

Abstract
The synaptic homeostasis hypothesis (SHY) proposes that sleep is an essential process needed by the brain to maintain the total amount of synaptic strength under control. SHY predicts that by the end of a waking day the synaptic connections of many neural circuits undergo a net increase in synaptic strength due to ongoing learning, which is mainly mediated by synaptic potentiation. Stronger synapses require more energy and supplies and are prone to saturation, creating the need for synaptic renormalization. Such renormalization should mainly occur during sleep, when the brain is disconnected from the environment and neural circuits can be broadly reactivated off-line to undergo a systematic but specific synaptic down-selection. In short, according to SHY sleep is the price to pay for waking plasticity, to avoid runaway potentiation, decreased signal-to-noise ratio, and impaired learning due to saturation. In this review we briefly discuss the rationale of the hypothesis and recent supportive ultrastructural evidence obtained in our laboratory. We then examine recent studies by other groups showing the causal role of cortical slow waves and hippocampal sharp waves/ripples in sleep-dependent down-selection of neural activity and synaptic strength. Finally, we discuss some of the molecular mechanisms that could mediate synaptic weakening during sleep.
Graphical Abstract
The synaptic homeostasis hypothesis (SHY): due to ongoing learning synaptic strength increases during wake in many brain circuits, leading to the strengthening of a majority of synapses. This net increase in synaptic strength is followed by synaptic down-selection during sleep, when the brain is disconnected from the environment, allowing the weakening of the majority of synapses. Learning during waking occurs because the brain adapts to an ever-changing environment, whether or not the subject is trained in a specific “task”.
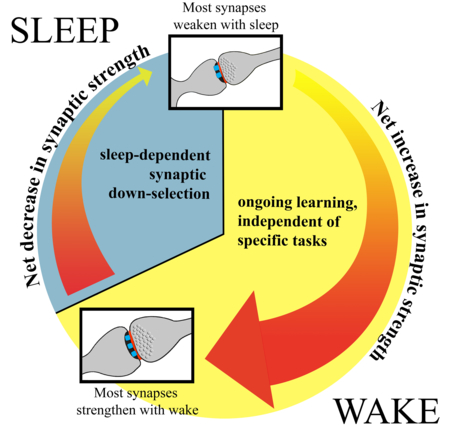
The synaptic homeostasis hypothesis of sleep
The overall rationale supporting SHY has been discussed in detail in 2014 (Tononi & Cirelli, 2014) and is only briefly summarized here. During wakefulness neurons operate under strict neurobiological constraints because firing is energetically expensive and most of the brain’s energy is spent to sustain spiking and synaptic activity (Karbowski, 2014). Neurons also face informational constraints: they receive thousands of different inputs but their output is more or less binary, either they fire or burst, or they do not. For this reason neurons tend to maintain low firing in natural conditions, and respond to selective and salient stimuli by an increase, rather than a decrease, in spiking activity. When neurons do fire, their signals are more likely to percolate throughout the brain if the connections to their target neurons are strong. SHY suggests that new, salient events should then be learned through the strengthening of synapses rather than their weakening. Several examples of learning paradigms that lead to synaptic potentiation within the relevant neural circuits have been previously reviewed (Tononi & Cirelli, 2014). Other recently characterized cases include the selective strengthening of the synapses between mossy fibers and CA3 pyramidal neurons during contextual learning experience (Weng et al., 2018), the potentiation of synapses in layers 2-3 of primary motor cortex after rotarod training (Hayashi-Takagi et al., 2015), and the potentiation of auditory inputs to the lateral amygdala in fear conditioning (Nabavi et al., 2014). As discussed previously, however, while SHY does claim that wake should result in a net increase in synaptic strength and sleep in a net decrease across many/most brain circuits, it certainly does not rule out the occurrence of some synaptic depression in wake (e.g. in reversal learning) or of some potentiation in sleep (see Tononi & Cirelli, 2014, contrary to some claims in Puentes-Mestril & Aton, 2017; Frank & Seibt, 2018).
Synaptic strengthening due to ongoing learning creates the need for synaptic renormalization. Indeed, scientists have long considered how neural circuits deal with the plasticity/stability dilemma, to avoid runaway potentiation and saturation of the ability to learn (von der Malsburg, 1973; Oja, 1982; Hinton et al., 1995; Chistiakova et al., 2015). It is implicitly assumed that the brain is able to maintain overall synaptic homeostasis at all times, implying that any increase in synaptic strength in a specific neural circuit is more or less immediately counterbalanced by synaptic weakening in other neural networks not actively engaged in learning. According to SHY, instead, the overall balance in total synaptic strength is maintained across a longer time scale, that of the 24-hour sleep/wake cycle (Figure 1). Thus, there is a preferred time to learn, when we are awake, which is poised to lead to an absolute increase in synaptic strength in many brain circuits, followed by a preferred time for synaptic renormalization, when we are asleep. The sleeping brain is disconnected from the world – the defining feature of sleep - but thalamic and cortical neurons are broadly activated, allowing a comprehensive sampling of an animal’s memories. Sensory disconnection therefore permits a systematic synaptic renormalization, and is the reason why this process should not happen during wake, when we are slave of the “here and now” and synaptic inputs are necessarily biased by a specific situation (Tononi & Cirelli, 2014). In this way, unlike models that attribute changes in synaptic plasticity to circadian time (Frank & Cantera, 2014), SHY offers a principled explanation for why sleep is needed (to renormalize the increase in synaptic strength during wake plasticity) and why it is characterized by disconnection from the environment (to ensure that the down-selection of synapses occurs in a smart, comprehensive manner).
