

Abstract
Motivation
Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) sequencing technologies can produce long-reads up to tens of kilobases, but with high error rates. In order to reduce sequencing error, Rolling Circle Amplification (RCA) has been used to improve library preparation by amplifying circularized template molecules. Linear products of the RCA contain multiple tandem copies of the template molecule. By integrating additional in silico processing steps, these tandem sequences can be collapsed into a consensus sequence with a higher accuracy than the original raw reads. Existing pipelines using alignment-based methods to discover the tandem repeat patterns from the long-reads are either inefficient or lack sensitivity.
Results
We present a novel tandem repeat detection and consensus calling tool, TideHunter, to efficiently discover tandem repeat patterns and generate high-quality consensus sequences from amplified tandemly repeated long-read sequencing data. TideHunter works with noisy long-reads (PacBio and ONT) at error rates of up to 20% and does not have any limitation of the maximal repeat pattern size. We benchmarked TideHunter using simulated and real datasets with varying error rates and repeat pattern sizes. TideHunter is tens of times faster than state-of-the-art methods and has a higher sensitivity and accuracy.
Availability and implementation
TideHunter is written in C, it is open source and is available at https://github.com/yangao07/TideHunter
1 Introduction
While Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) long-read sequencing technologies are capable of providing improved reference genomes, comprehensive structural variant identification, as well as a more complete view of transcriptomes, the relatively high error rates prevent their widespread adoption (Goodwin et al., 2016).
Several error correction methods have been developed to reduce the sequencing error in the long-reads. Such approaches can be classified into two types: hybrid correction using short-reads (Koren et al., 2012; Zimin et al., 2013; Salmela and Rivals, 2014; Goodwin et al., 2015) and self or non-hybrid correction using only long-reads (Chin et al., 2013, 2016; Salmela et al., 2017). Despite these methods providing better base-pair accuracy than the raw data, each method has drawbacks. Hybrid correction approaches could introduce systematic errors from the short-reads into the long-reads while the performance of non-hybrid correction relies heavily on the sequencing depth.
PacBio platforms generate high-accuracy circular consensus (CCS) reads from raw subreads through in silico processing (Weirather et al., 2017), with an error rate as low as 1%. However, the yield of CCS reads is much lower than subreads. ONT uses a similar strategy to call a relatively accurate consensus sequence, i.e. 2D or 1D2 reads, from the template and complement of 1D reads, but at the cost of lower throughput (de Lannoy et al., 2017).
In recent studies (Li et al., 2016; Volden et al., 2018; Calus et al., 2018), Rolling Circle Amplification (RCA) was used to amplify circularized template molecules in order to generate linear products containing multiple tandem copies of the templates. After long-read sequencing with PacBio or ONT, the resulting sequences can be used to generate accurate consensus sequences through additional computational processing steps, which can be considered as high-quality reconstructed reads of the templates.
Along with the RCA workflow, computational pipelines for amplified tandemly repeated sequences have been developed. INC-seq (Li et al., 2016) extracts subsequences from the raw long-reads using non-overlapping sliding windows. These subsequences serve as anchors which are then aligned back to the read. Anchors with the higher number of alignments are used to partition the reads into multiple segments, which are then used to construct a consensus sequence with pbdagcon (Chin et al., 2013). C3POa (Volden et al., 2018) uses a similar alignment-based strategy. Instead of extracting subsequences, C3POa uses a known splint sequence to determine a start point, then performs a self-to-self alignment to discover the tandem repeat signal embedded in the raw reads. For INC-seq, the choice of anchor length is non-trivial and has a significant impact on the result. For C3POa, self-to-self alignment may confuse the determination of repeat sizes, especially on data with a high error rate. Moreover, as exhaustive alignment-based methods, both methods are time consuming.
Tandem repeat detection in DNA sequences is a classical bioinformatics problem that motivated the development of numerous tools over the past 20 years (Lim et al., 2013). However, most of these tools are not suitable for processing RCA-based long-read data, as they only focus on short tandem repeats whose period size is generally less than 100 bp. Tandem Repeats Finder (TRF) (Benson et al., 1999) is one of the most widely used and robust tandem repeat detection tools. It calculates the possible repeat pattern size by short k-mer matches at adjacent locations on the sequence and uses statistically based recognition criteria to find candidate tandem repeats. TRF allows for the detection of imperfect repeats, which makes it suitable for noisy long-read data. The most significant issue for TRF is that it limits the maximal period size as 2000 bp. For whole genome or transcriptome studies, the length of the template molecule could easily exceed this limitation. With the increasing use of long-read sequencing technologies, we expect the new RCA-based protocol to be widely adopted. Thus, it is crucial to develop an efficient and sensitive tandem repeat detection and consensus calling tool to take full advantage of this type of data.
