 96 patients who had a typical CK, that is, CK containing abnormalities leading to loss of material from 5q, 7q and/or 17p. The remaining 40 patients had an atypical CK, in which no abnormality resulted in 5q, 7q or 17p loss.
Within the atypical CK category (n=40), we recognized two subtypes. Karyotypes of the first (n=15) contained numerical abnormalities, either exclusively (n=11) or predominantly (n=4); each of the four patients with predominantly numerical aberrations had only one unbalanced structural abnormality, which in three patients was found in only one of three or four abnormal clones. Thereafter, we refer to this subset as “atypical CK with numerical abnormalities”. The second atypical CK subset (n=25), which we thereafter refer to as “other atypical CK”, comprised patients with a mixture of structural and numerical aberrations, with the former usually being more numerous.
Finally, to perform exploratory analyses, we identified seven subtypes within the typical CK category (n=96), which were defined by combinations of 5q, 7q and 17p abnormalities (Figure 1).
Molecular analyses
Mononuclear cells were enriched through Ficoll-Hypaque gradient centrifugation and cryopreserved until use. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany). The mutational status of 80 protein-coding genes was established centrally at The Ohio State University by targeted amplicon sequencing using two separate gene panels on the MiSeq platform (Illumina, San Diego, CA). Details of the assay and genes included in the panels are provided in the Supplementary Information. Because of sample availability, the mutational status of the NF1 gene was determined in 90 patients. The data were analyzed using the MuCor program,38 followed by visual inspection and quality control of the BAM files. The gene mutation status was deemed to be not evaluable if <15 reads were present. Variants (missense, nonsense or frameshift) were considered to be mutations if they were not reported in the 1000 Genome or dbSNP142 databases. Only variants occurring with variant allele fraction (VAF) ≥0.10 were included in the analyses. In addition to 80 genes analyzed by NGS, the CEBPA mutational status was determined as previously described,39 thus bringing the total number of genes analyzed to 81. Only patients harboring biallelic CEBPA mutations were considered to be mutated.11 Testing for internal tandem duplication of the FLT3 gene (FLT3-ITD) was performed as described previously.40
The presence of copy number variations in the 17p chromosome arm was determined by genotyping of the available patient samples (n=121) using Illumina Omni-Express single nucleotide polymorphism (SNP) arrays, followed by analysis with Illumina GenomeStudio plugin cnvpartition v3.2.0. Copy-neutral loss of heterozygosity (CN-LOH) was set to 1Mb.
Statistical analyses
Baseline characteristics were compared using the two-sided Fisher’s exact test for categorical variables and two-sided Wilcoxon rank-sum test for continuous variables.41 All other P-values were from one-sided tests. A P-value of <0.05 was considered statistically significant. Outcome analyses included only patients who did not undergo allogeneic stem-cell transplantation in first complete remission (CR) (n=136). Disease-free survival (DFS) was measured from the CR date until the date of relapse or death from any cause, and relapse-free patients were censored at last follow-up. Overall survival (OS) was measured from the date on study until the date of death from any cause; patients alive at last follow-up were censored. For multivariable analyses, we used stepwise logistic regression for modeling CR achievement and Cox proportional hazard stepwise regression for modeling DFS and OS. The dataset was locked on July 27, 2017. Data collection and statistical analyses were performed by the Alliance Statistics and Data Center using SAS 9.4.
RESULTS
Mutational landscape of all patients with CK-AML
In the entire cohort of 160 patients, we detected 359 mutations in 59 of 81 genes analyzed, with a median of two mutations per patient (range, 0-7). No patient with CK-AML had a mutation in any of the following 22 genes analyzed: ARAF, ATM, BCL2, BRAF, CBL, CCND2, CSNK1A1, GATA1, IKZF3, JAK2, KLHL6, MAPK1, MYD88, PIK3CD, PLEKHG5, PRKD3, RAD21, SF3A1, TGM7, U2AF2, XPO1 or ZMYM3.
As expected, the most often mutated gene was TP53, whose mutations were detected in 81 of 160 patients (51%). Other recurrent mutations were less common, with the most frequent being mutations in the TET2 (n=24, 15% of the entire patient cohort), DNMT3A (n=19, 12%), NF1 (n=10, 11% of 90 patients analyzed), NRAS (n=15, 9%), IDH2 (n=12, 8%), RUNX1 (n=12, 8%), NPM1 (n=11, 7%), SRSF2 (n=10, 6%), ZRSR2 (n=10, 6%), ASXL1 (n=8, 5%) and PHF6 (n=8, 5%) genes, and FLT3-ITD (n=8, 5%). However, the aforementioned and other, less common mutations were not uniformly distributed among CK subsets, and major differences in frequencies of particular gene mutations, as well as in cytogenetic and clinical characteristics, emerged when we compared specific subsets of CK-AML patients.
Typical versus atypical complex karyotype
In addition to abnormalities of 5q, 7q and 17p, whose presence or absence have been used to define, respectively, typical and atypical CK-AML, these CK categories also differed with regard to the degree of karyotype complexity, with atypical CKs containing fewer cytogenetic abnormalities than typical CKs (median, 4 vs 9 abnormalities; P<0.001).
On the other hand, there was no significant difference in the number of gene mutations per patient between patients with atypical and those with typical CK-AML (P=0.23; Table 1). Regarding particular gene mutations, the most striking difference between the CK categories was the rarity of TP53 mutations in patients with atypical CK-AML (4 of 40 patients, 10%) as opposed to their presence in most patients with typical CK-AML (65 of 96 patients, 67%; P<0.001). Moreover, when present in atypical CK-AML patients, TP53 mutations had lower VAFs than TP53 mutations found in patients with typical CK-AML (Supplementary Figure S1). The difference between atypical and typical CK-AML was even more evident when we also considered genomic rearrangements of 17p encompassing the TP53 locus, mostly losses of 17p material but also CN-LOH. Among patients with a successful SNP array analysis (including 69 patients with typical and 33 with atypical CK-AML), an alteration of TP53 (i.e., TP53 mutation and/or loss of TP53 locus or CN-LOH) was detected in 88% of patients with typical but only in 15% of patients with atypical CK-AML (P<0.001; Figure 2).
Table 1.
