

Abstract
MicroRNAs (miRNAs) are small endogenous non-coding functional RNAs that post-transcriptionally regulate gene expression. They play essential roles in nearly all biological processes including cell development and differentiation, DNA damage repair, cell death as well as intercellular communication. They are highly involved in cancer, acting as tumor suppressors and/or promoters to modulate cell proliferation, epithelial-mesenchymal transition and tumor invasion and metastasis. Recent studies have shown that more than half of miRNAs are located within protein-coding or non-coding genes. Intragenic miRNAs and their host genes either share the promoter or have independent transcription. Meanwhile, miRNAs work as partners or antagonists of their host genes by fine-tuning their target genes functionally associated with host genes. This review outlined the complicated relationship between intragenic miRNAs and host genes. Focusing on miRNAs known as oncogenes or tumor suppressors in specific cancer types, it studied co-expression relationships between these miRNAs and host genes in the cancer types using TCGA data sets, which validated previous findings and revealed common, tumor-specific and even subtype-specific patterns. These observations will help understand the function of intragenic miRNAs and further develop miRNA therapeutics in cancer.
Keywords: miRNA, intragenic miRNA biogenesis, host genes, interplay, cancer
Introduction
MicroRNA (miRNA) is a class of small endogenous non-coding RNAs with a length between 20 and 24 nucleotides. They recognize and bind mRNA transcripts in a sequence-specific mode, resulting in degradation or translational repression of the corresponding mRNAs [1, 2]. The generation of miRNA from primary miRNA is a multi-step but fairly well understood process. After cleavage of primary transcripts to form precursor miRNAs in the nucleus, precursor miRNAs are exported into the cytoplasm for additional cleavage by an enzyme called Dicer to produce single-stranded mature miRNAs. Mature miRNAs are then loaded onto the RNA-induced silencing complex where they bind to 3′-UTR sequences of target genes. If the 3′-UTR of the target mRNA is fully complementary to the miRNA seed site, the mRNA is targeted for degradation. If it is partially complementary to the miRNA, the mRNA is targeted for translational inhibition [3–5]. In this way, an individual miRNA can post-transcriptionally regulate expression of hundreds of mRNAs, which profoundly alter gene expression and drive cells toward transformation.
Cancer is a complex disease involving multiple genes and pathways; therefore, it is not very useful to target one single gene for cancer treatment. A single miRNA can regulate expression of hundreds of genes involved in distinct cellular functions, which makes miRNAs the promising therapeutic targets. Since the miRNA dysregulation in cancer was first reported in 2002 [6], many studies have found miRNAs’ function in carcinogenesis including tumor suppressor miRNAs (ts-miRs) and oncogenic miRNAs (onco-miRs). The upregulated onco-miRs and downregulated ts-miRs lead to cancer initiation and progression [7, 8]. These miRNAs have attracted lots of attention as biomarkers and therapeutic targets. Controlling miRNAs transcriptional regulation by genome or RNA-editing tool has become a novel and promising approach [9, 10].
Although there is a general understanding of the generation of mature miRNA from primary miRNA, the transcriptional regulation of primary miRNAs is still unclear. The location of miRNAs is one major factor that determines how primary miRNAs are transcriptionally regulated. According to their genomic position, miRNAs can be classified into intragenic and intergenic miRNAs. Intragenic miRNAs, embedded within introns or exons of genes on the same strand, are believed to be co-regulated with their host genes by Pol II, whereas intergenic miRNAs, located between genes, are transcribed from their own Pol II or Pol III promoters. Recent studies, however, revealed that intragenic miRNAs, even intronic miRNAs, are not always co-transcribed with their host genes [11, 12]. Some intronic miRNAs have been found to interact with their host genes to affect their stability [13, 14]. More importantly, bioinformatics studies predicted that approximately 20% of intronic miRNAs target host mRNA transcripts in a feedback loop [13–15].
In this review, we outlined the complicated relationships between miRNAs and their host genes. Focusing on onco-miRs and ts-miRs in specific cancer types, we studied co-expression relationships between these miRNAs and host genes using TCGA data sets. We not only found common co-expression patterns, but also revealed tumor-specific and even subtype-specific relationships, which are very helpful for understanding transcriptional and post-transcriptional regulation of miRNAs and further developing miRNA therapeutics in cancer.
Definition of intragenic miRNAs and their host genes
Location of miRNAs is one of the hot topics in miRNA research since it is a major factor to determine how miRNAs are regulated. Based on their location, miRNAs can be classified into intragenic miRNAs and intergenic miRNAs. miRNAs located within protein-coding or non-coding genes are called intragenic miRNAs, while the genes in which the miRNAs are embedded are called host genes. Intragenic miRNAs can be further categorized into intronic miRNAs (miRNAs located in intronic regions), exonic miRNAs (miRNAs located in exonic regions), junction miRNAs (miRNAs located in gene intron–exon junctions) and antisense miRNAs (miRNAs located in the antisense strand of gene) (Figure 1). Intergenic miRNAs, located between genes, are believed to be transcribed from their own Pol II or Pol III promoters. However, recent studies revealed that there is a small percentage of intergenic miRNAs that are potentially co-transcribed with their neighboring genes through two mechanisms. One mechanism is called readthrough, where the transcription of one gene continues beyond the normal transcription termination site into intergenic regions. miRNAs located in the immediate (<4000 bp) downstream sense region of protein-coding or non-coding genes are likely to be transcribed by readthrough transcription [16] (Figure 1E). The other is called divergent transcription, defined as two polymerases transcribing on both sense and antisense directions from the same promoter. As a common feature for active promoters, divergent transcription generates long non-coding antisense transcripts coupled to active gene promoters [16–18] (Figure 1F). These two types of intergenic miRNAs (readthrough or divergent) can be classified into `intragenic’ category as well since they potentially share the same promoter with their neighboring genes, which are different from other intergenic miRNAs in transcriptional regulation. The neighboring genes that miRNAs are potentially co-transcribed with can also be called `host’ genes.
Figure 1.
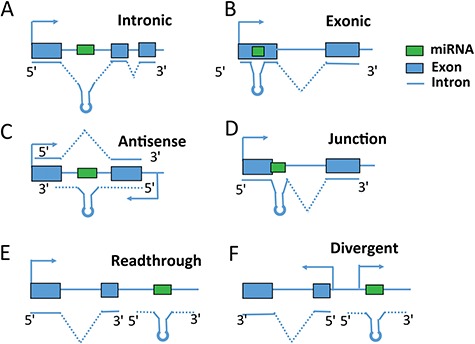
Intragenic miRNA classification. miRNA categories are defined based on their locations relative to exon, intron and gene regions.
Previous studies have reported that more than half of miRNAs are intragenic miRNAs. To facilitate the analysis of genomic and structure features of intragenic miRNAs and host genes, miRIAD (http://www.miriad-database.org) provides a comprehensive knowledge on intragenic miRNAs by collecting public data from multiple sources, such as host gene function, miRNA and host gene expression and miRNA targets [19]. As gene annotation is improved and new miRNAs are identified, more intragenic miRNAs are classified [20]. To date, there are 1881 miRNAs in miRBase v21 [21–25]. Based on their relative location to protein-coding or non-coding genes (Figure 1), 1434 can be classified as intragenic miRNAs, among which 918 (48.8%), 74 (3.9%), 45 (2.4%), 59 (3.1%), 50 (2.6%), 198 (10.5%) are intronic, exonic, junction, readthrough, divergent and antisense miRNAs, respectively (Figure 2). There are 90 miRNAs (4.8%), which can not be assigned into one specific category. For example, an miRNA is located within the intron region of one transcript and also embedded in exon regions of another transcript. In this case, this miRNA is assigned into a `mixed’ category (Figure 2).
Figure 2.
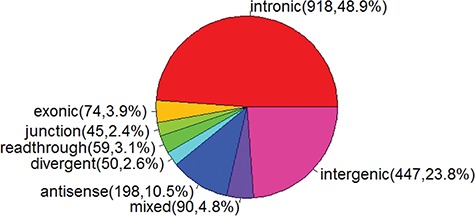
Pie chart of miRNA categories.
