

SUMMARY
NatA co-translationally acetylates the N-termini of over 40% of eukaryotic proteins and can associate with another catalytic subunit, Naa50, to form a ternary NatA/Naa50 dual enzyme complex (also called NatE). The molecular basis of association between Naa50 and NatA and the mechanism for how their association affects their catalytic activities in yeast and human are poorly understood. Here we determined the X-ray crystal structure of yeast NatA/Naa50 as a scaffold to understand coregulation of NatA/Naa50 activity in both yeast and human. We find that Naa50 makes evolutionarily conserved contacts to both the Naa10 and Naa15 subunits of NatA. These interactions promote catalytic crosstalk within the human complex, but do so to a lesser extent in the yeast complex, where Naa50 activity is compromised. These studies have implications for understanding the role of the NatA/Naa50 complex in modulating the majority of the N-terminal acetylome in diverse species.
Keywords: N-terminal acetylation, X-ray crystallography, protein complex, NatA, Naa50, NatE
Graphical abstract
In Brief.
The NatA/Naa50 complex contains two catalytic subunits and one auxiliary subunit for co-translational N-terminal acetylation. Deng et al. describe the X-ray crystal structure of yeast NatA/Naa50 and use it as a scaffold to uncover evolutionarily conserved catalytic crosstalk within the orthologous complexes in yeast and human.
INTRODUCTION
The majority of nascent peptides biosynthesized by the eukaryote cytosolic ribosome undergo several N-terminal co-translational modifications before they become fully functional and mature proteins. Along with initial methionine cleavage by methionine aminopeptidase (iMAP) and N-terminal myristoylation, N-terminal acetylation (Nt-acetylation), which transfers an acetyl moiety from acetyl-CoA to the N-terminal amino group of proteins, is an irreversible, highly conserved and abundant modification (Giglione et al., 2015). About 50–70% of S. cerevisiae proteins, 70%−75% of A. thaliana proteins, and 70–90% of H. sapiens proteins are subject to Nt-acetylation by a family of N-terminal acetyl transferases (NATs) (Starheim et al., 2012). Nt-acetylation is generally considered to be a co-translational process, with most NATs making direct interactions with both nascent peptide and the ribosome (Gautschi et al., 2003, Polevoda et al., 2008, Magin et al., 2017, Knorr et al., 2018).
Numerous studies have demonstrated that Nt-acetylation can affect diverse protein and cellular activities including gene regulation, apoptosis, protein folding, protein degradation, protein complex formation and subcellular localization (Aksnes et al., 2016, Holmes et al., 2014, Scott et al., 2011, Shemorry et al., Behnia et al., 2004, Yang et al., 2013, Dikiy and Eliezer, 2014, Hwang et al., 2010, Schiza et al., 2013, Starheim et al., 2009, Arnesen et al., 2006b, Setty et al., 2004, Pavlou and Kirmizis, 2016). Knockout of NATs frequently results in severe cellular defects including slow growth and reduced mating efficiency in yeast (Polevoda and Sherman, 2003). In higher eukaryotes, misregulation of N-terminal acetylation can lead to numerous developmental disorders and cancers (Kalvik and Arnesen, 2012, Myklebust et al., 2015, Lee et al., 2010, Yu et al., 2009, Arnesen et al., 2006a, Ametzazurra et al., 2008, Starheim et al., 2008, Starheim et al., 2009, Fluge et al., 2002, Hua et al., 2011). For example, the NatA catalytic subunit, Naa10, plays important roles in mammalian cells including cellular hypoxia, bone formation and DNA damage (Dorfel and Lyon, 2015, Jeong et al., 2002, Yoon et al., 2014, Yi et al., 2007). Mutation in human Naa10 causes the genetic disorder Ogden syndrome (Rope et al., 2011).
Several NATs, including NatA, NatB, NatC, NatD, NatE, and NatF are conserved across eukaryotes (Aksnes et al., 2016) (Aksnes et al., 2015, Aksnes et al., 2017, Stove et al., 2016). NatG is resident in chloroplasts of plant cells (Dinh et al., 2015), and the recently identified NatH/Naa80 exists only in animals to acetylate a subset of processed actins (Drazic et al., 2018). The presence of NatF, NatG, and NatH indicates that this modification can also occur post-translationally. NATs can acetylate the N-termini either when the initiator methionine (iMet) is cleaved or retained, mostly depending on the identity of the first two amino acids of the nascent peptide (Aksnes et al., 2016), which further enriches the complexity of the eukaryote proteome. Both NatA and NatD complete the process after iMet is cleaved. NatA shows specificity toward the N-termini of exposed A-, S-, T-, V-, C-, and sometimes G-, while NatD can only acetylate the N-terminal serine of histones H2A and H4 (Magin et al., 2015, Gottlieb and Marmorstein, 2018, Liszczak et al., 2013, Arnesen et al., 2009a, Song et al., 2003). Other NATs like NatB, NatC, NatE, and NatF modify peptides with the iMet retained. NatB prefers the Met-Asx/Glx type N-termini (MD-, ME-, MN-, and MQ-starting), whereas NatC, NatE, and NatF have some substrate overlap, and they acetylate peptides with “hydrophobic/amphipathic” residues like L, I, F, Y, K directly following iMet (Liszczak et al., 2011, Stove et al., 2016, Van Damme et al., 2016, Tercero et al., 1993, Evjenth et al., 2009, Van Damme et al., 2011b). NatA, B and C are the three major contributors to the Nt-acetylome, as they cover about 80% of all NAT substrates (Aksnes et al., 2016). Notably, NatA, B and C contain at least one auxiliary subunit to couple with the catalytic subunit for enzymatic activity towards cognate substrates and anchoring of the catalytic subunit to the ribosome, while NatD, F, G, H are each composed of only one single catalytic subunit. NatE is composed to two catalytic and one auxiliary subunit.
NatA is comprised of the catalytic subunit Naa10 and the auxiliary subunit Naa15 (Liszczak et al., 2013, Gottlieb and Marmorstein, 2018). Naa10 can also exist independently of Naa15, exhibiting altered substrate specificity and might also be involved in transcriptional activity inside the nucleus (Arnesen et al., 2005, Van Damme et al., 2011a). Alternatively, NatA can also form a complex with another catalytic subunit Naa50 to form a ternary NatA/Naa50 complex (also called NatE) (Gautschi et al., 2003), and NatA from some eukaryotic species including Chaetomium thermophilum and human exhibit tight binding affinity with a regulatory protein called HYPK (Gottlieb and Marmorstein, 2018, Weyer et al., 2017, Arnesen et al., 2010). Deletion of Naa50 in yeast shows no phenotype (Gautschi et al., 2003), while Naa50 knockout can affect sister chromatid cohesion in higher organisms like Drosophila and human (Hou et al., 2007, Chu et al., 2011, Ribeiro et al., 2016, Williams et al., 2003). Endogenous pulldown of NatA/Naa50 complexes from human cells indicates that over 80% of Naa50 is not associated with NatA in vivo (Hou et al., 2007), whereas the remaining ~20% forms a stable and stoichiometric complex. Furthermore, another recent study demonstrates that loss of Naa50 in Drosophila leads to a decrease of NatA in vivo acetylation activity (Rathore et al., 2016). A recent cryo-EM structure of a yeast NatA/Naa50-ribosome complex with an 8 Å NatA local resolution reveals that both Naa50 and the Naa15 regulatory subunit of NatA contribute to ribosome interaction, with the Naa10 active site about 50 Å from the ribosome exit tunnel, and the Naa50 active site about 2-times that distance (Knorr et al., 2018). The underlying molecular basis for the potential functional coupling and interplay between the catalytic subunits of the NatA/Naa50 complex is still poorly understood.
To understand the molecular basis for NatA/Naa50 co-regulation of N-terminal protein acetylation across species, we determined the X-ray crystal structure of a yeast NatA/Naa50 complex and used it as a scaffold to understand coregulation of NatA/Naa50 activity in yeast and human. We find that NatA/Naa50 forms a stable complex in yeast and human through conserved interactions: Naa50 makes contacts to both the Naa10 and Naa15 subunits of NatA. We demonstrate that NatA-Naa50 interactions can significantly promote the catalytic activities of the two catalytic subunits within the human complex relative to NatA or Naa50 alone. In contrast, in the yeast complex, Naa50 modestly increases the activity of NatA, and Naa50 is defective in catalytic activity, likely through compromised peptide binding. These studies have implications for understanding the role of the NatA/Naa50 complex in modulating the majority of the N-terminal acetylome in diverse species.