Frequencies of gene mutations in patients with acute myeloid leukemia with atypical complex karyotype and in those with typical complex karyotype
| Genea | Atypical CK n=40 | Typical CK n=96 | P-valueb |
|---|---|---|---|
| TP53, n (%) | <0.001 | ||
| Mutated | 4 (10) | 65 (67) | |
| Wild-type | 36 (90) | 31 (33) | |
| TET2, n (%) | 0.13 | ||
| Mutated | 10 (25) | 13 (14) | |
| Wild-type | 30 (75) | 83 (86) | |
| DNMT3A, n (%) | 0.41 | ||
| Mutated | 7 (18) | 11 (11) | |
| R882 | 2 | 5 | |
| Non-R882 | 5 | 6 | |
| Wild-type | 33 (83) | 85 (89) | |
| NF1, n (%) | 0.42 | ||
| Mutated | 1 (5) | 7 (14) | |
| Wild-type | 21 (95) | 44 (86) | |
| NRAS, n (%) | 0.18 | ||
| Mutated | 6 (15) | 6 (6) | |
| Wild-type | 34 (85) | 90 (94) | |
| RUNX1, n (%) | 0.75 | ||
| Mutated | 4 (10) | 8 (8) | |
| Wild-type | 36 (90) | 88 (92) | |
| ZRSR2, n (%) | 0.06 | ||
| Mutated | 6 (15) | 4 (4) | |
| Wild-type | 34 (85) | 92 (96) | |
| IDH2, n (%) | 0.48 | ||
| Mutated | 4 (10) | 6 (6) | |
| Wild-type | 36 (90) | 90 (94) | |
| NPM1, n (%) | 0.02 | ||
| Mutated | 6 (15) | 3 (3) | |
| Wild-type | 34 (85) | 90 (97) | |
| SRSF2, n (%) | 0.72 | ||
| Mutated | 3 (8) | 6 (6) | |
| Wild-type | 35 (92) | 88 (94) | |
| PHF6, n (%) | 0.008 | ||
| Mutated | 6 (15) | 2 (2) | |
| Wild-type | 34 (85) | 94 (98) | |
| FLT3-ITD, n (%) | 0.22 | ||
| Present | 4 (11) | 4 (5) | |
| Absent | 31 (89) | 84 (95) | |
| ASXL1, n (%) | 0.42 | ||
| Mutated | 3 (8) | 4 (4) | |
| Wild-type | 37 (93) | 92 (96) | |
| IDH1, n (%) | 0.36 | ||
| Mutated | 3 (8) | 3 (3) | |
| Wild-type | 37 (92) | 93 (97) | |
| BCOR, n (%) | 0.67 | ||
| Mutated | 1 (3) | 5 (5) | |
| Wild-type | 39 (98) | 91 (95) | |
| FLT3-TKD, n (%) | 0.02 | ||
| Present | 4 (11) | 1 (1) | |
| Absent | 34 (89) | 93 (99) | |
| PTPN11, n (%) | 0.63 | ||
| Mutated | 2 (5) | 3 (3) | |
| Wild-type | 38 (95) | 93 (97) | |
| PRKCB, n (%) | 0.32 | ||
| Mutated | 0 (0) | 4 (4) | |
| Wild-type | 40 (100) | 92 (96) | |
| SMARCA2, n (%) | 0.32 | ||
| Mutated | 0 (0) | 4 (4) | |
| Wild-type | 40 (100) | 92 (96) | |
| ETV6, n (%) | 0.58 | ||
| Mutated | 2 (5) | 2 (2) | |
| Wild-type | 38 (95) | 94 (98) | |
| STAG2, n (%) | 0.58 | ||
| Mutated | 2 (5) | 2 (2) | |
| Wild-type | 38 (95) | 94 (98) | |
| TYK2, n (%) | 0.58 | ||
| Mutated | 2 (5) | 2 (2) | |
| Wild-type | 38 (95) | 94 (98) | |
| WT1, n (%) | 0.58 | ||
| Mutated | 2 (5) | 2 (2) | |
| Wild-type | 38 (95) | 94 (98) | |
| KIT, n (%) | 1.00 | ||
| Mutated | 1 (3) | 3 (3) | |
| Wild-type | 36 (97) | 91 (97) | |
| KMT2A, n (%) | 1.00 | ||
| Mutated | 1 (3) | 3 (3) | |
| Wild-type | 39 (98) | 93 (97) | |
| PLCG2, n (%) | 1.00 | ||
| Mutated | 1 (3) | 3 (3) | |
| Wild-type | 39 (98) | 93 (97) | |
| MED12, n (%) | 0.02 | ||
| Mutated | 3 (8) | 0 (0) | |
| Wild-type | 37 (93) | 96 (100) | |
| BRD4, n (%) | 1.00 | ||
| Mutated | 1 (3) | 2 (2) | |
| Wild-type | 39 (98) | 94 (98) | |
| NOTCH1, n (%) | 1.00 | ||
| Mutated | 1 (3) | 2 (2) | |
| Wild-type | 39 (98) | 94 (98) | |
| PIK3CG, n (%) | 1.00 | ||
| Mutated | 1 (3) | 2 (2) | |
| Wild-type | 39 (98) | 94 (98) | |
| SMC3, n (%) | 1.00 | ||
| Mutated | 1 (3) | 2 (2) | |
| Wild-type | 39 (98) | 94 (98) | |
| KRAS, n (%) | 0.08 | ||
| Mutated | 2 (5) | 0 (0) | |
| Wild-type | 37 (95) | 96 (100) | |
| CCND1, n (%) | 0.50 | ||
| Mutated | 1 (3) | 1 (1) | |
| Wild-type | 39 (98) | 95 (99) | |
| GSK3B, n (%) | 0.50 | ||
| Mutated | 1 (3) | 1 (1) | |
| Wild-type | 39 (98) | 95 (99) | |
| IKZF1, n (%) | 0.50 | ||
| Mutated | 1 (3) | 1 (1) | |
| Wild-type | 39 (98) | 95 (99) | |
| SMC1A, n (%) | 0.50 | ||
| Mutated | 1 (3) | 1 (1) | |
| Wild-type | 39 (98) | 95 (99) | |
| AKT1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 40 (100) | 94 (98) | |
| BRINP3, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 40 (100) | 94 (98) | |
| CEBPA, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 34 (100) | 83 (98) | |
| HIST1H1E, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 40 (100) | 94 (98) | |
| SETBP1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 40 (100) | 94 (98) | |
| U2AF1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 2 (2) | |
| Wild-type | 40 (100) | 94 (98) | |
| BCORL1, n (%) | 0.29 | ||
| Mutated | 1 (3) | 0 (0) | |
| Wild-type | 39 (98) | 96 (100) | |
| HNRNPK, n (%) | 0.29 | ||
| Mutated | 1 (3) | 0 (0) | |
| Wild-type | 39 (98) | 96 (100) | |
| IL7R, n (%) | 0.29 | ||
| Mutated | 1 (3) | 0 (0) | |
| Wild-type | 38 (97) | 96 (100) | |
| JAK1, n (%) | 0.29 | ||
| Mutated | 1 (3) | 0 (0) | |
| Wild-type | 39 (98) | 96 (100) | |
| SF3B1, n (%) | 0.29 | ||
| Mutated | 1 (3) | 0 (0) | |
| Wild-type | 39 (98) | 96 (100) | |
| AXL, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| BTK, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| FBXW7, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 39 (100) | 95 (99) | |
| GATA2, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| JAK3, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| MAPK3, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| PTEN, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| RAF1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| SAMHD1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| SF1, n (%) | 1.00 | ||
| Mutated | 0 (0) | 1 (1) | |
| Wild-type | 40 (100) | 95 (99) | |
| Total number of mutations | 0.23 | ||
| Median | 3 | 2 | |
| Range | 0-7 | 0-6 | |
Abbreviations: CK, complex karyotype; FLT3-ITD, internal tandem duplication of the FLT3 gene; FLT3-TKD, tyrosine kinase domain mutation in the FLT3 gene.
