

Abstract
Motivation
Polyadenylation is a critical step for gene expression regulation during the maturation of mRNA. An accurate and robust method for poly(A) signals (PASs) identification is not only desired for the purpose of better transcripts’ end annotation, but can also help us gain a deeper insight of the underlying regulatory mechanism. Although many methods have been proposed for PAS recognition, most of them are PAS motif- and human-specific, which leads to high risks of overfitting, low generalization power, and inability to reveal the connections between the underlying mechanisms of different mammals.
Results
In this work, we propose a robust, PAS motif agnostic, and highly interpretable and transferrable deep learning model for accurate PAS recognition, which requires no prior knowledge or human-designed features. We show that our single model trained over all human PAS motifs not only outperforms the state-of-the-art methods trained on specific motifs, but can also be generalized well to two mouse datasets. Moreover, we further increase the prediction accuracy by transferring the deep learning model trained on the data of one species to the data of a different species. Several novel underlying poly(A) patterns are revealed through the visualization of important oligomers and positions in our trained models. Finally, we interpret the deep learning models by converting the convolutional filters into sequence logos and quantitatively compare the sequence logos between human and mouse datasets.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Polyadenylation, as a critical and pervasive process (Proudfoot, 1991) during the maturation of mRNA, is essentially composed of two coupled steps: a cleavage at the poly(A) site and an addition of an adenosine tail. Studies have found a great number of poly(A) signals (PASs) in most eukaryotes (Shen et al., 2008a,b). It is well accepted that the recognition of polyadenylation sites requires a signal residing in the ∼10–30 nt upstream region of the cleavage site (Proudfoot, 2011), which consists of 6 nt known as PAS motifs.
Studies on PAS motifs and their surrounding regions are crucial, as they provide insights on how transcription is ended thereby determining the fate of the RNA transcripts (Shaw and Kamen, 1986) and the association of their mutations with diseases (Lin et al., 2012; Pastrello et al., 2006). In the past few decades, numerous works have been done for this purpose, to characterize the sequence information around PASs and identify core elements and their roles in polyadenylation. It has been shown that the surrounding sequences of a poly(A) site are U-rich in general (Tian et al., 2005). Similarly, GU-rich elements found in the downstream of the cleavage site are also believed to regulate polyadenylation and have been regarded as informative patterns to specify the true PASs (Zarudnaya et al., 2003).
Previous works have made great efforts in the prediction of PAS in mRNA and genomic DNA sequences. Specifically, most methods aim to identify true PAS from pseudo ones which have the same hexamer (e.g. ATTAAA) as true PAS but do not involve in polyadenylation. Earlier studies (Helden et al., 2000; Matis et al., 1996; Tabaska and Zhang, 1999) focus on exploring the statistical information of PAS surrounding sequences. Based on prior knowledge of DNA sequences, many carefully hand-crafted features have been proposed by experts which formed the basis of most PAS recognition models (Akhtar et al., 2010; Cheng et al., 2006; Hu et al., 2005; Liu et al., 2003; Salamov and Solovyev, 1997; Tabaska and Zhang, 1999).
Recently, several methods have greatly advanced the accuracy of human poly(A) recognition. Kalkatawi et al. (2012) provided a benchmark (denoted as the Dragon human data) containing 14 740 sequences for 12 human PAS motif variants. Based on many expert-crafted features, they proposed an artificial neural network-based method and a random forest (RF) based method. After that, Xie et al. (2013) derived a new set of latent features for DNA sequences with hidden Markov model (HMM) and fed these features to an SVM model for classification (referred as HSVM hereafter), which significantly increased the accuracy on the Dragon human data. Very recently, Magana-Mora et al. (2017) developed a new set of hand-crafted features in combination with multiple classification models, including decision tree, RF etc. Their model, Omni-PolyA, improved the results of HSVM.
However, methods like RF, HSVM and Omni-PolyA have two limitations. First, they are all feature-based methods which require a large amount of prior knowledge and cannot cope with the rapidly increasing size of data. Second and more importantly, they are not robust or generic, in the sense that they require training a separate model for each of the 12 human motif variants. And due to the features that are hand crafted for human PAS motifs, they cannot be extended to different species.
In this article, we propose an accurate and robust deep learning model, i.e. Deep Regulatory Code and Tools for Polyadenylation (DeeReCT-PolyA), for PAS recognition, which is PAS motif variant agnostic. For example, PAS motifs are slightly different between human and mouse. We train one single generic convolutional neural network (CNN) that can deal with all 12 human PAS motif variants which still significantly reduces the error rate on both two standard human benchmarks, compared with the state-of-the-art methods which require training 12 separate models. Further experiments on C57BL/6J (BL) and SPRET/EiJ (SP) mouse data demonstrate that our model can consistently perform very well across different species. Moreover, we adopt transfer learning in PAS identification by transferring a deep neural network pre-trained with one dataset to recognize PAS motifs on a new dataset of a different species and show transfer learning can further improve the accuracy and more importantly, address the problem of insufficient training data. We propose several methods to visualize our model and investigate the biological significance of the model. We reveal some novel patterns in poly(A) regulation and the similarities of such patterns across different species including human, BL and SP mouse.
