

Abstract
Motivation
Hi-C is a genome-wide technology for investigating 3D chromatin conformation by measuring physical contacts between pairs of genomic regions. The resolution of Hi-C data directly impacts the effectiveness and accuracy of downstream analysis such as identifying topologically associating domains (TADs) and meaningful chromatin loops. High resolution Hi-C data are valuable resources which implicate the relationship between 3D genome conformation and function, especially linking distal regulatory elements to their target genes. However, high resolution Hi-C data across various tissues and cell types are not always available due to the high sequencing cost. It is therefore indispensable to develop computational approaches for enhancing the resolution of Hi-C data.
Results
We proposed hicGAN, an open-sourced framework, for inferring high resolution Hi-C data from low resolution Hi-C data with generative adversarial networks (GANs). To the best of our knowledge, this is the first study to apply GANs to 3D genome analysis. We demonstrate that hicGAN effectively enhances the resolution of low resolution Hi-C data by generating matrices that are highly consistent with the original high resolution Hi-C matrices. A typical scenario of usage for our approach is to enhance low resolution Hi-C data in new cell types, especially where the high resolution Hi-C data are not available. Our study not only presents a novel approach for enhancing Hi-C data resolution, but also provides fascinating insights into disclosing complex mechanism underlying the formation of chromatin contacts.
Availability and implementation
We release hicGAN as an open-sourced software at https://github.com/kimmo1019/hicGAN.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Chromatin inside nucleus adopts an intricate three-dimensional (3D) organization which is critical for regulating the genome function and nuclear processes such as DNA replication and transcription. Previous studies have revealed that such a form of genome organization is interconnected with nuclear architecture and highly dynamic across different cell states (Quinodoz et al., 2018; Schmitt et al., 2016; Uhler and Shivashankar, 2017).
In the past decade, the development of the chromosome conformation capture (3C) technique (Dekker et al., 2002) and its derivatives (Dostie et al., 2006; Simonis et al., 2006) have emerged as a proliferation of data measuring towards genome architecture. The advent of Hi-C technology (Lieberman-Aiden et al., 2009) has further enabled the measurement of chromatin contacts genome wide. Hi-C technology has greatly facilitated the discoveries of topologically associating domains (TADs), chromatin loops and A/B compartment (Dixon et al., 2012; Nora et al., 2012; Rao et al., 2014). Although both the experimental and computational methodologies for 3D genome analysis have been rapidly increased over the past few years, the current comprehension of how the organization of genome influence cell function and fate in disease and physiology is still limited. One major concern is that our understanding of genome architecture remains relatively poor at kilobase pair (Kbp) to megabase pair (Mbp) scale, which impedes the further analysis of more refined chromatin structure. For an instance, the resolution of Hi-C data is usually defined as the finest scale that one can reliably discern local features. It also refers to the bin size for dividing the genome when constructing a Hi-C contact matrix. Most available Hi-C data have relatively low resolution ranging from 25 kb to 1 Mb.
On the one hand, high resolution Hi-C data that require massive sequencing reads and high sequencing cost are only available in several tissues or cell lines. On the other hand, the resolution of Hi-C data largely affects the downstream analysis such as identifying topologically associating domains (TADs) and chromatin loops (Dixon et al., 2012; Rao et al., 2014; Sexton et al., 2012). Typically, the deeper the sequencing depth is, the higher the resolution will be. High resolution Hi-C data can not only help us identity more clear TADs, but also makes it possible for uncovering sub-TADs or contact domains at a finer genomic scale, which are believed to be more variable across cell types and species (Phillips-Cremins et al., 2013; Yu and Ren, 2017). Based on the above considerations, it is urgent to develop computational approaches for enhancing Hi-C data resolution by learning the mapping relationship between low resolution Hi-C data and high resolution Hi-C data.
Recently, deep learning technologies have achieved unprecedented advancements in many fields, including but not limited to computer vision (CV) and natural language processing (NLP) (LeCun et al., 2015). In the field of computational biology, deep neural networks have been successfully applied to the prediction of functional genomic sequence (Alipanahi et al., 2015; Li et al., 2019; Liu et al., 2018; Min et al., 2017; Zhou and Troyanskaya, 2015), gene expression patterns (Singh et al., 2016) and protein structures (Heffernan et al., 2015). Nevertheless, applications of deep learning technologies to 3D genome data analysis is still rare. HiCPlus is the first work that applies a convolutional neural network in enhancing the resolution of Hi-C data by minimizing the mean squared error (MSE) between real high resolution Hi-C data and generated Hi-C data (Zhang et al., 2018). However, it tends to generate Hi-C images with limited dynamic details and can only predict Hi-C matrix with fixed window size. Most importantly, it is very sensitive to the sequencing depth of the Hi-C data.
Considering the limitations of previous work, we take advantage of generative adversarial networks (GANs), a hot deep learning technology that has attracted a lot of attention in producing high quality synthesized images (Goodfellow et al., 2014). We present hicGAN, as an open-sourced computational framework, for inferring high resolution Hi-C data from low resolution Hi-C data with generative adversarial networks (GANs). Instead of using a single neural network for generating Hi-C data, hicGAN is composed of two competitive neural networks and adopts an adversarial training strategy. We demonstrate that hicGAN can effectively generate matrices that are highly consistent to high resolution Hi-C matrices while only adopts as few as 1/16 of original sequencing reads. We systematically evaluate the quality of generated samples by hicGAN through comparison between high resolution samples and generated samples in several perspectives, including image similarity and identified chromatin interactions. After model training in one cell type, hicGAN can even enhance the resolution of insufficient sequenced Hi-C samples in other cell types, which implies that some local patterns are shared across different cell types. We finally summarize hicGAN, as an effective prediction tool for enhancing resolution of Hi-C data, could shed light on the understanding of genome organization built on pair-wise interactions.