Figure 1.
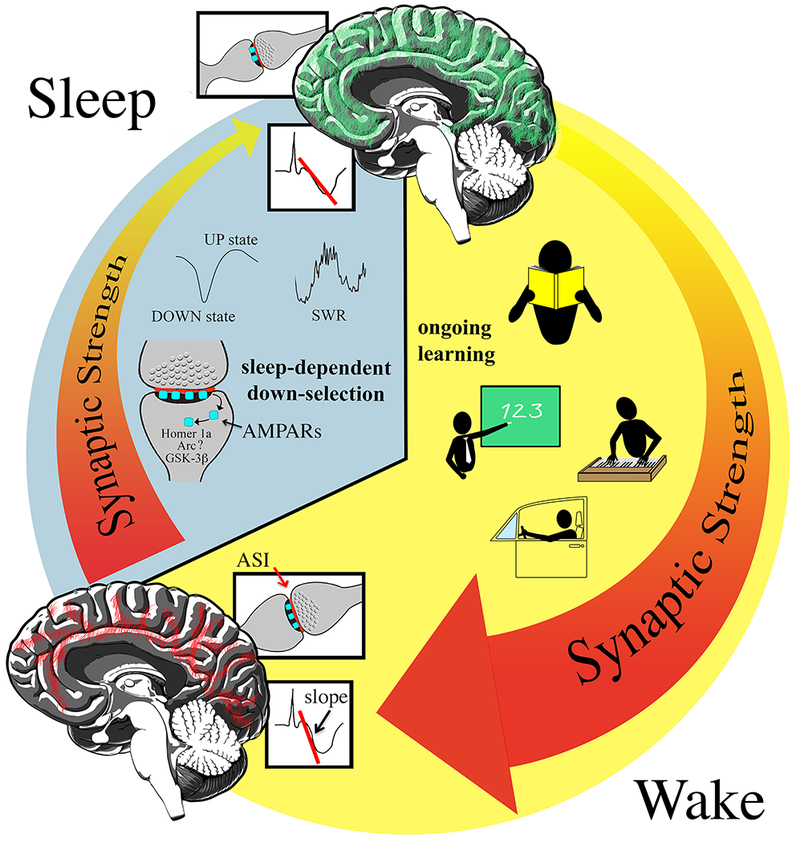
Schematic diagram describing the synaptic homeostasis hypothesis (SHY). Due to ongoing learning synaptic strength increases during wake (the light phase in humans and diurnal animals) in many brain circuits (red lines in the sagittal view of the human brain). During sleep, when the brain is disconnected from the environment, most, if not all circuits undergo synaptic down-selection (green lines). Sleep-dependent renormalization depends on specific patterns of activity during NREM sleep, the UP states of the cortical slow waves and hippocampal sharp waves/ripples (SWR). At the synaptic level, the accumulation in the spine of Homer1a, Arc, GSK-3ß and other proteins likely controls the endocytosis of glutamatergic receptors, and thus synaptic weakening. ASI, axon spine interface (red line); AMPARs, AMPA receptors. The slope used to measure cortical evoked responses is indicated (steeper slope indicates greater response).
While synaptic strength and firing rate are related, they can also be dissociated from each other. It is important to emphasize that according to SHY, the key regulated variable is synaptic strength, not firing rate (Tononi & Cirelli, 2014). This contrasts with the view that synaptic homeostatic mechanisms have the ultimate goal of maintaining neuronal output within a physiological range (for a recent discussion, see (Styr & Slutsky, 2018)). This view has been challenged by experiments with different models of visual deprivation, which leads to an early decrease in spiking activity in visual cortex followed by an increase. Several studies found that these changes in firing rates are fast and may precede synaptic changes, with one of the key underlying mechanisms being the disinhibition of excitatory pyramidal neurons in response to decreased activity of inhibitory interneurons (Aton et al., 2013; Kuhlman et al., 2013; Huang et al., 2015). Other recent experiments also found that changes in firing rate are not the ultimate consequence of synaptic plasticity, as is often assumed, but rather a necessary condition for the occurrence of synaptic homeostasis (Bridi et al., 2018).