2 Materials and methods
2.1 The proposed CNN for PAS recognition
We propose a deep neural network-based method, DeeReCT-PolyA, for automatic feature extraction and PAS identification. Figure 1 illustrates the architecture of DeeReCT-PolyA. The first layer is a convolutional layer consisting of 16 filters, which are essentially motif detectors. When processing a raw DNA sequence (encoded with one-hot encoding), each motif detector will search sequence patterns that can discriminate true PASs from pseudo ones. The outputs of the convolutional layer are divided into several groups and normalized within each group by a subsequent filter-group normalization layer (GN). We propose such a novel normalization layer with the motivation that some motif detectors are correlated and show it improves the PAS recognition accuracy comparing to other normalization techniques in deep learning (e.g. batch normalization). More importantly, results show that comparing to batch normalization; GN enables the model to be naturally transferrable from a pre-trained task to a new target dataset (Section 2.5). Details and results for GN can be found in Supplementary Material S3.
Fig. 1.
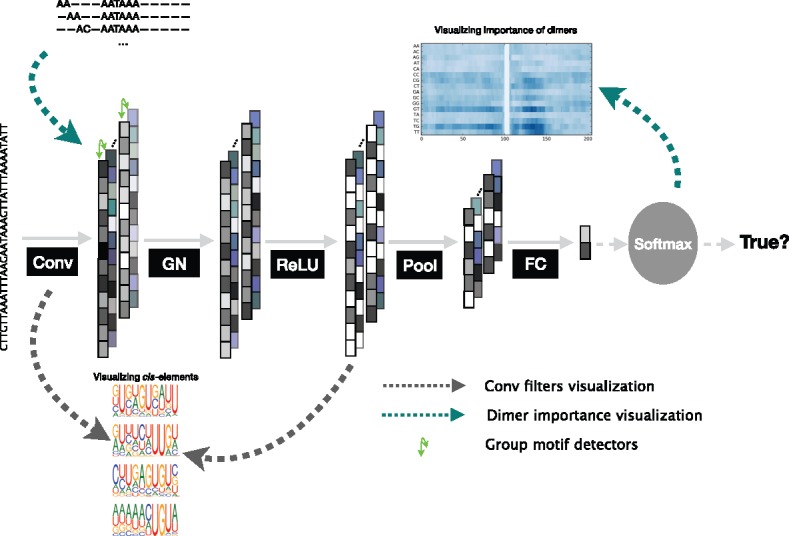
The architecture of the proposed DeeReCT-PolyA network. The output feature channels (shown as a column) of the conv layer is divided into groups (green arrows) and each group is jointly normalized by the group normalization layer. After tunable parameters are learned from the data, two visualization methods (shown as dashed lines in green and gray) are applied to the model without normalization to extract cis-elements and variants for the regulation of polyadenylation (Color version of this figure is available at Bioinformatics online.)
A rectified linear unit is applied to the normalized results as the activation function. After a max-pooling layer, all feature vectors are concatenated together and fed to the fully connected (FC) network followed by a softmax function which normalizes the prediction to values between 0 and 1. In fact, we have examined several other deep learning architectures (Supplementary Material S1) and finally chose the proposed one.
In addition to the proposed normalization layer, we faciliate the proposed CNN with several other techniques. Dropout is applied to the hidden neurons in the FC network (Srivastava et al., 2014) to alleviate overfitting. Namely, at each training iteration, some hidden neurons will be randomly set to 0 to force the model to make predictions with a subset of the parameters and the training will then yield a more robust model. Besides, our empirical experiment shows that dropout will also prevent the optimization of the model from being stuck at a local optimum. As for the initialization of the network, we find that the standard Xavier initializer (Glorot and Bengio, 2010) often leads to unsatisfactory results. Thus we propose to address this problem by sampling the initialized value of each parameter from a normal distribution with zero mean and a random variance. Then we do a random search on the validation dataset to get the best initialization variance for each layer. Our loss function comprises two terms: a cross-entropy loss of the prediction against true labels and a regularization term of the weights in the convolutional layer and FC layers, which is known as weight decay. The loss function is minimized by stochastic gradient descent with momentum. We also apply an exponential decay to the learning rate every 3000 iterations to make the training process more stable.
2.2 Cross validation and hyper-parameter search
To be consistent with previous studies, we use the standard 5-fold cross validation in all experiments we conduct. Specifically, we randomly partition the data into five equally sized folds and use three of them for training, one for validation and the remaining one for testing.
In the DeeReCT-PolyA model, there are several hyper-parameters we need to specify for the training process, including the number of groups in GN, the learning rate etc. Thus, the validation fold is used for hyper-parameter search. The search is implemented in a random sampling manner as Alipanahi et al. (2015) which randomly samples a set of hyper-parameters and tests its performance on the validation data. The one with the best accuracy on the validation fold is reserved as the final model and evaluated on the test set. The convolutional layer is set to have 16 filters with a length of 10. The number of hidden nodes in the FC network is searched within {32, 64, 128} and finally fixed to 64. We sample the number of groups from and the keep probability of dropout layer from . Details can be found in Supplementary Material S2 and Supplementary Table S1.
2.3 Evaluation metric
To measure the performance of our model, we adopt the classification error rate as the evaluation metric. The error rate is defined as
where TP, TN, FP and FN stands for the number of predictions that are true positive, true negative, false positive and false negative.
2.4 Datasets
We comprehensively evaluate our model on four different datasets, including two human PAS motif benchmarks. The first benchmark, Dragon human poly(A) dataset, is proposed in Kalkatawi et al. (2012), containing 14 740 sequences for the 12 main human PAS motif variants. Many previously proposed methods have used this dataset as a standard benchmark to carry out comparative analysis. The second benckmark (referred as the Omni human poly(A) dataset) is proposed very recently by Magana-Mora et al. (2017). Omni human poly(A) is a much larger dataset that consists of 18 786 positive true PAS sequences for 12 human PAS motif variants. An equal number of pseudo-PAS sequences are extracted from human Chromosome 21 after excluding all the true PAS sequences.