2 Materials and methods
2.1 Overview of hicGAN
The overall framework of hicGAN is illustrated in Figure 1A. The design of hicGAN is inspired by Game Theory. Instead of using a single neural network, hicGAN is composed of two competitive neural networks, which are called the generator network (Fig. 1B) and the discriminator network (Fig. 1C), respectively. The generator takes low resolution Hi-C samples as input and tries to produce pseudo high resolution Hi-C (or called super resolution) samples. The discriminator works as a classifier to discern between real high resolution Hi-C data and super resolution Hi-C data. After adversarial training, the generator tends to generate real-like data underlying almost the same distribution of high resolution Hi-C data. Given paired low resolution and high resolution Hi-C samples, of which low resolution data are usually obtained from 1/16 down sampled sequencing reads of original high resolution Hi-C experiment, hicGAN is able to conduct an adversarial training process. Then we only use generator network for enhancing the resolution of Hi-C data after training converges.
Fig. 1.
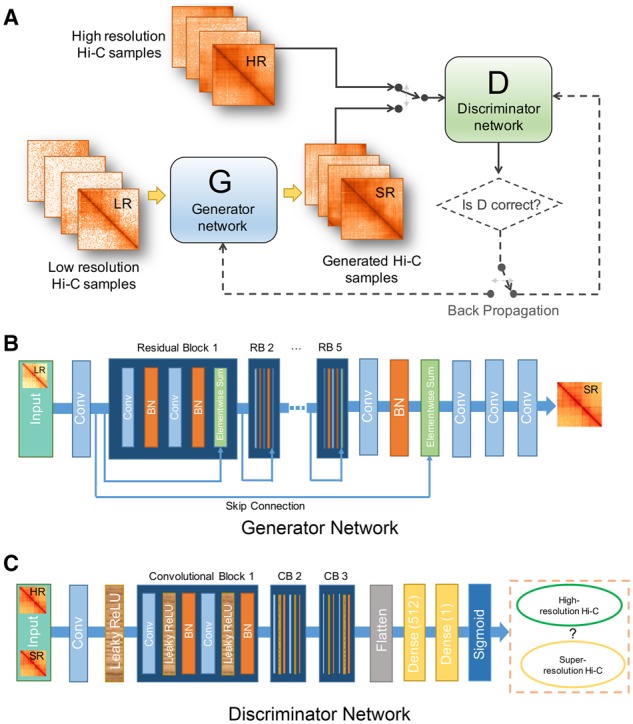
The overall schematic of hicGAN. (A) hicGAN consists of two competitive networks. G tries to generate super resolution samples that are highly similar to real high resolution samples while D tries to discriminate generated super resolution samples from real high resolution Hi-C samples. Parameters of G and D are updated through an adversarial training process. (B) The architecture of the generator network. Generator network adopts a novel dual-stream residual architecture which contains five inner residual blocks (RBs) and an outer skip connection. Rectangles with different colors represent different functional layers. Blue denotes convolutional layer, orange denotes batch normalization layer and green denotes an operation of element-wise summation of the previous layer’s output and the output of the skipped layer. It outputs a super resolution Hi-C sample given an insufficient sequenced Hi-C sample as input. (C) The architecture of discriminator network. Discriminator network is a typical deep convolutional neural network. The convolutional part has been modularized as three convolutional blocks. It outputs the estimated probability that the input is a high resolution Hi-C sample
The generator network (Fig. 1B) adopts a novel dual-stream residual architecture which contains five inner residual blocks (RBs) and an outer skip connection. Each residual block consists of two convolutional layers with filters and 64 feature maps. Each convolutional layer is followed up by a batch normalization layer, which is proved to effectively prevent overfitting (Ioffe and Szegedy, 2015). Residual architecture has shown superior performance in various computer vision tasks and has been proved to alleviate gradient vanishing problem as it contains additional shortcuts compared to traditional deep convolutional neural networks (He et al., 2016; Ledig et al., 2017). Note that the generator network is a typical fully convolutional network (without dense layer), thus can handle any size of input low resolution Hi-C samples in a prediction task. The discriminator network is a deep convolutional neural network, which has been modularized as three convolutional blocks (CBs). The size of input data will reduce by half when going through each CB. High-level features after CBs will be flatted before entering two dense layers and transformed by a sigmoid function. The detailed hyper-parameters of both the generator and discriminator networks can be found in Supplementary Tables S1 and S2.
2.2 Adversarial training
We apply an adversarial training strategy for training two competitive neural network simultaneously. We define generator network as , parametrized by , which outputs the pseudo high resolution sample (or super resolution) given an low resolution Hi-C sample. We define discriminator as , parametrized by , which outputs the probability that the input of discriminator is a high resolution Hi-C sample. We further denote the data distributions of low resolution and high resolution Hi-C sample are and . Then the training process can be regarded as an min-max problem:
The general idea of the above formulation is to train a generative model with the goal of generating samples to fool the discriminator, which has the goal of discerning real high resolution samples from super resolution samples. Towards this goal, generator network gradually learns to generate samples that are highly similar to real high resolution samples by learning the mapping relationship from to . Different from HiCPlus (Zhang et al., 2018), we do not consider minimizing any pixel-wise error measurements, such as MSE, which is believed to generate over-smoothing results (Xu et al., 2017). We finally summarize the detailed adversarial training process in Table 1.
Table 1.
The adversarial training of hicGAN model
| Algorithm Adversarial training of hicGAN |
|---|
| Require: for initial parameters of generator network, for initial parameters of discriminator network, batch size and learning rate . |
| While has not converged, do |
| Sample as a batch high resolution Hi-C data |
| Sample as a batch low resolution Hi-C data |
| end while |
Note: The default settings in all the experiments are as follows. We use Adam optimizer for gradient descent and parameters updating. , , and are initialized with a normal distribution where mean equals 0 and standard deviation equals 0.02.
We implement hicGAN algorithm with the TensorFlow framework (Abadi et al., 2016) and all the computational experiments were carried on a Linux platform equipped with 10 NVIDIA GeForce RTX 2080 Ti GPU cards, which can significantly accelerate the training process.