In this article, we present a novel tandem repeat detection and consensus calling tool, TideHunter, which is specifically designed for RCA-based long-read data. TideHunter uses a fast seed-and-chain algorithm to efficiently recognize the underlying repeat pattern size, and then partition the original long-read into multiple repeat units. High-quality consensus sequences are generated using a Single Instruction Multiple Data (SIMD) accelerated Partial Order Alignment (POA) (Lee et al., 2002) on the partitioned segments. TideHunter does not have any limitation of the maximal repeat pattern size and is able to tolerate high error rates. We benchmarked TideHunter using simulated and real datasets with varying error rates and repeat pattern lengths. TideHunter is tens of times faster than state-of-the-art methods and has a higher sensitivity and accuracy.
2 Materials and Methods
2.1 Overview
The reconstruction of template sequences from RCA-based long-reads has two main steps: tandem repeat detection and consensus calling. During the tandem repeat detection, both the repeat unit length and the copy number need to be determined in an ab initio manner. Furthermore, all the tandem copies could be divergent from each other due to the high error rate of long-reads, making it more difficult to discover the repeat signal. After tandem repeat detection, every detected repeat unit is expected to represent one copy of the template sequence. Consensus calling can be accomplished based on the multiple sequence alignment of all detected repeat units. The generated consensus sequence is considered a reconstructed template sequence with a lower error rate than the original long-read. Moreover, for some specific sequencing libraries, additional adapter information is available and can be utilized to convert the consensus to a full-length template sequence (Volden et al., 2018).
TideHunter is inspired by several existing tandem repeat detection tools (Benson et al., 1999; Pellegrini et al., 2010) and noisy long-read alignment approaches (Liu et al., 2017; Li, 2018). It adopts a specifically designed seed-and-chain algorithm to efficiently recognize the underlying repeat pattern size and implements a SIMD accelerated POA to generate high-quality consensus sequences. TideHunter collects seeds of long-reads which consist of hash values and locations of short substrings (k-mers). Collected seeds are sorted by both the hash value and the location, then stored in a linear table. Tandem repeat hit is identified for each pair of seeds that have identical hash values and are adjacent to each other in the sorted table. The hit distance, i.e. the location distance of two seeds having a tandem repeat hit, is usually close to the true repeat pattern size or its multiples. TideHunter considers all such hits as anchors and attempts to find an optimal chain of colinear anchors using dynamic programming. The optimal chain is expected to consist of anchors that have a hit distance close to the repeat pattern size. TideHunter partitions the original long-read into multiple segments based on the optimal chain. A SIMD accelerated POA of these segments is then applied to generate an accurate consensus sequence.
2.2 Collecting seeds
TideHunter takes a two-tuple (v, c) as a seed to represent every k-mer of a read, where v is the hash value and c is the coordinate of the last base for each k-mer. Seeds are collected based on two parameters: k and s, where k is the k-mer length and s is the collecting step size. By default, we have k = 8 and s = 1, meaning we exhaustively collect all the short substrings of 8 bp from the read. A hash value is assigned to each k-mer with a simple hash function:
where . This hash function enables TideHunter to avoid collision as distinct k-mers always have different hash values. After collecting all the seeds, TideHunter sorts them by both the hash value and the coordinate using radix sorting. Sorted seeds are then stored in a linear table for further use.
2.3 Identifying tandem repeat anchors
A tandem repeat hit is a match of two identical k-mers which are adjacent to each other in the seeds linear table. In general, n identical k-mers will result in n − 1 tandem repeat hits.
TideHunter takes another set of two-tuples (e, d) as anchors to represent all the tandem repeat hits, where e is the ending position, which is the coordinate of the last base in the following k-mer; and d is the hit distance, which is the coordinate distance between the two identical k-mers. All the tandem repeat anchors are sorted by the ending position e using radix sorting again.