To assess the performance of our model beyond human, we apply it on two additional poly(A) datasets from C57BL/6J (BL) and SPRET/EiJ (SP) mouse strains, respectively. Overall, there are 46 224 genomic sequences in the BL mouse dataset and 40 230 sequences in the SP dataset. Both datasets consist of an equal number of true PAS and pseudo-PAS sequences. The positive sequences are generated based on the poly(A) sites identified in the previous study, which applies a well-established genome-wide experimental approach to capture the poly(A) sites (Xiao et al., 2016). In brief, the poly(A) containing RNAs, which are extracted from the fibroblast in C57BL/6J and SPRET/EiJ mouse, are fragmented and only the 3’-end is captured for high throughput sequencing. The 200 nt genomic sequences flanking the poly(A) sites are abstracted from the BL and SP genome, respectively. We obtain the pseudo-PAS sequences by scanning the genomic sequences of the transcripts expressed in the same cell lines and selecting those that are not close to any annotated transcription end in GENCODE or any poly(A) sites identified by the experimental data. The same number of pseudo-PAS sequences, which also contain the same PAS motifs, are then randomly retrieved. Note that for the mouse data, there are 13 potential PAS motif variants instead of 12 for the human poly(A) data.
2.5 Transfer pre-trained models to new datasets
Transfer learning has been widely used (Do and Ng, 2006; Li et al., 2018; Yosinski et al., 2014). The idea of transfer learning is that one can solve a problem with the knowledge gained by solving another similar problem. There are two major advantages of applying the transfer learning idea in the PAS recognition problem. First, integrating the information from two related datasets with transfer learning is likely to further boost the performance as we will show in Section 3.3. Most importantly, in many species there are only a limited amount of annotated poly(A) data (Ji et al., 2014), which poses great challenges to most machine-learning methods, especially the data hungry ones, such as deep learning. One possible solution is to transfer an available model learned from a larger dataset (e.g. human poly[A] data and mouse poly[A] data) to the other species, and then fine-tune the network with a small set of new data (Section 3.3).
To investigate the efficacy of pre-training and transfer learning in PAS identification, we adopt transfer learning in two different scenarios. In the first scenario, we incorporate a different dataset to increase the classification accuracy. Specifically, we transfer a model pre-trained on one dataset and then fine-tune it with the whole target dataset (cross-species or not). The fine-tuned model is then evaluated on the target dataset in comparison with baseline models which is trained only on target data without pre-training.
In the second scenario, we evaluate whether transfer learning can address the problem of insufficient training dataset, in which we are limited by the number of annotated PAS sequences in the target species. The experiment is conducted on the dataset of a new species, a rat poly(A) dataset proposed very recently by Wang et al. (2018). Models are pre-trained on human or mouse data and tested on the new rat dataset. We investigated scenarios where we have different numbers of training sequences to fine-tune the pre-trained model. We split one fifth of the rat dataset as the test dataset, on which all pre-trained and fine-tuned models are evaluated. Different numbers of rat sequences are sampled from the rest of the dataset for fine-tuning. The detailed procedure of transfer learning is presented in Supplementary Material S4.
2.6 Visualization of oligomer importance at different positions
A particular advantage of our model is that it can provide a characterization of critical features in the sequence context flanking the poly(A) motif. Following Xie et al. (2013), we investigate the importance of all possible 2 nt subsequences at different locations. As shown by the green arrows in Figure 1, the method we use is to construct a special sequence to feed the trained model and take the outputs for visualization (Supplementary Material S5). We have also visualized the importance of positions by examing where the convolutional filters are mostly ‘looking at’ (Supplementary Material S5 and Supplementary Fig. S2).
2.7 Convolutional filter visualization and similarity measurement
Convolutional filters, which directly scan through the whole genomic sequence, are believed to carry abundant information for polyadenylation. We generate sequence logos for the interpretation of such information. Specifically, for each particular filter of DeeReCT-PolyA, we derive a position frequency matrix (PFM) by inputting all genomic sequences in the validation dataset to that filter. The filter will assign a score to every fixed-length subsequence of the input sequence. The subsequence with the maximum score (if larger than 0) is considered as an instance of the cis-element captured by that filter from the input sequence. Instances captured by a filter for all validation data are aligned to generate a PFM and transformed into a sequence logo subsequently (Supplementary Material S5). We empirically find that only 11 filters are active in the model trained with the Dragon human data. This is probably due to the limited size of the dataset.
Furthermore, we quantify the similarity of the extracted cis-elements by measuring the correlation between convolutional layers in different models. This can be directly computed with the PFM of each filter. There are many works on measuring similarity of PFMs. We adopt the one proposed in Pape et al. (2008). Since each model has 16 filters, when comparing two models (e.g. A and B), for each filter in model A, we compute its similarity against all 16 filters in model B and take the maximum similarity score as the score for this filter. Then we average the score for each filter in A.
3 Results
We comprehensively evaluated the performance of DeeReCT-PolyA on the Dragon and Omni human datasets, and the SP and BL mouse datasets. We first showed that DeeReCT-PolyA outperforms the state-of-the-art PAS recognition methods on both Dragon and Omni human datasets (Section 3.1). We then evaluated the robustness of DeeReCT-PolyA by evaluating it on the SP and BL mouse datasets, challenging it to recognize unseen motifs by cross-motif validation and testing it on noisy regions in sequencing data (Section 3.2). Transfer learning across datasets and species was evaluated under two scenarios where we had sufficient and insufficient number of sequences for training in Section 3.3. Results showed that with transfer learning, DeeReCT-PolyA could achieve significantly better performance than models without pre-training when there are insufficient training data. The importance of dimers at different positions is measured in Section 3.4. We finally interpreted the deep learning model by constructing the sequence logos from the convolutional filters and measuring the similarities of the logos between human and mouse species (Section 3.5).