Interplay between intragenic miRNAs and their host genes
Early evidence proposed a co-expression model suggesting that intronic miRNAs are derived from the same primary transcripts as their host genes. Intronic miRNAs use the transcriptional start sites (TSSs) of their host genes to initiate transcription. The primary transcript is then processed into mRNA and miRNAs by microprocessor and splicing. There is no interplay between microprocessor cleavage and splicing, resulting in the co-expression between miRNA and mRNA [26–28]. For example, the expression levels of miR-335 significantly correlated with MEST in 21 HCC cell lines, supporting the notion that the intronic miRNA is co-expressed with its host gene under the regulation of the host gene promoter [29]. Strongly correlated expression profiles between 175 miRNAs and their host genes across 24 different human organs have been reported [26]. An extreme case of coordination between intronic miRNA processing and splicing happens in very short introns, where the entire intron is so short that it is already a pre-miRNA without the involvement of microprocessor. These short introns, called mirtrons, are highly co-expressed with their host genes [30].
Increasing evidence, however, has shown that intronic miRNAs and host genes are not co-expressed as frequently as we previously expected due to two main reasons [31, 32]. One reason is that miRNAs have their own independent promoters. It has been found that over one-third of intronic miRNAs have their own promoters (Polymerase II or III), whose transcription is independent from their host genes [11, 33]. For example, it has been validated that miR-26b has an independent TSS, promoter signal and transcriptional factor from its host gene CTDSP [31]. Kim et al. [34] revealed that the splicing factor SRSF3 regulates the expression of miR-1908 independently from its host gene FADS1, suggesting that miR-1908 could be affected by its own transcription unit. Ozsolak et al. [35] reported that roughly 30% of intragenic miRNAs may be initiated independently especially if they are resided in large-sized (>5 kbp) intronic segments. The intronic regions are long enough to carry their own transcription units of pre-miRNA genes [32]. The other reason is the crosstalk between microprocessor cleavage and splicing [36]. In addition, the evolution conservation also contributes to the expression discordance between miRNAs and their hosts. He et al. [11] reported that evolutionary non-conserved intragenic miRNAs are less co-expressed with host genes than conserved ones. They suggest that most non-conserved miRNAs are expressed serendipitously and weakly but not necessarily always from the same transcripts as the host genes and some are co-expressed owing to shared chromatin dynamics. They then conjecture that for some of these their co-expression is beneficial and selection favors them to be what call ‘embedded’, meaning they evolve stronger co-expression, which in turn favors conservation of miRNA target sites on 3′-UTRs of desirable target genes [11].
The transcriptional regulation of exonic miRNAs is different from intronic miRNAs since Drosha processing of an exonic miRNA will inhibit production of the spliced host mRNA [37, 38]. Sundaram et al. [37] showed a clear inverse correlation between the expression pattern of miR-198 and FSTL1 (host gene), which highlights the importance of this regulatory switch in controlling context-specific gene expression to orchestrate wound re-epithelialization.
The biogenesis of junction miRNAs is most complicated since there is competition between spliceosome and the microprocessor. When the splicing machinery does not recognize the internal exon, and the microprocessor components, rather than the spliceosome complex, bind to the RNA transcript at the pre-miRNA region to generate pre-miRNA. When the internal exon is recognized through interaction with specific splicing factors, the RNA secondary structure is released and splicing of the internal exon is carried out by the spliceosome before DGCR8 and Drosha bind to the pre-miRNA region. The spliced variant containing the alternative exon is the product and no pre-miRNA is made [39]. That is to say, junction miRNA is negatively regulated by splicing. Melamed et al. [39] studied an miRNA cluster including one exonic miRNA miR-410, one intronic miRNA miR-541 and one junction miRNA miR-412. They confirmed that the splicing and the processing of pre-miRNAs located in exon-intron junctions are mutually exclusive and alternative splicing is inversely correlated with levels of miR-412.
Readthrough and divergent miRNAs are special whose transcription is a byproduct of their neighboring genes. Readthrough miRNA is generated by a continuous transcription beyond a normal stop sign from an active gene (Figure 1E). For example, miR-21 is the most commonly over-expressed miRNA in cancer and a proven oncogene. miR-21 is located immediately downstream of the vacuole membrane protein-1 (VMP1) gene, which was reported to bypass the polyadenylation signals to include miR-21, thus providing a novel and independently regulated source of miR-21, termed VMP1–miR-21 [38]. Divergent miRNA is a product of bi-directional transcription of active promoters. In humans, some bidirectional promoter regions transcribing an miRNA and protein-coding gene simultaneously have been experimentally identified, e.g. the BTG4 promoter upstream of miR-34b and miR-34c [40], and the POLR3D promoter upstream of miR-320a on chromosome 8 [41].
In addition to the transcriptional regulation of intragenic miRNAs by their host genes, host genes are also subject to the regulation of miRNAs. The complicated and mutual regulation between miRNAs and host genes form a negative or positive feedback loop, where miRNAs play an antagonistic or synergetic role as a competitor or partner of a host gene [13, 14]. Bioinformatics studies have demonstrated that approximately 20% of intronic miRNAs were predicted to target their host genes [13]. One example is miR-338-3p, which inhibits the transcription of its host gene AATK by binding to its 3′-UTR [42]. Another example is miR-26b, which controls neuronal differentiation by repressing its host transcript, ctdsp2 [43]. Such an interaction forms the `first-order’ negative-feedback circuit for fine-turning host gene expression. Besides direct regulation, an miRNA can negatively regulate its host gene function by targeting a gene whose product is a downstream effector of the host gene product. Such regulation could be termed `second-order’ negative feedback. For example, miR-641 is not predicted to target its host, AKT2, while its predicted targets, which include PI3K, EGFR, PTEN, PDK1, RAS, MEK and ERK, are in functional synergy with its host, giving credence to the second-order negative-feedback circuitry line of evidence [44].
Association between intragenic onco/ts-miRNAs and their host genes in cancer
Disruption of miRNA expression is frequently observed in cancer. They can act as oncogenes and/or tumor suppressors. Study of a large collection of chronic lymphocytic leukemias (CLLs) showed knockdown or knock out of miR-15a and miR-16-1 in approximately 69% of CLLs. This is the first evidence that miRNAs might be involved in the pathogenesis of human cancer [6]. Frixa et al. [45] reported that miR-128-3p, upregulated in lung cancer, targets Drosha and Dicer, two key enzymes of miRNAs processing, leading to the widespread down-regulation of miRNA expression that promotes lung cancer cell migration [46]. The capability of targeting a number of genes with diverse functions makes miRNAs ideal candidates to regulate core carcinogenesis process [47–50].
Most onco-miRs or ts-miRs are intragenic miRNAs. Increasing evidences demonstrated links between intragenic miRNAs and host genes in cancer. They act in synergistic or antagonistic ways, which is achieved by miRNA-mediated fine-tuning target gene expression functionally related to host genes [51]. Due to the close functional connection between intragenic miRNAs and host genes, it is necessary and important to interpret the function of these onco-miRs and ts-miRs in the context of host genes. TCGA provides a rich source of multi-dimensional omics data, which gives a unique opportunity to study the expression of intragenic miRNAs and their host genes simultaneously across multiple cancer types. A positively correlated expression patterns would indicate a co-regulation and/or synergistic relationship between host and intragenic miRNAs, while anti-correlated or uncorrelated expression patterns would suggest an independent or antagonistic process.