It is worth noting that only tandem repeat hits within a specific range of hit distance (default: 30–100 000) will be collected by TideHunter. Specifically, for each seed TideHunter attempts to find its first valid hit in a loop starting with the nearest identical k-mer. By doing this, TideHunter avoids meaningless hits which are unlikely to have a distance close to the true repeat pattern size.
2.4 Chaining of tandem repeat anchors
2.4.1 Chaining
Among all tandem repeat anchors with different hit distances, TideHunter attempts to find an optimal chain of anchors all having a hit distance close to the true repeat pattern size (Fig. 1).
Fig. 1.
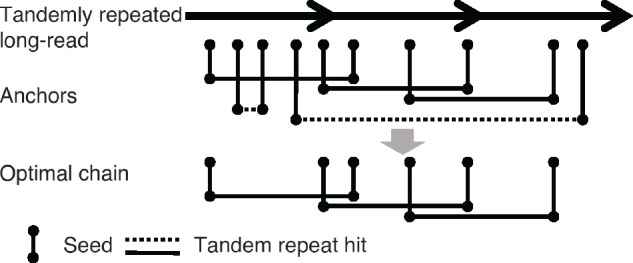
Chaining of tandem repeat anchors. Three arrows represent three copies of a template sequence. Vertical line represents seed for each k-mer. The same height between seeds indicates identical k-mers. Horizontal line represents tandem repeat hit of identical k-mers. Solid and dashed lines indicate their hit distances are likely and unlikely, respectively, to be the true repeat pattern size. After the dynamic programming, the optimal chain is expected to consist of anchors that have hit distances close to the repeat pattern size
Given a list of anchors sorted by the hit ending position, TideHunter identifies the optimal chain by performing dynamic programming with a specifically designed scoring function.
Here, we use S(i) to denote the maximal chaining score up to the ith anchor in the list. The recurrence equation of the scoring function is:
where is the number of additional matching bases when anchor i and j are chained together. Here, . is the chaining cost for any two anchors:
here, . Moreover, an initial score, i.e. the number of bases in the k-mers: , is directly assigned to the anchor that does not have any precursors. By assigning positive infinity to the chaining cost, TideHunter avoids chaining any two non-collinear anchors, i.e. and ei > ej. The optimal precursor of each anchor is chosen based on the maximal chaining score during the calculation of chaining scores.
The cost function consists of two parts: the square of two anchors’ hit distance difference and the logarithm of the distance between two anchors’ endpoints. By using this cost function, TideHunter tends to chain together two anchors that have a similar hit distance and are close to each other as their chaining cost is low (Fig. 1). As such, all the anchors coming from any two consecutive copies in the long-read are likely to be chained together to form the optimal chain. The quadratic and logarithmic functions are chosen to make sure the hit distance difference weighs more than the anchor distance during the precursor determination. Theoretically, other functions could also be applied. However, on our simulated and real datasets, the quadratic and logarithmic functions achieve the best performance in practice.
2.4.2 Backtracking
To obtain an optimal chain, TideHunter starts with the anchor having the maximal chaining score, and then recursively performs backtracking to find the best precursor for each anchor. The best precursor is determined based on the chaining score during the dynamic programming. All tandem repeat anchors in the optimal chain are expected to have a hit distance that is close to the true repeat pattern size.
In most cases, the optimal chain will cover almost the whole read. However, as long-read sequencing may go through abnormal molecular ligation and template switching (Li et al., 2016), chimeric tandem repeats can potentially exist. To solve this issue, TideHunter performs additional backtracking beginning with the remaining maximal score anchor, which has not been included in any existing chains. The backtracking stops whenever the precursor has already been tracked to ensure every anchor only shows up in one chain.
For a set of obtained chains, TideHunter discards a chain when it overlaps with any other higher score chains by at least 50%. Thus, TideHunter is able to collect a set of local optimal chains for all the chimeric regions of the read.
2.5 Partitioning read
2.5.1 Selecting a medoid anchor
Given a chain of tandem repeat anchors, , TideHunter selects a medoid anchor M by calculating the summation of the distance between one anchor and all other anchors. Here, the anchor distance is defined as:
The medoid anchor is selected by:
If multiple optimal medoids exist, TideHunter arbitrarily chooses the anchor with the smallest coordinate.