Only genes mutated in at least one patient are listed, and they are arranged according to the frequency of mutations, from most to least frequent. No mutation was detected in the following genes tested: ARAF, ATM, BCL2, BRAF, CBL, CCND2, CSNK1A1, CTNNB1, EZH2, GATA1, IKZF3, JAK2, KLHL6, MAPK1, MYD88, PIK3CD, PLEKHG5, PRKD3, RAD21, SF3A1, SYK, TGM7, U2AF2, XPO1 and ZMYM3.
P-values for categorical variables are from Fisher’s exact test, P-values for continuous variables are from Wilcoxon rank sum test.
Figure 2.
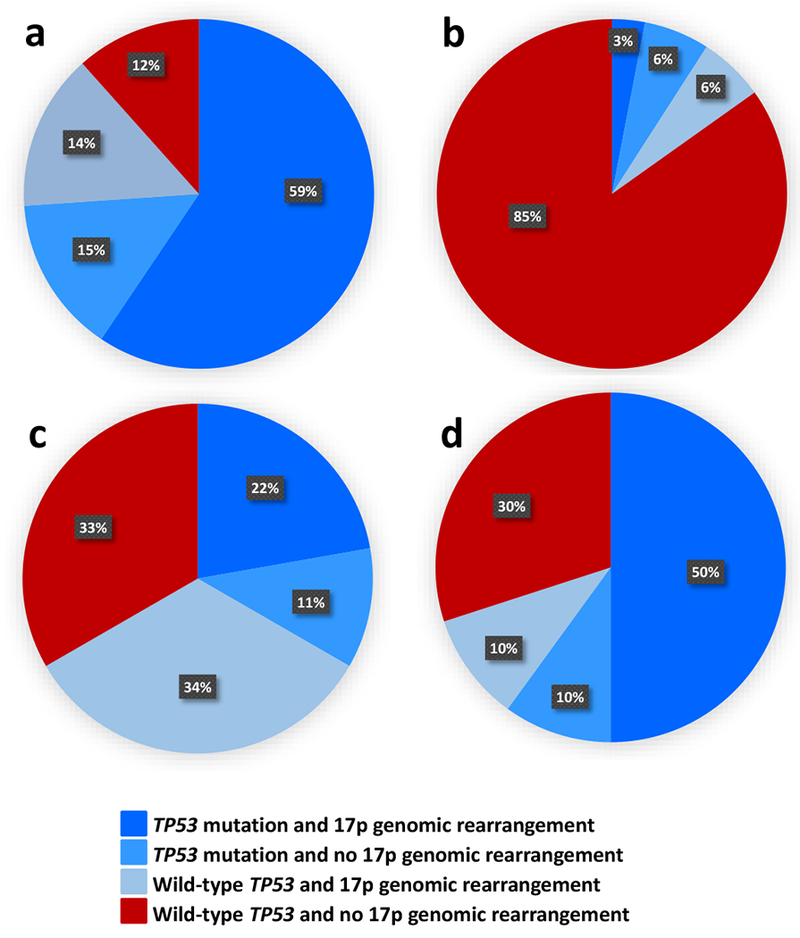
Distribution of the combinations of TP53 mutations and 17p genomic rearrangements (determined using SNP arrays) in subsets of patients with acute myeloid leukemia (AML) and a complex karyotype (CK): (a) typical CK (n=69), (b) atypical CK (n=33), (c) CK with rare recurrent balanced chromosome abnormalities (n=9), (d) CK with unique balanced chromosome abnormalities (n=10). Dark blue color denotes patients with both TP53 mutation and 17p genomic rearrangement; lighter blue, patients with TP53 mutation and no 17p genomic rearrangement; light blue, patients with wild-type TP53 and 17p genomic rearrangement present, and red color indicates patients with wild-type TP53 and no 17p genomic rearrangement. All patients in the first three subsets combined, indicated by the various shades of blue, are considered to harbor an alteration of TP53 (i.e., TP53 mutation, deletion of 17p resulting in loss of TP53 locus and/or copy-neutral loss of heterozygosity encompassing TP53 locus). Typical CK-AML (a) clearly differs from atypical CK-AML (b) with regard to the frequency of TP53 alterations (88% vs 15%; P<0.001).
In contrast, PHF6, another tumor suppressor gene,18 was more frequently mutated in patients with atypical than typical CK-AML (15% vs 2%; P=0.008), as were the MED12 (8% vs 0%; P=0.02) and NPM1 (15% vs 3%; P=0.02) genes and FLT3-TKD (11% vs 1%; P=0.02; Table 1, Figure 3). Interestingly, all mutations in the MED12 gene (Supplementary Figure S2), which is located at Xq13.1, had high VAFs (0.96, 0.74, 0.51), which in two patients (no. 14 and 29) were markedly higher than VAFs of other detected mutations, suggesting that MED12 mutations may represent early mutational events, which might be associated with the FAB M0 morphology (patients no. 5 and 14; Supplementary Table S1). We also compared involvement of genes categorized into the major AML-associated functional groups18 and found additional differences (Supplementary Table S2). Beside lower incidence of mutations in tumor suppressor genes (28% vs 71%; P<0.001), mostly driven by paucity of TP53 mutations, patients with atypical CKs carried mutations in genes encoding kinases (30% vs 13%; P=0.03) and in RAS pathway genes (25% vs 9%; P=0.03) more often.
Figure 3.