Known onco-miRs and ts-miRs, specific cancer(s) where they are involved in, their cognate host genes and their expression correlation coefficients in specific cancer(s) are listed in Table 1. Most intragenic onco-miRs and ts-miRs are embedded within intron regions of genes. They exhibited a strong co-expression relationship with their cognate host genes, suggesting co-transcription and/or potential cooperation in regulating cancer development. The results derived from TCGA data sets further validated previous findings. For example, miR-335 harbored within an intron of a protein-coding gene MEST, was known to be co-regulated with MEST by promoter hypermethylation in breast cancer cells [119], HCC [29] and gastric cancer [120]. Consistently, the expression of miR-335 and MEST are highly correlated in TCGA HCC (R = 0.88; FDR = 9.87e-05), breast (R = 0.78; FDR = 0) and gastric cancer (R = 0.79; FDR = 0), further validating of the co-regulation relationship between miR-335 and MEST is needed. MiR-106b-25 cluster, composed of the highly conserved miR-106b, miR-93 and miR-25, is embedded within an intron 13 of MCM7. Petrocca et al. [121] reported that the MCM7 and miR-106b-25 cooperate in exerting their oncogenic function through different complementary mechanisms. Consistently, miR-106b-25 and MCM7 are highly co-expressed in TCGA breast cancer (R = 0.68, 0.57, 0.66; FDR = 0, 0, 0), further demonstrating that they are derived from the same primary transcript. Meanwhile, intronic miR-224/452 cluster showed significant co-expression with their host gene GABRE in prostate cancer (R = 0.58, 0.63; FDR = 0, 0) [122].
Table 1.
Summary of co-expression patterns between onco/ts-miRs and their host genes in specific cancer types
| miR cluster | miRNA | Host | Loc | Type | Cancer | Correlation in TCGA | Ref. |
|---|---|---|---|---|---|---|---|
| miR-1 | miR-1-1 | MIR1-1HG | Intron | ts-miR | Thyroid; bladder cancer | – | [52, 53] |
| miR-9 | miR-9-1 | C1orf61 | Intron | onco-miR | Breast cancer | 0.39** | [54–56] |
| miR-9-3 | MIR9-3HG | Mix | ts-miR | Gastric cancer | 0.25** | [56] | |
| miR-15/16–1 | miR-15a | DLEU2 | Intron | ts-miR | CLL | – | [57, 58] |
| miR-16-1 | – | ||||||
| miR-17-92 | miR-17 | C13ORF25 | Mix | onco-miR | Lymphomas; lung cancer; renal cell cancer | –, 0.49**, 0.07 | [57–59] |
| miR-18a | –, 0.48**, 0.01 | ||||||
| miR-19a | –, 0.50**, 0.11 | ||||||
| miR-20a | –, 0.55**, 0.07 | ||||||
| miR-19b1 | –, 0.48**, 0.16 | ||||||
| miR-92a1 | –, 0.51**, 0.19 | ||||||
| miR-21 | miR-21 | VMP1 | Read through | onco-miR | Breast cancer | 0.52** | [60–65] |
| Lung cancer | 0.36* | ||||||
| Pancreas cancer | 0.61** | ||||||
| Liver cancer | 0.40 | ||||||
| Gastric cancer | 0.54** | ||||||
| Cervical cancer | 0.39** | ||||||
| Hematopoietic cancer | – | ||||||
| miR-23b | miR-23b | C9ORF3 | Intron | ts-miR | Gastric cancer | 0.65** | [66] |
| miR-26a | miR-26a-1 | CTDSPL | Intron | ts-miR | Osteosarcoma; colorectal cancer | –,– | [67–69] |
| miR-26a-2 | CTDSP2 | –,– | |||||
| miR-26b | miR-26b | CTDSP1 | Intron | ts-miR | Colorectal cancer | – | [67, 69] |
| miR-27b | miR-27b | C9ORF3 | Intron | ts-miR | Prostate cancer | 0.62** | [60, 70] |
| miR-30c | miR-30c-1 | NFYC | Intron | ts-miR | Prostate cancer | 0.06 | [71] |
| miR-33a | miR-33a | SREBF2 | Intron | ts-miR | Prostate cancer | 0.24** | [72] |
| miR-34b | miR-34b | BTG4 | Divergent | ts-miR | Breast cancer | −0.05 | [73] |
| Prostate cancer | – | ||||||
| miR-95 | miR-95 | ABLIM2 | Intron | onco-miR | Liver cancer | 0.75** | [74] |
| miR-101 | miR-101-2 | RCL1 | Intron | ts-miR | Gastric cancer | 0.17* | [75] |
| miR-106b/25 | miR-106b | MCM7 | Intron | onco-miR | Breast cancer | 0.68** | [76] |
| miR-93 | 0.57** | ||||||
| miR-25 | 0.66** | ||||||
| miR-107 | miR-107 | PANK1 | Intron | ts-miR | Pancreatic cancer | 0.22* | [77] |
| miR-125b | miR-125b-1 | MIR100HG | Intron | onco-miR | Lung cancer | 0.69** | [63] |
| miR-125b-2 | MIR99AHG | 0.77** | |||||
| miR-126 | miR-126 | EGFL7 | Intron | ts-miR | Pancreatic cancer | 0.56** | [78] |
| Breast cancer | 0.13* | ||||||
| Lung cancer | 0.28 | ||||||
| miR-128 | miR-128-1 | R3HDM1 | Intron | ts-miR | AML | – | [79–81] |
| Esophageal cancer | – | ||||||
| Glioblastoma | – | ||||||
| miR-137 | miR-137 | MIR137HG | Exon | ts-miR | Neuroblastoma | – | [82, 83] |
| miR-139 | miR-139 | PDE2A | Intron | ts-miR | Endometrial; colorectal cancer | – | [67, 84, 85] |
| miR-149 | miR-149 | GPC1 | Intron | ts-miR | Breast cancer | 0.37** | [86] |
| miR-152 | miR-152 | COPZ2 | Intron | ts-miR | Prostate cancer | 0.31** 0.70** |
[67] |
| Endometrial cancer | |||||||
| miR-153 | miR-153-1 | PTPRN | Intron | ts-miR | Ovarian cancer | – | [87] |
| Glioblastoma | – | ||||||
| Gastric cancer | −0.007 | ||||||
| miR-153-2 | PTPRN2 | Ovarian cancer | – | [87] | |||
| Glioblastoma | – | ||||||
| Gastric cancer | 0.45** | ||||||
| miR-155 | miR-155 | MIR155HG | Exon | onco-miR | B-cell cancers | – | [88–91] |
| Glioblastoma | 0.68** | ||||||
| miR-181a | miR-181a-1 | MIR181A1HG | Intron | onco-miR | Cervical cancer | – | [92, 93] |
| Breast cancer | – | ||||||
| miR-181a-2 | MIR181A2HG | Cervical cancer | – | ||||
| Breast cancer | – | ||||||
| miR-185 | miR-185 | TANGO2 | Intron | ts-miR | Colorectal cancer | – | [94] |
| Lung cancer | 0.49** | ||||||
| Breast cancer | 0.36** | ||||||
| miR-186 | miR-186 | ARANB2 | Intron | ts-miR | Glioblastoma | 0.4** | [95] |
| miR-198 | miR-198 | FSTL1 | Exon | ts-miR | Breast cancer | −0.04 | [96] |
| miR-210 | miR-210 | MIR210HG | Intron | onco-miR | Pancreatic cancer | – | [97] |
| miR-218 | miR-218-1 | SLIT2 | Intron | ts-miR | Breast cancer | 0.17** | [98] |
| Gastric cancer | 0.66** | ||||||
| miR-218-2 | SLIT3 | Breast cancer | 0.33** | 118 | |||
| miR-224/452 | miR-224 | GABRE | Intron | onco-miR | Gastric cancer; lung cancer | 0.69**, 0.83** 0.75**, 0.80** |
[99] |
| miR-452 | ts-miR | Prostate cancer | 0.58**, 0.63** | ||||
| miR-326 | miR-326 | ARRB1 | Intron | ts-miR | Lung cancer | 0.41** | [67, 100] |
| miR-335 | miR-335 | MEST | Intron | onco-miR | Ovarian cancer | 0.66** | [101, 102] |
| ts-miR | Gastric cancer | 0.79** | [103, 104] | ||||
| Liver cancer | 0.88** | ||||||
| Breast cancer | 0.78** | ||||||
| miR-338 | miR-338 | AATK | Intron | ts-miR | Breast cancer | 0.61** | [105] |
| miR-340 | miR-340 | RNF130 | Intron | onco-miR | Gastric cancer | 0.35** | [99] |
| ts-miR | Breast cancer | 0.28** | [106] | ||||
| miR-342 | miR-342 | EVL | Intron | ts-miR | Colorectal cancer | – | [107] |
| miR-346 | miR-346 | GRID1 | Intron | ts-miR | Liver cancer | 0.10 | [108] |
| miR-449a/b | miR-449a | CDC20B | Intron | ts-miR | Neuroblastoma | 0.23**, 0.61** | [109] |
| miR-449b | Prostate cancer | 0.09, −0.25** | [110] | ||||
| miR-486 | miR-486 | ANK1 | Intron | ts-miR | Esophageal cancer | 0.31** | [111] |
| miR-488 | miR-488 | ASTN1 | Intron | ts-miR | Gastric cancer | −0.2* | [112] |
| miR-489 | miR-489 | CALCR | Intron | ts-miR | Esophageal cancer | 0.02 | [113] |
| miR-491 | miR-491 | FOCAD | Intron | ts-miR | Colorectal cancer | – | [114] |
| miR-504 | miR-504 | FGF13 | Intron | onco-miR | HSCC | – | [115–117] |
| Neuroblastoma | – | ||||||
| ts-miR | Glioblastoma | 0.67** | |||||
| miR-634 | miR-634 | PRKCA | Intron | ts-miR | Breast cancer | – | [118] |
| miR-1908 | miR-1908 | FADS1 | Intron | onco-miR | Glioblastoma | −0.12* | [57, 58] |
| Ovarian cancer | 0.01 |
Cells with `–’ means correlation coefficients were not available since the expression of miRNA and/or its host gene were undetected in this cancer type.