2.5.2 Determining repeat unit boundary
TideHunter uses the selected medoid anchor as a starting point to repeatedly determine the boundaries for all repeat units. Figure 2 provides an example of searching for the next repeat unit boundary on the right side.
Fig. 2.

Searching for repeat unit boundary based on the global alignment. s and e are the current repeat boundaries. Two anchors and are selected as their starting positions are the closest to e. Two subsequences starting from s1 to s2 and from e1 to e2 are extracted to perform an end-to-end global alignment. The next repeat unit boundary can be calculated based on the alignment result. In this example, the base G of e is matched with the G in subsequence , whose coordinate is then considered as the putative next boundary
For each tandem repeat anchor, let the starting position s and ending position e be the two coordinates of the last base in two k-mers (Fig. 2). Given a pair of current repeat unit boundaries s and e, TideHunter first searches for two anchors and around e, where s1 and s2 are the closest to e and . Specifically, when s2 is equal to e, the next repeat unit boundary is directly set to e2. In most cases, where , TideHunter performs an end-to-end global sequence alignment between the two subsequences of the read, and . Based on the alignment result, the putative next boundary within e1 and e2 can be calculated. In more detail, we first locate the corresponding base of e in the subsequence . Then, based on the global sequence alignment of and , the matched base of e in can be derived using the alignment CIGAR. We further calculate the coordinate of the matched base and consider it as the putative next boundary (Fig. 2).
As such, TideHunter collects a set of repeat unit boundaries, then uses them to partition the original long-read into multiple segments.
To avoid extending the tandem repeat to a very high error rate or chimeric region, TideHunter stops searching for the boundaries when the ratio of identical nucleotides based on the global alignment is lower than a threshold of 0.75 by default.
2.6 Generating consensus sequence
TideHunter performs a POA (Lee et al., 2002) on the partitioned multiple segments and calls the consensus sequence using the heaviest-bundling algorithm described in Lee (2003). The POA is accelerated by using a SIMD implementation (Vaser et al., 2017).
In more detail, POA performs multiple sequence alignment by iteratively aligning a query sequence to a target directed acyclic graph (DAG) and adding the query to the DAG based on the alignment result (Lee et al., 2002). For the DAG, the node represents individual sequence base and the edge represents two consecutive bases in the sequence (Fig. 3). Same as the traditional sequence-to-sequence alignment, a dynamic programming matrix needs to be filled out for the sequence-to-graph alignment, where each row represents one node of the DAG and each column represents one base of the query. To fill out the matrix, three operations need to be considered for each cell of the matrix: match (diagonal), deletion (vertical) and insertion (horizontal) (Fig. 3). The first two operations, match and deletion, only rely on the information of cells in the previous rows, thus multiple cells can be processed simultaneously by using a SIMD vector. However, parallelization cannot be accomplished for the insertion operation as it depends on the left cell, which is in the same row. Thus, with the SIMD parallelization, the overall time complexity is decreased from to roughly , where np is the average number of precursors, is the number of nodes in the DAG, n is the length of the query sequence and k is the number of variables that fit in a SIMD vector, which is generally 16 or 8.
Fig. 3.
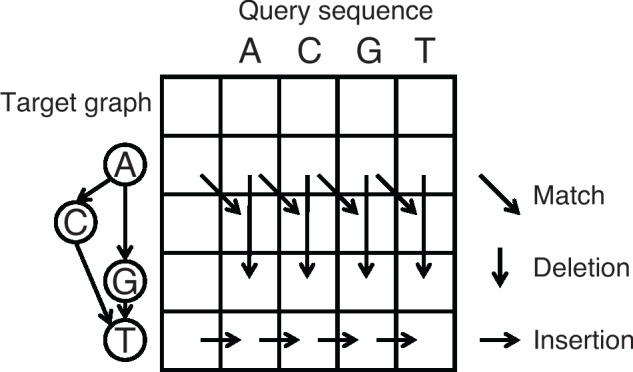
Dynamic programming matrix of sequence-to-graph alignment and three types of operations. SIMD parallelization is applicable for the match and deletion operations as they only rely on the previous rows. Insertion operation must be processed linearly as it depends on the left cell, which is in the same row
After the iterative sequence-to-graph alignment, the final DAG is used to generate a consensus sequence with the heaviest-bundling algorithm (Lee, 2003). Then, TideHunter takes the consensus sequence as the query to perform an extension alignment on each side of the repeat region in order to incorporate the non-full copies of the repeat into the final result.