Supporting evidence and recent ultrastructural studies
Early experiments tested the key prediction of SHY that overall synaptic strength should increase during wake and decrease during sleep. There are several markers of synaptic strength, some more direct than others (Cirelli, 2017), but the overall emerging picture is consistent with SHY’s prediction. For instance, the expression of excitatory glutamatergic AMPA (Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid) receptors, when measured at the synaptic level in the rat cerebral cortex and hippocampus of adult rats (Vyazovskiy et al., 2008) and in the post-synaptic densities taken from the whole forebrain of post-adolescence mice (Diering et al., 2017), is higher after wakefulness than after sleep (cytoplasmic presence/absence of immunoreactivity for these receptors, as reported in (Del Cid-Pellitero et al., 2017), is not a measure of synaptic strength). Evoked responses and field excitatory postsynaptic potentials (fEPSPs) recorded in vivo also increase with wake duration and decrease during sleep in cortex and hippocampus of adult rodents and humans (Vyazovskiy et al., 2008; Huber et al., 2013; Norimoto et al., 2018), although cortical evoked responses are also modulated by circadian time (Ly et al., 2016). In both rodents and humans it is more difficult to induce long-term potentiation (LTP)-like plasticity after prolonged wake (Vyazovskiy et al., 2008; Kuhn et al., 2016; Fattinger et al., 2017). Larger evoked responses and fEPSPs during wake reflect increased cortical excitability, which may result from increased synaptic strength due to greater expression of AMPA receptors, or increased intrinsic excitability, due to changes in voltage- and calcium-dependent ion channels. However, it is increasingly appreciated that synaptic plasticity and intrinsic excitability often change in the same direction and support each other during learning, under the control of the same set of neurotransmitters and neuromodulators (Kim & Linden, 2007; Titley et al., 2017). For a recent review that summarizes sleep/wake changes in both synaptic strength and neuronal excitability see (Niethard et al., 2017).
Since stronger synapses are also bigger, a stringent prediction of SHY is that the majority of synapses should grow with wake and shrink with sleep. Using serial block face electron microscopy we recently reconstructed ~ 7,000 synapses in the cortex of adolescent mice and found that the axon-spine interface, the direct area of contact between pre-synapse and post-synapse, decreases on average by 18% after 6-8 hours of sleep as compared to 6-8 hours of wake, independent of whether wake occurs during the day or the night ((de Vivo et al., 2017); similar results were found after measuring the size of the spine head in all ~ 7,000 synapses). Sleep-dependent synaptic renormalization was found in small and medium size synapses, which together represent ~80% of all synapses, but not in the largest synapses. At the population level the decrease in synapse size during sleep could be described as size-dependent downscaling, i.e. small and medium size synapses shrank during sleep in a multiplicative manner, proportional to their size. Another study in post-adolescence mice imaged cortical spines twice a day for 3 consecutive days using two-photon microscopy, averaging all time points and focusing on the stable spines that persisted for at least 2 days. It found that the majority of spines were smaller 4 hours into the light phase, when mice are likely to be asleep (but animals were not implanted for electroencephalographic (EEG) recording)), as compared to 4 hours into the dark phase, after mice had been mostly awake (Diering et al., 2017). The changes were specific for the larger spines that had a higher content of AMPA receptors. As mentioned before, according to SHY, the goal of sleep is to renormalize total synaptic strength, but total does not mean that all synapses need to be renormalized. The two cited studies (de Vivo et al., 2017; Diering et al., 2017) agree that in the brain region tested most, but not all, synapses shrink with sleep but disagree on whether sleep-dependent renormalization preferentially targets small or large synapses, an important point to be addressed in future experiments. As we discuss below, it will also be useful to assess whether sleep-dependent renormalization, which can be described as multiplicative downscaling at the population level, remains so at the level of individual synapses. The de Vivo et al. study (de Vivo et al., 2017) was performed in layer 2 of two primary cortical areas (motor and sensory cortex) of adolescent mice; it remains to be investigated whether similar changes occur earlier or later in life, in other layers and areas of the cerebral cortex, and in other brain regions.
Role of neuronal activity in sleep-dependent down-selection: the UP states of cortical slow waves
An important goal for the future is to identify the mechanisms underlying the synaptic renormalization afforded by sleep. The comprehensive and yet selective nature of this process must account for the many diverse effects of sleep at the cognitive level, from memory consolidation to forgetting (Feld et al., 2016). Bridging the gap between molecular, ultrastructural and cellular changes on one hand, and behavioral and cognitive effects on the other hand, remains a challenge. Even more so since it is increasingly appreciated that all these processes, from the turnover of spines (Berry & Nedivi, 2017) to “memory” (Kukushkin & Carew, 2017) are highly dynamic, change constantly due to ongoing stimulation, and include multiple overlapping windows that span from minutes to days and weeks. Yet, the past few years have seen significant progress and two general principles have emerged that will be discussed below. First, neuronal activity during sleep is crucial for down-selection to occur. Second, sleep-dependent renormalization seems to spare those neurons and/or synapses that are most active during sleep. These principles were established in the mammalian brain and it is still unknown whether they apply to non-mammalian animal species.