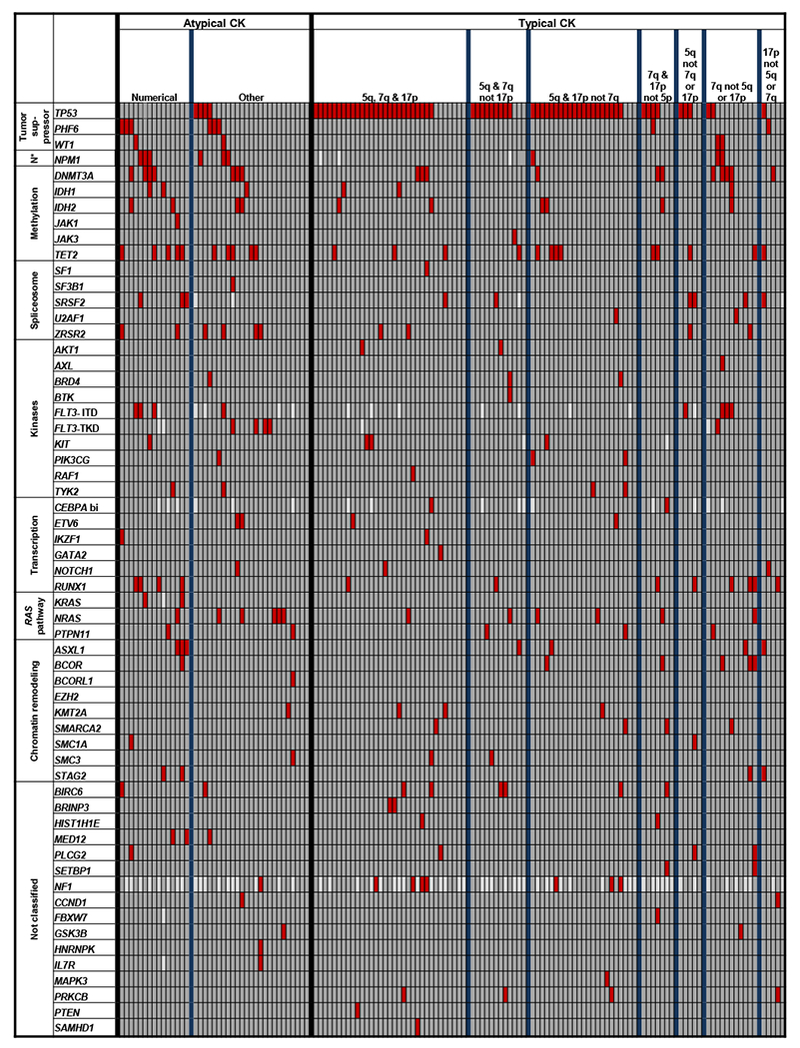
Oncoprint of mutations detected in acute myeloid leukemia patients with atypical and those with typical complex karyotype (CK). Subsets identified within both atypical and typical CK are separated by thicker blue vertical lines. Each column represents an individual patient, and each row represents a single gene. The genes are clustered into the previously reported functional groups,18 with N* indicating “NPM1”. “CEBPA bi” indicates biallelic mutations of the CEBPA gene. Red color denotes gene mutation, darker grey, wild-type status of the gene, and lighter grey, mutation status not determined.
Clinically, patients with atypical CK-AML were younger (P=0.007; median age, 53 vs 59 years), had higher white blood cell (WBC) counts (P=0.001; 23.8 vs 6.0×109/l), percentages of BM (76% vs 46%; P<0.001) and blood (59% vs 28%; P=0.006) blasts, and tended to have extramedullary involvement more often (24% vs 11%; P=0.06; Table 2).
Table 2.
Comparison of pretreatment characteristics between patients with acute myeloid leukemia with typical and those with atypical complex karyotype
| Characteristic | Atypical CK n=40 | Typical CK n=96 | P-valuea |
|---|---|---|---|
| Age, years | 0.007 | ||
| Median | 53 | 59 | |
| Range | 18-77 | 30-84 | |
| Sex, n (%) | 0.23 | ||
| Male | 30 (75) | 61 (64) | |
| Female | 10 (25) | 35 (36) | |
| Race, n (%) | 0.78 | ||
| White | 34 (85) | 79 (88) | |
| Non-white | 6 (15) | 11 (12) | |
| Hemoglobin, g/dl | 0.14 | ||
| Median | 9.5 | 9.0 | |
| Range | 5.5-14.4 | 6.0-14.7 | |
| Platelet count, ×109/l | 0.96 | ||
| Median | 54 | 49 | |
| Range | 6-376 | 4-323 | |
| WBC count, ×109/l | 0.001 | ||
| Median | 23.8 | 6.0 | |
| Range | 0.8-225.3 | 0.7-137.6 | |
| Bone marrow blasts, % | <0.001 | ||
| Median | 76 | 46 | |
| Range | 20-92 | 13-97 | |
| Blood blasts, % | 0.006 | ||
| Median | 59 | 28 | |
| Range | 0-98 | 0-99 | |
| Extramedullary involvement, n (%) | 9 (24) | 10 (11) | 0.06 |
| Number of chromosome abnormalities | <0.001 | ||
| Median | 4 | 9 | |
| Range | 3-15 | 3-33 | |
Abbreviations: CK, complex karyotype; WBC, white blood cell.
P-values for categorical variables are from Fisher’s exact test, P-values for continuous variables are from Wilcoxon rank sum test.
Importantly, although treatment outcomes were generally poor, atypical CK-AML patients had higher CR rates (59% vs 35%; P=0.02), a trend towards longer DFS (P=0.08; 3-year rates, 10% vs 0%; Figure 4a) and longer OS (P<0.001; 3-year rates, 24% vs 1%; Figure 4b) than typical CK-AML patients. In multivariable analyses, the type of CK remained prognostically significant for all outcome endpoints (Table 4). The differences in clinical outcome were also present when we considered patients aged <60 years and those aged ≥60 years separately. In both age groups, OS of patients with atypical CK-AML was longer than that of patients with typical CK-AML (younger patients: P=0.005; 3-year rates, 25% vs 3%; older patients: P=0.002; 3-year rates, 21% vs 0%), and there were trends for patients with atypical CK-AML to have higher CR rates (younger patients: 60% vs 38%, P=0.11; older patients: 57% vs 33%, P=0.12). Younger patients with atypical CK-AML had a longer DFS (P=0.05; 3-year rates, 17% vs 0%), whereas DFS did not differ significantly between the CK-AML groups among older patients (Table 3; Supplementary Figures S3 and S4).
Figure 4.
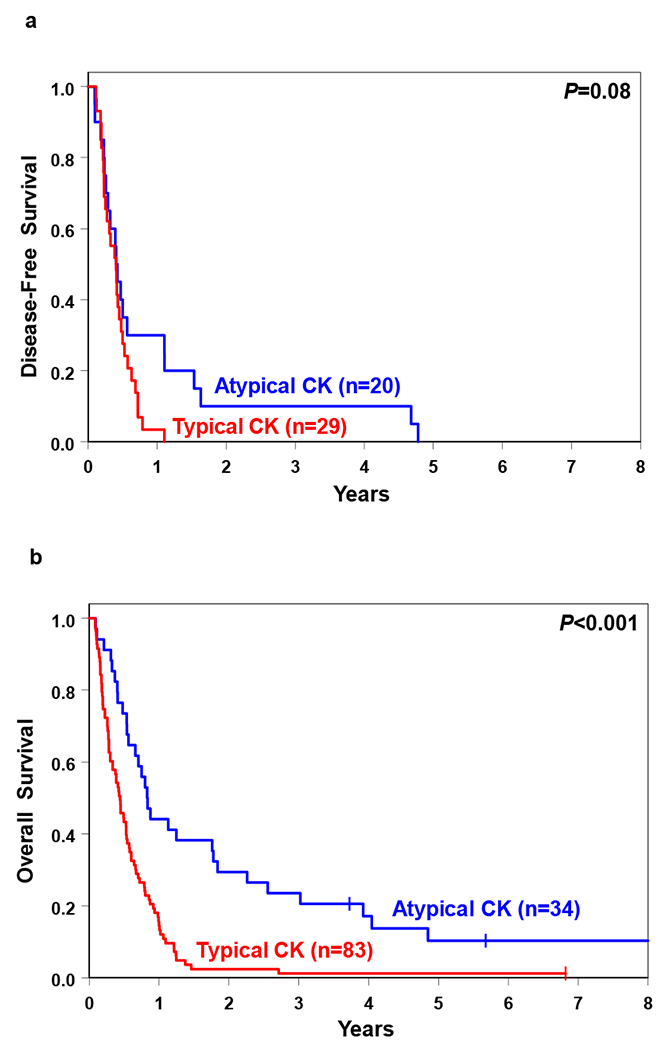
Comparison of (a) disease-free survival and (b) overall survival of acute myeloid leukemia patients with typical and atypical complex karyotype (CK).