3.1 Performance comparison on human PAS prediction
We first evaluated our model, DeeReCT-PolyA, on two human poly(A) benchmarks. We reported the average error rates over the 5-fold cross-validation. As shown in Table 1, our model gives a significantly higher accuracy than previous state-of-art methods on the Dragon human poly(A) dataset (Kalkatawi et al., 2012) with a 2.86% improvement. Note that while RF, HSVM and Omni-PolyA each trained 12 PAS variant-specific models for the 12 PAS motif variants of human, DeeReCT-PolyA uses a single generic model that deals with all variants simultaneously which is much more challenging. Our results show that in spite of this, our variant-agnostic model still outperforms variant-specific models of Omni-PolyA on most PAS motif variants (11 out of 12).
Table 1.
Error rate comparison between RF, HSVM, Omni-PolyA and our model (DeeReCT-PolyA) on the Dragon human poly(A) data
| Variants | Size | Error Rate (%) |
||||
|---|---|---|---|---|---|---|
| RF | HSVM | Omni- PolyA | DeeReCT- PolyA | Rel | ||
| AATAAA | 5190 | 20.06 | 18.59 | 14.02 | 11.81 | 2.21 |
| ATTAAA | 2400 | 18.42 | 16.21 | 12.50 | 9.00 | 3.50 |
| AAAAAG | 1250 | 16.64 | 9.36 | 10.80 | 5.77 | 3.59 |
| AAGAAA | 1230 | 11.06 | 5.45 | 4.87 | 7.76 | −2.89 |
| TATAAA | 880 | 19.55 | 15.34 | 13.52 | 7.69 | 5.83 |
| AATACA | 780 | 19.36 | 11.15 | 13.85 | 10.45 | 0.70 |
| AGTAAA | 690 | 27.83 | 16.96 | 14.49 | 9.55 | 4.94 |
| ACTAAA | 670 | 22.09 | 14.33 | 13.13 | 10.72 | 2.41 |
| GATAAA | 460 | 20.00 | 9.57 | 8.48 | 8.04 | 0.44 |
| CATAAA | 410 | 18.54 | 9.27 | 13.41 | 9.02 | 0.25 |
| AATATA | 410 | 24.88 | 12.68 | 14.39 | 8.78 | 3.90 |
| AATAGA | 370 | 18.38 | 5.14 | 11.62 | 4.59 | 0.55 |
| Average | – | 19.19 | 14.42 | 12.43 | 9.57 | 2.86 |
Note: Rel denotes the improvement of DeeReCT-PolyA with respect to the best of the other three methods. Bold indicates the error rate of the best model for each PAS motif variant. Average is the weighted average of all motif variants with the size as weights. While results of all three previous methods are reported for 12 variant-specific models, the results of DeeReCT-PolyA are the performance of one single generic model that deals with all 12 variants.
Similar conclusions can be drawn from Table 2 in which we evaluated our model on the recent Omni human poly(A) dataset (Magana-Mora et al., 2017). Results for RF and HSVM were reported by Magana-Mora et al. (2017). Result shows that our generic model consistently performs better than variant-specific models over the majority of the PAS motif variants, which leads to a clear improvement for the average error rate. Although our model is agnostic to PAS motif variants, we still tried to investigate the differences between different PASs. Specifically we visualized if the model ‘looks for’ different cis-elements and ‘looks at’ different positions for different PAS motifs (Supplementary Material S6).
Table 2.
Error rate comparison between RF, HSVM, Omni-PolyA and our model (DeeReCT-PolyA) on the Omni human poly(A) data
| Variants | Size | Error Rate (%) |
||||
|---|---|---|---|---|---|---|
| RF | HMM | Omni- PolyA | DeeReCT- PolyA | Rel | ||
| AATAAA | 24310 | 25.49 | 27.91 | 23.96 | 21.99 | 1.97 |
| ATTAAA | 7098 | 25.59 | 33.48 | 24.20 | 23.01 | 1.09 |
| AAAAAG | 1640 | 26.52 | 36.83 | 25.86 | 27.76 | −1.90 |
| AAGAAA | 1306 | 26.67 | 34.77 | 23.07 | 26.80 | −3.73 |
| TATAAA | 682 | 30.88 | 38.38 | 26.91 | 23.60 | 3.31 |
| AATACA | 634 | 24.41 | 36.98 | 22.06 | 22.00 | 0.06 |
| AGTAAA | 528 | 28.11 | 37.31 | 23.26 | 20.21 | 3.05 |
| ACTAAA | 368 | 32.97 | 33.89 | 24.72 | 25.79 | −1.07 |
| GATAAA | 342 | 31.18 | 41.76 | 29.41 | 22.15 | 7.26 |
| CATAAA | 314 | 28.89 | 39.03 | 24.51 | 25.54 | −1.03 |
| AATATA | 250 | 31.60 | 36.00 | 26.80 | 17.82 | 8.98 |
| AATAGA | 100 | 34.00 | 40.00 | 23.00 | 20.00 | 3.00 |
| Average | – | 25.93 | 30.43 | 24.15 | 22.64 | 1.51 |
Note: Rel denotes the improvement of DeeReCT-PolyA with respect to the best of the other three methods. Bold indicates the error rate of the best model for each PAS motif variant. Average is the weighted average of all motif variants with the size as weights.