Neuronal activity during non-rapid eye movement (NREM) sleep, which in mammals accounts for 70-80% of total sleep, is important for sleep-dependent down-selection. More specifically, new studies point to the key role of spiking activity during cortical slow waves and hippocampal sharp waves/ripples (Gulati et al., 2017; Norimoto et al., 2018). During NREM sleep thalamic and cortical neurons are bistable, oscillating roughly every second between an UP/ON period of firing and a DOWN/OFF period of neuronal silence (Steriade et al., 2001). This slow oscillation occurs more or less synchronously across all neurons, allowing their pooled activity to be detected at the cortical surface as large slow waves. In a recent series of experiments the link between ON states and sleep-dependent down-selection of neuronal activity was demonstrated by studying how sleep affects neuroprosthetic learning (Gulati et al., 2017). In this paradigm adult rats are trained to control the angular velocity of a feeding tube by directly pairing behavior with the firing rate of a few neurons in primary motor cortex, only two cells in each animal. At the beginning of the training session the experimenter selects a single pair of “direct trained” units whose activity will causally control the movement of the tube. Successful learning occurs in a 2-hour session and leads to a task-dependent increase in firing in the trained neurons, as well as in many “indirect” units whose activity is modulated by the task but does not control the tube. It was found that rats performed better if they were allowed to sleep after learning. Moreover, after sleep task-dependent activity changed in opposite direction in direct and indirect neurons: most direct units (67%) showed a slight but significant (7%) increase in peak firing during the task relative to baseline firing, while the great majority of indirect units (90%) showed a substantial decrease in peak activity (−32%). Furthermore, lack of firing during sleep predicted down-selection, suggesting how sleep can identify which neurons should undergo down-selection and which should be spared. According to the authors, this is how sleep can solve the credit assignment problem: it decreases non-causal activity of the many indirect units while at the same time promoting the sparse activation pattern of the few neurons that are causally linked to behavior. The same study also used closed-loop optogenetic inhibition to reduce firing during the ON/UP states of the slow oscillation. This manipulation prevented both the post-sleep improvement in neuroprosthetic learning and the task-related decline in non-causal firing, even if the duration of NREM sleep was not affected. The same optogenetic manipulation during the DOWN/OFF periods had no effect, showing that spiking activity during the UP state of the slow oscillation is required for down-selection to occur. In summary, the study shows that post-learning sleep leads to a slight increase in firing in a small set of neurons whose activity was causally linked to learning the task, and to a greater decrease in firing in a larger set of indirect neurons.
Role of neuronal activity in sleep-dependent down-selection: hippocampal sharp waves/ripples
Sharp waves/ripples (SWRs) are large high-frequency field potentials recorded from the hippocampus. They occur mainly during NREM sleep, as well as during quiet, non-exploratory wake. It is well established that neurons activated during exploration and learning are preferentially reactivated with a similar sequential pattern of firing during SWRs (the so-called “replay” (Buhry et al., 2011)). Moreover, disruption of SWRs impairs memory (Girardeau et al., 2009), suggesting that they play an important role in memory consolidation (Rasch & Born, 2013). The replayed neuronal activity during SWRs has long been considered a likely candidate to induce synaptic potentiation (Buzsaki, 1989). A recent study showed instead that SWRs promote synaptic weakening (Norimoto et al., 2018). The authors first replicated a previous finding that SWRs become more frequent after spatial learning. Then they showed in vivo in adult mice that closed-loop optogenetic inhibition of SWRs prevents the decline in the slope of hippocampal fEPSPs that normally occurs in sleeping mice. The authors also took advantage of an in vitro model of SWRs, the obliquely cut hippocampal slices, which spontaneously emit SWRs. As in vivo in adult mice, in these slices taken from adolescent mice the occurrence of SWRs led to a progressive decline in the fEPSPs slope that could be blocked by optogenetic inhibition of SWRs. By contrast, the fEPSPs did not decline in horizontal slices that lacked SWRs. Two-photon imaging also showed that the head size of most CA1 spines decreases with time in spontaneously emitting SWRs slices. The decrease occurs in thin and stubby spines but not in mushroom spines, which are the largest, consistent with the findings in mouse cortex (de Vivo et al., 2017). The study also confirmed, both in vitro and in vivo, that hippocampal place cells recently activated during exploration were more likely to be reactivated during subsequent SWRs. These cells maintained their firing rate in the course of sleep, while neurons not engaged in previous learning decreased their firing during sleep. This last finding is consistent with the results after neuroprosthetic learning, when trained neurons showed the strongest increase in task-related firing and did not decrease their activity in the course of sleep (Gulati et al., 2017).
In another recent study pyramidal neurons in the CA3 and CA1 hippocampal regions were recorded for 10 minutes while adult rats explored a familiar track, and during the first 5 minutes after the animals were moved to a rest box (Sadowski et al., 2016). As expected some place cells that fired while the rat was on the track were reactivated in the early post-run quiescent period. The specific spike train reactivation pattern of these neurons was then used to stimulate CA3 and CA1 cells in hippocampal slices of naïve rats. This stimulation in most cases led to synaptic potentiation of CA3 to CA1 synapses, provided that the ripple pathway was also stimulated to match the level of depolarization associated with SWRs in vivo. Thus, synaptic potentiation can be elicited in vitro using spike train patterns and depolarization levels that match those associated with SWRs in vivo. The slice preparation used in this study lacks SWRs, and is unknown whether in vivo, or in slices spontaneously emitting SWRs, the reactivated CA3 to CA1 synapses would show net synaptic potentiation, net synaptic depression or no net change (lack of synaptic depression). As discussed by the authors, the reactivation pattern used in vitro was recorded during the first 5 minutes after the end of the run, when the animals are settling down but are most likely awake rather than in NREM sleep. A related previous study found that CA3-CA1 spike train reactivation patterns could not induce synaptic potentiation unless a cholinergic agonist was added to the slices, to match the high cholinergic tone present during exploration (Isaac et al., 2009). Thus, age differences (adolescent vs. adult animals) or the fact that Norimoto and colleagues studied spontaneously emitting SWRs slices, while Sadowski and colleagues did not, may explain the different results. Another possibility however, which would be consistent with the findings in vivo and in vitro (Sadowski et al., 2016; Norimoto et al., 2018) is that SWRs lead to net potentiation of specific synapses in the early wake immobility immediately after the run, when the cholinergic tone is high (Teles-Grilo Ruivo et al., 2017). The same reactivated synapses would only maintain, but do not increase, their strength during NREM sleep, when cholinergic levels are low.