RESULTS
NatA and Naa50 form stable complexes in yeast and human
We used recombinant proteins to characterize the interaction between Naa50 and NatA from human (h) and S. pombe (Sp) in vitro. Recombinant SpNatA, SpNaa50, and human Naa50 were overexpressed and purified from E. coli, while hNatA was prepared from baculovirus-infected Sf9 insect cells. Consistent with previous data reporting a direct interaction between NatA and Naa50 (Gautschi et al., 2003, Hou et al., 2007), we found that N-terminal GST-tagged Naa50 can pull-down NatA. We also found that human and S. pombe NatA and Naa50 co-migrate by size-exclusion chromatography to form stochiometric complexes under near native salt concentration (200 mM NaCl) (Figure 1A). Interestingly, SpNaa50 maintained the ability to co-migrate with SpNatA in sizing buffer with NaCl concentration as high as 1 M (Figure 1A), suggesting that this interaction in S. pombe is robust and not dominated by electrostatic interactions, while hNatA and hNaa50 failed to form a stochiometric complex in the high salt buffer (Figure 1A) suggesting some divergence of the NatA-Naa50 interaction between the yeast and human complexes. In addition, we observed that SpNaa50 displays similar comigration with ScNatA under both native and high NaCl concentrations (Figure 1A), strongly indicating that the NatA-Naa50 interaction within yeast is highly conserved.
Figure 1. NatA and Naa50 form stable complexes in yeast and human.

(A) Gel filtration elution profiles of Naa50 with NatA in S. pombe (top),human (middle), and cross-species between yeast S. pombe and S. cerevisiae (bottom), using a Superdex S200 column, with either 200 mM or 1M NaCl in sizing buffer. Coomassie-stained SDS–PAGE of peak fractions are shown to the right of the chromatograms.
(B) Fluorescence polarization assays with NatA titrated into fluorescein-5-maleimide labeled Naa50 in both S. pombe (left) and human (right) systems. The data is fit to calculate a dissociation constant between NatA and Naa50.
(C) Differential scanning fluorimetry assays of NatA alone or with Naa50 in both yeast and human systems. Recorded melting temperature transitions are indicated.
To more quantitatively asses the interaction between NatA and Naa50, we carried out a Fluorescence Polarization (FP) assay, using Naa50 labeled with Fluorescein-5-Maleimide via exposed surface cysteines. We observed that Naa50 tightly binds to NatA in both the S. pombe and human systems with Kd values of 17 ± 3.4 nM and 29 ± 5.6 nM, respectively (Figure 1B). These data, together with the high degree of sequence conservation of the interaction interfaces between Naa50 and NatA in the two species (Figures S1–S3), suggest that Naa50 and NatA from yeast and human are very likely to have similar association mechanisms. However, we found that GST-hNaa50 failed to pull down SpNatA, and that hNaa50 and SpNatA could not form a stoichiometric complex in size-exclusion chromatography (data not shown), indicating that the details of the hNatA/Naa50 interaction have diverged over evolutionary time. This was further validated using differential scanning fluorimetry experiments, where we found that the addition of SpNaa50 to SpNatA and hNaa50 to hNatA had thermal melting temperature increases of 37.8 °C to 40.4 °C and 44 °C to 46.6 °C, respective ly; while the addition of hNaa50 to SpNatA had a thermal melting temperature of 37.5 °C, very close to SpNatA alone (Figure 1C). Notably, SpNaa50 significantly extended the melting temperature of ScNatA by 7.4°C, further con firming the existence of a conserved robust interaction across these two yeast species (Figure 1C).
Crystal structure of the ternary NatA/Naa50 complex shows that Naa50 contacts both subunits of NatA
To reveal the molecular basis for the association and regulation mechanism of the NatA/Naa50 complex, we determined its crystal structure. We were unable to obtain crystals of the human complex, and crystals of the S. pombe complex diffracted X-rays poorly. However, we found that full-length ScNaa15 (residues 1–854) from S. cerevisiae (Sc) with a C-terminal truncation of ScNaa10 (1–226 out of 238 total residues) and full-length ScNaa50 (residues 1–176) produced crystals with good diffraction quality, in the presence of inositol hexaphosphate (IP6) and bi-substrate analogs for both Naa50 and Naa10. The crystals formed in the P212121 space group with one ternary complex in the asymmetric unit. The diffraction data set was collected to 2.7 Å resolution, and the structure was determined by molecular replacement with an unpublished ScNatE complex (Protein Data Bank code: 4XNH) as an initial search model. The final structure was refined to Rwork and Rfree values of 22.21% and 25.03%, respectively. Refinement statistics can be found in Table 1.
Table 1.
Data Statistics for ScNatA/Naa50 Crystal Structure (PDB: 6O07)
| Crystal Parameters | |
|---|---|
| Space group | P212121 |
| Unit cell dimension | 84.44(90) |
| a, b, c (Å) | 125.73(90) |
| α, β, γ (°) | 145.92(90) |
| Data Collection | |
| Resolution (Å) | 50–2.7 (3.0–2.7a) |
| Unique reflections | 41,731 |
| Rmerge | 0.11 (0.18a) |
| l/σ | 13.11(1.923a) |
| Completeness (%) | 99.3 (99.1a) |
| Redundancy | 6.1 (5.7a) |
| Refinement | |
| Rwork/Rfree | 0.2221/0.2503 |
| Root-mean-square deviation | |
| Bonds (Å) | 0.002 |
| Angels (°) | 0.424 |
| Average B factors (Å2) | |
| Protein | 58.26 |
| Solvent | 60.44 |
| Ligand | 83.58 |
| Ramachandran statistics (%) | |
| Favored | 94.3% |
| Allowed | 5.7% |
| Outliers | 0 |
Value in parentheses are for highest-resolution shell.
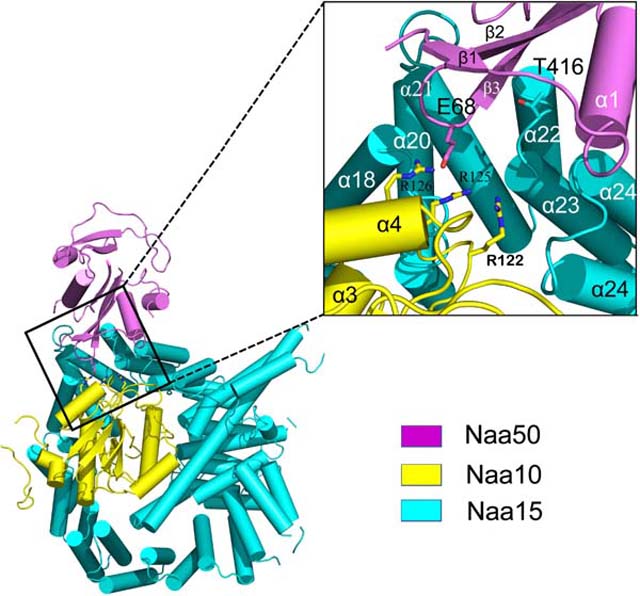
Not surprisingly, the ScNaa15 auxiliary subunit of NatA displays a high degree of structure conservation with SpNaa15 and hNaa15, with root-mean square deviation of 1.409 Å (over 577 common Cα atoms) and 1.537 Å (over 640 common Cα atoms), respectively. Shaped like a horseshoe, ScNaa15 is comprised of 15 TPR motifs, which often mediate protein-protein interactions. The electron density for the first two alpha helices (N terminal 1–53 residues) is poorly resolved and not built in our model (Figure S1). This auxiliary subunit, consisting of a total 42 α-helices, serves as the binding scaffold for both catalytic subunits. ScNaa10 is completely wrapped by the Naa15 helices (from α11 to α30, encompassing residues Lys198- Gly595) with extensive interactions, with an overall contact surface area of 3555.5 Å2 (Figure 2A). The longest alpha helix is α29, expanding to be as long as 70 Å across almost the entire length of the protein. This extremely long helix bridges the overall ring-like tertiary structure of Naa15 and locks Naa10 into the well-formed cradle made by the surrounding helices. Naa50 sits adjacent to Naa10 and interacts mostly with several Naa15 helices (α21, α22, α23, α24, encompassing residues Pro380-Asp448), and more modestly with the b2–b3 loop of Naa10, with an overall contact surface area of only 632.3 Å2 (Figure 2B).
Figure 2. Crystal structure of the ternary NatA/Naa50 complex shows that Naa50 contacts both subunits of NatA.

(A) ScNaa10 (yellow), ScNaa15(cyan), and ScNaa50 (violet) are shown in cartoon. The N-terminal two alpha helices (residues 1–53) of Naa15 are not resolved and not shown in the structure. Several alpha helices of Naa15 that contribute to Naa10 and Naa50 binding are labeled.
(B) Zoom-in view of the interface between Naa50, Naa10 and Naa15. Residues that contribute to interactions between Naa10 and Naa50 are shown.
(C) Zoom-in view showing key residues involved in interactions with IP6.
(D) Representative isothermal titration calorimetry (ITC) of curve of IP6 titrated into SpNatA with the calculated dissociation constant indicated.
The significantly less contact surface between Naa50 and Naa15, relative to between Naa10 and Naa15, is probably one reason that Naa50 in this ternary complex displays surprising flexibility, as indicated by its high average B factor (87.6 Å2) compared to Naa10 (48.9 Å2) and Naa15 (56.2 Å2). Several Naa50 residues away from the Naa50-Naa15 interface have poor side chain density and are therefore modeled as alanine residues and some parts of α helices and β strands are built as loops. Interestingly, the NatA bisubstrate analog used in the cocrystallization could not be resolved due to its poor electron density and was therefore omitted from the model, while only the CoA portion of the Naa50 bisubstrate inhibitor was well resolved and are therefore included in the model.