2.3 Data preparation and preprocessing
The high resolution Hi-C datasets across four cell types (GM12878, K562, IMR90 and NHEK) were downloaded from the GEO database with accession number GSE63525. For cell type that contains multiple Hi-C experiments, we first pooled all the aligned Hi-C reads together (Supplementary Table S3), then we used the Juicer toolbox (Durand et al., 2016) for generating HIC file and further extracted raw chromatin contacts for constructing Hi-C matrix. Note that we only considered intra-chromosomal interactions and removed chromosome X and Y for each cell type to eliminate sex effect. A ChIA-PET dataset with CTCF target from K562 cell type was downloaded from https://4dgenome.research.chop.edu as ground truth to verify the chromatin loops and interactions predicted by our model. Note that the ChIA-PET dataset has already been processed and we directly used the ChIA-PET chromatin contact text file. ChIP-seq data with CTCF target from K562 was download from the ENCODE project (Consortium, 2012) with accession number ENCSR000AKO. The narrowPeak file was used in Section 3.3 for generating background pairs of genomic regions.
Let be the original raw contact Hi-C matrix. We first normalized the sequencing depth by the following formulation:
where is the total aligned reads of chromosome k and . Then we further transformed the normalized Hi-C matrix to the range [-1, 1] by performing the linear transformation , where . As for low resolution Hi-C data, we randomly down-sampled the original aligned reads to 1/16 for simulating low resolution Hi-C data. Then we constructed Hi-C matrix with the same bin size (e.g. 10 kb) and performed the 2-step data normalization just the same as high resolution Hi-C data.
Before training hicGAN, we divided normalized high resolution and low resolution Hi-C matrices into multiple non-overlapping small patches with size . Each patch contains pixels with 10 kb resolution. Considering the fact that the average genomic distance of TADs is usually less than 1 Mb, we only kept patches of which genomic distance between two loci is less than 2 Mb, thus filtering too far off-diagonal patches as there are few meaningful contacts outside 2 Mb. Then, both high resolution and low resolution Hi-C samples were fed to hicGAN for model training. Particularly, Hi-C samples extracted from chromosomes 1–17 were for training and Hi-C samples extracted from chromosomes 18–22 were for testing.
In the process of prediction, there is no restriction on the input size of low resolution Hi-C sample as the generator network is a fully convolutional neural network. The generator network can make a prediction given one or multiple insufficient sequenced Hi-C samples of arbitrary size at one time. While the adversarial training process usually takes hours, the prediction process is ultra-fast and usually done in seconds.
2.4 Model evaluation
We evaluate the quality of generated samples by hicGAN in multiple perspectives. We first compare the pixel-wise error measurement such as MSE between high resolution samples and super resolution samples generated by generator network, which is calculated by
where and denote a high resolution Hi-C sample and a super resolution Hi-C sample, respectively. is the sample size which is set to 40.
We then calculate the peak signal-to-noise ratio (PSNR) and structure similarity index (SSIM) (Wang et al., 2004) which are two commonly used metrics in natural images compression, synthesis and super-resolution.
where denotes the data range of high resolution sample in our case.
SSIM is an overall measurement, which considers both the brightness, contrast and structure similarity of the two comparing images. SSIM ranges from 0 to 1. The closer to 1, the higher similarity the two images have.
where and denote the mean value of a high resolution Hi-C sample and corresponding super resolution Hi-C sample, and denote the standard deviation of a high resolution Hi-C sample and corresponding super resolution Hi-C sample. denotes covariance of a high resolution Hi-C sample and a super resolution Hi-C sample. and are two constants which are set to and , respectively.
2.5 Evaluating pairs of genomic regions with Hi-C chromatin loops
We proposed a strategy for scoring any pair of genomic regions with existing Hi-C chromatin interactions to give an estimation of whether the two genomic regions have contact with each other. Note that only intra-chromosomal interactions are retained in Hi-C data. Assuming a pair of genomic regions is denoted as and . Hi-C chromatin interactions are represented as . While , the estimated score is calculated as
where is the decay parameter which is set to 0.0001 in our experiments and are the indexes that ensure . In other cases, score is set to 0. The higher the score is, the stronger the estimated interaction strength will be.
2.6 Baseline models
In order to evaluate the performance of hicGAN, we designed a series of systematical experiments and compared hicGAN to baseline models. HiCPlus (Zhang et al., 2018) is a recent approach that applies a convolutional neural network in enhancing Hi-C resolution. It was downloaded from https://github.com/zhangyan32/HiCPlus and the default hyper-parameters were used in all experiments. Besides, we discuss in detail the limitations of HiCPlus in Supplementary Note S1.
Another baseline model 2D Gaussian is an unsupervised approach which applies a sliding Gaussian window for smoothing the input matrix. To determine the best parameter of 2D Gaussian, the deviation Sigma is finally set to 1 after performing a grid search strategy.
3 Results
3.1 hicGAN recovers high resolution Hi-C data from insufficient sequenced samples
We first designed a series of systematical experiments to verify the performance of hicGAN by multiple perspectives. We downloaded high resolution datasets of GM12878 from GEO database with accession number GSE63525. 10 kb resolution Hi-C matrix was constructed by all the aligned sequencing reads. Then we randomly down-sampled all the reads to 1/16 and constructed the interaction Hi-C matrix with the same resolution. Note we only kept intra-chromosomal interactions in a reasonable genomic distance and then cropped the matrices into non-overlapping patches with size (See Methods). Each patch was treated as an individual sample which contains pixels in the aspect of image. We used Hi-C samples from chromosomes 1–17 for training and Hi-C samples from chromosomes 18–22 for test. Then we compared the samples generated by hicGAN to high resolution Hi-C samples from test set in three different metrics (Fig. 2A–C). We grouped the test samples based on the genomic distance of the diagonal interactions of a given sample. The three measurements show the same trend that Hi-C samples containing closer interactions are easier to be predicted. At the genomic distance of 0, Hi-C samples were extracted from diagonal of original Hi-C matrix, hicGAN on average achieves an MSE of 0.0078, a PSNR of 23.90 dB and an SSIM of 0.731. At the genomic distance of 1.2 Mb, the performance of hicGAN drops to an acceptable level with an MSE of 0.042, a PSNR of 13.07 dB and an SSIM of 0.209 on average. We also note that hicGAN outperforms HiCPlus under the above three measurements under different genomic distance by a large margin (Supplementary Figs S1–S3).