Down-scaling or down-selection?
The studies just discussed point to being co-active during sleep as a key mechanism that allows neurons to escape the down-selection. But while these studies looked at overall spiking activity, future experiments will need to test whether, and to which extent, sleep-dependent renormalization is synapse-specific. For the synaptic data that we collected in cortex using electron microscopy, we first tested whether a scaling mechanism could account for the sleep-dependent decline in synaptic size that we observed after sleep, and it did. Then, we applied Monte Carlo simulations on bootstrapped data to test which kind of scaling could best explain the results. The answer was size-dependent multiplicative scaling. Size-dependent, because a minority of synapses, the largest ones, did not scale. Multiplicative, because the great majority of synapses that did scale (80% of all synapses) shrank by an amount proportional to their size. This statistical analysis could only be done at the population level, because we could not follow the same synapse across the sleep/wake cycle. The results therefore do not mean that the renormalization afforded by sleep should be multiplicative at the level of each individual synapse. In fact, according to SHY, only a specific process of down-selection can explain how sleep at the same time promotes new learning, memory consolidation, integration of new facts into the overall body of previous knowledge, gist extraction, and forgetting (Tononi & Cirelli, 2014). As previously discussed (Tononi & Cirelli, 2014) several different synaptic rules are in theory compatible with renormalization as predicted by SHY, and several of them were successfully tested in our laboratory using computer simulations. They included a downscaling rule where all synapses decrease in strength proportionally but those that end up below a minimal threshold become virtually ineffective (Hill et al., 2008); a modified spike-timing-dependent synaptic plasticity (STDP) rule by which stronger synapses are depressed less than weaker ones (Olcese et al., 2010); and a ‘‘protection from depression’’ mechanism (Hashmi et al., 2013; Nere et al., 2013). With this last rule, when a neuron fires strongly during sleep because many of its inputs are co-activated, then its synapses are protected from depression rather than being potentiated as during wake. The end result of this protection from depression is not an absolute increase in synaptic strength but a relative potentiation, relative to all other synapses that depress during sleep. At the time of these computer simulations there were no experimental data that could suggest which, if any, of these rules would work in vivo. A new study, however, shows that a protection from depression rule applies in vivo, at least under urethane anesthesia that is believed to closely mimic the UP and DOWN states of NREM sleep (Gonzalez-Rueda et al., 2018). The experiments were performed in the mouse barrel cortex and involved the optogenetic stimulation of layer 4 presynaptic afferents coupled with whole-cell recordings of the target pyramidal neurons in layer 2/3. According to the canonical STDP rules, presynaptic activation can lead to either no change in synaptic strength, synaptic depression or synaptic potentiation depending on the absence, presence, and exact timing of postsynaptic spiking. The study found that these conventional rules apply when neurons were stimulated during the DOWN states when they are usually silent. During the UP states instead, when neurons fire spontaneously, the STDP rules were strongly biased toward synaptic depression. Specifically, after stimulation of layer 4 to layer 2/3 connections synaptic strength never significantly increased, it remained unchanged when presynaptic activation was quickly followed by postsynaptic activity, and it decreased when postsynaptic activity either preceded presynaptic activation or followed it at long intervals. This synaptic depression associated with the UP states was mainly characterized in young animals and persisted at significant but smaller levels in older mice (mean depression in %, P (postnatal day)16-P21 = 63%, P30-P50 = 71%)). Neurons rarely if ever fire during the DOWN states, and during sleep UP states last longer than DOWN states. Thus, the authors reasonably concluded that if the results hold during natural sleep, and in both young and adult animals, this rule could explain how sleep can lead to diffuse but synapse-specific synaptic renormalization: synapses strengthened during wake, as well as those that are most coherently reactivated during sleep, would be more likely to show coincident firing during the UP states and thus be protected from synaptic depression (Gonzalez-Rueda et al., 2018). It would be, indeed, a smart synaptic down-selection (Tononi & Cirelli, 2014).