Table 4.
Multivariable analyses of patients with acute myeloid leukemia and complex karyotype
| Variable | Complete remission | Disease-free survival | Overall survival | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value | |
| Type of complex karyotype, typical vs atypical | 0.38 (0.17-0.85) | 0.02 | 2.03 (1.03-4.01) | 0.04 | 1.86 (1.11-3.12) | 0.02 |
| Hemoglobin, continuous, per 1-unit increase | 1.27 (1.05-1.53) | 0.01 | ||||
| TP53, mutated vs wild-type | 1.75 (1.11-2.75) | 0.02 | ||||
| Age, continuous, per 10-year increase | 1.16 (1.02-1.32) | 0.02 | ||||
Abbreviations: CI, confidence interval; HR, hazard ratio; OR, odds ratio. An odds ratio <1 means a lower CR rate for the first category listed for the categorical variables. A hazard ratio >1 corresponds to a higher risk of an event for higher values of continuous variables and the first category listed of a dichotomous variable. Variables were considered for inclusion in the multivariable models if they had a univariable P-value of ≤0.20. Variables considered for inclusion in the model for achievement of CR were: the type of complex karyotype (typical vs atypical), sex (male vs female), race (white vs nonwhite), white blood cell count [(WBC) as a continuous variable, in 50-unit increments], hemoglobin (as a continuous variable, in 1-unit increments), the number of chromosome abnormalities (as a continuous variable), NPM1 mutational status (mutated vs wild-type), NRAS mutational status (mutated vs wild-type) and TP53 mutational status (mutated vs wild-type). In the model for disease-free survival, we considered the type of complex karyotype (typical vs atypical), hemoglobin (as a continuous variable, in 1-unit increments), extramedullary involvement (present vs absent), the number of chromosome abnormalities (as a continuous variable), DNMT3 mutational status (mutated vs wild-type), PHF6 mutational status (mutated vs wild-type), TET2 mutational status (mutated vs wild-type) and TP53 mutational status (mutated vs wild-type). Variables considered for inclusion in the model for overall survival were the type of complex karyotype (typical vs atypical), age (as a continuous variable, in 10-year increments), sex (male vs female), the number of chromosome abnormalities (as a continuous variable), FLT3-ITD (present vs absent), NF1 mutational status (mutated vs wild-type), PHF6 mutational status (mutated vs wild-type) and TP53 mutational status (mutated vs wild-type). See the Supplementary Information for a full list of variables evaluated in univariable analyses for all three outcome endpoints.
Table 3.
Outcomes of acute myeloid leukemia patients with typical and those with atypical complex karyotype
|
All patients | |||
| Endpoint | Atypical CK n=34 | Typical CK n=83 | P-valuea |
| Complete remission, n (%) | 20 (59) | 29 (35) | 0.02 |
| Disease-free survival | 0.08 | ||
| Median, years | 0.4 | 0.4 | |
| % Disease-free at 1 year (95% CI) | 30 (12-50) | 3 (0-15) | |
| % Disease-free at 3 years (95% CI) | 10 (2-27) | 0 | |
| Overall survival | <0.001 | ||
| Median, years | 0.8 | 0.4 | |
| % Alive at 1 year (95% CI) | 44 (27-60) | 14 (8-23) | |
| % Alive at 3 years (95% CI) | 24 (11-39) | 1 (0-6) | |
| Patients aged <60 years | |||
| Endpoint | Atypical CK n=20 | Typical CK n=40 | P-valuea |
| Complete remission, n (%) | 12 (60) | 15 (38) | 0.11 |
| Disease-free survival | 0.05 | ||
| Median, years | 0.5 | 0.4 | |
| % Disease-free at 1 year (95% CI) | 42 (15-67) | 0 | |
| % Disease-free at 3 years (95% CI) | 17 (3-41) | 0 | |
| Overall survival | 0.005 | ||
| Median, years | 0.9 | 0.6 | |
| % Alive at 1 year (95% CI) | 45 (23-65) | 23 (11-36) | |
| % Alive at 3 years (95% CI) | 25 (9-45) | 3 (0-11) | |
| Patients aged ≥60 years | |||
| Endpoint | Atypical CK n=14 | Typical CK n=43 | P-valuea |
| Complete remission, n (%) | 8 (57) | 14 (33) | 0.12 |
| Disease-free survival | 0.87 | ||
| Median, years | 0.4 | 0.3 | |
| % Disease-free at 1 year (95% CI) | 13 (1-42) | 7 (0-28) | |
| % Disease-free at 3 years (95% CI) | 0 | 0 | |
| Overall survival | 0.002 | ||
| Median, years | 0.8 | 0.4 | |
| % Alive at 1 year (95% CI) | 43 (18-66) | 7 (2-17) | |
| % Alive at 3 years (95% CI) | 21 (5-45) | 0 | |
Abbreviations: CK, complex karyotype; CI, confidence interval.
P-values for categorical variables are from Fisher’s exact test, P-values for the time to event variables are from the log-rank test.
TP53 mutations and TP53 alterations in typical complex karyotype
Although TP53 mutations are regarded as a molecular hallmark of patients with CK-AML,8,13,15–17 they do not occur in all patients and, as shown above, are most frequent in patients with typical, but rare in those with atypical, CK-AML. However, even among typical CK-AML patients, approximately one-third do not harbor TP53 mutations. Thus, we compared cytogenetic, molecular and clinical features between typical CK-AML patients with and without TP53 mutations.