Fig. 2.
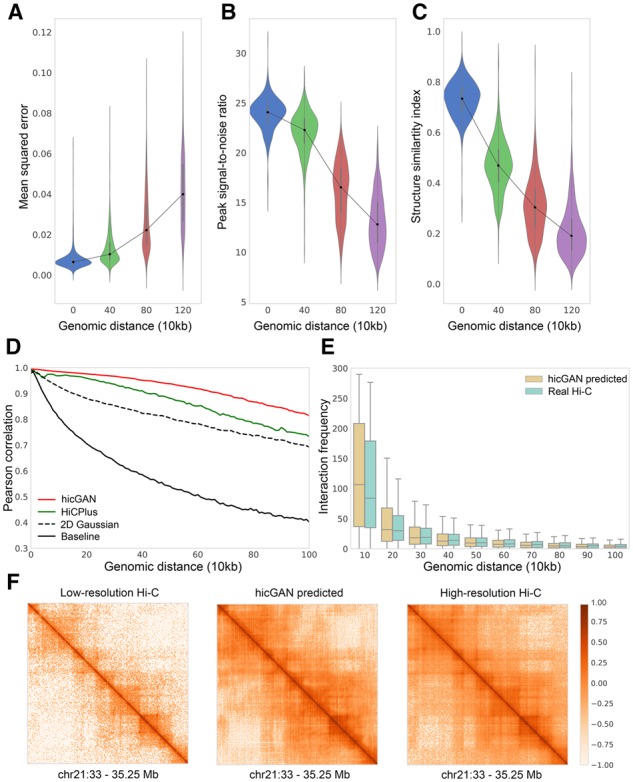
Evaluation of Hi-C data generated by hicGAN in GM12878 cell type. Model training was performed on chromosomes 1–17, model evaluation was performed on chromosomes 18–22. (A-C) Mean squared error (MSE), peak signal-to-noise ratio (PSNR) and structure similarity index (SSIM) measurements between high resolution Hi-C samples and super resolution samples generated by hicGAN. (D) The Pearson correlation coefficients (PCCs) between real Hi-C data and predicted Hi-C data under different genomic distance. hicGAN significantly outperforms other methods in different genomic distance. (E) The distribution of interaction frequency within different genomic ranges (e.g. 0–10 kb, 10–20 kb, etc.) observed from real high resolution Hi-C data and Hi-C data predicted by hicGAN. The distribution of interaction frequency captured by hicGAN is highly consistent with real Hi-C data. (F) An example shows the low resolution Hi-C sample (left), Hi-C sample predicted by hicGAN (middle) and high resolution Hi-C sample (right). Hi-C sample generated by hicGAN is highly similar to high resolution Hi-C sample
We then compared hicGAN to three other approaches (HiCPlus, 2D Gaussian and baseline) by measuring the Pearson correlation at different genomic distance. The interactions with the same genomic distance were grouped and the Pearson correlation coefficient (PCC) between real interactions and interactions predicted by different methods are calculated (Fig. 2D). HiCPlus and 2D Gaussian are described in Section 2. The baseline method is directly using interactions from down-sampled Hi-C matrix and compared to the real interactions from high resolution Hi-C matrix. Our hicGAN model outperforms HiCPlus (P-values<, Supplementary Table S4) and other two methods at any genomic distance ranging from 0 to 1 Mb. At the genomic distance of 200 kb, hicGAN achieves a Pearson correlation coefficient of 0.976, compared to 0.958 of HiCPlus, 0.881 of 2D Gaussian and 0.705 of baseline. At the genomic distance of 1 Mb, hicGAN outperforms HiCPlus by a large margin (0.814 versus 0.732). We note that 2D Gaussian shows a decent performance as it can effectively reduce the noise level by a smoothing process.
To verify whether hicGAN can effectively capture the interaction frequency at different genomic distance, we compared the distribution of interaction frequency within different genomic ranges, such as 0–kb, 100–200 kb, etc. We first transformed the predicted normalized Hi-C data into interaction frequency count. Then we observed that the interaction frequency predicted by hicGAN has highly similar distribution as real Hi-C data (Fig. 2E). In the aspect of a visualized image, samples generated by hicGAN tend to have domain boundaries as clear as high resolution Hi-C data. We demonstrated vivid examples of visualized Hi-C samples generated by hicGAN model (Fig. 2F and Supplementary Fig. S4). Leveraging only a small fraction of the original sequencing reads, hicGAN captures the features of high resolution Hi-C data across chromosomes.
3.2 hicGAN recovers high resolution Hi-C data across different cell types
In the previous section, hicGAN has demonstrated the powerful ability of recovering super resolution Hi-C data that are highly similar to real high resolution data. We then ask whether hicGAN can be used for enhancing the resolution of Hi-C matrix across different cell types. Towards this goal, we downloaded Hi-C data across 4 cell types (GM12878, K562, IMR90 and NHEK) from GEO database with accession number GSE63525. The same data preprocessing was performed to each cell type. With the hicGAN model trained in GM12878 in the previous section, we then applied it to enhance insufficient sequenced Hi-C matrices in three test cell types. We selected a genomic region (chr17: 70.5–75.75 Mb) that contains highly dynamic chromatin contacts across three test cell types, experiments show that hicGAN can well capture the differences of domain boundaries across different cell types and generate super resolution Hi-C samples that are highly similar to original high resolution Hi-C samples (Fig. 3A).
Fig. 3.