Molecular candidates for sleep-dependent synaptic down-selection
Recent studies are also starting to identify specific proteins that could play a major role in synaptic weakening during sleep. In excitatory synapses, increases and decreases in strength ultimately depend, respectively, on the insertion or removal of glutamatergic AMPA receptors. Molecules involved in the trafficking of these receptors to and from the synaptic surface are therefore likely candidates to mediate sleep-dependent synaptic down-selection. One of these proteins is Homer1a, a dominant negative short variant form of Homer induced in response to neuronal activation. Homer1a was one of the first immediate early genes whose expression was found to increase in many brain regions during wake relative to sleep, and genetic and electrophysiological studies had linked its expression to the homeostatic regulation of sleep (Franken et al., 2001; Cirelli et al., 2004; Maret et al., 2007; Mackiewicz et al., 2008). A recent study (Diering et al., 2017) confirmed that Homer1a levels are higher in wake than in sleep when measured in whole brain tissue. It also found, however, that the opposite was true in the post-synaptic density, where Homer1a accumulated during sleep. The authors further showed that inside the synapse Homer1a displaces the long forms of Homer that normally link Group I metabotropic glutamate receptors to ine (inositol trisphosphate) receptors. The disassembly of this signaling complex ultimately leads to the removal of AMPA receptors from the synaptic surface. It was also shown that the entry of Homer1a in the spine is prevented by noradrenaline, which is high during active wake, and promoted by adenosine, which accumulates with wake duration and promotes sleep. Moreover, the entry of Homer1a in the spine occurs in sleepy mice, which were sleep deprived using just mild cage shaking that presumably is associated with low noradrenaline levels, but does not happen in mice treated with amphetamines (Diering et al., 2017), nor in mice kept awake by exploration of novel objects (Dr. Graham Diering, personal communication). Thus, these results suggest a possible gating mechanism that accounts for why Homer1a is induced during wake but accumulates in the synapses during sleep (Diering et al., 2017).
Arc is another immediate early gene whose expression in the cerebral cortex is higher after wake than after sleep (Cirelli & Tononi, 2000). Arc is upregulated in excitatory glutamatergic neurons when synaptic activity increases, and recent experiments showed that like for Homer1a, the accumulation of Arc inside the spines promotes the endocytosis of AMPA receptors and synaptic weakening. More specifically, after increased activity and LTP-inducing stimuli, Arc avoids the potentiated synapses and accumulates in the less activated spines, resulting in their weakening (Okuno et al., 2012; El-Boustani et al., 2018). Arc is therefore an intriguing candidate to reverse-tag synapses destined to depression. However, whether this synapse-specific mechanism is at work during sleep remains to be tested (Honjoh et al., 2017).
Finally, another recent study identified a potential candidate to tag the synapses that need to be protected from down-selection, the opposite of what Arc would do. The experiments were performed in acute slices of adolescent mouse medial entorhinal cortex, which maintain UP/DOWN states like those seen during NREM sleep (Bartram et al., 2017). Using whole-cell recordings from pyramidal neurons of layer 3 it was found that inputs arriving during the UP states underwent synaptic weakening unless they were paired with postsynaptic spiking. When pre- and post-firing co-occurred, synaptic strength did not change, consistent with the protection from depression rule identified in vivo under urethane anesthesia (Gonzalez-Rueda et al., 2018). Inputs arriving during the DOWN states were also unchanged. The UP state-dependent synaptic weakening required postsynaptic NMDA receptors and the activity of glycogen synthase kinase-3ß (GSK-3ß), an enzyme involved in long-term synaptic depression. The induction of LTP leads to GSK-3ß phosphorylation at Serine 9, which blocks its activity and prevents the induction of long-term depression in the same synapses (Peineau et al., 2007). Of note, in adult rats levels of phosphorylated GSK-3ß are higher in wake than in sleep (Vyazovskiy et al., 2008). Thus, the phosphorylation of GSK-3ß could be a mechanism to tag synapses potentiated during wake and protect them from weakening during the following sleep period.
Conclusions
In this review we focused on some recent experiments in cortex and hippocampus, both in vitro and in vivo, which show how specific patterns of neuronal activity during NREM sleep play an essential role in sleep-dependent down-selection (Figure 1). Neurons that are co-active during learning are more likely to co-fire during subsequent NREM sleep and thus be protected from down-selection. After sleep these neurons maintain or slightly increase the overall level of activity that they had after learning, but we do not know what happens to their synapses. Are they all protected or only the synapses engaged in learning? To answer this question one will need to comprehensively measure both synaptic activity and spiking activity in vivo in neurons engaged in a specific task, as well as in control neurons not involved in learning. We also reviewed studies that identified a few candidate molecules, such as Homer1a, Arc, and GSK-3ß, whose accumulation inside the spine either triggers or blocks the endocytosis of glutamatergic receptors. It is likely that other candidates will be discovered. The challenge for the future will be to show which of these mechanisms are active in vivo under conditions of physiological sleep, and to link changes at the synaptic level with behavioral and cognitive outcomes. Despite the difficulty of these experiments there has been great progress in the past few years, and one can start seeing how the complicated process of sleep-dependent renormalization could lead to cognitive benefits.
Acknowledgements:
This work was supported by NIH Grants DP 1OD579 (G.T.), 1R01MH091326 (G.T.), 1R01MH099231 (G.T., C.C.), and 1P01NS083514 (G.T., C.C.).