*Represents the FDR ≤ 0.01.
**Represents the FDR ≤ 0.001.
Although most intronic onco/ts-miRs are co-expressed with their host genes in cancers, there are some exceptions. For example, expression of miR-30c-1 is not correlated with its host gene NFYC (R = 0.06; FDR = 0.23) in prostate cancer, suggesting that it does not originate from the NFYC transcription unit. Several cryptic promoters and possible transcription factor binding sites have been detected in the intron regions of NFYC where miR-30c-1 is located, indicating miR-30c-1 is transcribed independent from NFYC [123]. Consistent with previous findings that miR-1908 is regulated by its own transcription unit rather than co-regulated with its host gene FADS1 [34], we found that expressions of miR-1908 and FADS1 are negatively correlated/uncorrelated in glioblastoma and ovarian cancer (R = −0.12, 0.01; FDR = 0.01, 0.87). In addition to known independent transcriptional relationships, TCGA data sets revealed novel uncorrelated/negatively correlated patterns between some intronic miRNAs and their host genes—for example, miR-346 and GRID1 in liver cancer (R = 0.1; FDR = 0.81), miR-488 and ASTN1 in gastric cancer (R = −0.2; FDR = 0.006) and miR-489 and CALCR in esophageal cancer (R = 0.02; FDR = 0.72). The uncorrelated/negative correlation indicated these miRNAs might have their own transcription mechanism rather than share the promoter as their host genes.
There are three onco/ts-miRs harbored within exonic regions, miR-137, miR-155 and miR-198. MiR-198 is located in the 3′-UTR region of protein-coding gene FSTL1, while the other two miRNAs are encoded in the exon of non-coding genes, MIR137HG and MIR155HG. MiR-198 is not co-expressed with its host gene FSTL1 (R = −0.04 in breast cancer; FDR = 0.52), which validates previous finding about their mutually exclusive expression from a single transcript [37]. In contrast, miR-155 is co-transcribed with MIR155HG possibly because the gene itself is to generate the miRNA rather than produce other functional unit, leading to the high co-expression pattern in glioblastoma (R = 0.68; FDR = 0).
MiR-21, involved in many cancers, has been reported to be a readthrough miRNA from VMP1. The host gene, VMP1, was recently found to be involved in the process of tumor metastasis and it plays a vital role in balancing apoptosis and autophagy [38, 124]. miR-21 and VMP1 are co-expressed across multiple cancer types (R = 0.52, 0.36, 0.61, 0.39; FDR = 0, 0.003, 0, 4.18e-12, in breast, lung, pancreas, and cervical cancers, respectively) (Figure 3). These findings further validated that miR-21 is the byproduct of transcription of VMP1.
Figure 3.
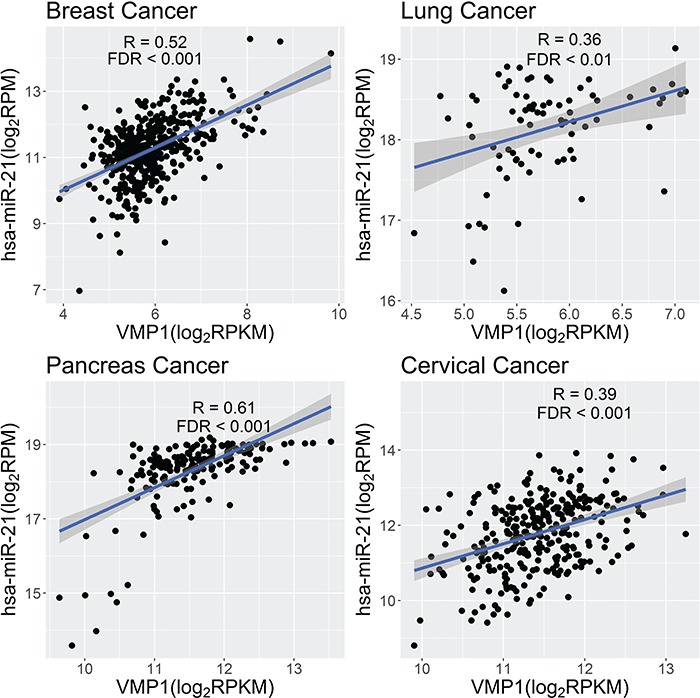
Scatter plot of expression abundance of miR-21 and its host gene VMP1 in multiple cancers.
MiR-34b and BTG4 are co-regulated by a bidirectional promoter [40]. It has been revealed that miR-34b has tumor suppressive ability in breast and prostate cancer [73]. However, TCGA data indicated that there is no correlation between expression of miR-34b and BTG4. Further research is needed to investigate whether this is caused by independent transcription or whether the co-transcription is disrupted by regulation feed-back between miRNAs and host genes.
Most interestingly, 15 out of 46 miRNA clusters show cancer-specific co-expression patterns with their host genes, suggesting the common involvement of cancer-specific post-transcription regulation—for example, miR-17-92 cluster, encoded the miR-17, miR-18a, miR-19a/b1, miR-20a and miR-92a1. miR-17-92 cluster, regarded as oncogene, was frequently overexpressed in lymphomas, lung cancer and renal cancer [57–59]. All miRNAs in the cluster showed strong correlation with the host gene C13orf25 in lung cancer (R = 0.49, 0.48, 0.58, 0.55, 0.48, 0.51; FDR = 2.26e-5, 2.89e-5, 1.35e-7, 1.08e-6, 2.82e-5, 9.90e-6), but weak/discordant correlation in renal cell cancer (R = 0.07, 0.01, 0.11, 0.07, 0.16, 0.19; FDR = 0.47, 0.93, 0.22, 0.47, 0.06, 0.02). These results suggest that miRNAs and host genes are co-transcribed from one transcript but renal cancer-specific post-transcription interferes the co-transcriptional regulation. In addition to cancer-specific pattern, some miRNAs even exhibit subtype-specific co-expression with their host genes in one cancer. For example, miR-338 is strongly co-expressed with its host gene AATK in ER/PR/HER2-positive breast cancers (R = 0.63, 0.65, 0.63; FDR = 0, 0, 2.49e-7), but this positive correlation is lost in triple-negative breast cancer (R = −0.27, FDR = 0.07) (Figure 4).
Figure 4.