Previous studies have revealed that human NatA contains a bound IP6 molecule that bridges interactions between helices α19, α24, α25 of hNaa15 and the β2-loop-β3 of hNaa10 (Gottlieb and Marmorstein, 2018). To identify whether a similar IP6 binding pocket exists in ScNatA, IP6 was added to the crystallization of the ternary ScNatA/Naa50 complex. Clear electron density for IP6 was visible between Naa10 and Naa15 (Figure S4), in almost the same position identified in the corresponding human NatA complex (data not shown). Extensive electropositive and hydrogen-bond interactions contribute to this interaction including K80, Y85, and K91 from Naa10, and K429, K457, R464, R426, H430 and Y461 from Naa15 (Figure 2C). All these residues in Naa15 are conserved from yeast to human (Figure S1), which suggests that this binding mode is structurally conserved. We validated the conserved intimate binding of IP6 by also showing its robust binding to SpNatA using isothermal titration calorimetry (ITC), measuring a dissociation constant of ~130 nM, with a 1:1 stoichiometry (Figure 2D).
Hydrophobic interactions dominate the ScNatA-ScNaa50 binding interface
The NatA/Naa50 structure reveals that Naa15 is the primary docking site for Naa50. The contact interface is mediated primarily by the Naa50 β2-β3 and β4-α2 loops with the Naa15 α22-α23 loop. These regions form a network of hydrophobic interactions mediated by several non-polar residues, including Met18, Leu22, Tyr65, Pro70, and Val71 from Naa50 and Leu447, Val418, Thr416, Leu417 from Naa15 (Figure 3A). A few H-bonds are observed: the Naa15 Thr414 sidechain bridges and interaction with the side chain of Naa50 Tyr102 and Naa15 His413, and the Naa15 Thr416 sidechain interacts with the backbone carbonyl group of Naa50 Pro70 (Figure 3A). The largely hydrophobic nature of the interface is consistent with our biochemical observation that the SpNatA and Naa50 interaction can be maintained under high salt. Importantly, we found that the TPTLXE (where X represents either V or I) motif in the Naa15 α20-α21 loop is highly conserved in both yeast and human, thus suggesting that yeast and human share a similar NatA-Naa50 binding mode (Figure S1).
Figure 3. Hydrophobic interactions dominate the ScNatA-ScNaa50 binding interface.

(A) Zoom-in view of the major hydrophobic binding interface between Naa15 and Naa50 with residues that participate in interaction shown.
(B) Zoom-in view of the contacts between Naa10 and Naa50 with residues that participate in interaction shown.
Unlike Naa10, which is surrounded on all sides by interactions with Naa15 helical segments, thereby locking Naa10 in place, contacts between Naa50 and Naa10 are more modest and mediated by the α3-β4 loop of Naa10. Specifically, contact between Naa10 and Naa50 involves several basic residues within the α3-β4 loop of Naa10 (Arg122, Arg125 and Arg126) and Glu68 within the β2-β3 of Naa50, which make a network of electrostatic interactions (Figure 3B). The distance of Glu68 to Arg122, Arg125 and Arg126 is 2.3 Å,3.8 Å, and 6.2 Å, respectively. The relative paucity of Naa50-NatA interactions likely allows Naa50 to be conformationally flexible to allow it to impose regulatory effects on Naa10 activity. Supporting this notion is the fact that Naa10 Arg125 is the first conserved residue of the acetyl-CoA binding motif Q/RxxGxG/A (Figure S2).
The NatA/Naa50 complex promotes catalytic crosstalk
Since deletion of ScNaa50 shows no phenotype (Gautschi et al., 2003), while Naa50 knockout in higher organisms has been shown to perturb sister chromatid cohesion (Hou et al., 2007, Chu et al., 2011, Ribeiro et al., 2016) and NatA acetylation activity in vivo (Rathore et al., 2016), we hypothesized that there might be fundamental functional differences in the catalytic properties of Naa50 within the NatA/Naa50 complexes in yeast and human. To test this hypothesis, we took advantage of the fact that NatA and Naa50 have distinct substrate preferences. Canonical NatA substrates include peptides with the first four amino acids of SASE (Arnesen et al., 2009b, Liszczak et al., 2013) (hereinafter called “SASE peptide”) and Naa50 substrates include peptides with the first four amino acids MLGP (Liszczak et al., 2011, Evjenth et al., 2009) (hereinafter called “MLGP peptide”). These peptides were used because of their high specificity by their cognate NATs. For our kinetic analysis, we ignored any background activity contribution of NatA toward MLGP peptide and Naa50 toward SASE peptide, based on our data that NatA has negligible activity towards an MLGP peptide (Figure 7) and previously published data showing that Naa50 has negligible activity towards non-methionine containing N-termini (Evjenth et al., 2009).
Figure 7. NatA TY mutants disrupt NatA/Naa50 complex interactions.

(A) Fluorescence polarization assays of NatA TY mutants and Naa50 in S. pombe and human.
(B) Differential scanning fluorimetry assays of NatA TY mutants with and without Naa50. Recorded melting temperature transitions are indicated.
(C) Comparison of acetylation activity of NatA wild-type and TY mutants. Activities are normalized to WT protein activity level.
We found that hNaa50, either alone or in complex with hNatA, harbored robust in vitro acetylation activity toward the Naa50 cognate peptide, MLGP (Figure 4A). Unlike hNaa50, we found that both SpNaa50 alone or with SpNatA, and ScNaa50 alone or with ScNatA, were enzymatically inactive toward the MLGP peptide (Figure 4A), consistent with the reported lack of in vivo phenotype of ScNaa50 deletion (Gautschi et al., 2003). Notably, we found that that hNaa50 activity was significantly promoted within the ternary hNatA/Naa50 complex (Figure 4B). Indeed, kinetic analysis as a function of peptide substrate demonstrated that hNaa50 harbored a 14-fold decrease in Km value and 11-fold increase of catalytic efficiency when bound to hNatA (Figure 4B, Table 2). The dramatic change in Km value suggested that either hNaa15 or hNaa10 can affect hsNaa50 activity by increasing its substrate binding affinity.
Figure 4. The NatA/Naa50 complex promotes catalytic crosstalk.

(A) Time course acetylation activity of SpNaa50, SpNatA/Naa50, hNaa50, hNatA/Naa50, ScNaa50 and ScNatA/Naa50 against the MLGP peptide substrate.
(B) Michaelis–Menten kinetic curve of hNaa50 and hNatA/Naa50 against the MLGP peptide substrate.
(C) Michaelis–Menten kinetic curve of hNatA and hNatA/Naa50 against the SASE peptide substrate.
(D) Michaelis–Menten kinetic curve of SpNatA and SpNatA/Naa50 against the SASE peptide substrate.
Table 2.
Catalytic Parameters for NatA, Naa50, and NatA/Naa50 against peptide substrate
| ╱ | Normalized Km | Normalized Vmax | Normalized catalytic efficiency |
|---|---|---|---|
| SpNatA (SASE peptide) | 1 | 1 | 1 |
| SpNatA/Naa50 (SASE peptide) | 0.41 | 0.63 | 1.54 |
| hNatA (SASE peptide) | 1 | 1 | 1 |
| hNatA/Naa50 (SASE peptide) | 0.43 | 0.57 | 1.33 |
| hNaa50 (MLGP peptide) | 1 | 1 | 1 |
| hNatA/Naa50 (MLGP peptide) | 0.071 | 0.77 | 10.8 |
We also investigated the effect of Naa50 binding on hNatA activity towards its cognate SASE peptide in both the yeast and human systems. We observed that hNatA showed about a 2.29-fold decrease of Km value and 1.75-fold decrease of Vmax when it bound to hNaa50 (Figure 4C, Table 2). Similarly, we found that SpNatA showed about a 2.45-fold decrease of Km value and 1.60-fold decrease of Vmax when bound to SpNaa50 (Figure 4D, Table 2). Overall, SpNatA and hNatA showed comparably significant but modest increases in catalytic efficiencies of 1.53-fold and 1.31-fold, respectively. It is noteworthy that both the Naa10 catalytic subunit of the NatA complex and hNaa50 display a lower Km for peptide when in the ternary complex, while SpNaa10 also displays a lower Km in the yeast complex. Taken together, we conclude that the acetylation activities of both catalytic subunits- Naa10 and Naa50 are promoted in this ternary human NatA/Naa50 complex and Naa10 activity is more modestly promoted in the S. pombe complex.