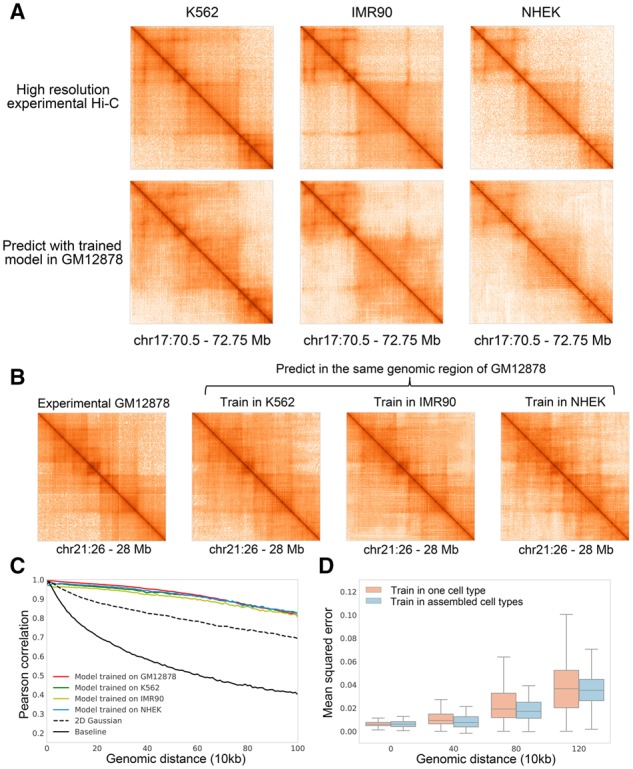
Cross-cell-type experiments by hicGAN. Training data is obtained from chromosomes 1–17 and test data is obtained from chromosomes 18–22. (A) hicGAN model trained in GM12878 cell type was used for generating super resolution Hi-C samples (chr17: 70.5–72.75Mb) in three other cell types (K562, IMR90 and NHEK). The generated Hi-C samples are highly similar to the high resolution Hi-C samples in the corresponding cell type. (B) hicGAN model was trained in three cell types (K562, IMR90 and NHEK), respectively. Then the trained hicGAN model in each cell type was used for predicted in the same genomic region of GM12878 (chr21: 26–28 Mb). No matter which cell type the hicGAN was trained in, it can generate Hi-C sample that is highly consistent to the original high resolution Hi-C sample in GM12878. (C) We trained four hicGAN models on chromosomes 1–17 of GM12878, K562, IMR90 and NHEK, respectively. Then we used the four trained hicGAN model to predict Hi-C samples on chromosomes 18–22 of GM12878. The performance of model trained in K562, IMR90 and NHEK drops slightly compared to model trained in GM12878. The four hicGAN models outperform 2D Gaussian and baseline by a large margin. (D) We pooled the Hi-C samples on chromosomes 1–17 of four cell types (K562, IMR90, GM12878 and NHEK) together and trained a hicGAN with the constructed assembled Hi-C dataset. For comparison, we trained a baseline hicGAN model on chromosomes 1–17 of K562 cell types. We used the above two hicGAN models to predict Hi-C samples on chromosomes 18–22 of K562 cell type. Model trained in assembled cell types achieves slightly lower mean squared error and lower variance, especially at long genomic distance
To further verify our observation, we then designed an experiment in which we trained hicGAN model in different cell types while predicting the same Hi-C sample. We first trained hicGAN on chromosomes 1–17 of K562, IMR90 and NHEK, respectively. Then we used trained hicGAN model to make a prediction given the same insufficient sequenced Hi-C sample from GM12878 (chr21: 26–28 M). The results predicted by hicGAN model across three test cell types show high consistence to the high resolution Hi-C sample in GM12878 (Fig. 3B). We further evaluated the cross-cell-type generated Hi-C samples by measuring the Pearson correlation similar to the previous section. We trained four hicGAN models on chromosomes 1–17 of GM12878, K562, IMR90 and NHEK, respectively. Then we used the four trained hicGAN model to predict Hi-C samples on chromosomes 18–22 of GM12878. 2D Gaussian applies a Gaussian smoothing to insufficient sequenced Hi-C samples on Chromosomes 18–22 of GM12878. The baseline directly shows the Pearson correlation coefficient (PCC) between insufficient sequenced Hi-C samples and high resolution Hi-C samples on chromosomes 18–22 of GM12878. Compared to training and test in the same cell type, the performance of cross-cell-type experiments only decreases slightly (Fig. 3C). At the genomic distance of 200 kb, hicGAN trained in GM12878 achieves a Pearson correlation coefficient of 0.976, compared to 0.961 in K562, 0.952 in IMR90 and 0.967 in NHEK. The performance of four hicGAN models outperform 2D Gaussian and baseline model by a large margin (P-values < , Supplementary Table S5). The superior results of cross-cell-type experiments suggest that different cell types may share some local patterns in the 3D genome contacts map so hicGAN can borrow information of local patterns in one cell type when making prediction in another cell type.
We further investigate whether hicGAN can borrow information from multiple cell types. We first pooled all the Hi-C samples from chromosomes 1–17 of four cell types together to construct an assembled training set. We then trained hicGAN model under the assembled training data and compared the performance to another hicGAN model trained on chromosomes 1–17 of K562 cell type. We finally used the above trained hicGAN model to make a prediction on chromosomes 18–22 of K562 cell type (Fig. 3D). The hicGAN model trained in assembled cell types has a slightly better performance than the model trained in a single cell type at different genomic distance (P-values < , Supplementary Table S6). At the genomic distance of 400 kb, hicGAN trained in assembled cell types can reduce the mean squared error (MSE) by 0.0039. Moreover, hicGAN model trained in assembled cell types tends to be more robust at a long genomic distance as achieves obviously lower variance. The standard deviation of MSE achieved by hicGAN with assembled training cell types at 800 kb is 0.0117, compared to 0.0156 of single cell type trained hicGAN model. This experiment again suggests that the local patterns or features among different cell types may have some common properties. Once a hicGAN model is trained in one or multiple cell types, it can be applied to make prediction in new cell types.