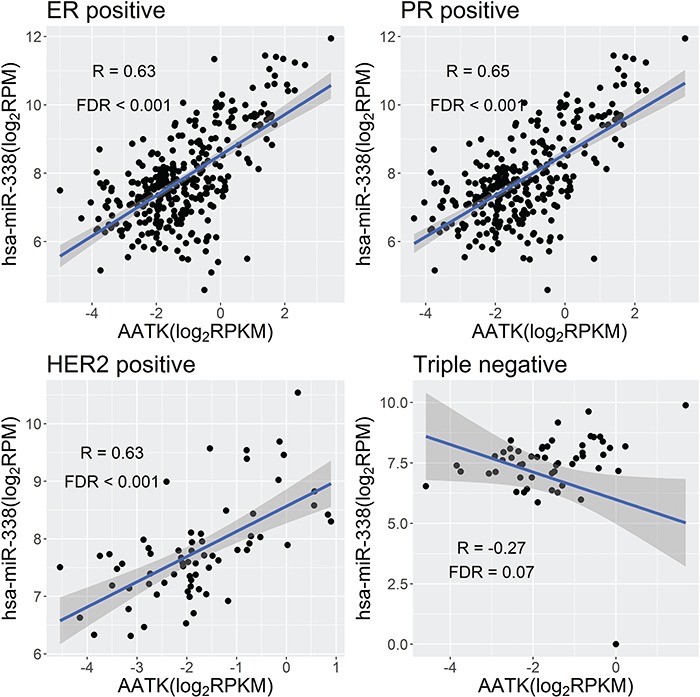
Scatter plot of expression abundance of miR-338 and its host gene AATK in ER/PR/HER2-positive and triple-negative breast cancers.
Potential as the therapeutic targets in cancer
As master regulators of cellular processes, miRNAs have become promising therapeutic targets for cancer treatment. Upregulation of the expression of a ts-miR can induce apoptosis and senescence, block cell proliferation, self-renewal, invasion, metastasis, angiogenesis and drug resistance by selectively downregulating multiple genes [125]. Conversely, inhibiting the function or blocking the expression of an onco-miR in cancer cells can reactivate multiple tumor-suppressor genes, leading to tumor regression or even to tumor eradication [126].
Since most onco-miRs or ts-miRs are intragenic miRNAs, the observation that they are often co-expressed with host genes (Table 1), provides a new strategy to regulate miRNAs expression for cancer therapy. When miRNAs are derived from the transcription of host genes rather than have their own independent promoters, disruption of the expression of host genes or especially epigenetic modification of promoters of host genes is highly likely to change the miRNA expression. For example, miR-335, known to suppress breast cancer cell migration, is highly co-expressed with its host gene in breast cancer (Table 1). Furthermore, miR-335 expression is significantly negatively correlated with the MEST promoter DNA methylation level (R = −0.32; P = 0.0002), suggesting that promoter DNA demethylation would activate miR-335.
Since epigenetic modification is potentially reversible, it has been aggressively pursued as therapeutic strategy. Grady et al. [107] showed that there is synchronous epigenetic silencing of the intronic ts-miR miR-342 and its host gene EVL in colorectal cancer. Reconstitution of miR-342 activity induces apoptosis in colorectal cancer cells. Daniunaite et al. [127] analyzed the promoter methylation level of host genes of miR-155, miR-152, miR-137 and miR-31 in prostate cancer. They found that aberrant promoter methylation of the host genes is prostate cancer-specific. Downregulation of miR-155-5p significantly correlates with the promoter methylation level, which can be used as the promising diagnostic and/or prognostic biomarker of prostate cancer. The methylation status of host genes of particular miRNA as independent variables or in combinations with other signals might assist physicians in identifying prostate cancer patients with poor prognosis preoperatively. Besides, miR-152 and COPZ2 were both silenced by hypermethylation in endometrial cancer. They are highly co-expressed in TCGA endometrial cancer data (R = 0.70; FDR = 0; Table 1). Tsuruta et al. [67] investigated the relationship between DNA hypermethylation and downregulation of miR-152 and COPZ2 expression. They found that DNA hypermethylation-mediated silencing of miR-152 and COPZ2 was a relatively frequent molecular event in endometrial cancer and inhibited cell growth in endometrial cancer cell lines, suggesting the epigenetic silencing of these genes to contribute to endometrial carcinogenesis. The restoration of miR-152 expression in endometrial cancer cell lines was sufficient to inhibit tumor cell growth in vitro and in vivo.
For the specific miRNA with dual identity that either onco-miR or ts-miR, there are different methylation pattern in different cancers. miR-224 and miR-452 are located in the intron 6 of the host gene γ-aminobutyric acid A receptor (GABRE). There are significant positive correlations between expressions of miR-224, miR-452 and GABRE in previous studies [122] and TCGA data sets across cancer types (R = 0.58~0.83; FDR = 0; Table 1), demonstrating that these three genes are transcriptionally co-regulated under the same promoter. Cui et al. [128] found that miR-224 was significantly upregulated and the CpG island in the promoter region of miR-224 was hypomethylated in lung cancer, suggesting the potent oncogenic role in lung cancer. In contrast, GABRE~miR-452~miR-224 locus is downregulated and hypermethylated in prostate cancer, suggesting tumor-suppressive role in prostate cancer [122]. Silencing/activating these miRNAs by epigenetic alterations of cognate host genes can be a potential way for cancer therapy.
Besides DNA methylation, histone acetylation is another way of epigenetic regulation. The ts-miRs of miR-15a/16–1, encoded within an intron region of the non-coding DLEU2 gene, are downregulated in multiple tumor types while being frequently deleted in CLL. miR-15a/16–1 is believed to be transcribed from the DLEU2 promoter. Kasar et al. [129] revealed that the transcription factor BSAP (B-cell-specific activator protein) directly interacts with DLEU2, resulting in repression of miR-15a/16–1 expression by negative regulation of the DLEU2 promoter. Derepression of the DLEU2 promoter via inhibition of histone deacetylation in combination with BSAP knockdown increased miR-15a/16–1 expression, leading to malignant B-cell death. Thus, therapy targeting enhanced host gene DLEU2 transcription may augment CLL therapy. miR-126, harbored within the intron of the EGFL7 gene, is downregulated in cancer cell lines and also in primary bladder and prostate tumors. miR-126 and one of the transcripts of EGFL7 are concomitantly upregulated in cancer cell lines by inhibitors of DNA methylation and histone deacetylation. All these findings demonstrated that epigenetic changes can control the expression of onco/ts-miRs by directly controlling their host genes [130]. The knowledge of common and cancer-specific co-transcription patterns between miRNAs and host genes will help design therapeutic methods, targeting miRNA expression efficiently.
The advantage of miRNAs as therapeutic agents is based on their ability to concurrently target a large number of genes/pathways. However, this ability also causes a major concern on their toxicity due to potential off-target effects. While miRNAs target oncogenic pathways, they modulate other biological processes, which would cause far stronger side effects than generally appreciated [131]. Additionally, most existing strategies only focus on single miRNA or a family of miRNAs. Since miRNAs act cooperatively in tumor initiation and progression, modulating single miRNA expression has a limited effect. Tumor proliferation can be easily restored through bypassing the miRNA. Strategies to reprogram aberrant miRNA networks in cancer would be more effective [132]. Other challenges for developing miRNA therapeutics include low bioavailability, limited effective delivery and long-term safety [133].
Conclusion
Dysregulation of miRNAs expression plays a critical role in human diseases, especially in cancer. There is a close functional connection and complicated interplay between miRNAs and host genes; therefore, it is essential to study the miRNA regulation in the context of their cognate host genes. Since most onco/ts-miRs are intragenic miRNAs, we studied the co-expression relationship between these miRNAs and host genes based on TCGA data sets. The common and cancer-specific co-expression relationships will facilitate the design of therapeutic methods to silence/activate onco/ts-miRs efficiently by epigenetic alterations of cognate host genes.
Key Points
Although there is a general understanding of the generation of mature miRNA from primary miRNA, the transcriptional regulation of primary miRNAs is still unclear.
Recent studies have shown that more than half of miRNAs are located within protein-coding or non-coding genes. Intragenic miRNAs and their host genes either share the promoter or have independent transcription.
Most ts-miRs and onco-miRs are intragenic miRNAs; therefore, it is necessary and important to interpret their function in the context of host genes.