3.2 Robustness of DeeReCT-PolyA to PAS variants and different species
To demonstrate the robustness of our model, we validated DeeReCT-PolyA on two mouse poly(A) datasets, i.e. SP and BL (Section 2.4), which cannot be handled by conventional human-feature-based methods. Table 3 shows that our model achieved an error rate of 24.11 and 23.49% on SP and BL mouse data, respectively. While there is no existing method for poly(A) identification of SP and BL data, for the purpose of comparison we trained a linear regression (LR) model. The LR model yielded an average error rate of 27.26 and 26.98% on SP and BL mouse data, which is significantly worse than DeeReCT-PolyA. We have also tried adding some hand-crafted features to our DeeReCT-PolyA model; however, the improvement is marginal. Detailed results can be found in Supplementary Material S7.
Table 3.
Error rate of DeeReCT-PolyA on SP and BL mouse poly(A) data
| Variants | SP |
BL |
||
|---|---|---|---|---|
| Size | Error Rate (%) | Size | Error Rate (%) | |
| AATAAA | 17 708 | 26.50 | 20 250 | 25.48 |
| ATTAAA | 7550 | 25.30 | 9056 | 24.89 |
| TTTAAA | 2336 | 19.95 | 2688 | 18.19 |
| TATAAA | 2178 | 22.91 | 2518 | 22.44 |
| AGTAAA | 2224 | 22.88 | 2376 | 21.63 |
| CATAAA | 1432 | 20.53 | 1760 | 19.77 |
| AATATA | 1334 | 23.55 | 1528 | 23.23 |
| AATACA | 1210 | 21.40 | 1326 | 22.55 |
| GATAAA | 1032 | 17.84 | 1176 | 18.54 |
| AAGAAA | 1022 | 15.07 | 1126 | 15.81 |
| AATGAA | 982 | 18.84 | 1108 | 18.86 |
| ACTAAA | 728 | 19.37 | 776 | 20.24 |
| AATAGA | 494 | 18.64 | 536 | 21.24 |
| Average | – | 24.11 | – | 23.49 |
Although results from four benchmarks show that our model can generalize over PAS motif variants, we want to ask a further question: is the model able to recognize a PAS motif that it has never seen before? If so, then it would be an interesting idea to use DeeReCT-PolyA to process non-annotated DNA sequences which may help us find PASs that are currently unknown. We answered this question by evaluating our model with a leave-one-motif-out validation on the Dragon human data. Specifically, we trained a DeeReCT-PolyA model with human poly(A) data that contains only 11 types of PAS variants. All data for the remaining left-out variant, which our model did not see during training, was used as the test set. For each PAS motif variant, Table 4 reports the prediction error rate for two models evaluated on the data of this variant: one was trained with the regular 5-fold cross-validation (Section 3.1) and the other model was trained with data of all PAS motif variants excluding this held-out variant. The results show that our model works surprisingly well even on PAS motif variants that were not included in the training dataset. Notably, for most PAS motif variants (8 out of 12), the model trained with leave-one-motif-out data surpasses the model in 5-fold cross validation. The reason is that although the network could not see any sequence containing the test motif, it did see more training examples because all the data from the remaining 11 variants were included in the training. As an example, when testing the motif AATACA, the leave-one-motif-out model was trained with 14 740 – 780 = 13 960 sequences while the 5-fold cross-validation model only had 14 740 × 4/5 = 11 792 examples for training. The similarity of these twelve leave-one-motif-out models is visualized in Supplementary Material S5 and Supplementary Figure S3.
Table 4.
DeeReCT-PolyA with leave-one-motif-out test on the Dragon human dataset
| Variants | Size | Error Rate (%) |
|
|---|---|---|---|
| 5-fold cross-validation | leave-one-motif-out | ||
| AATAAA | 5190 | 11.81 | 14.20 |
| ATTAAA | 2400 | 9.00 | 8.17 |
| AAAAAG | 1250 | 5.77 | 5.45 |
| AAGAAA | 1230 | 7.76 | 7.52 |
| TATAAA | 880 | 7.69 | 7.18 |
| AATACA | 780 | 10.45 | 8.86 |
| AGTAAA | 690 | 9.55 | 7.46 |
| ACTAAA | 670 | 10.72 | 11.16 |
| GATAAA | 460 | 8.04 | 8.04 |
| CATAAA | 410 | 9.02 | 11.46 |
| AATATA | 410 | 8.78 | 8.54 |
| AATAGA | 370 | 4.59 | 4.05 |
| Average | – | 9.57 | 10.08 |
Note: For the leave-one-motif-out test, for each PAS variant, a DeeReCT-PolyA model was trained with data of all the other motif variants and then test only on this variant. Bold indicates the error rate of best model for each PAS variant.
We further investigated if the proposed model is robust to noise by introducing mismatches in the whole sequence or certain sub-regions. Results show that in general DeeReCT-PolyA is robust to mismatches as even if there are 40 mismatches in the whole sequence; DeeReCT-PolyA still only has an around 18% error rate on the Dragon human data (Supplementary Material S8).