3.3 hicGAN facilitates identifying meaningful chromatin interactions
Previous experiments have demonstrated that hicGAN is able to enhance the resolution of insufficient sequenced Hi-C samples. We then investigated that whether hicGAN can help facilitate the identification of chromatin contacts or chromatin loops. Towards this purpose, we applied Fit-Hi-C software (Ay et al., 2014), as a chromatin loop caller, for identifying the significant chromatin loops given high resolution Hi-C data and predicted Hi-C data. Similar to the previous experiments, we first trained a hicGAN model on chromosomes 1–17 of GM12878 cell type, then we used the trained hicGAN model to predict Hi-C samples on chromosomes 18–22. As not all the chromatin interactions are of equal importance, what really made us interested is the interactions that are enriched for regulatory elements, such as promoters and enhancers. We applied the Fit-Hi-C tool to predicted Hi-C data and real high resolution Hi-C data in GM12878 cell type for calling significant chromatin loops with a strict threshold (q-value < 1e−06), respectively. Then we filtered the called chromatin loops and only kept significant chromatin loops within the genomic distance from 30 to 300 kb, which resulted in 124 321 significant chromatin loops of high resolution Hi-C data and 147 890 significant chromatin loops of hicGAN predicted Hi-C data. We observed that 90.86% of the significant chromatin loops from high resolution Hi-C data were successfully recovered by hicGAN. 76.34% of the significant chromatin loops from hicGAN predicted Hi-C data were also identified in high resolution Hi-C data (Fig. 4A). We also noticed that down-sampled Hi-C matrix can only recover 13.36% of the significant chromatin loops from the high resolution Hi-C data, which suggests that hicGAN can effectively promote the identification of significant chromatin loops give insufficient sequenced Hi-C samples. We also conducted similar experiments in other cell lines to further support our conclusion (Supplementary Fig. S5).
Fig. 4.
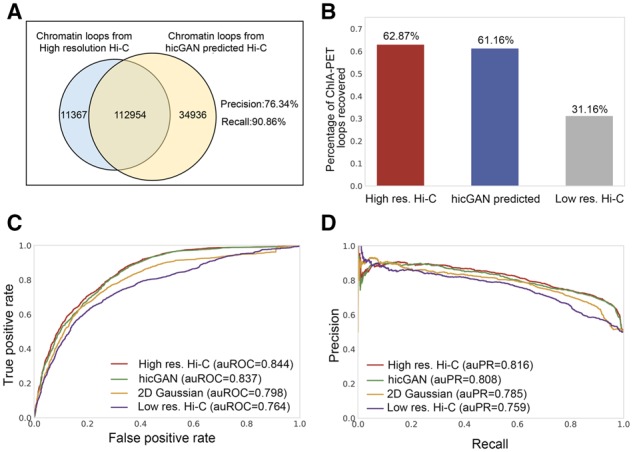
Evaluation of significant chromatin loop inferred from Hi-C data predicted by hicGAN model. (A) The Venn plot of the significant chromatin loops from high resolution Hi-C data and Hi-C data predicted by hicGAN model in GM12878 cell type using Fit-Hi-C software with a strict threshold (q-value < 1e−06). More than 90 percent of the significant chromatin loops from real high resolution Hi-C data can be also identified in Hi-C data predicted by hicGAN. (B) High resolution Hi-C data and Hi-C data predicted recovers comparable percentage of ChIA-PET chromatin loops while down-sampled low resolution Hi-C data recovers much less ChIA-PET chromatin loops. (C) The receiver operating characteristic (ROC) curve in discerning ChIA-PET chromatin loops from random pairs of CTCF ChIP-seq peaks. High resolution H-C data and Hi-C data predicted by hicGAN model achieve comparable results with the areas under ROC curve (auROCs) 0.844 versus 0.837, which outperform 2D Gaussian and down-sampled low resolution Hi-C. (D) The precision-recall curve in discerning ChIA-PET chromatin loops from random pairs of CTCF ChIP-seq peaks. The areas under precision-recall curve (auPRs) implicates the consistent conclusion
Next, we introduced another type of chromatin contacts data from ChIA-PET, a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, paired-end tagging and high-throughput sequencing (Wei et al., 2006). ChIA-PET can accurately help us identity de novo long-range chromatin interactions genome-wide with targeted protein. We downloaded ChIA-PET datasets of K562 cell type with CTCF target, a key transcription factor that involves in the regulation of chromatin architecture (Phillips and Corces, 2009). We first investigated whether hicGAN can recover CTCF chromatin interaction from ChIA-PET data. Similar to the experiments settings previously, we used Fit-Hi-C for identifying significant chromatin loops from high resolution Hi-C data, down-sampled Hi-C data and Hi-C data predicted by hicGAN with a strict threshold (q-value < 1e−06) in K562 cell type. We then calculated how many CTCF chromatin interactions were covered by significant Hi-C chromatin loops. We observed that high resolution Hi-C data and Hi-C data predicted by hicGAN model can recover 62.87% and 61.16% of the ChIA-PET chromatin interactions, respectively. While the down-sampled low resolution Hi-C data can only recover 31.16% of the ChIA-PET chromatin interactions (Fig. 4B), which implicated that hicGAN can significantly help recover ChIA-PET chromatin loops given down-sampled low resolution Hi-C data.
We further investigated whether significant Hi-C loops can help discriminate ChIA-PET chromatin loops from random pairs of genomic regions. We treated ChIA-PET chromatin interactions with CTCF target as ground truth or true positives, then we selected the same number of random pairs from CTCF ChIP-seq peaks, which have no overlaps with any of the ChIA-PET chromatin loops, as negative samples. Finally, we used Hi-C chromatin interactions to score each of the above positive and negative samples (see Methods). It showed that Hi-C chromatin interactions from high resolution Hi-C data and Hi-C data predicted by hicGAN can discern ChIA-PET chromatin loops from random pairs of CTCF ChIP-seq peaks at a comparable level (Fig. 4C–D). Using high resolution Hi-C data achieves an auROC of 0.844, compared to 0.837 of hicGAN, 0.798 of 2D Gaussian and 0.764 of using down-sampled low resolution Hi-C data. The precision-recall curves also showed that the high resolution Hi-C data and hicGAN predicted Hi-C data can achieve similar performance, which outperform 2D Gaussian and down-sampled low resolution Hi-C data by a relatively large margin.