The knowledge of common and tumor-specific co-expression patterns between miRNAs and host genes is very helpful for understanding transcriptional and post-transcriptional regulation of miRNAs and further developing miRNA therapeutics in cancer.
Supplementary Material
Baohong Liu got her PhD in bioinformatics from Tongji University in 2011 and worked in State Key Laboratory of Veterinary Etiological Biology, Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences from 2014. In 2017–18, she was a visiting scholar in Vanderbilt University.
Yu Shyr received his PhD in biostatistics from the University of Michigan in 1994. Now he is the Chair of Department of Biostatistics in Vanderbilt University.
Jianping Cai is the director of the laboratory of poultry research in State Key Laboratory of Veterinary Etiological Biology, Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences. He is interested in the study of the genome, protein function for chicken parasites.
Qi Liu received her PhD at Shanghai Jiaotong University in 2003, completed her postdoc at Vanderbilt University and is now the assistant professor in Vanderbilt University.
Funding
This work was supported by the Innovative Special Project of Agricultural Sci-Tech (Grant No. CAASASTIP-2014-LVRI-09).
Reference
- 1. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136(2):215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nozawa M, Miura S, Nei M. Origins and evolution of microRNA genes in plant species. Genome Biol Evol 2012;4(3):230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ohtsuka M, Ling H, Doki Y, et al. MicroRNA processing and human cancer. J Clin Med 2015;4(8):1651–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perron MP, Provost P. Protein interactions and complexes in human microRNA biogenesis and function. Front Biosci 2008;13:2537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- 6. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002;99(24):15524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oliveto S, Mancino M, Manfrini N, et al. Role of microRNAs in translation regulation and cancer. World J Biol Chem 2017;8(1):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M, Matyunina LV, Walker LD, et al. Evidence for the importance of post-transcriptional regulatory changes in ovarian cancer progression and the contribution of miRNAs. Sci Rep 2017;7(1):8171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jing W, Zhang X, Sun W, et al. CRISPR/CAS9-mediated genome editing of miRNA-155 inhibits proinflammatory cytokine production by RAW264.7 cells. Biomed Res Int 2015;2015:1–7,326042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huo W, Zhao G, Yin J, et al. Lentiviral CRISPR/Cas9 vector mediated miR-21 gene editing inhibits the epithelial to mesenchymal transition in ovarian cancer cells. J Cancer 2017;8(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He C, Li Z, Chen P, et al. Young intragenic miRNAs are less coexpressed with host genes than old ones: implications of miRNA-host gene coevolution. Nucleic Acids Res 2012;40(9):4002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao X, Qiao Y, Han D, et al. Enemy or partner: relationship between intronic micrornas and their host genes. IUBMB Life 2012;64(10):835–40. [DOI] [PubMed] [Google Scholar]
- 13. Ma N, Wang X, Qiao Y, et al. Coexpression of an intronic microRNA and its host gene reveals a potential role for miR-483-5p as an IGF2 partner. Mol Cell Endocrinol 2011;333(1):96–101. [DOI] [PubMed] [Google Scholar]
- 14. Li G, Wu X, Qian W, et al. CCAR1 5' UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res 2016;26(6):655–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinske LC, Galante PA, Kuo WP, et al. A potential role for intragenic miRNAs on their hosts' interactome. BMC Genomics 2010;11:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Georgakilas G, Vlachos IS, Paraskevopoulou MD, et al. microTSS: accurate microRNA transcription start site identification reveals a significant number of divergent pri-miRNAs. Nat Commun 2014;5:5700. [DOI] [PubMed] [Google Scholar]
- 17. Seila AC, Calabrese JM, Levine SS, et al. Divergent transcription from active promoters. Science 2008;322(5909):1849–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seila AC, Core LJ, Lis JT, et al. Divergent transcription: a new feature of active promoters. Cell Cycle 2009;8(16):2557–64. [DOI] [PubMed] [Google Scholar]
- 19. Hinske LC, Franca GS, Torres HA, et al. miRIAD-integrating microRNA inter- and intragenic data. Database (Oxford) 2014;2014:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lutter D, Marr C, Krumsiek J, et al. Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genomics 2010;11:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42(Database issue):D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011;39(Database issue):D152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffiths-Jones S. miRBase: microRNA sequences and annotation. Curr Protoc Bioinformatics 2010;Chapter 12, Unit 12.9;1–10. [DOI] [PubMed] [Google Scholar]
- 24. Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol 2006;342:129–38. [DOI] [PubMed] [Google Scholar]
- 25. Griffiths-Jones S, Saini HK, Van Dongen S, et al. , miRBase: tools for microRNA genomics. Nucleic Acids Res 2008;36(Database issue):D154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005;11(3):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J 2007;26(3):775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corcoran DL, Pandit KV, Gordon B, et al. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One 2009;4(4):1–10,e5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dohi O, Yasui K, Gen Y, et al. Epigenetic silencing of miR-335 and its host gene MEST in hepatocellular carcinoma. Int J Oncol 2013;42(2):411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barron N, Sanchez N, Sanchez P, et al. MicroRNAs: tiny targets for engineering CHO cell phenotypes? Biotechnol Lett 2011;33(1):11–21. [DOI] [PubMed] [Google Scholar]
- 31. Sun Y, Ji F, Kumar MR, et al. Transcriptome integration analysis in hepatocellular carcinoma reveals discordant intronic miRNA-host gene pairs in expression. Int J Biol Sci 2017;13(11):1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li SC, Tang P, Lin WC. Intronic microRNA: discovery and biological implications. DNA Cell Biol 2007;26(4):195–207. [DOI] [PubMed] [Google Scholar]
- 33. Schanen BC, Li X. Transcriptional regulation of mammalian miRNA genes. Genomics 2011;97(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim HR, Shin CH, Lee H, et al. MicroRNA-1908-5p contributes to the oncogenic function of the splicing factor SRSF3. Oncotarget 2017;8(5):8342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ozsolak F, Poling LL, Wang Z, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev 2008;22(22): 3172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agranat-Tamir L, Shomron N, Sperling J, et al. Interplay between pre-mRNA splicing and microRNA biogenesis within the supraspliceosome. Nucleic Acids Res 2014;42(7):4640–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sundaram GM, Common JE, Gopal FE, et al. ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing. Nature 2013;495(7439):103–6. [DOI] [PubMed] [Google Scholar]
- 38. Ribas J, Ni X, Castanares M, et al. A novel source for miR-21 expression through the alternative polyadenylation of VMP1 gene transcripts. Nucleic Acids Res 2012;40(14):6821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melamed Z, Levy A, Ashwal-Fluss R, et al. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Mol Cell 2013;50(6):869–81. [DOI] [PubMed] [Google Scholar]
- 40. Toyota M, Suzuki H, Sasaki Y, et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008;68(11):4123–32. [DOI] [PubMed] [Google Scholar]
- 41. Kim DH, Saetrom P, Snove O, et al. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A 2008;105(42):16230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kos A, Olde Loohuis F, Wieczorek ML, et al. A potential regulatory role for intronic microRNA-338-3p for its host gene encoding apoptosis-associated tyrosine kinase. PLoS One 2012;7(2):e31022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dill H, Linder B, Fehr A, et al. Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev 2012;26(1):25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmitt D, Tan M. The enemy within: regulation of host genes by intronic microRNAs. Chemotherapy, 2014. 3(e126):1–2. [Google Scholar]