3.3 Transfer learning for PAS recognition
To validate the idea of transfer learning in the PAS recognition problem, we tested its performance in two cases in which we have sufficient and insufficient target training sequences. In the first case, we evaluated transfer learning on three similarly sized datasets, Omni human and the SP and BL mouse dataset. Dragon human dataset was not considered in this scenario since its size is relatively small. Table 5 shows the performance of transferred models evaluated on SP dataset. We first pre-train the model on Omni human or BL mouse dataset and then fine-tune it with all SP training data. Note that the model before fine-tuning has not seen any SP data which the model is evaluated on. Yet, even without fine-tuning the transferred model still gets a comparative result comparing to the baseline model, which is trained only on the target dataset with no pre-training, especially when we transfer the model between close species. After fine-tuning, the transferred model yields superior performance than the model without pre-training. Similar conclusions can also be drawn from Tables 6 and 7 where we transfer models to BL data and Omni human data, respectively. The consistent results in these tables illustrate that while we already have sufficient data for training, transferring the pre-trained model from a relative dataset can further improve the prediction accuracy, even when the transferring is cross-species (human to mouse or mouse to human).
Table 5.
Evaluation of transferred DeeReCT-PolyA models on SP mouse poly(A) data before and after fine-tuning
| Average Error Rate (%) |
|||
|---|---|---|---|
| Pre-trained on | None | Omni | BL |
| Before fine-tuning | – | 30.23 | 23.67 |
| After fine-tuning | 24.11 | 24.04 | 22.57 |
Note: None denotes a model of no pre-training and trained with SP mouse data. Models respectively pre-trained on Omni and BL dataset are evaluated on SP mouse dataset before and after fine-tuning with SP data. Average error rate over all PAS motif variants is reported.
Table 6.
Evaluation of transferred DeeReCT-PolyA models on BL mouse poly(A) data before and after fine-tuning
| Average Error Rate (%) |
|||
|---|---|---|---|
| Pre-trained on | None | Omni | SP |
| Before fine-tuning | – | 29.75 | 23.13 |
| After fine-tuning | 23.49 | 23.38 | 22.08 |
Table 7.
Evaluation of transferred DeeReCT-PolyA models on Omni human poly(A) data before and after fine-tuning
| Average Error Rate (%) |
|||
|---|---|---|---|
| Pre-trained on | None | SP | BL |
| Before fine-tuning | – | 29.58 | 29.07 |
| After fine-tuning | 22.64 | 22.40 | 22.44 |
In the second case, we addressed the problem of insufficient training data by transferring a pre-trained model to a new species. Specifically, models are pre-trained on human or mouse data and tested on the new rat dataset. We investigated scenarios where we have different numbers of rat sequences to fine-tune the pre-trained model. The baseline is a model without any pre-training. Results (Table 8) show that without pre-training, CNN fails when there are insufficient training data. However, by pre-training on a different species, even if the pre-trained model itself (e.g. model pre-trained on the Dragon human data) is not suitable for prediction on the new dataset, after fine-tuning with a very small number of sequences the model is able to achieve much better performance. As expected, as the number of training data increases, the error rate of fine-tuned models becomes smaller.
Table 8.
Transfer learning for insufficient amount of sequences in the rat poly(A) dataset
|
Average Error Rate (%)
|
|||||
|---|---|---|---|---|---|
| Pre-trained on | None | Dragon | Omni | SP | BL |
| n = 0 | – | 40.55 | 29.30 | 22.11 | 22.40 |
| n = 100 | 50.00±0.00 | 39.32±1.84 | 28.94±0.31 | 22.65±0.74 | 22.27±0.12 |
| n = 500 | 48.90±1.47 | 29.72±3.79 | 25.61±0.36 | 22.63±0.37 | 22.22±0.22 |
| n = 1000 | 49.71±0.76 | 26.44±0.68 | 24.77±0.22 | 22.10±0.16 | 22.03±0.18 |
| n = 2000 | 49.06±1.40 | 25.26±0.54 | 24.35±0.23 | 22.04±0.20 | 22.43±0.33 |
| n = 5000 | 26.88±8.74 | 24.25±0.22 | 23.65±0.16 | 21.91±0.21 | 21.90±0.25 |
| n = 10 000 | 22.63±0.16 | 23.13±0.36 | 22.68±0.08 | 21.67±0.34 | 21.40±0.18 |
| n = 42 233 | 20.23 | 20.48 | 20.38 | 19.82 | 19.99 |
Note: n denotes the number of rat sequences used for fine-tuning. For every n except 0 and 42 233 (the total size of rat training data), n sequences are randomly sampled from the rat training dataset and used to fine-tune the pre-trained model. Such step is repeated 10 times for every n. The table shows the average error rate of these 10 repeats with the standard deviation on the rat test set. None indicates a model without any pre-training.
We also observed that if the model is pre-trained on a less similar species, e.g. pre-trained on human data and tested on rats, fine-tuning is crucial as it can adapt the pre-trained model to the new data even when we only have very limited data for fine-tuning. While for similar species, i.e. mice to rats, the pre-trained model is already quite good. Note that here we did not perform any hyper-parameter (such as learning rate, dropout rate) search for fine-tuning because of insufficient data, instead we just used the best set of hyper-parameters found during pre-training. In general, the results not only demonstrate that our proposed DeeReCT-PolyA model can generalize across species, but also offer us an effective method for solving the issue of the shortage of PAS annotation in some species.
3.4 Visualizing importance of dimers and positions
One advantage of our method is it can extract important patterns of polyadenylation by learning from data. Thus it provides us a direct way to understand the underlying regulatory elements of polyadenylation through the visualization of the trained model.
Figure 2 presents the importance of all 16 dimers at different positions surrounding the PAS motif. Several interesting patterns can be observed here. For the Dragon human poly(A) dataset, dimer AA is found to be an informative subsequence in Xie et al. (2013) within 30 nt downstream of the candidate PAS motif. The same observation was also obtained by our deep learning model. Another finding is that when GT or TG appears in the downstream region of the PAS motif, it strongly suggests a true PAS motif. This finding coincides with Hu et al. (2005), where their result shows that there are many GU-rich elements in the downside of poly(A) sites.