The above results demonstrate that the chromatin loops enriched in down-sampled low resolution Hi-C data are only a small portion of the chromatin loops from high resolution Hi-C data. However, with our powerful hicGAN model, one can significantly improve the number of enriched chromatin loops, thus reaching a comparable level with the high resolution Hi-C data.
3.4 hicGAN facilitates identifying cell-type specific contact domain boundaries
We finally applied hicGAN in inferring super resolution Hi-C data in two differential cell types GM12878 and K562 with a hicGAN model trained in NHEK cell type. Previous experiments have demonstrated the ability of cross-cell-type prediction of hicGAN. We now focus on the exploring the relationship between domain boundaries inferred from Hi-C data and the implicating functions. We extracted a 1 Mbp genomic region (chr9: 36.5–37.5 M), which contains Pax5, a master regulator of B cell development (Medvedovic et al., 2011). At the same time, we collected several annotation tracks using WU Epigenome Browser (Zhou et al., 2011).We note that Pax5 regulator was only expressed in GM12878 cell type according to the RNA-seq annotation track. The down-sampled low resolution Hi-C data across two cell types contained blurry contact domain boundaries while Hi-C data predicted by hicGAN showed as clear TADs or sub-TADs as high resolution Hi-C data (Fig. 5A–B). Interestingly, hicGAN identified several promoter-enhancer interactions which are consistent with the annotations by two histone modifications. Compared to K562 cell type, GM12878 contains more sub-domain boundaries, which are enriched in signals from two histone modifications and ChIA-PET with CTCF. More importantly, except for the common contact domain boundaries (yellow dots), we also observed that GM12878 contained cell type specific domain boundaries (blue dots) that were not discovered in K562 cell types. Previous studies have shown that such cell type specific domain boundaries are crucial to the relevant chromatin architecture and the underlying gene regulations (Smith et al., 2016). A great portion of the cell type specific contact domain boundaries may not be uncovered in insufficient sequenced Hi-C data. With the powerful hicGAN model, one can significantly enhance the resolution of Hi-C data and identity more refined contact domain boundaries.
Fig. 5.

Three types of Hi-C data extracted from a differential genomic region (chr9: 36.5–37.5 M) between GM12878 and K562 cell type. Several annotation tracks, including RNA-seq, ChIP-seq with CTCF, ChIP-seq with two histone modifications and ChIA-PET with CTCF target across two cell types were also shown below the Hi-C data. The high resolution Hi-C data and Hi-C data predicted by hicGAN have significantly clearer chromatin contact boundaries compared to down-sampled low resolution Hi-C data. We observed that a B cell important regulator, Pax5, only expressed in GM12878 cell type. Hi-C data also reveals promoter-enhancer interactions which is highly consistent with the signals from the two histone markers (a promoter P and three potential enhancers E1-E3 were denoted in GM12878). More importantly, we noticed that the above two cell types contain common contact domain boundaries and cell-type specific contact domain boundaries (GM12878 specific contact boundaries were shown with blue dots and the common domain boundaries were shown with yellow dots). (A) Hi-C maps and annotation tracks in GM12878 cell type. (B) Hi-C maps and annotation tracks in K562 cell type
3.5 Hyperparameter settings for hicGAN
As hicGAN contains two neural networks with complicated architectures, we modularized both generator network and discriminator network into functional layers or blocks, which makes it easy for us to determine the best hyperparameters. We considered learning rate , batch size , the number of residual blocks (RBs) and the number of convolutional blocks (CBs) as the major tuning hyperparameters. We directly used a grid search strategy for the best combination of the above four hyperparameters. The learning rate was chosen from {0.001, 0.0001, 0.00001}, batch size was chosen from {32, 64, 128}, the number of RBs was chosen from {1, 5, 10} and the number of CBs was chosen from {1, 2, 3, 4}. The best hyperparameters were finally determined as described in Supplementary Table S1 and S2.
We also evaluated whether the batch normalization will help improve the performance of hicGAN. We tried adding batch normalization after each convolutional layer and removing batch normalization layer of both generative network and discriminator network. Interestingly, batch normalization can indeed help improve the performance of generative network, but we did not observe difference when applying to discriminator network. We implemented hicGAN with the best hyperparameters we have obtained so far in the software.
4 Discussion
We proposed hicGAN, an open-sourced computational framework, for inferring high resolution Hi-C data from low resolution Hi-C data with adversarial generative networks (GANs). To the best of our knowledge, hicGAN is the first work to apply GANs in the generation of 3D genome data. We designed a series of systematical experiments to verify the quality of generated Hi-C samples by hicGAN model given insufficient sequenced Hi-C samples. Experimental results show that Hi-C samples generated by hicGAN (super resolution Hi-C samples) are highly similar to the original high resolution Hi-C data. With the powerful learning ability, hicGAN effectively enhances the resolution of low resolution Hi-C data, which is typically constructed by down-sampling the original aligned sequencing reads to as few as 1/16. More importantly, hicGAN can help us identify significant chromatin interactions or loops where a portion of the boundary information is already missing in the low resolution Hi-C data. A typical scenario of using hicGAN is applying it to low resolution Hi-C samples where the corresponding high resolution Hi-C data is not available. Some meaningful interactions such as promoters-enhancers might not be detected in the low resolution Hi-C data, but we have the potential to recover such interactions with our hicGAN model. We expect to see wide application of hicGAN to both public or in-house Hi-C data in the identification of chromatin interactions across various cell types.