- 45. Frixa T, Sacconi A, Cioce M, et al. MicroRNA-128-3p-mediated depletion of Drosha promotes lung cancer cell migration. Carcinogenesis 2018;39(2):293–304. [DOI] [PubMed] [Google Scholar]
- 46. Bi L, Sun L, Jin Z, et al. MicroRNA-10a/b are regulators of myeloid differentiation and acute myeloid leukemia. Oncol Lett 2018;15(4):5611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang B, Pan X, Cobb GP, et al. microRNAs as oncogenes and tumor suppressors. Dev Biol, 2007. 302(1):1–12. [DOI] [PubMed] [Google Scholar]
- 48. Palma Flores C, Garcia-Vazquez R, Gallardo Rincon D, et al. MicroRNAs driving invasion and metastasis in ovarian cancer: opportunities for translational medicine (Review). Int J Oncol 2017;50(5):1461–76. [DOI] [PubMed] [Google Scholar]
- 49. Yan J, Ma C, Gao Y. MicroRNA-30a-5p suppresses epithelial-mesenchymal transition by targeting profilin-2 in high invasive non-small cell lung cancer cell lines. Oncol Rep 2017;37(5):3146–54. [DOI] [PubMed] [Google Scholar]
- 50. Cheng AM, Byrom MW, Shelton J, et al. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005. 33(4):1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hinske LC, Heyn J, Galante PA, et al. Setting up an intronic miRNA database. Methods Mol Biol 2013;936:69–76. [DOI] [PubMed] [Google Scholar]
- 52. Leone V, D'angelo D, Rubio I, et al. MiR-1 is a tumor suppressor in thyroid carcinogenesis targeting CCND2, CXCR4, and SDF-1alpha. J Clin Endocrinol Metab 2011;96(9):E1388–98. [DOI] [PubMed] [Google Scholar]
- 53. Letelier P, Garcia P, Leal P, et al. miR-1 and miR-145 act as tumor suppressor microRNAs in gallbladder cancer. Int J Clin Exp Pathol 2014;7(5):1849–67. [PMC free article] [PubMed] [Google Scholar]
- 54. Chen D, Sun Y, Wei Y, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med 2012;18(10):1511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma L, Young J, Prabhala H, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010;12(3):247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meng Q, Xiang L, Fu J, et al. Transcriptome profiling reveals miR-9-3p as a novel tumor suppressor in gastric cancer. Oncotarget 2017;8(23):37321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics 2010;11(7):537–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 2006;25(46):6188–96. [DOI] [PubMed] [Google Scholar]
- 59. Chow TF, Mankaruos M, Scorilas A, et al. The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol 2010;183(2):743–51. [DOI] [PubMed] [Google Scholar]
- 60. Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007;133(2):647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther 2010;10(12):1224–32. [DOI] [PubMed] [Google Scholar]
- 62. Zhu S, Si ML, Wu H, et al. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 2007;282(19):14328–36. [DOI] [PubMed] [Google Scholar]
- 63. Peralta-Zaragoza O, Deas J, Meneses-Acosta A, et al. Relevance of miR-21 in regulation of tumor suppressor gene PTEN in human cervical cancer cells. BMC Cancer 2016;16:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang X, Gee H, Rose B, et al. Regulation of the tumour suppressor PDCD4 by miR-499 and miR-21 in oropharyngeal cancers. BMC Cancer 2016;16:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reis ST, Pontes-Junior J, Antunes AA, et al. miR-21 may acts as an oncomir by targeting RECK, a matrix metalloproteinase regulator, in prostate cancer. BMC Urol 2012;12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Campos-Viguri GE, Jimenez-Wences H, Peralta-Zaragoza O, et al. miR-23b as a potential tumor suppressor and its regulation by DNA methylation in cervical cancer. Infect Agent Cancer 2015;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsuruta T, Kozaki K, Uesugi A, et al. miR-152 is a tumor suppressor microRNA that is silenced by DNA hypermethylation in endometrial cancer. Cancer Res 2011;71(20):6450–62. [DOI] [PubMed] [Google Scholar]
- 68. Tan X, Fan S, Wu W, et al. MicroRNA-26a inhibits osteosarcoma cell proliferation by targeting IGF-1. Bone Res 2015;3:15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li Y, Sun Z, Liu B, et al. Tumor-suppressive miR-26a and miR-26b inhibit cell aggressiveness by regulating FUT4 in colorectal cancer. Cell Death Dis 2017;8(6):1–11,e2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Goto Y, Kojima S, Nishikawa R, et al. The microRNA-23b/27b/24-1 cluster is a disease progression marker and tumor suppressor in prostate cancer. Oncotarget 2014;5(17):7748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang J, Wang X, Wang Y, et al. Low expression of microRNA-30c promotes prostate cancer cells invasion involved in downregulation of KRAS protein. Oncol Lett 2017;14(1):363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Karatas OF, Wang J, Shao L, et al. miR-33a is a tumor suppressor microRNA that is decreased in prostate cancer. Oncotarget 2017;8(36):60243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Majid S, Wang J, Shao L, et al. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin Cancer Res 2013;19(1):73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ye J, Yao Y, Song Q, et al. Up-regulation of miR-95-3p in hepatocellular carcinoma promotes tumorigenesis by targeting p21 expression. Sci Rep 2016;6:1–12,34034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Riquelme I, Tapia O, Leal P, et al. miR-101-2, miR-125b-2 and miR-451a act as potential tumor suppressors in gastric cancer through regulation of the PI3K/AKT/mTOR pathway. Cell Oncol (Dordr) 2016;39(1):23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu Q, Wang J, Zhao Y, et al. Identification of active miRNA promoters from nuclear run-on RNA sequencing. Nucleic Acids Res 2017;45(13):e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Imamura T, Komatsu S, Ichikawa D, et al. Depleted tumor suppressor miR-107 in plasma relates to tumor progression and is a novel therapeutic target in pancreatic cancer. Sci Rep 2017;7(1):5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hamada S, Satoh K, Fujibuchi W, et al. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol Cancer Res 2012;10(1):3–10. [DOI] [PubMed] [Google Scholar]
- 79. Mets E, Van Peer G, Van Deer Meulen J, et al. MicroRNA-128-3p is a novel oncomiR targeting PHF6 in T-cell acute lymphoblastic leukemia. Haematologica 2014;99(8):1326–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhao L, Li R, Xu S, et al. Tumor suppressor miR-128-3p inhibits metastasis and epithelial-mesenchymal transition by targeting ZEB1 in esophageal squamous-cell cancer. Acta Biochim Biophys Sin (Shanghai) 2018;50(2):171–80. [DOI] [PubMed] [Google Scholar]
- 81. Papagiannakopoulos T, Friedmann-Morvinski D, Neveu P, et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene 2012;31(15):1884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kozaki K, Imoto I, Mogi S, et al. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 2008;68(7):2094–105. [DOI] [PubMed] [Google Scholar]
- 83. Althoff K, Beckers A, Odersky A, et al. MiR-137 functions as a tumor suppressor in neuroblastoma by downregulating KDM1A. Int J Cancer 2013;133(5):1064–73. [DOI] [PubMed] [Google Scholar]
- 84. Liu J, Li C, Jiang Y, et al. Tumor-suppressor role of miR-139-5p in endometrial cancer. Cancer Cell Int 2018;18:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Song M, Li C, Jiang Y, et al. MiR-139-5p inhibits migration and invasion of colorectal cancer by downregulating AMFR and NOTCH1. Protein Cell 2014;5(11):851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bischoff A, Huck B, Keller B, et al. miR149 functions as a tumor suppressor by controlling breast epithelial cell migration and invasion. Cancer Res 2014;74(18):5256–65. [DOI] [PubMed] [Google Scholar]
- 87. Zhou J, Xie M, Shi Y, et al. MicroRNA-153 functions as a tumor suppressor by targeting SET7 and ZEB2 in ovarian cancer cells. Oncol Rep 2015;34(1):111–20. [DOI] [PubMed] [Google Scholar]
- 88. Yang L, Li C, Liang F, et al. MiRNA-155 promotes proliferation by targeting caudal-type homeobox 1 (CDX1) in glioma cells. Biomed Pharmacother 2017;95:1759–64. [DOI] [PubMed] [Google Scholar]
- 89. Due H, Svendsen P, Bodker JS, et al. miR-155 as a biomarker in B-cell malignancies. Biomed Res Int 2016;2016:1–14,9513037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tili E, Svendsen P, Bodker JS, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 2007;179(8):5082–9. [DOI] [PubMed] [Google Scholar]