Narrow substrate binding sites likely contribute to the catalytic inactivity of yeast Naa50s
Since we were unable to observe catalytic activity for SpNaa50 and ScNaa50, we were surprised to observe electron density in the ScNaa50 active site that could be used to model the acetyl-CoA portion of the acetyl-CoA-MLGP bisubstrate inhibitor that was used in the cocrystallization. Moreover, sequence alignment of Naa50 in human and yeast reveals that SpNaa50 and ScNaa50 do not contain an optimal Q/RxxGxG/A consensus acetyl-CoA binding motif, where in both SpNaa50 and ScNaa50 the first glycine residue is replaced with a serine residue (Figure S3). Using ITC, we were able to detect acetyl-CoA binding to hNaa50 (Figure 5A) but not to SpNaa50 (Figure 5B). We also observed that the SpNatA/Naa50 complex bound acetyl-CoA about 4.5-fold more strongly than SpNatA alone (Figures 5C and 5D). Given that SpNaa50 alone binding is not detected in ITC, we propose that the 4.5-fold increase in acetyl-CoA binding affinity for SpNatA/Naa50 relative to SpNatA alone suggests that SpNaa50 binding to SpNatA facilitates acetyl-CoA binding activity to SpNatA. This is consistent with our structural observation that ScNaa50 sits closely to the Q/RxxGxG/A acetyl-CoA binding motif of ScNaa10, and thereby able to modulate the acetyl-CoA binding activity of ScNatA. Surprisingly, binding signal was detected by ITC between GST-ScNaa50 and acetyl-CoA (Figure 5E), but not the free GST or GST-SpNaa50 controls (Figure 5F and 5G). This result suggests that the lack of acetyltransferase activity of ScNaa50 is unlikely due to a defect in acetyl CoA binding. Consistent with the catalytic inactivity of SpNaa50 and ScNaa50, we found that the α1-α2 and β6-β7 loops of ScNaa50 that typically flank the peptide substrate, form a much narrower groove compared to hNaa50 for peptide binding, with the most intimate distance between the loops of less than 2.8 Å (Figure 5H). In addition, both SpNaa50 and ScNaa50 lack the characteristic YY motif found in the b6–b7 loop which is important for substrate binding in all NATs except for Naa40 and Naa80 (Figure S3)(Liszczak et al., 2013, Liszczak et al., 2011, Magin et al., 2015, Goris et al., 2018, Hong et al., 2017, Aksnes et al., 2015). Taken together, it appears that the acetyl-CoA and N-terminal peptide substrate binding sites of SpNaa50 are not properly configured for catalytic activity, while ScNaa50 binds acetyl-CoA but likely does not bind peptide substrate.
Figure 5. Weak acetyl-CoA binding activity and a narrow substrate binding grove contributes to the catalytic inactivity of yeast Naa50.

(A) Representative ITC curve of acetyl-CoA titrated into hNaa50.
(B) Representative ITC curve of acetyl-CoA titrated into SpNaa50.
(C) Representative ITC curve of acetyl-CoA titrated into SpNatA. The calculated dissociation constant is indicated.
(D) Representative ITC curve of acetyl-CoA titrated into SpNatA/Naa50. The calculated dissociation constant is indicated.
(E) Representative ITC curve of acetyl-CoA titrated into GST-ScNaa50.
(F) Representative ITC curve of acetyl-CoA titrated into GST-SpNaa50.
(G) Representative ITC curve of acetyl-CoA titrated into free GST.
(H) Superimposition of ScNaa50 and hNaa50 (PDB: 3TFY) structures.
NatA-Naa50 from yeast and human make conserved interactions
To evaluate the degree of evolutionary conservation of the NatA-Naa50 interface, we first used the ScNatA/Naa50 complex as a scaffold to carry out mutagenesis and binding studies of the yeast NatA/Naa50 complex, using S. pombe for mutagenesis for consistency with the earlier biochemical studies in S. pombe. Superimposition of the ScNatA/Naa50 and SpNatA structures highlighted a few potential key S. pombe candidate residues for interaction, including SpNaa10 R117, SpNaa15 T412, and SpNaa50 Y49 and V53 (Figures 3 and S1-S3). Using pull-down assays, we demonstrated that SpNaa10-R117E had an insignificant effect on SpNatA binding, while SpNaa50-V53K and -Y49A displayed some decrease in SpNatA binding (Figure 6A, left). As expected, the double mutation, SpNaa50- Y49A/V53K was significantly defective in SpNatA and ScNatA binding (Figure 6A). As these two residues are conserved from yeast to human, it was not surprising that the double mutation of hNaa50-Y50A/I54K displayed similar defects in hNatA binding. The ScNatA/Naa50 crystal structure reveals that ScNatA-T412 sits roughly in the middle position of the conserved Naa15 TPTLXE motif and is located in the center of the binding interface within Naa15 and Naa50 (Figure 3A). Interestingly, we found that a single SpNaa15-T412 mutation to either tyrosine or lysine completely disrupted complex formation with SpNaa50 (Figure 6A). As expected, sequence alignment demonstrates that SpNaa15-T412 is well conserved in both human and yeast (Figure S1). We further observed that both ScNaa15-T416Y and hNaa15-T406Y (hereinafter referred to as TY mutants) lost their ability to association with Naa50 in pull down assays (Figure 6A). Consistent with the pull-down results (Figure 6A), the Naa15 TY mutant from S. pombe, S. cerevisiae and human did not co-migrate with Naa50 by size-exclusion chromatography (Figure 6B). For further studies, we focused on SpNatA T412Y and the conserved hNatA T406Y and characterized their roles in NatA-Naa50 association. While the addition of Naa50 to NatA showed robust binding using a FP assay (Figure 1A) and increased thermal melting temperature of NatA by about 2.5 °C using DSF assay (Figure 1B), addition of Naa50 to the NatA TY mutants did not show detectable binding (Figure 7A) or increase in NatA TY mutant thermal melting temperature (Figure 7B). We further tested the effect of the NatA TY mutants on NatA acetylation activity and found that they maintained acetylation activity towards cognate substrates although the hNatA TY mutant showed an apparent increase in acetylation activity relative to WT (Figure 7C). The reason for this is unclear. Taken together, the Naa15 TY mutants can disrupt the association between NatA and Naa50 in yeast and human. These studies highlight the important roles of this conserve threonine residue in the association between Naa50 and NatA.
Figure 6. NatA TY mutants are unable to pull-down Naa50 and co-migrate with Naa50.

(A) GST pull-down assay with NatA/Naa50 mutants to interrogate the contribution of residues in Naa50-NatA association.
(B) Gel filtration elution profiles of Naa50 with either wild-type (dotted red line taken from Figure 1A) or TY mutants (blue line) of NatA using a Superdex S200 column. Coomassie-stained SDS–PAGE of peak fractions of Naa50 with NatA TY mutants shown below the chromatograms.
DISCUSSION
While the molecular basis for acetylation activity by either NatA or Naa50 independently has been previously described, the underlying molecular mechanism of how these enzymes work in complex was poorly understood before this study. Although similar to an unpublished structure of ScNatE that has been deposited in the PDB (4XNH), our reported crystal structure of the ScNatA/Naa50 complex in combination with related biochemical data provides a peer reviewed and comprehensive analysis of the structure and mechanism of acetylation by the N-terminal dual enzyme NatA/Naa50 complex across evolution. Overall, the Naa50 electron density is poorly resolved relative to NatA, which suggested that its association with NatA is likely dynamic. Indeed, the major contact region between Naa50 and Naa15 is mediated by loop-loop interactions, unlike the extensive interactions made between Naa10 and Naa15 within the NatA complex. We also found that ScNaa50 makes direct electrostatic interactions with Naa10, providing a path of contact for Naa10 and Naa50 to influence each other’s catalytic functions.
Because of their conserved association motif, the hNaa50 docking site on hNaa15 could be identified, and was further supported by mutational analysis. We propose that the proper distance between Naa10 and Naa50 that is required for cognate acetylation of N-terminal protein substrates by the respective NATs, coupled to the less rigid positioning of Naa50, relative to Naa10, may allow Naa50 to adopt multiple conformations within the NatE complex to facilitate both Naa50 and Naa10 cognate substrate binding. Indeed, we found that hNaa50 and hNatA displayed significant enhancement in substrate affinity and catalytic efficiency in the context of the complex, although such enhancement was not observed in the yeast complex, where we found that Naa50 is catalytically defective. This is consistent with our structural data showing that unlike Naa10, Naa50 harbors a narrow substrate binding site that is incompatible with N-terminal protein substrate binding. However, we cannot exclude the possibility that yeast Naa50 is enzymatically active against another substrate. Regardless, our findings suggest some evolutionary divergence between yeast and human NatE complexes. Nonetheless, we also found that SpNaa50 does not bind human NatA and vice versa, yet from conservation analysis there is no obvious reason why this should be the case. There may be subtle differences in the yeast and human systems that have co evolved, perhaps to reflect the regulatory role of the HYPK protein in the human but not S. pombe and S. cerevisiae yeast systems(Arnesen et al., 2009a).
Based on our in vitro activity data, it is noteworthy that both human Naa10 and Naa50 catalytic subunits display lower Km values for their respective peptide substrates and increased catalytic efficiencies, when they are in the NatA/Naa50 relative to when they are not in complex. Meanwhile, we observed that the other substrate, acetyl-CoA, while not showing detectable binding to SpNaa50, binds to SpNatA/Naa50 about 4.5-fold tighter than SpNatA alone. It is likely that SpNaa50 facilitates acetyl-CoA binding to SpNatA and the structural observation that ScNaa50 sits closely to the Q/RxxGxG/A acetyl-CoA binding motif of ScNaa10 provides a plausible explanation for how SpNaa50 may modulate the acetyl-CoA binding activity of SpNatA. Taken together, we conclude that in human, NatA and Naa50 have cooperative effects on their acetylation activities when associating together, while in yeast, the inactive Naa50 still contributes to NatA activity, which is consistent with previous result that reduction of some NatA type N-termini was detected in yeast Naa50 deletion mutants (Van Damme et al., 2015) We note that the cooperative effects between NatA and Naa50 that we observed in vitro are based only on single cognate NatA and Naa50 peptide substrates. It is currently unknown if the degree of cooperation varies with different peptide substrates.