The cross-cell-type experiments implicate that Hi-C datasets consist of many local patterns that are shared across different cell types. The information of local structures or patterns can be borrowed when making a prediction in other cell types. However, due to the ‘black box’ property of neural network model, it is still somehow tough to interpret the learned features. One thing for sure is that the features should be highly related to the crucial functions of 3D genome organization, such as TADs, sub-TADs and contact domain. So a major future task is for us to visualize and interpret the features learned in hicGAN. Perhaps we can investigate the kernel weights in the convolutional layers to see if they contain low-level structure information that corresponds to chromatin boundaries.
Another possible improvement of our work is to consider the potential noise in Hi-C data. Although we regard high resolution Hi-C data as the gold standard, it still contains multiple sources of noises such as the random ligations generated by Hi-C protocol (Lajoie et al., 2015). So variations exist even between high resolution Hi-C data from two biological replicates (Dixon et al., 2012). One feasible solution might be modifying hicGAN model by using random noise as input and low resolution Hi-C data as prior information. More experiments need to be conducted to validate our assumption and shed light on how to further improve the performance of current model. The current evaluation of generated Hi-C data mainly focusses on image-based measurements. We can further take Hi-C specialized measurement, such as HiCRep (Yang et al., 2017), into consideration.
Theoretically, hicGAN can be applies to any type of chromatin interaction data such as Capture Promoter Hi-C and ChIA-PET. But the current public datasets are not as abundant as Hi-C datasets. We can investigate the performance of hicGAN applied in other types of chromatin interaction data as more datasets across various tissues or cell types become available.
To sum up, hicGAN presents an end-to-end solution for enhancing the resolution of insufficient sequenced Hi-C data. Our study not only provides a novel approach for inferring high resolution Hi-C data, but also implies fascinating insights into deciphering complex mechanism underlying the 3D genome organization.
Supplementary Material
Acknowledgements
We thank Xianglin Zhang for his helpful instructions about Hi-C data preprocessing and normalization. We also thank Fengling Chen for her useful comments on Hi-C data chromatin loops calling software. Rui Jiang is a RONG professor at the Institute for Data Science, Tsinghua University.
Funding
This research was partially supported by the National Key Research and Development Program of China (No. 2018YFC0910404), the National Natural Science Foundation of China (Nos. 61873141, 61721003, 61573207) and the Tsinghua-Fuzhou Institute for Data Technology.
Conflict of Interest: none declared.
References
- Abadi M. et al. (2016) Tensorflow: a system for large-scale machine learning In: OSDI, Savannah, GA, USA, pp. 265–283. USENIX. [Google Scholar]
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. [DOI] [PubMed] [Google Scholar]
- Ay F. et al. (2014) Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res., 24, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. et al. (2002) Capturing chromosome conformation. Science, 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dixon J.R. et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J. et al. (2006) Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res., 16, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand N.C. et al. (2016) Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst., 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I. et al. (2014) Generative adversarial nets. In: Advances in Neural Information Processing Systems, Curran Associates, Inc. Montreal, Quebec, Canada, Dec 8, 2014 – Dec 13, 2014, pp. 2672–2680. [Google Scholar]
- He K. et al. (2016) Deep residual learning for image recognition In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, June 27–30, 2016, Las Vegas, NV, USA. pp. 770–778. [Google Scholar]
- Heffernan R. et al. (2015) Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep., 5, 11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe S., Szegedy C. (2015) Batch normalization: accelerating deep network training by reducing internal covariate shift In: Proceedings of the 32nd International Conference on International Conference on Machine Learning, July 6–11, 2015, Vol. 37, Lille, France, pp. 448–456. [Google Scholar]
- Lajoie B.R. et al. (2015) The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods, 72, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y. et al. (2015) Deep learning. Nature, 521, 436.. [DOI] [PubMed] [Google Scholar]
- Ledig C. et al. (2017) Photo-realistic single image super-resolution using a generative adversarial network In: 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, Hawaii, July 21 to July 26, 2017, pp. 105–114. IEEE. [Google Scholar]
- Li W. et al. (2019) DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res., doi.org/10.1093/nar/gkz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E. et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. (2018) Chromatin accessibility prediction via a hybrid deep convolutional neural network. Bioinformatics, 34, 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J. et al. (2011) Pax5: a master regulator of B cell development and leukemogenesis Adv. Immunol., 111, 179–206. [DOI] [PubMed] [Google Scholar]
- Min X. et al. (2017) Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics, 33, i92–i101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P. et al. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature, 485, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins J.E. et al. (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell, 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.E., Corces V.G. (2009) CTCF: master weaver of the genome. Cell, 137, 1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz S.A. et al. (2018) Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell, 174, 744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A.D. et al. (2016) Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol., 17, 743.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T. et al. (2012) Three-dimensional folding and functional organization principles of the Drosophila genome. Cell, 148, 458–472. [DOI] [PubMed] [Google Scholar]
- Simonis M. et al. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet., 38, 1348. [DOI] [PubMed] [Google Scholar]
- Singh R. et al. (2016) Deepchrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics, 32, i639–i648. [DOI] [PubMed] [Google Scholar]
- Smith E.M. et al. (2016) Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet., 98, 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler C., Shivashankar G. (2017) Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol., 18, 717.. [DOI] [PubMed] [Google Scholar]
- Wang Z. et al. (2004) Image quality assessment: from error visibility to structural similarity. IEEE Trans. Image Process., 13, 600–612. [DOI] [PubMed] [Google Scholar]
- Wei C.-L. et al. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell, 124, 207–219. [DOI] [PubMed] [Google Scholar]
- Xu X. et al. (2017) Learning to super-resolve blurry face and text images In: 2017 IEEE International Conference on Computer Vision (ICCV), October 22–29, 2017, Venice, Italy, pp. 251–260. IEEE. [Google Scholar]
- Yang T. et al. (2017) HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res., 27, 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Ren B. (2017) The three-dimensional organization of mammalian genomes. Annu. Rev. Cell Dev. Biol., 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2018) Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat. Commun., 9, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2011) The human epigenome browser at Washington University. Nat. Methods, 8, 989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.