- 91. O'Connell RM, Chaudhuri AA, Rao DS, et al. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A 2009;106(17):7113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yue F, Cheng Y, Breschi A, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014;515(7527):355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Taylor MA, Sossey-Alaoui K, Thompson CL, et al. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest 2013;123(1):150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dong-Xu W, Jia L, Su-Juan Z. MicroRNA-185 is a novel tumor suppressor by negatively modulating the Wnt/beta-catenin pathway in human colorectal cancer. Indian J Cancer 2015;52(Suppl 3):E182–5. [DOI] [PubMed] [Google Scholar]
- 95. Hui X, Jian J, Li Z, et al. MiR-186 acts as a tumor suppressor by targeting TWIST1/2 and regulating the epithelial-mesenchymal transition in glioblastoma multiforme. Int J Clin Exp Pathol 2016;9(9):9706–14. [Google Scholar]
- 96. Hu Y, Tang Z, Jiang B, et al. miR-198 functions as a tumor suppressor in breast cancer by targeting CUB domain-containing protein 1. Oncol Lett 2017;13(3):1753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ren D, Yang Q, Dai Y, et al. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-kappaB signaling pathway. Mol Cancer 2017;16(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liu B, Tian Y, Li F, et al. Tumor-suppressing roles of miR-214 and miR-218 in breast cancer. Oncol Rep 2016;35(6):3178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hashimoto Y, Akiyama Y, Yuasa Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One 2013;8(5):1–11,e62589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sun C, Huang C, Li S, et al. miR-326 targets CCND1 and inhibits non-small cell lung cancer development. Oncotarget 2016;7(7):8341–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Scarola M, Schoeftner S, Schneider C, et al. miR-335 directly targets Rb1 (pRb/p105) in a proximal connection to p53-dependent stress response. Cancer Res 2010;70(17):6925–33. [DOI] [PubMed] [Google Scholar]
- 102. Cao J, Cai J, Huang D, et al. miR-335 represents an invasion suppressor gene in ovarian cancer by targeting Bcl-w. Oncol Rep 2013;30(2):701–6. [DOI] [PubMed] [Google Scholar]
- 103. Sandoval-Borquez A, Polakovicova I, Carrasco-Veliz N, et al. MicroRNA-335-5p is a potential suppressor of metastasis and invasion in gastric cancer. Clin Epigenetics 2017;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu H, Li W, Chen C, et al. MiR-335 acts as a potential tumor suppressor miRNA via downregulating ROCK1 expression in hepatocellular carcinoma. Tumour Biol 2015;36(8):6313–9. [DOI] [PubMed] [Google Scholar]
- 105. Jin Y, Zhao M, Xie Q, et al. MicroRNA-338-3p functions as tumor suppressor in breast cancer by targeting SOX4. Int J Oncol 2015;47(4):1594–602. [DOI] [PubMed] [Google Scholar]
- 106. Wu ZS, Wu Q, Wang CQ, et al. miR-340 inhibition of breast cancer cell migration and invasion through targeting of oncoprotein c-Met. Cancer 2011;117(13):2842–52. [DOI] [PubMed] [Google Scholar]
- 107. Grady WM, Parkin RK, Mitchell PS, et al. Epigenetic silencing of the intronic microRNA miR-342 and its host gene EVL in colorectal cancer. Oncogene 2008;27(27):3880–8. [DOI] [PubMed] [Google Scholar]
- 108. Zhu W, Qing J, Ma L, et al. MiR-346 suppresses cell proliferation through SMYD3 dependent approach in hepatocellular carcinoma. Oncotarget 2017;8(39):65218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li F, Liang J, Bai L. MicroRNA-449a functions as a tumor suppressor in pancreatic cancer by the epigenetic regulation of ATDC expression. Biomed Pharmacother 2018;103:782–9. [DOI] [PubMed] [Google Scholar]
- 110. Budd W. Combinatorial analysis of tumorigenic microRNAs driving prostate cancer. PhD diss., Virginia Commonwealth University, 2012.
- 111. Yi Y, Lu X, Chen J, et al. Downregulated miR-486-5p acts as a tumor suppressor in esophageal squamous cell carcinoma. Exp Ther Med 2016;12(5):3411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhao Y, Lu G, Ke X, et al. miR-488 acts as a tumor suppressor gene in gastric cancer. Tumour Biol 2016;37(7):8691–8. [DOI] [PubMed] [Google Scholar]
- 113. Zhang B, Ji S, Ma F, et al. miR-489 acts as a tumor suppressor in human gastric cancer by targeting PROX1. Am J Cancer Res 2016;6(9):2021–30. [PMC free article] [PubMed] [Google Scholar]
- 114. Nakano H, Miyazawa T, Kinoshita K, et al. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-X(L) in colorectal cancer cells. Int J Cancer 2010;127(5):1072–80. [DOI] [PubMed] [Google Scholar]
- 115. Hu W, Chen CS, Wu R, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell 2010;38(5):689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kikkawa N, Kinoshita T, Nohata N, et al. microRNA-504 inhibits cancer cell proliferation via targeting CDK6 in hypopharyngeal squamous cell carcinoma. Int J Oncol 2014;44(6):2085–92. [DOI] [PubMed] [Google Scholar]
- 117. Cui R, Guan Y, Sun C, et al. A tumor-suppressive microRNA, miR-504, inhibits cell proliferation and promotes apoptosis by targeting FOXP1 in human glioma. Cancer Lett 2016;374(1):1–11. [DOI] [PubMed] [Google Scholar]
- 118. Leivonen SK, Salhberg KK, Makela R, et al. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol Oncol 2014;8(1):93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Png KJ, Yoshida M, Zhang XH, et al. MicroRNA-335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev 2011;25(3):226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhang JK, Li YS, Zhang CD, et al. Up-regulation of CRKL by microRNA-335 methylation is associated with poor prognosis in gastric cancer. Cancer Cell Int 2017;17:28. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121. Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res 2008;68(20):8191–4. [DOI] [PubMed] [Google Scholar]
- 122. Kristensen H, Haldrup C, Strand S, et al. Hypermethylation of the GABRE~miR-452~miR-224 promoter in prostate cancer predicts biochemical recurrence after radical prostatectomy. Clin Cancer Res 2014;20(8):2169–81. [DOI] [PubMed] [Google Scholar]
- 123. Patel N, Tahara SM, Malik P, et al. Involvement of miR-30c and miR-301a in immediate induction of plasminogen activator inhibitor-1 by placental growth factor in human pulmonary endothelial cells. Biochem J 2011;434(3):473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Guo XZ, Ye XL, Xiao WZ, et al. Downregulation of VMP1 confers aggressive properties to colorectal cancer. Oncol Rep 2015;34(5):2557–66. [DOI] [PubMed] [Google Scholar]
- 125. Gambari R, Brognara E, Spandidos DA, et al. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: new trends in the development of miRNA therapeutic strategies in oncology (Review). Int J Oncol 2016;49(1):5–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nguyen DD, Chang S. Development of novel therapeutic agents by inhibition of oncogenic microRNAs. Int J Mol Sci 2017;19(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Daniunaite K, Dubikaityte M, Gibas P, et al. Clinical significance of miRNA host gene promoter methylation in prostate cancer. Hum Mol Genet 2017;26(13):2451–61. [DOI] [PubMed] [Google Scholar]
- 128. Cui R, Meng W, Sun HL, et al. MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci U S A, 2015;112(31):E4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kasar S, Underbayev C, Yuan Y, et al. Therapeutic implications of activation of the host gene (Dleu2) promoter for miR-15a/16-1 in chronic lymphocytic leukemia. Oncogene 2014;33(25):3307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Saito Y, Friedman JM, Chihara Y, et al. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun 2009;379(3):726–31. [DOI] [PubMed] [Google Scholar]
- 131. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov 2010;9(10):775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Li X, Su Y, Sun B, et al. An artificially designed interfering lncRNA expressed by oncolytic adenovirus competitively consumes oncomiRs to exert antitumor efficacy in hepatocellular carcinoma. Mol Cancer Ther 2016;15(7):1436–51. [DOI] [PubMed] [Google Scholar]
- 133. Price C, Chen J. MicroRNAs in cancer biology and therapy: current status and perspectives. Genes Dis 2014;1(1):53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.