Cytogenetically, TP53-mutated patients (n=65) had more complex karyotypes (P=0.02; median, 10 vs 7 chromosome abnormalities), and more often harbored 5q (89% vs 48%, P<0.001) and 17p (78% vs 55%, P=0.03), but not 7q (63% vs 71%, P=0.50), abnormalities than patients with wild-type TP53 (n=31). Molecularly, patients with wild-type TP53 more often carried BCOR mutations (13% vs 2%; P=0.04), and were the only ones with mutations in SMARCA2 and PLCG2 (Supplementary Table S3). Clinically, TP53-mutated patients were older (P=0.008; median age, 63 vs 54 years) and tended to be less frequently male (57% vs 77%; P=0.07). Although their CR rates and DFS were similar, patients with TP53 mutations had shorter OS than those without (P=0.003; Supplementary Table S4).
TP53 mutations often coexist with genomic abnormalities of 17p, such as deletions of a normal copy of TP53, resulting in the hemizygous TP53 mutation, or a homologous recombination involving 17p that results in uniparental disomy and homozygous TP53 mutations.8 TP53 mutations and/or genomic losses (hereafter named TP53 alterations) were shown to be the most important prognostic factor in patients with CK-AML.8 However, Rücker et al.8 did not distinguish between typical and atypical CK-AML. Our analysis has revealed that TP53 alterations are also associated with shorter OS (P<0.001; 3-year rates, 0% vs 13%; Supplementary Table S5) when only typical CK-AML patients are analyzed. Moreover, patients without TP53 alterations had less complex karyotypes (P<0.001; median, 4 vs 10 chromosome abnormalities) and more often harbored mutations in NPM1, WT1, BCOR and FLT3-ITD (Supplementary Table S6).
Atypical complex karyotypes with numerical abnormalities versus other atypical complex karyotypes
Within the atypical CK-AML category (n=40), patients with only numerical abnormalities (n=15) tended to carry fewer chromosome abnormalities than 25 patients with other atypical CK-AML (median, 3 vs 4 abnormalities; P=0.07). In the latter subset, most frequent were +8 (found in 32% of patients) and loss of 1p (28%), 12p (24%) and 9q (20%), whereas in patients with only numerical abnormalities most common were +8 (60%), +4 (40%), +13 (33%) and +21 (33%).
Molecularly, patients with numerical abnormalities had more mutations than patients with other atypical CK-AML (median, 3 vs 2; P=0.03), and they were the only ones harboring mutations in RUNX1, ASXL1, SRSF2, KRAS and STAG2. In contrast, TP53 mutations, generally infrequent in atypical CK-AML, were found exclusively in patients with other atypical CK-AML (Figure 3, Supplementary Table S7).
There were no significant differences in pretreatment characteristics between the subsets, except for lower platelet counts in patients with numerical abnormalities (P=0.009; median 32 vs 69 × 109/l). Likewise, CR rates, DFS and OS of patients in both subsets of atypical CK-AML were not significantly different (Supplementary Table S8).
Typical CK with abnormalities of 7q, but not of 5q or 17p, differs from typical CKs with other combinations of 5q, 7q and 17p abnormalities
We performed exploratory analyses of seven subtypes of typical CK-AML delineated by combinations of 5q, 7q and 17p abnormalities. Although most of these subtypes were similar, patients with only 7q abnormalities (n=11) turned out to constitute a subtype that differed cytogenetically, molecularly and clinically from the remaining patients combined (n=85; Supplementary Tables S9 and S10). Karyotypes of patients with only 7q abnormalities contained fewer chromosome abnormalities (P<0.001; median, 5 vs 10), but several gene mutations were more frequent in these patients. Specifically, more common were FLT3-ITD and mutations in BCOR, WT1, DNMT3A, NPM1 and RUNX1. In contrast, TP53 mutations were much less frequent in this typical CK-AML subtype than in patients with other combinations of 5q, 7q and 17p abnormalities (18% vs 74%; P<0.001; Supplementary Table S9). Additionally, among patients with SNP array data available, none of eight patients with only 7q abnormalities had submicroscopic genomic alterations of 17p involving the TP53 locus. In contrast, among the remaining typical CK-AML patients, seven of 12 (58%; P=0.01) patients without cytogenetically detectable 17p abnormalities carried submicroscopic 17p alterations uncovered by SNP array analysis.
Moreover, patients with only 7q abnormalities were younger (P=0.04; median age, 49 vs 61 years), had lower platelet counts (P=0.03; median, 32 vs 53 × 109/l) and higher WBC counts (P=0.05; median, 15.7 vs 5.5 × 109/l) and percentages of BM blasts (68% vs 44%; P=0.02; Supplementary Table S10). Although CR rates were not significantly different (50% vs 33%; P=0.31), OS of patients with only 7q abnormalities was longer than OS of the remaining patients with typical CK-AML (P<0.001; 3-year rates, 10% vs 0%; Supplementary Figure S5).
Complex karyotypes containing balanced chromosome abnormalities: rare recurrent versus unique abnormalities
The most striking feature of a small group of patients with CK-AML with a rare recurrent balanced chromosome abnormality was a paucity of gene mutations. Four of nine patients in this group did not carry a single mutation in any of the 81 genes analyzed, and the number of mutations was lower than that in 15 patients with CK-AML with a unique (i.e., not previously reported) balanced abnormality (median, 1 vs 2; Supplementary Figure S6 and Table S11). Only the TP53 mutation was recurrent, detected in three (33%) patients. Although there were no significant differences in outcome or most pretreatment features (data not shown) between CK-AML patients with a rare recurrent balanced abnormality and those with unique balanced aberrations, the latter were older (median, 61 vs 39 years; P=0.05). Their karyotypes tended to be more complex (median, 9 vs 5 abnormalities), and contained abnormalities of 5q, 7q and/or 17p in almost three-fourths of these patients.
When we compared clinical outcome (Supplementary Table S12) and the distribution of gene mutations (Supplementary Table S13) between CK-AML patients with unique balanced aberrations and patients with typical CK-AML with exclusively unbalanced abnormalities, we found no significant differences (with a single exception of SF3B1 mutations being more frequent in CK-AML with unique balanced aberrations). These data suggest that patients with a unique balanced aberration should not be prevented from being classified in the typical CK-AML category.
DISCUSSION
The salient finding of our study is the demonstration that patients with typical and atypical CK-AML differ considerably with regard to mutation patterns, pretreatment characteristics and clinical outcome. Remarkably, TP53 mutations, considered to constitute a hallmark of complex karyotypes in AML,8,13–18 were virtually absent in patients with atypical CK-AML, and, when present, had relatively low VAFs. Instead, atypical CK-AML patients carried more often PHF6, NPM1 and MED12 mutations and FLT3-TKD, which were either very rare (PHF6, NPM1, FLT3-TKD) or not observed at all (MED12) in patients with typical CK-AML. Moreover, patients with atypical CK-AML had more frequently mutations in genes comprising such functional groups as kinases and the RAS pathway, thus underscoring the fact that atypical and typical CKs differ profoundly with respect to their genetic make-up.