List of abbreviations:
- AMPA
Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid
- ASI
axon spine interface
- EEG
electroencephalographic
- fEPSPs
field excitatory postsynaptic potentials
- GSK-3ß
glycogen synthase kinase-3ß
- IP3
inositol trisphosphate
- LTP
long-term potentiation
- NREM sleep
non-rapid eye movement sleep
- P
postnatal day
- SHY
synaptic homeostasis hypothesis
- STDP
spike-timing-dependent synaptic plasticity
- SWRs
sharp waves/ripples
Footnotes
Disclosure of potential conflicts of interest. The authors declare no competing financial or non-financial interests.
References
- Aton SJ, Broussard C, Dumoulin M, Seibt J, Watson A, Coleman T & Frank MG (2013) Visual experience and subsequent sleep induce sequential plastic changes in putative inhibitory and excitatory cortical neurons. Proc Natl Acad Sci U S A, 110, 3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartram J, Kahn MC, Tuohy S, Paulsen O, Wilson T & Mann EO (2017) Cortical Up states induce the selective weakening of subthreshold synaptic inputs. Nat Commun, 8, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP & Nedivi E (2017) Spine Dynamics: Are They All the Same? Neuron, 96, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridi MCD, de Pasquale R, Lantz CL, Gu Y, Borrell A, Choi SY, He K, Tran T, Hong SZ, Dykman A, Lee HK, Quinlan EM & Kirkwood A (2018) Two distinct mechanisms for experience-dependent homeostasis. Nat Neurosci, 21, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhry L, Azizi AH & Cheng S (2011) Reactivation, replay, and preplay: how it might all fit together. Neural Plast, 2011, 203462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G (1989) Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience, 31, 551–570. [DOI] [PubMed] [Google Scholar]
- Chistiakova M, Bannon NM, Chen JY, Bazhenov M & Volgushev M (2015) Homeostatic role of heterosynaptic plasticity: models and experiments. Front Comput Neurosci, 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C (2017) Sleep, synaptic homeostasis and neuronal firing rates. Curr Opin Neurobiol, 44, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Gutierrez CM & Tononi G (2004) Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron, 41, 35–43. [DOI] [PubMed] [Google Scholar]
- Cirelli C & Tononi G (2000) Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci, 20, 9187–9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G & Cirelli C (2017) Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science, 355, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Cid-Pellitero E, Plavski A, Mainville L & Jones BE (2017) Homeostatic Changes in GABA and Glutamate Receptors on Excitatory Cortical Neurons during Sleep Deprivation and Recovery. Front Syst Neurosci, 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A & Huganir RL (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science, 355, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Boustani S, Ip JPK, Breton-Provencher V, Knott GW, Okuno H, Bito H & Sur M (2018) Locally coordinated synaptic plasticity of visual cortex neurons in vivo. Science, 360, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattinger S, de Beukelaar TT, Ruddy KL, Volk C, Heyse NC, Herbst JA, Hahnloser RHR, Wenderoth N & Huber R (2017) Deep sleep maintains learning efficiency of the human brain. Nat Commun, 8, 15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld GB, Weis PP & Born J (2016) The Limited Capacity of Sleep-Dependent Memory Consolidation. Front Psychol, 7, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG & Cantera R (2014) Sleep, clocks, and synaptic plasticity. Trends Neurosci, 37, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG & Seibt J (2018) Sleep and plasticity: Waking from a fevered dream. Sleep Med Rev, 39, 1–2. [DOI] [PubMed] [Google Scholar]
- Franken P, Chollet D & Tafti M (2001) The homeostatic regulation of sleep need is under genetic control. J Neurosci, 21, 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsaki G & Zugaro MB (2009) Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci, 12, 1222–1223. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rueda A, Pedrosa V, Feord RC, Clopath C & Paulsen O (2018) Activity-Dependent Downscaling of Subthreshold Synaptic Inputs during Slow-Wave-Sleep-like Activity In Vivo. Neuron, 97, 1244–1252 e1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati T, Guo L, Ramanathan DS, Bodepudi A & Ganguly K (2017) Neural reactivations during sleep determine network credit assignment. Nat Neurosci, 20, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmi A, Nere A & Tononi G (2013) Sleep Dependent Synaptic Down-Selection (II): Single Neuron Level Benefits for Matching, Selectivity, and Specificity. Frontiers in Sleep and Chronobiology, 4, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM & Kasai H (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature, 525, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S, Tononi G & Ghilardi MF (2008) Sleep improves the variability of motor performance. Brain Res Bull, 76, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton GE, Dayan P, Frey BJ & Neal RM (1995) The “wake-sleep” algorithm for unsupervised neural networks. Science, 268, 1158–1161. [DOI] [PubMed] [Google Scholar]