One surprising aspect of the ScNatA/Naa50 structure is the identification of Inositol hexaphosphate (IP6), which bridges interactions between the Naa10 and Naa15 subunits of NatA with a dissociation constant in the sub-mircomolar range. A similar role for IP6 was identified in the recently reported hNatA/HYPK complex (Gottlieb and Marmorstein, 2018), suggesting an evolutionarily conserved role of IP6 in stabilizing the NatA complex. Since IP6 has long been appreciated to have many signaling properties in diverse organisms including yeast and mammals (Abul Kalam and Sanchita, 2012), it is also possible that IP6 may have a yet unidentified signaling role in NatA function.
HYPK is an important regulatory protein that binds to hNatA but is not conserved in S. pombe or S. cerevisiae. Recent structural studies suggest that HYPK was a nanomolar dissociation constant NatA binding partner and that HYPK binding likely inhibits Naa50 binding (Weyer et al., 2017, Gottlieb and Marmorstein, 2018). Here we have demonstrated that hNaa50 can promote hNatA acetylation activity. We propose that the slightly divergent association mechanism between NatA and Naa50 in yeast and human may be related to the presence of HYPK in human and higher eukaryotes but not in most yeast. It will be of interest to further investigate the interplay among these binding partners within the tetrameric human NatA/Naa50/HYPK complex.
We find in our study that SpNaa50 is catalytically inactive, and this is likely due to its reduced affinity for acetyl-CoA and the relative narrowness of the peptide substrate binding site of ScNaa50 observed in the ScNatA/Naa50 structure. Surprisingly, we observed binding between ScNaa50 and acetyl-CoA, although the optimal acetyl-CoA binding motif is missing in both ScNaa50 and SpNaa50. Nonetheless, both ScNaa50 and SpNaa50 are catalytically inactive in out acetyltransferase assays. We propose that the yeast lineage lost catalytically active Naa50 at least due to the lack of peptide substrate binding. Indeed, unlike metazoans, ScNaa50 knock out displays no phenotype (Gautschi et al., 2003). In another study, no impact on Met-starting yeast N-termini was observed where yeast Naa50 was deleted (Van Damme et al., 2015). Meanwhile, Naa50 has functional roles in sister chromatic cohesion in higher organism, independent of the NatA complex, which is not observed in yeast (Rong et al., 2016, Ribeiro et al., 2016, Williams et al., 2003). Thus, yeast Naa50 is very likely enzymatically inactive, only providing extra stability and regulatory roles for NatA, including participation in ribosome binding (see below). We propose that Naa50 substrates are processed by NatC, which recognizes a similar cognate sequence.
A recent cryo-EM structure of a native ScNatA/Naa50-ribosome structure, where the ribosome is resolved to 3.4 Å and the NatA/Naa50 portion is resolved to about 8 Å resolution, indicates that NatA/Naa50 associates with the ribosome through interaction with the ribosomal RNA expansion segments involving predominantly Naa15, but also Naa50 (Knorr et al., 2018). While Naa10 is positioned proximal to the ribosome peptide exit tunnel, Naa50 is remote from the peptide exit tunnel. A docking of our 2.7 Å resolution NatE structure onto the cryo-EM structure (PDB: 6HD7) provides a pseudo-3.4 Å resolution structure of the NatE-ribosome complex (Figure 8). This superposition reveals that Naa50 is positioned ~80 Å away from the peptide exit tunnel of the ribosome, consistent with the finding of Knorr et al. that approximates this distance to be ~ 85 Å (Knorr et al., 2018). As this observation suggests that peptides emerging from the ribosome exit tunnel encounter the Naa10 active site prior to the Naa50 active site, we speculate that it is likely that Naa50 requires a longer nascent chain for acetylation. However, we found that SpNaa50 binding slightly promotes SpNatA acetylation catalytic efficiency. This effect could be more significant in the context of the ribosome, given that Knorr et al. observe that Naa50 makes contact to the ribosome RNA expansion segment, thus also playing a role in NatA recruitment to the ribosome. Such a contribution to NatA-ribosome recruitment by Naa50 is consistent with a recent observation that D. melanogaster Naa50 knockout impairs NatA acetylation activity in vivo (Rathore et al., 2016). We placed the Naa10 acetyl CoA binding site by overlapping our structure to the SpNatA structure in complex with bi-substrate analogue (PDB:4KVM) and found that the acetyl-CoA binding sites for both Naa10 and Naa50 are oriented in the same direction and facing the ribosome nascent peptide exit tunnel and separated by a distance of only 26.6 Å (Figure 8). It is likely that the relative position of these two catalytic subunits, Naa10 and Naa50, might change on the ribosome to accommodate the cognate peptide substrate that emerges out of the ribosome exit tunnel. We propose that the flexibility of the hNatA-Naa50 interface (as suggested by the relatively high B-factor of Naa50 relative to NatA) may also facilitate movement of Naa50 closer to the ribosome exit tunnel once its cognate substrate is in proximity for N-terminal acetylation. This might be facilitated further if human Naa50 did not participate in NatA anchoring to the ribosome as it does in the yeast system. Additional interactions might be compensated through the binding of HYPK.
Figure 8. Docking of our ScNatA/Naa50 crystal structure onto the ribosome-NatA/Naa50 Cryo-EM structure.

The ScNatA/Naa50 crystal structure is aligned to the NatA/Naa50-ribosome structure (PDB: 6HD7). The magnified view shows that Naa10 is most proximal to the ribosome peptide exit tunnel, while the acetyl-CoA binding sites of Naa10 and Naa50 are facing the same side.
Taken together, our biochemical and structural characterization of ternary NatA/Naa50 complexes in yeast and human in this study has provided new and significant insights into the association and regulatory mechanism of this unusual dual enzyme system. These studies set the stage for future studies to decipher the molecular mechanism of the human NatA/Naa50/HYPK complex. In addition, our identification of mutants that prevent NatA/Naa50 association in yeast and humans will facilitate analysis of the uncoupling of co- and post-translational protein acetylation by Naa50.
STAR Methods text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronen Marmorstein (marmor@upenn.edu).
EXPERIMENTAL MODEL
We used E.coli Rosetta (DE3)pLysS cells for recombinant expression of ScNatA/Naa50, SpNatA, SpNaa50, and hNaa50 for biochemical and biophysical experiments. The cells were cultured using standard practices in LB media.
We used Spodoptera frugiperda (Sf9) cells cultured in SFM II medium for the recombinant expression of hNatA for biochemical, biophysical, and X-ray crystallography experiments.
METHOD DETAILS
NatA and Naa50 expression and purification
Human NatA(hNatA), human Naa50(hNaa50), and Schizosaccharomyces pombe NatA(SpNatA) were expressed and purified as described previously(Liszczak et al., 2013, 2018, Liszczak et al., 2011). Full-length Schizosaccharomyces pombe Naa50(spNaa50) was cloned into pRSF with a N-terminal GST fusion, and expressed and purified with the same method as described for spNatA(Liszczak et al., 2013). Full-length Saccharomyces cerevisiae Naa15(ScNaa15) was cloned into pRSF_Duet vector with a N terminal PolyHis tag and a Tobacco Etch Virus (TEV) cleavage site. C-terminal truncated ScNaa10(1–226) with no tag was cloned into pET_Duet, and un-tagged full-length ScNaa50 was cloned into pCDF-Duet. To express the ternary ScNaa10-ScNaa15-ScNaa50 complex (ScNatA/Naa50 complex), these three plasmids were co-transformed into Rosetta (DE3)pLysS competent E. coli cells, which were grown to an OD600 of 1 and induced with 0.5 mM of Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C for ~16 h. ScNatA/Naa50 complex purification procedure was modified from SpNatA as described (Liszczak et al., 2013). Cells were isolated by centrifugation for 20 mins at 4500 rpm and lysed by sonication in a buffer containing 25 mM Tris, pH 8.0, 500 mM NaCl, 0.1 mg/mL PMSF. After centrifugation, the supernatant was isolated and passed over Ni-resin (Thermo Scientific), which was subsequently washed with 10 column volumes of lysis buffer supplemented with 25 mM imidazole and 10 mM 2-mercaptoethanol. Protein was eluted with 300 mM imidazole and dialyzed into buffer containing 25 mM sodium citrate monobasic, pH 5.5, 10 mM NaCl and 10 mM 2-mercaptoethanol. Protein was purified with a 5-mL HiTrap SP ion-exchange column and eluted in a salt gradient (10–700 mM NaCl). Peak fractions were concentrated to ~ 0.5 mL with a 100-kDa concentrator (Amicon Ultra, Millipore), and loaded onto an S200 gel-filtration column (GE Healthcare) in a buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, 5% glycerol and 1 mM TCEP. Proteins were aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C for further u se. Full-length ScNaa50 was cloned into pRSF with a N-terminal GST tag and a TEV cleavage site, to express and purify ScNaa50 alone. We observed that ScNaa50 was not stable in the absence of the GST tag and absorbs minimally at 280 nm. We therefore only worked with GST-ScNaa50 in biochemical experiments. GST-ScNaa50 was purified using the same method as described above. Concentrations of hNatA, hNaa50, SpNatA, SpNaa50, ScNatA/Naa50 and GST-ScNaa50 were determined by absorbance at 280 nm and using their extinction coefficient of 132,480 cm−1M−1,13,410 cm−1M−1, 120,000 cm−1M−1, 14,440 cm−1M−1, 151,800 cm−1M−1, and 58,790 cm−1M−1, respectively. All protein mutants were generated using the QuikChange protocol from Stratagene and obtained following the expression and purification protocols described above.