Somatic mutations in the MED12 gene, which encodes a subunit of the Mediator complex that plays a vital role in regulation of transcription of most protein-coding genes, have been reported to be recurrent in and associated with markers of poor prognosis in chronic lymphocytic leukemia,42 but, to date, rarely detected in AML.14 Although MED12 mutations were infrequent, their VAFs were higher than VAFs of all other mutations found in two patients and higher than the VAF of the TP53 mutation in the third, indicating that MED12 mutations represent early and pathogenetically important mutational events. Identification of more patients with atypical CK-AML carrying MED12 mutations is necessary to confirm their potential association with FAB M0 morphology (detected in two of three patients) or to determine whether MED12 mutations have prognostic significance.
In addition to dissimilar cytogenetic and molecular characteristics, patients with atypical CK-AML had better clinical outcome than those with typical CK-AML in the entire patient cohort, both in univariable and multivariable analyses, and also when we considered younger and older patients separately. This is likely associated with a marked difference in the incidence of TP53 alterations between CK-AML types, and further supports the notion that atypical and typical CK-AML constitute distinct disease subtypes, which should be considered independently in search of potential therapeutic targets. It is thus possible that patients with atypical CK-AML are less likely to respond to treatment regimens shown to be more efficacious in AML patients with TP53 mutations.43,44
Within the atypical CK-AML category, two subsets (i.e., numerical and other CK-AML) differed in frequencies of specific mutations, with RUNX1, ASXL1, SRSF2, KRAS and STAG2 mutations occurring exclusively in patients with atypical CK with numerical abnormalities, but infrequent TP53 mutations found only in patients with the other atypical CK-AML. However, there were essentially no clinical differences between the subsets.
This was not the case for a small subset of typical CK-AML characterized cytogenetically by the presence of 7q but not 5q or 17p abnormalities. Patients in this subset had longer OS than other patients with typical CK-AML, which may have been in part associated with their younger age and the presence of favorable molecular findings such as a lower incidence of TP53 mutations, absence of submicroscopic genomic alterations involving the TP53 locus or a higher frequency of NPM1 mutations. These patients also had adverse features such as lower platelet and higher WBC counts, and higher frequency of FLT3-ITD and WT1 and RUNX1 mutations. However, since the number of patients with only 7q abnormalities was very small, these results require corroboration.
We grouped CK-AML patients harboring balanced abnormalities such as reciprocal translocations or inversions, either known to be recurrent or unique, i.e., hitherto not reported in the literature, into a separate category. Our preliminary results seem to support this approach for most CK-AML patients with rare recurrent balanced abnormalities, who either do not harbor any or have very few gene mutations in addition to their balanced chromosome abnormality, and infrequently carry TP53 mutations. This resembles findings of others18,45 and ours14,46 in patients with balanced rearrangements involving 11q23/KMT2A(MLL), who have very few gene mutations. It is possible that once more cases with such currently rare recurrent balanced abnormalities as t(8;16)(p11.2;p13.3), t(12;22)(p13;q12) or t(10;11)(p13;q21), each present in one patient in our series, are reported in the literature, these chromosome abnormalities will be recognized by the WHO classification as denoting specific clinico-pathologic entities of AML. When this happens, the presence of the aforementioned recurrent balanced abnormalities in patients with ≥3 abnormalities will automatically exclude such patients from the CK-AML category akin to the current WHO-designated reciprocal translocations or inversions.11
In contrast, patients with CK-AML with a unique reciprocal translocation or inversion had both cytogenetic and molecular findings resembling those found in typical CK-AML patients (including a high frequency of TP53 alterations), suggesting that the presence of unique balanced abnormalities should not exclude these patients from the typical CK-AML category.
In summary, we show that CK-AML is heterogeneous cytogenetically, clinically and at the molecular level. Our data provide rationale for separating patients with typical from those with atypical CK-AML since their outcomes differ when current chemotherapeutic regimens are used, and potential targeted therapies will likely be different because of their vastly dissimilar molecular genetic backgrounds.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients who participated in clinical trials and the families who supported them; Donna Bucci and the CALGB/Alliance Leukemia Tissue Bank at The Ohio State University Comprehensive Cancer Center, Columbus, OH, for sample processing and storage services and Lisa J. Sterling and Chris Finks for data management. This work was supported in part by the National Cancer Institute (grants CA101140, CA140158, CA180861, CA196171, CA016058, CA180821, CA180882, and CA077658), the Leukemia Clinical Research Foundation, the Warren D. Brown Foundation, the Pelotonia Fellowship Program (to A-KE), and by an allocation of computing resources from The Ohio Supercomputer Center. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no conflicts of interest.
Supplementary Information for this article is available at Leukemia’s website.
REFERENCES
- 1.Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev 2004; 18: 115–136. [DOI] [PubMed] [Google Scholar]
- 2.Byrd JC, Mrózek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002; 100: 4325–4336. [DOI] [PubMed] [Google Scholar]
- 3.Schoch C, Haferlach T, Haase D, Fonatsch C, Löffler H, Schlegelberger B et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: a study of 90 patients. Br J Haematol 2001; 112: 118–126. [DOI] [PubMed] [Google Scholar]
- 4.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group study. Blood 2000; 96: 4075–4083. [PubMed] [Google Scholar]
- 5.Farag SS, Archer KJ, Mrózek K, Ruppert AS, Carroll AJ, Vardiman JW et al. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: results from Cancer and Leukemia Group B 8461. Blood 2006; 108: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fröhling S, Schlenk RF, Kayser S, Morhardt M, Benner A, Döhner K et al. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: results from AMLSG trial AML HD98-B. Blood 2006; 108: 3280–3288. [DOI] [PubMed] [Google Scholar]
- 7.Schoch C, Haferlach T, Bursch S, Gerstner D, Schnittger S, Dugas M et al. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: a detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Genes Chromosomes Cancer 2002; 35: 20–29. [DOI] [PubMed] [Google Scholar]
- 8.Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012; 119: 2114–2121. [DOI] [PubMed] [Google Scholar]
- 9.Haferlach C, Alpermann T, Schnittger S, Kern W, Chromik J, Schmid C et al. Prognostic value of monosomal karyotype in comparison to complex aberrant karyotype in acute myeloid leukemia: a study on 824 cases with aberrant karyotype. Blood 2012; 119: 2122–2125. [DOI] [PubMed] [Google Scholar]