Fig. 2.

Visualization of the importance of different dimers at different positions for models trained with four datasets. The colors denote the contribution of the dimer at that position to determining a true PAS motif. The darker blue, the more contribution the dimer at that position has to determining a true PAS motif. The more white, the less contribution. The x-axis shows the positions of the dimer in the sequence, where Position 0 is the first base of the PAS motif. The y-axis lists all possible dimers
On the other hand, our model also suggested some novel patterns that have not been observed before. Dimer CT, as an interesting subsequence, is very informative when occurring 20–nt downstream the PAS especially for the BL mouse data. And in general, the downstream region contains more information than the upstream one which makes sense since the cleavage site usually resides 10–30 nt downstream the PAS motif. More importantly, it is clearly shown in Figure 2 that human, BL mouse and SP mouse share many similar poly(A) patterns. The importance of positions can be found in Supplementary Figure S2. We have also visualized the importance of some known motifs, i.e. RNA-binding protein motifs (Supplementary Material S5 and Supplementary Fig. S1).
3.5 Interpreting convolutional filters and measuring similarities between human and mouse
Deep learning models are often criticized to be ‘black-box’ models, which lack interpretability. Here we tried to overcome this bottleneck by interpreting the learned convolutional filters as cis-elements and visualizing them as sequence logos (Section 2.7). We presented sequence logos for each filter of the four DeeReCT-PolyA models trained with different datasets in Figure 3.
Fig. 3.

Sequence logos of cis-elements identified by each convolutional filter in four DeeReCT-PolyA models trained with different datasets. Thymine (T) is replaced by uracil (U) for the purpose of comparison with previous works on statistics of PASs in mRNA sequences. Many subsequences, such as U/GU-rich elements, UC elements and UGUA, are shown to have great influences on polyadenylation in both human and mouse
As shown in Figure 3, our model reveals many cis-elements, including some subsequences that have been found and validated before. Evidently, U-rich elements are essential for PAS motif recognition for both human and mouse, which is in line with many previous studies. Other elements, like GU-rich elements which are shown to have great importance in Figure 2 and Hu et al. (2005), can also be found in the sequence logos. Specifically, the UGUA element, which is implicated by Hu et al. (2005) and Venkataraman et al. (2005) in human poly(A) site recognition, is also shown to be an informative cis-element in both human and mouse datasets by our model. Our results also indicate some novel elements that have not been reported before, such as the UC and AAUA elements.
Our results in Figure 3 clearly demonstrate that there are high similarities among the cis-elements in human, BL mouse and SP mouse. To quantitatively measure the similarity of patterns extracted from different datasets, we adopted the measurement method proposed in Pape et al. (2008). For the purpose of baseline comparison, we generated two additional sets of sequence logos with two randomly initialized deep neural networks. Similarity scores are reported in Table 9. As expected, the similarity of human and mouse poly(A) cis-elements are much higher than that of the randomly initialized models. In addition, the similarities of cis-elements between the human datasets and between the mouse datasets are often higher than that between human and mouse datasets. Interestingly, the similarity between the sequence logos of the Omni human model and that of the BL mouse model is higher than that between the Omni human model and the SP model, which is consistent with the visual similarity between the 2-mer importance distributions of the Omni human and the BL mouse (Fig. 2).
Table 9.
Similarity of sequence logos generated for different models
| Models | Dragon-vs-Omni | Dragon-vs-SP | Dragon-vs-BL | Omni-vs-SP | Omni-vs-BL | SP-vs-BL | random1-vs-random2 |
|---|---|---|---|---|---|---|---|
| Similarity (×10–3) | 1.15 | 1.03 | 1.02 | 1.07 | 1.24 | 1.40 | 0.36 |
Note: random1 and random2 denote two randomly initialized CNNs.
4 Discussions and conclusions
Here we highlight the main differences of our method to a recent work (Leung et al., 2018) which also makes use of deep neural networks for human poly(A) code inference. The problem they tried to solve is also a binary classification problem, but a different one, i.e. to determine which of the two true poly(A) sites has a higher usage. Additional supervision is required for training their model as it is trained on sequences containing true poly(A) sites and the strength of each site, while our model is trained with only the sequences. They also did an experiment for poly(A) site discovery, but the discovery task is indeed a much simpler problem than the PAS recognition problem studied in this paper, because in the former, to identify a false sequence one often only needs to search if there is a PAS hexamer (e.g. AATAAA) upstream the center of the sequence. In addition, their study focused on human data only, whereas we propose a robust PAS recognition method across different motifs and different species.
In this study, we proposed a deep learning model, DeeReCT-PolyA, along with a novel filter-GN method for automatic feature extraction and PAS recognition that is applicable to multiple species. The proposed method is generic, PAS variant agnostic and outperforms the state-of-art variant-specific methods on two standard human benchmarks.
We obtained two additional poly(A) datasets for BL and SP mouse and showed that DeeReCT-PolyA can consistently achieve high accuracy across species. Furthermore, our results demonstrate that transfer learning can further improve the PAS recognition accuracy on each individual dataset. In particular, by transferring a pre-trained model, DeeReCT-PolyA can outperform the previous state-of-the-art method while using much less training data in the target dataset, which also provides us a way to address the problem of insufficient data in many species.
Visualization methods were applied to DeeReCT-PolyA which revealed some interesting features including several novel cis-elements. We visualize the convolutional filters as sequence logos and quantitatively measure the similarity of trained models to show that human, BL mouse and SP mouse share a number of similar features in polyadenylation.