- Honjoh S, de Vivo L, Okuno H, Bito H, Tononi G & Cirelli C (2017) Higher Arc Nucleus-to-Cytoplasm Ratio during Sleep in the Superficial Layers of the Mouse Cortex. Front Neural Circuits, 11, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Hokenson K, Bandyopadhyay S, Russek SJ & Kirkwood A (2015) Brief Dark Exposure Reduces Tonic Inhibition in Visual Cortex. J Neurosci, 35, 15916–15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Maki H, Rosanova M, Casarotto S, Canali P, Casali AG, Tononi G & Massimini M (2013) Human cortical excitability increases with time awake. Cereb Cortex, 23, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Buchanan KA, Muller RU & Mellor JR (2009) Hippocampal place cell firing patterns can induce long-term synaptic plasticity in vitro. J Neurosci, 29, 6840–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski J (2014) Constancy and trade-offs in the neuroanatomical and metabolic design of the cerebral cortex. Frontiers in Neural Circuits, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ & Linden DJ (2007) Ubiquitous plasticity and memory storage. Neuron, 56, 582–592. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X & Trachtenberg JT (2013) A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature, 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, Burklin J, Maywald S, Mall V, Jung NH, Reis J, Spiegelhalder K, Kloppel S, Sterr A, Eckert A, Riemann D, Normann C & Nissen C (2016) Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun, 7, 12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukushkin NV & Carew TJ (2017) Memory Takes Time. Neuron, 95, 259–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly JQM, Gaggioni G, Chellappa SL, Papachilleos S, Brzozowski A, Borsu C, Rosanova M, Sarasso S, Middleton B, Luxen A, Archer SN, Phillips C, Dijk DJ, Maquet P, Massimini M & Vandewalle G (2016) Circadian regulation of human cortical excitability. Nature Communications, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz M, Paigen B, Naidoo N & Pack AI (2008) Analysis of the QTL for sleep homeostasis in mice: Homer1a is a likely candidate. Physiol Genomics, 33, 91–99. [DOI] [PubMed] [Google Scholar]
- Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O’Hara BF, Franken P & Tafti M (2007) Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A, 104, 20090–20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY & Malinow R (2014) Engineering a memory with LTD and LTP. Nature, 511, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nere A, Hashmi A, Cirelli C & Tononi G (2013) Sleep-dependent synaptic down-selection (I): modeling the benefits of sleep on memory consolidation and integration. Front Neurol, 4, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethard N, Burgalossi A & Born J (2017) Plasticity during Sleep Is Linked to Specific Regulation of Cortical Circuit Activity. Front Neural Circuits, 11, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norimoto H, Makino K, Gao M, Shikano Y, Okamoto K, Ishikawa T, Sasaki T, Hioki H, Fujisawa S & Ikegaya Y (2018) Hippocampal ripples down-regulate synapses. Science, 359, 1524–1527. [DOI] [PubMed] [Google Scholar]
- Oja E (1982) A simplified neuron model as a principal component analyzer. J Math Biol, 15, 267–273. [DOI] [PubMed] [Google Scholar]
- Okuno H, Akashi K, Ishii Y, Yagishita-Kyo N, Suzuki K, Nonaka M, Kawashima T, Fujii H, Takemoto-Kimura S, Abe M, Natsume R, Chowdhury S, Sakimura K, Worley PF & Bito H (2012) Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIbeta. Cell, 149, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese U, Esser SK & Tononi G (2010) Sleep and synaptic renormalization: a computational study. J Neurophysiol, 104, 3476–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT & Collingridge GL (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron, 53, 703–717. [DOI] [PubMed] [Google Scholar]
- Puentes-Mestril C & Aton SJ (2017) Linking Network Activity to Synaptic Plasticity during Sleep: Hypotheses and Recent Data. Front Neural Circuits, 11, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch B & Born J (2013) About sleep’s role in memory. Physiol Rev, 93, 681–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski JH, Jones MW & Mellor JR (2016) Sharp-Wave Ripples Orchestrate the Induction of Synaptic Plasticity during Reactivation of Place Cell Firing Patterns in the Hippocampus. Cell Rep, 14, 1916–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I & Grenier F (2001) Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol, 85, 1969–1985. [DOI] [PubMed] [Google Scholar]
- Styr B & Slutsky I (2018) Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat Neurosci, 21, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles-Grilo Ruivo LM, Baker KL, Conway MW, Kinsley PJ, Gilmour G, Phillips KG, Isaac JTR, Lowry JP & Mellor JR (2017) Coordinated Acetylcholine Release in Prefrontal Cortex and Hippocampus Is Associated with Arousal and Reward on Distinct Timescales. Cell Rep, 18, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titley HK, Brunel N & Hansel C (2017) Toward a Neurocentric View of Learning. Neuron, 95, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G & Cirelli C (2014) Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron, 81, 12–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Malsburg C (1973) Self-organization of orientation sensitive cells in the striate cortex. Kybernetik, 14, 85–100. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy V, Cirelli C, Pfister-Genskow M, Faraguna U & Tononi G (2008) Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci, 11, 200–208. [DOI] [PubMed] [Google Scholar]
- Weng FJ, Garcia RI, Lutzu S, Alvina K, Zhang Y, Dushko M, Ku T, Zemoura K, Rich D, Garcia-Dominguez D, Hung M, Yelhekar TD, Sorensen AT, Xu W, Chung K, Castillo PE & Lin Y (2018) Npas4 Is a Critical Regulator of Learning-Induced Plasticity at Mossy Fiber-CA3 Synapses during Contextual Memory Formation. Neuron, 97, 1137–1152 e1135. [DOI] [PMC free article] [PubMed] [Google Scholar]