Fluorescence polarization (FP) assays
There are five cysteine residues present in SpNaa50 (Cys22, Cys54, Cys60, and Cys96) and four cysteine residues in hNaa50 (Cys60, Cys61, Cys79, and Cys100). We found that we did not have to make mutations to obtain the fluorescein labeled Naa50, which suggested that at least one cysteine in each is exposed to solvent. 25-fold excess of Fluorescein-5-Maleimide (ThermoFisher) was added into about 100 L of 1 mg/ml purified hNaa50 or SpNaa50 in sizing buffer. The reaction was quenched with 2-mercaptoethanol after overnight incubation at 4°C. Excess Fluoresce in-5-Maleimide was removed by S75 gel-filtration chromatography. To run the assays,10 nM of labeled Naa50 was used in all reactions, and NatA concentrations were varied to determine the dissociation constant (Kd). 5 mg/mL BSA and 0.2% v/v Tween were added into the reaction buffer (25 mM HEPES, pH 7.0, 200 mM NaCl and 10 mM DTT) to prevent non-specific binding. FP readings were recorded with a Perkin Elmer EnVision and each curve was repeated in triplicate. GraphPad Prism, version 5.01, was used for all data fitting. Errors were reported in Standard Deviation (SD) with n = 3.
Acetyltransferase activity assays
All the acetyltransferase assays were performed essentially as previously described (Liszczak et al., 2013, Liszczak et al., 2011) and carried out at room temperature in the reaction buffer containing 75 mM HEPES, pH 7.0, 120 mM NaCl, 1 mM DTT. The SASE substrate peptide (NH2-SASEAGVRWGRPVGRRRRP-COOH; GenScript) and the MLGP substrate peptide (NH2-MLGPEGGRWGRPVGRRRRP-COOH; GenScript) were used to determine the enzymatic activity of NatA and Naa50, respectively. For measuring Naa50 activity, a time course of activity was carried out with either 500 nM of SpNaa50, SpNatA/SpNaa50, hNaa50, hNatA/hNaa50, ScNaa50, and ScNatA/Naa50 mixed with 300 M of C14 labeled acetyl-CoA (4 mCi mmol−1; PerkinElmer Life Sciences), and 500 M MLGP peptide in a 100 L reaction volume. To quench the reaction at specific times, 15 μL of the reaction mixture was added to P81 paper discs (Whatman), and the paper discs were immediately placed in wash buffer with 10 mM HEPES, pH 7.5. Unreacted acetyl-CoA is removed by washing at least three times. Paper discs were dried with acetone and mixed with 4 mL scintillation fluid for signal measurement by a Packard Tri-Carb 1500 liquid scintillation analyzer. Data was plotted with GraphPad Prism, version 5.01. To determine steady-state catalytic parameters, enzymes were mixed with 500 μM C14 labeled acetyl-CoA and the corresponding peptide substrate concentration was varied. A 20 μL reaction were performed but only 15 μL of the reaction mixture was quenched onto the paper discs after specific times. For assaying hNaa50 and hNatA/hNaa50 against MLGP peptide, 300 nM enzyme was used in 40-minute reactions. For assaying hNatA and hNatA/hNaa50 against SASE peptide, 100 nM enzyme was used in 12-minute reactions. For assaying SpNatA and SpNatA/SpNaa50 against SASE peptide, 50 nM enzyme was used in 10-minute reactions. Paper disc washes and signal reading steps were performed in the same manner as mentioned above. GraphPad Prism, version 5.01, was used for all kinetic data fitting to the Michaelis–Menten equation. Each curve was repeated at least 3 times. For NatA TY mutant activity tests, 100 nM of SpNatA, SpNatA-T412Y, hNatA, and hNatA-T406Y were mixed with 300 M of C14 labeled acetyl-CoA and 500 M of peptide substrate for 30-minutes reactions. Signals were normalized to the WT activity, and errors were reported in Standard Error of the Mean (SEM) with n = 3.
Size exclusion chromatography assays
A total of 500 l of purified NatA (4.2 μM) and purified Naa50 (12.6 μM) was injected onto a S200 gel-filtration column (GE Healthcare). UV absorbance values were normalized to the maximum value of the first elution peak, then plotted against the elution volume in GraphPad Prism, version 5.01. Only elution volumes around the NatA, Naa50 and NatA/Naa50 elution peaks are shown in the figures. Corresponding peak fractions were run on SDS-PAGE and stained with colloidal Coomassie blue for imaging.
GST pull-down assays
1 μM of GST tagged Naa50 and 5 μM of NatA was incubate with 50 μL of glutathione agarose resin at 4 °C for 1 hour in rea ction buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl and 1 mM TCEP. Resin was then washed extensively with the same buffer and eluted with glutathione. Samples were collected for analysis on SDS-PAGE.
Differential scanning fluorimetry assays
Sypro Orange (5000X stock, ThermoFisher Scientific) was diluted 1:200 and 4 μL was mixed with 16 μL solution with 5 μM NatA and 5 μM Naa50 in sizing buffer. Fluorescent readings were recorded using a qPCR (ABI 7900 RealTime PCR) with a 2% ramp rate, while heated from 20 °C t o 95 °C. Melting curves were generated from these readings and melting temperatures were determined by taking the first derivative of the curves. DSF scans of all samples were performed in triplicate. Error bars in the figure indicated the Standard Deviation (SD) of each sample.
ITC measurements
Measurements were recorded on a MicroCal iTC200 at 20 °C. Samples were dialyzed into buffer containing 25 mM HEPES pH 7.0, 200 mM NaCl, 1 mM DTT. Protein samples (SpNaa50, SpNatA, SpNatA/Naa50, GST-SpNaa50, GST-ScNaa50, and GST) with concentrations of 50 M in the cell and 1mM of acetyl-CoA in the syringe were used in the experiments. For hNaa50, 100 M protein and 0.5 mM of acetyl-CoA was used. The raw data was analyzed with MicroCal ITC analysis software.
ScNatA/Naa50 crystallization and data collection
Ternary ScNatA/Naa50 complex was mixed with 3 molar excess of NatA bisubstrate inhibitor (CoA-SASEA), Naa50 bisubstrate inhibitor (CoA-MLGPE) and IP6. Bisubstrate inhibitors were prepared as previously described (Liszczak et al., 2013). Crystals were grown within ~3 days by hanging drop vapor diffusion at 20 °C at 7.5 mg/ml, using one to one drop ratios against reservoir solution containing 10% PEG 3350, 0.1 M Sodium Malonate (pH 5.0). Crystals were transferred into mother liquor supplemented with 25% glycerol and flash-frozen in liquid nitrogen. Data were collected at the Advanced Photon Source (beamline 24-ID-C) and processed using HKL2000 (Otwinowski and Minor, 1997).
ScNatA/Naa50 structure determination and refinement
The collected diffraction data showed strong anisotropy and ellipsoidal truncation and anisotropic scaling was performed (Strong et al., 2006). The resolution limit along a*, b*, c* are 2.7 Å, 3.4 Å, 2.7 Å, respectively. The crystal structure of ScNatA/Naa50 was determined by molecular replacement using a structure of unpublished ScNatE (PDB ID Code: 4XNH) without ligand or solvent molecules as a search model. Molecular replacement was done using Phaser in Phenix(Adams et al., 2010). Initial Manual model building was done in Coot (Emsley and Cowtan, 2004) and all subsequent rounds of refinement were performed using Phenix refine and Coot interchangeably. Refinement statistics can be found in Table 1. The final model and structure factors were submitted to the Protein Data Bank with code 6O07 (Research Collaboratory for Structural Bioinformatics).
Distance calculations, as well as three-dimensional alignment r.m.s. deviations and graphics were generated in PyMOL (www.pymol.org/)
Sequence alignments in the manuscript were performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and visualized using ESPript 3.0(Robert and Gouet, 2014) (http://espript.ibcp.fr/ESPript/ESPript/)
All surface area calculations were performed using PDBePISA (Proteins, Interfaces, Structures and Assemblies) (http://www.ebi.ac.uk/pdbe/pisa/)
QUANTIFICATION AND STATISTICAL ANALYSIS
In fluorescence polarization assays, Kd errors in the figure were generated by GraphPad Prism with a one-site binding model with n = 3 and value errors were reported as SD.