Supplementary Material
Acknowledgements
We would like to thank Jeffery Jung and Min Zhang for insightful discussion.
Funding
This work was supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under [Awards No. FCC/1/1976-04, URF/1/2602-01, URF/1/3007-01, URF/1/3412-01, URF/1/3450-01 and URF/1/3454-01.] Y.H. was supported by the International Cooperation Research Grant [No. GJHZ20170310161947503] from Science and Technology Innovation Commission of Shenzhen Municipal Government. W.C. was supported by Basic Research Grant [JCYJ20170307105752508] from Science and Technology Innovation Commission of Shenzhen Municipal Government.
Conflict of Interest: none declared.
References
- Akhtar M.N. et al. (2010) Polyar, a new computer program for prediction of poly (a) sites in human sequences. BMC Genomics, 11, 646.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831.. [DOI] [PubMed] [Google Scholar]
- Cheng Y. et al. (2006) Prediction of mrna polyadenylation sites by support vector machine. Bioinformatics, 22, 2320–2325. [DOI] [PubMed] [Google Scholar]
- Do C.B., Ng A.Y. (2006) Transfer learning for text classification. In: Weiss Y.et al (eds) Advances in Neural Information Processing Systems 18, MIT Press, pp. 299–306. [Google Scholar]
- Glorot X., Bengio Y. (2010). Understanding the difficulty of training deep feedforward neural networks. In: Teh Y.W., Titterington M. (eds) Proceedings of the Thirteenth International Conference on Artificial Intelligence and statistics, PMLR, pp. 249–256.
- Helden J.V. et al. (2000) Statistical analysis of yeast genomic downstream sequences reveals putative polyadenylation signals. Nucleic Acids Res., 28, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. et al. (2005) Bioinformatic identification of candidate cis-regulatory elements involved in human mrna polyadenylation. RNA, 11, 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G. et al. (2015) Genome-wide identification and predictive modeling of polyadenylation sites in eukaryotes. Brief. Bioinform., 16, 304–313. [DOI] [PubMed] [Google Scholar]
- Kalkatawi M. et al. (2012) Dragon polya spotter: predictor of poly (a) motifs within human genomic DNA sequences. Bioinformatics, 28, 127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M.K.K. et al. (2018) Inference of the human polyadenylation code. Bioinformatics, 34, 2889–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2018) Deepre: sequence-based enzyme ec number prediction by deep learning. Bioinformatics, 34, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. et al. (2012) An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res., 40, 8460–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2003) An in-silico method for prediction of polyadenylation signals in human sequences. Genome Inform., 14, 84–93. [PubMed] [Google Scholar]
- Magana-Mora A. et al. (2017) Omni-PolyA: a method and tool for accurate recognition of Poly (A) signals in human genomic DNA. BMC Genomics, 18, 620.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matis S. et al. (1996) Detection of RNA polymerase II promoters and polyadenylation sites in human DNA sequence. Comput. Chem., 20, 135–140. [DOI] [PubMed] [Google Scholar]
- Pape U.J. et al. (2008) Natural similarity measures between position frequency matrices with an application to clustering. Bioinformatics, 24, 350–357. [DOI] [PubMed] [Google Scholar]
- Pastrello C. et al. (2006) Stability of bat26 in tumours of hereditary nonpolyposis colorectal cancer patients with msh2 intragenic deletion. Eur. J. Hum. Genet., 14, 63.. [DOI] [PubMed] [Google Scholar]
- Proudfoot N. (1991) Poly (a) signals. Cell, 64, 671–674. [DOI] [PubMed] [Google Scholar]
- Proudfoot N.J. (2011) Ending the message: poly (a) signals then and now. Genes Dev., 25, 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamov A.A., Solovyev V.V. (1997) Recognition of 3’-processing sites of human mrna precursors. Bioinformatics, 13, 23–28. [DOI] [PubMed] [Google Scholar]
- Shaw G., Kamen R. (1986) A conserved AU sequence from the 3’ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell, 46, 659–667. [DOI] [PubMed] [Google Scholar]
- Shen Y. et al. (2008a) Genome level analysis of rice mRNA 3’-end processing signals and alternative polyadenylation. Nucleic Acids Res., 36, 3150–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. et al. (2008b) Unique features of nuclear mRNA Poly (A) signals and alternative polyadenylation in chlamydomonas reinhardtii. Genetics, 179, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N. et al. (2014) Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res., 15, 1929–1958. [Google Scholar]
- Tabaska J.E., Zhang M.Q. (1999) Detection of polyadenylation signals in human dna sequences. Gene, 231, 77–86. [DOI] [PubMed] [Google Scholar]
- Tian B. et al. (2005) A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res., 33, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K. et al. (2005) Analysis of a noncanonical Poly (A) site reveals a tripartite mechanism for vertebrate Poly (A) site recognition. Genes Dev., 19, 1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2018) A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res., 28, 1427–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M.-S. et al. (2016) Global analysis of regulatory divergence in the evolution of mouse alternative polyadenylation. Mol. Syst. Biol., 12, 890.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B. et al. (2013) Poly (A) motif prediction using spectral latent features from human dna sequences. Bioinformatics, 29, i316–i325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosinski J. et al. (2014). How transferable are features in deep neural networks? In: Ghahramani Z.et al (eds) Advances in Neural Information Processing Systems 27, Curran Associates, Inc., pp. 3320–3328.
- Zarudnaya M.I. et al. (2003) Downstream elements of mammalian pre-mrna polyadenylation signals: primary, secondary and higher-order structures. Nucleic Acids Res., 31, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.