- 10.Stölzel F, Mohr B, Kramer M, Oelschlägel U, Bochtler T, Berdel WE et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood Cancer J 2016; 6: e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129: 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mrózek K, Heinonen K, Theil KS, Bloomfield CD. Spectral karyotyping in patients with acute myeloid leukemia and a complex karyotype shows hidden aberrations, including recurrent overrepresentation of 21q, 11q, and 22q. Genes Chromosomes Cancer 2002; 34: 137–153. [DOI] [PubMed] [Google Scholar]
- 13.Cytogenetic Mrózek K., molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol 2008; 35: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisfeld A-K, Mrózek K, Kohlschmidt J, Nicolet D, Orwick S, Walker CJ et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia 2017; 31: 2211–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenaux P, Jonveaux P, Quiquandon I, Laï JL, Pignon JM, Loucheux-Lefebvre MH et al. P53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood 1991; 78: 1652–1657. [PubMed] [Google Scholar]
- 16.Laï J-L, Preudhomme C, Zandecki M, Flactif M, Vanrumbeke M, Lepelley P et al. Myelodysplastic syndromes and acute myeloid leukemia with 17p deletion. An entity characterized by specific dysgranulopoiesis and a high incidence of P53 mutations. Leukemia 1995; 9: 370–381. [PubMed] [Google Scholar]
- 17.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol 2001; 19: 1405–1413. [DOI] [PubMed] [Google Scholar]
- 18.Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A et al. Genomic and epigenomic landscapes of adult de novo myeloid leukemia. N Engl J Med 2013; 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016; 128: 686–698. [DOI] [PubMed] [Google Scholar]
- 20.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374: 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolitz JE, George SL, Marcucci G, Vij R, Powell BL, Allen SL et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients under age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood 2010; 116: 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL et al. Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: A phase 2 Cancer and Leukemia Group B study (CALGB 10503). Leukemia 2017; 31: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baer MR, George SL, Caligiuri MA, Sanford BL, Bothun SM, Mrózek K et al. Low-dose interleukin-2 immunotherapy does not improve outcome of patients age 60 years and older with acute myeloid leukemia in first complete remission: Cancer and Leukemia Group B study 9720. J Clin Oncol 2008; 26: 4934–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baer MR, George SL, Sanford BL, Mrózek K, Kolitz JE, Moore JO et al. Escalation of daunorubicin and addition of etoposide in the ADE regimen in acute myeloid leukemia patients aged 60 years and older: Cancer and Leukemia Group B Study 9720. Leukemia 2011; 25: 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcucci G, Moser B, Blum W, Stock W, Wetzler M, Koltiz JE et al. A phase III randomized trial of intensive induction and consolidation chemotherapy ± oblimersen, a pro-apoptotic Bcl-2 antisense oligonucleotide in untreated acute myeloid leukemia patients >60 years old. J Clin Oncol 2007; 25: 360s [Google Scholar]
- 26.Kolitz JE, George SL, Dodge RK, Hurd DD, Powell BL, Allen SL et al. Dose escalation studies of cytarabine, daunorubicin, and etoposide with and without multidrug resistance modulation with PSC-833 in untreated adults with acute myeloid leukemia younger than 60 years: final induction results of Cancer and Leukemia Group B study 9621. J Clin Oncol 2004; 22: 4290–4301. [DOI] [PubMed] [Google Scholar]
- 27.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol 2012; 31: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med 1994; 331: 896–903. [DOI] [PubMed] [Google Scholar]
- 29.Moore JO, George SL, Dodge RK, Amrein PC, Powell BL, Kolitz JE et al. Sequential multiagent chemotherapy is not superior to high-dose cytarabine alone as postremission intensification therapy for acute myeloid leukemia in adults under 60 years of age: Cancer and Leukemia Group B study 9222. Blood 2005; 105: 3420–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore JO, Dodge RK, Amrein PC, Kolitz J, Lee EJ, Powell B et al. Granulocyte-colony stimulating factor (filgrastim) accelerates granulocyte recovery after intensive postremission chemotherapy for acute myeloid leukemia with aziridinyl benzoquinone and mitoxantrone: Cancer and Leukemia Group B study 9022. Blood 1997; 89: 780–788. [PubMed] [Google Scholar]
- 31.Lee EJ, George SL, Caligiuri M Szatrowski TP, Powell BL, Lemke S et al. Parallel phase I studies of daunorubicin given with cytarabine and etoposide with or without the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age or older with acute myeloid leukemia: Results of Cancer and Leukemia Group B study 9420. J Clin Oncol 1999; 17: 2831–2839. [DOI] [PubMed] [Google Scholar]
- 32.Stone RM, Berg DT, George SL, Dodge RK, Paciucci PA, Schulman P et al. Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. N Engl J Med 1995; 332: 1671–1677. [DOI] [PubMed] [Google Scholar]
- 33.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017; 377: 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roboz GJ, Mandrekar SJ, Desai P, Laumann K, Walker AR, Wang ES et al. A randomized trial of 10 days of decitabine alone or with bortezomib in previously untreated older patients with acute myeloid leukemia: CALGB 11002 (Alliance). Blood Adv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitelman F (ed). ISCN (1995): An International System for Human Cytogenetic Nomenclature. Karger: Basel, Switzerland, 1995. [Google Scholar]
- 36.Mrózek K, Carroll AJ, Maharry K, Rao KW, Patil SR, Pettenati MJ et al. Central review of cytogenetics is necessary for cooperative group correlative and clinical studies of adult acute leukemia: the Cancer and Leukemia Group B experience. Int J Oncol 2008; 33: 239–244. [PMC free article] [PubMed] [Google Scholar]
- 37.Mitelman F, Johansson B, Mertens F (eds). Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available at: http://cgap.nci.nih.gov/Chromosomes/Mitelman. Accessed: 5 June, 2018. [Google Scholar]
- 38.Kroll KW, Eisfeld A-K, Lozanski A, Bloomfield CD, Byrd JC, Blachly JS. MuCor: mutation aggregation and correlation. Bioinformatics 2016; 32: 1557–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol 2008; 26: 5078–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res 2001; 61: 7233–7239. [PubMed] [Google Scholar]
- 41.Vittinghoff E, Glidden DV, Shiboski SC, McCulloch CE. Regression Methods in Biostatistics: Linear, Logistic, Survival and Repeated Measures Models. Springer: New York, NY, USA, 2005. [Google Scholar]
- 42.Kämpjärvi K, Järvinen TM, Heikkinen T, Ruppert AS, Senter L, Hoag KW et al. Somatic MED12 mutations are associated with poor prognosis markers in chronic lymphocytic leukemia. Oncotarget 2015; 6: 1884–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016; 375: 2023–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 2018; 18: 89–102. [DOI] [PubMed] [Google Scholar]
- 45.Gröschel S, Schlenk RF, Engelmann J, Rockova V, Teleanu V, Kühn MWM et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J Clin Oncol 2013; 31: 95–103. [DOI] [PubMed] [Google Scholar]
- 46.Bhatnagar B, Blachly JS, Kohlschmidt J, Eisfeld A-K, Volinia S, Nicolet D et al. Clinical features and gene- and microRNA-expression patterns in adult acute leukemia patients with t(11;19)(q23;p13.1) and t(11;19q23;p13.3). Leukemia 2016; 30: 1586–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.