In acetyltransferase activity assays, kinetic curve error bars were generated by GraphPad Prism with Michaelis–Menten equation fitting, with n = 3. In WT and TY mutant activities comparison test, error bars in the figure were reported as SEM with n = 3.
In differential scanning fluorimetry assays, error bars in melting temperatures were indicated as SD with n = 3.
In ITC measurements, Kd errors were generated by MicroCal ITC software with a single-site binding mode fitting analysis.
DATA AND SOFTWARE AVAILABILITY
Coordinates of the structure described in this article have been deposited in the PDB with accession numbers PDB: 6O07
Supplementary Material
Highlights:
NatA/Naa50 forms a stable complex through evolutionarily conserved interactions
NatA-Naa50 interactions promote catalytic crosstalk between Naa10 and Naa50
Yeast Naa50 is defective in activity due to compromised substrate binding
Evolutionarily conserved Naa15 TY mutants can disrupt NatA-Naa50 association
ACKNOWLEDGMENTS
This work was supported by NSF grant CHE-1708759 to E.J.P. and NIH grant R35 GM118090 awarded to R.M. We acknowledge the support of the Perelman School of Medicine, University of Pennsylvania DNA Sequencing Core Facility and E. Dean from the High Throughput Screening Core Facility for providing the Sf9 cells containing human NatA/Naa50 for this study. We thank Adam Olia as well as Dan Ricketts for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interest.
REFERENCES
- ABUL KALAM S & SANCHITA B 2012. IP6 (Inositol Hexaphosphate) as a Signaling Molecule. Current Signal Transduction Therapy, 7, 289–304. [Google Scholar]
- ADAMS PD, AFONINE PV, BUNKOCZI G, CHEN VB, DAVIS IW, ECHOLS N, HEADD JJ, HUNG L-W, KAPRAL GJ,GROSSE-KUNSTLEVE RW, MCCOY AJ, MORIARTY NW, OEFFNER R, READ RJ, RICHARDSON DC, RICHARDSON JS, TERWILLIGER TC & ZWART PH 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKSNES H, DRAZIC A, MARIE M & ARNESEN T 2016. First Things First: Vital Protein Marks by N-Terminal Acetyltransferases. Trends in Biochemical Sciences, 41, 746–760. [DOI] [PubMed] [Google Scholar]
- AKSNES H, GORIS M, STROMLAND O, DRAZIC A, WAHEED Q, REUTER N & ARNESEN T 2017. Molecular determinants of the N-terminal acetyltransferase Naa60 anchoring to the Golgi membrane. J Biol Chem, 292, 6821–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKSNES H, VAN DAMME P, GORIS M, STARHEIM KK, MARIE M, STOVE SI, HOEL C, KALVIK TV, HOLE K, GLOMNES N, FURNES C, LJOSTVEIT S, ZIEGLER M, NIERE M, GEVAERT K & ARNESEN T 2015. An organellar nalpha-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell Rep, 10, 1362–74. [DOI] [PubMed] [Google Scholar]
- AMETZAZURRA A, LARREA E, CIVEIRA MP, PRIETO J & ALDABE R 2008. Implication of human N-alpha-acetyltransferase 5 in cellular proliferation and carcinogenesis. Oncogene, 27, 7296–306. [DOI] [PubMed] [Google Scholar]
- ARNESEN T, ANDERSON D, BALDERSHEIM C, LANOTTE M, VARHAUG JE & LILLEHAUG JR 2005. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem J, 386, 433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNESEN T, GROMYKO D, PENDINO F, RYNINGEN A, VARHAUG JE & LILLEHAUG JR 2006a. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene, 25, 4350–60. [DOI] [PubMed] [Google Scholar]
- ARNESEN T, GROMYKO D, PENDINO F, RYNINGEN A, VARHAUG JE & LILLEHAUG JR 2006b. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-α-acetyltransferase complex. Oncogene, 25, 4350. [DOI] [PubMed] [Google Scholar]
- ARNESEN T, STARHEIM KK, VAN DAMME P, EVJENTH R, DINH H, BETTS MJ, RYNINGEN A, VANDEKERCKHOVE J, GEVAERT K & ANDERSON D 2010. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol Cell Biol, 30, 1898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNESEN T, VAN DAMME P, POLEVODA B, HELSENS K, EVJENTH R, COLAERT N, VARHAUG JE, VANDEKERCKHOVE J, LILLEHAUG JR, SHERMAN F & GEVAERT K 2009a. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proceedings of the National Academy of Sciences, 106, 8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNESEN T, VAN DAMME P, POLEVODA B, HELSENS K, EVJENTH R, COLAERT N, VARHAUG JE, VANDEKERCKHOVE J, LILLEHAUG JR, SHERMAN F & GEVAERT K 2009b. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proceedings of the National Academy of Sciences, 106, 8157–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEHNIA R, PANIC B, WHYTE JRC & MUNRO S 2004. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nature Cell Biology, 6, 405. [DOI] [PubMed] [Google Scholar]
- CHU CW, HOU F, ZHANG J, PHU L, LOKTEV AV, KIRKPATRICK DS, JACKSON PK, ZHAO Y & ZOU H 2011. A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell, 22, 448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIKIY I & ELIEZER D 2014. N-terminal Acetylation Stabilizes N-terminal Helicity in Lipid- and Micelle-bound α-Synuclein and Increases Its Affinity for Physiological Membranes. Journal of Biological Chemistry, 289, 3652–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINH TV, BIENVENUT WV, LINSTER E, FELDMAN-SALIT A, JUNG VA, MEINNEL T, HELL R, GIGLIONE C & WIRTZ M 2015. Molecular identification and functional characterization of the first Nalpha-acetyltransferase in plastids by global acetylome profiling. Proteomics, 15, 2426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORFEL MJ & LYON GJ 2015. The biological functions of Naa10-From amino-terminal acetylation to human disease. Gene, 567, 103–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRAZIC A, AKSNES H, MARIE M, BOCZKOWSKA M, VARLAND S, TIMMERMAN E, FOYN H, GLOMNES N, REBOWSKI G, IMPENS F, GEVAERT K, DOMINGUEZ R & ARNESEN T 2018. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMSLEY P & COWTAN K 2004. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- EVJENTH R, HOLE K, KARLSEN OA, ZIEGLER M, ARNESEN T & LILLEHAUG JR 2009. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J Biol Chem, 284, 31122–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLUGE Ø, BRULAND O, AKSLEN LA, VARHAUG JE & LILLEHAUG JR 2002. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene, 21, 5056–5068. [DOI] [PubMed] [Google Scholar]
- GAUTSCHI M, JUST S, MUN A, ROSS S, RüCKNAGEL P, DUBAQUIé Y, EHRENHOFER-MURRAY A & ROSPERT S 2003. The Yeast Nα-Acetyltransferase NatA Is Quantitatively Anchored to the Ribosome and Interacts with Nascent Polypeptides. Molecular and Cellular Biology, 23, 7403–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIGLIONE C, FIEULAINE S & MEINNEL T 2015. N-terminal protein modifications: Bringing back into play the ribosome. Biochimie, 114, 134–146. [DOI] [PubMed] [Google Scholar]
- GORIS M, MAGIN RS, FOYN H, MYKLEBUST LM, VARLAND S, REE R, DRAZIC A, BHAMBRA P, STøVE SI, BAUMANN M, HAUG BE, MARMORSTEIN R & ARNESEN T 2018. Structural determinants and cellular environment define processed actin as the sole substrate of the N-terminal acetyltransferase NAA80. Proceedings of the National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOTTLIEB L & MARMORSTEIN R 2018. Structure of Human NatA and Its Regulation by the Huntingtin Interacting Protein HYPK. Structure, 26, 925–935.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLMES WM, MANNAKEE BK, GUTENKUNST RN & SERIO TR 2014. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nature Communications, 5, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONG HY, CAI YF, ZHANG SJ, DING HY, WANG HT & HAN AD 2017. Molecular Basis of Substrate Specific Acetylation by N-Terminal Acetyltransferase NatB. Structure, 25, 641–+. [DOI] [PubMed] [Google Scholar]
- HOU F, CHU C-W, KONG X, YOKOMORI K & ZOU H 2007. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. The Journal of Cell Biology, 177, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUA KT, TAN CT, JOHANSSON G, LEE JM, YANG PW, LU HY, CHEN CK, SU JL, CHEN PB, WU YL, CHI CC, KAO HJ, SHIH HJ, CHEN MW, CHIEN MH, CHEN PS, LEE WJ, CHENG TY, ROSENBERGER G, CHAI CY, YANG CJ, HUANG MS, LAI TC, CHOU TY, HSIAO M & KUO ML 2011. N-alpha-acetyltransferase 10 protein suppresses cancer cell metastasis by binding PIX proteins and inhibiting Cdc42/Rac1 activity. Cancer Cell, 19, 218–31. [DOI] [PubMed] [Google Scholar]
- HWANG C-S, SHEMORRY A & VARSHAVSKY A 2010. N-Terminal Acetylation of Cellular Proteins Creates Specific Degradation Signals. Science, 327, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEONG JW, BAE MK, AHN MY, KIM SH, SOHN TK, BAE MH, YOO MA, SONG EJ, LEE KJ & KIM KW 2002. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell, 111, 709–20. [DOI] [PubMed] [Google Scholar]
- KALVIK TV & ARNESEN T 2012. Protein N-terminal acetyltransferases in cancer. Oncogene, 32, 269. [DOI] [PubMed] [Google Scholar]
- KNORR AG, SCHMIDT C, TESINA P, BERNINGHAUSEN O, BECKER T, BEATRIX B & BECKMANN R 2018. Ribosome–NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nature Structural & Molecular Biology. [DOI] [PubMed] [Google Scholar]
- LEE CF, OU DS, LEE SB, CHANG LH, LIN RK, LI YS, UPADHYAY AK, CHENG X, WANG YC, HSU HS, HSIAO M, WU CW & JUAN LJ 2010. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J Clin Invest, 120, 2920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LISZCZAK G, ARNESEN T & MARMORSTEIN R 2011. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J Biol Chem, 286, 37002–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LISZCZAK G, GOLDBERG JM, FOYN H, PETERSSON EJ, ARNESEN T & MARMORSTEIN R 2013. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol, 20, 1098–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGIN RS, DENG S, ZHANG H, COOPERMAN B & MARMORSTEIN R 2017. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLOS ONE, 12, e0186278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGIN RS, LISZCZAK GP & MARMORSTEIN R 2015. The Molecular Basis for Histone H4-and H2A-Specific Amino-Terminal Acetylation by NatD. Structure, 23, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MYKLEBUST LM, STOVE SI & ARNESEN T 2015. Naa10 in development and disease. Oncotarget, 6, 34041–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OTWINOWSKI Z & MINOR W 1997. Processing of X-ray diffraction data collected in oscillation mode Methods in Enzymology. Academic Press. [DOI] [PubMed] [Google Scholar]
- PAVLOU D & KIRMIZIS A 2016. Depletion of histone N-terminal-acetyltransferase Naa40 induces p53-independent apoptosis in colorectal cancer cells via the mitochondrial pathway. Apoptosis, 21, 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLEVODA B, BROWN S, CARDILLO TS, RIGBY S & SHERMAN F 2008. Yeast N-alpha-terminal acetyltransferases are associated with ribosomes. Journal of Cellular Biochemistry, 103, 492–508. [DOI] [PubMed] [Google Scholar]
- POLEVODA B & SHERMAN F 2003. Composition and function of the eukaryotic N-terminal acetyltransferase subunits. Biochemical and Biophysical Research Communications, 308, 1–11. [DOI] [PubMed] [Google Scholar]
- RATHORE OS, FAUSTINO A, PRUDêNCIO P, VAN DAMME P, COX CJ & MARTINHO RG 2016. Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Scientific Reports, 6, 21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIBEIRO AL, SILVA RD, FOYN H, TIAGO MN, RATHORE OS, ARNESEN T & MARTINHO RG 2016. Naa50/San-dependent N-terminal acetylation of Scc1 is potentially important for sister chromatid cohesion. Scientific Reports, 6, 39118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERT X & GOUET P 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Research, 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RONG Z, OUYANG Z, MAGIN RS, MARMORSTEIN R & YU H 2016. Opposing Functions of the N-terminal Acetyltransferases Naa50 and NatA in Sister-chromatid Cohesion. Journal of Biological Chemistry, 291, 19079–19091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROPE AF, WANG K, EVJENTH R, XING J, JOHNSTON JJ, SWENSEN JJ, JOHNSON WE, MOORE B, HUFF CD, BIRD LM, CAREY JC, OPITZ JM, STEVENS CA, JIANG T, SCHANK C, FAIN HD, ROBISON R, DALLEY B, CHIN S, SOUTH ST, PYSHER TJ, JORDE LB, HAKONARSON H, LILLEHAUG JR, BIESECKER LG, YANDELL M, ARNESEN T & LYON GJ 2011. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Genet, 89, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIZA V, MOLINA-SERRANO D, KYRIAKOU D, HADJIANTONIOU A & KIRMIZIS A 2013. N-alpha-terminal Acetylation of Histone H4 Regulates Arginine Methylation and Ribosomal DNA Silencing. PLOS Genetics, 9, e1003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCOTT DC, MONDA JK, BENNETT EJ, HARPER JW & SCHULMAN BA 2011. N-Terminal Acetylation Acts as an Avidity Enhancer Within an Interconnected Multiprotein Complex. Science, 334, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETTY SRG, STROCHLIC TI, TONG AHY, BOONE C & BURD CG 2004. Golgi targeting of ARF-like GTPase Arl3p requires its Nα-acetylation and the integral membrane protein Sys1p. Nature Cell Biology, 6, 414. [DOI] [PubMed] [Google Scholar]
- SHEMORRY A, HWANG C-S & VARSHAVSKY A Control of Protein Quality and Stoichiometries by N-Terminal Acetylation and the N-End Rule Pathway. Molecular Cell, 50, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG OK, WANG X, WATERBORG JH & STERNGLANZ R 2003. An Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem, 278, 38109–12. [DOI] [PubMed] [Google Scholar]
- STARHEIM KRISTIANK, ARNESEN T, GROMYKO D, RYNINGEN A, VARHAUG JANE & LILLEHAUG JOHANR 2008. Identification of the human Nα-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochemical Journal, 415, 325–331. [DOI] [PubMed] [Google Scholar]
- STARHEIM KK, GEVAERT K & ARNESEN T 2012. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci, 37, 152–61. [DOI] [PubMed] [Google Scholar]
- STARHEIM KK, GROMYKO D, EVJENTH R, RYNINGEN A, VARHAUG JE, LILLEHAUG JR & ARNESEN T 2009. Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol Cell Biol, 29, 3569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOVE SI, MAGIN RS, FOYN H, HAUG BE, MARMORSTEIN R & ARNESEN T 2016. Crystal Structure of the Golgi-Associated Human N alpha-Acetyltransferase 60 Reveals the Molecular Determinants for Substrate-Specific Acetylation. Structure, 24, 1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRONG M, SAWAYA MR, WANG S, PHILLIPS M, CASCIO D & EISENBERG D 2006. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A, 103, 8060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERCERO JC, DINMAN JD & WICKNER RB 1993. Yeast MAK3 N-acetyltransferase recognizes the N-terminal four amino acids of the major coat protein (gag) of the L-A double-stranded RNA virus. Journal of bacteriology, 175, 3192–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DAMME P, EVJENTH R, FOYN H, DEMEYER K, DE BOCK P-J, LILLEHAUG JR, VANDEKERCKHOVE J, ARNESEN T & GEVAERT K 2011a. Proteome-derived Peptide Libraries Allow Detailed Analysis of the Substrate Specificities of Nα-acetyltransferases and Point to hNaa10p as the Post-translational Actin Nα-acetyltransferase. Molecular & Cellular Proteomics, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DAMME P, HOLE K, GEVAERT K & ARNESEN T 2015. N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics, 15, 2436–2446. [DOI] [PubMed] [Google Scholar]
- VAN DAMME P, HOLE K, PIMENTA-MARQUES A, HELSENS K, VANDEKERCKHOVE J, MARTINHO RG, GEVAERT K & ARNESEN T 2011b. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet, 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DAMME P, KALVIK TV, STARHEIM KK, JONCKHEERE V, MYKLEBUST LM, MENSCHAERT G, VARHAUG JE, GEVAERT K & ARNESEN T 2016. A Role for Human N-alpha Acetyltransferase 30 (Naa30) in Maintaining Mitochondrial Integrity. Molecular & cellular proteomics: MCP, 15, 3361–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEYER FA, GUMIERO A, LAPOUGE K, BANGE G, KOPP J & SINNING I 2017. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nature communications, 8, 15726–15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS BC, GARRETT-ENGELE CM, LI Z, WILLIAMS EV, ROSENMAN ED & GOLDBERG ML 2003. Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr Biol, 13, 2025–36. [DOI] [PubMed] [Google Scholar]
- YANG D, FANG Q, WANG M, REN R, WANG H, HE M, SUN Y, YANG N & XU R-M 2013. Nα-acetylated Sir3 stabilizes the conformation of a nucleosome-binding loop in the BAH domain. Nature Structural &Amp; Molecular Biology, 20, 1116. [DOI] [PubMed] [Google Scholar]
- YI CH, SOGAH DK, BOYCE M, DEGTEREV A, CHRISTOFFERSON DE & YUAN J 2007. A genome-wide RNAi screen reveals multiple regulators of caspase activation. The Journal of cell biology, 179, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOON H, KIM HL, CHUN YS, SHIN DH, LEE KH, SHIN CS, LEE DY, KIM HH, LEE ZH, RYOO HM, LEE MN, OH GT & PARK JW 2014. NAA10 controls osteoblast differentiation and bone formation as a feedback regulator of Runx2. Nat Commun, 5, 5176. [DOI] [PubMed] [Google Scholar]
- YU M, MA M, HUANG C, YANG H, LAI J, YAN S, LI L, XIANG M & TAN D 2009. Correlation of Expression of Human Arrest-Defective-1 (hARD1) Protein With Breast Cancer. Cancer Investigation, 27, 978–983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates of the structure described in this article have been deposited in the PDB with accession numbers PDB: 6O07