3 Result
TideHunter is implemented in the C programming language. It takes long-read sequencing data as input and outputs consensus sequences of tandem repeats in FASTA format by default. TideHunter supports multi-threading to achieve faster running speed on multi-core computers.
To demonstrate the efficiency and sensitivity of TideHunter, we evaluated it on both simulated and real datasets, along with TRF (version 4.09), INC-seq (git commit #0ab4ac81) and C3POa (git commit #fe370036).
As INC-seq and C3POa only output one consensus sequence for each read, to ensure the consistency of the evaluation, we only retained the repeat covering the longest read sequence for TideHunter and TRF when multiple repeats were found.
3.1 Simulation study
To simulate tandemly repeated reads, we first randomly extracted a sequence from GRCh37 human reference genome and directly copy the sequence multiple times. A 100 bp random flanking sequence was appended to each side. We then used PBSIM (Ono et al., 2013) to generate a simulated read from the tandemly repeated sequence. In total, five error rates with different substitution: insertion: deletion ratios (13%, 41:23:36, 15%-a, 37:42:21, 15%-b, 11:60:29, 16%, 28:24:48 and 20%, 48:15:37), five repeat pattern sizes (100, 500, 1000, 2000 and 3000) and five copy numbers (2, 3, 5, 10 and 20) were used to generate 15 simulated datasets (Tables 1–3). The five error rates and error distributions come from five public real datasets (PacBio: 15%-a, 15%-b and ONT: 13%, 16%, 20%) (Weirather et al., 2017; Harris et al., 2018). For each simulated dataset, 1000 reads were generated.
Table 1.
Performance on datasets with five error rates and distributions (1000 reads for each error rate and distribution, repeat pattern size is 1000 bp, copy number is 10)
| Error rate (sub.:ins.:del.) | Tool | Accuracy (%) | Ave. copy number | Ave. identical base | Run time (CPU min) |
|---|---|---|---|---|---|
| TideHunter | 99.9 | 9.6 | 983.1 | 0.8 | |
| 13% (41:23:36) | INC-seq | 99.7 | 8.7 | 969.6 | 95.7 |
| TRF | 71.4 | 9.6 | 960.6 | 3.7 | |
| TideHunter | 100.0 | 9.5 | 988.2 | 0.8 | |
| 15%-a (37:42:21) | INC-seq | 99.9 | 7.9 | 975.2 | 96.3 |
| TRF | 58.1 | 7.4 | 925.9 | 3.5 | |
| TideHunter | 99.9 | 9.2 | 988.0 | 0.9 | |
| 15%-b (11:60:29) | INC-seq | 96.4 | 5.1 | 958.0 | 85.0 |
| TRF | 88.8 | 9.1 | 970.1 | 2.7 | |
| TideHunter | 99.9 | 9.6 | 963.0 | 0.8 | |
| 16% (28:24:48) | INC-seq | 83.5 | 4.0 | 887.0 | 72.0 |
| TRF | 39.2 | 6.2 | 886.0 | 3.7 | |
| TideHunter | 99.8 | 7.6 | 965.8 | 0.9 | |
| 20% (48:15:37) | INC-seq | 99.1 | 5.8 | 939.0 | 92.2 |
| TRF | 0.0 | 0.0 | 0.0 | 0.5 |
Note: The best performance regarding each specific feature on each dataset.
On all datasets, TideHunter was run with default settings: kmer_length = 8, step_size = 1, min_copy = 2, min_period = 30, max_period = 100 000, max_diverg = 0.25. For TRF, recommended parameters were used: match = 2, mismatch = 7, indel = 7, match_frac = 80, indel_frac = 10, min_score = 100, max_period = 2000. INC-seq was run with default settings except: minReadLength = 1000, anchor_seg_step = 50, anchor_length = 50, copy_num_thre = 2. C3POa was excluded from the simulation study because it requires additional splint sequence information as input.
We consider a detected tandem repeat as correct only if the consensus length and the true repeat pattern size have a difference less than 20%, i.e.:
The accuracy is defined as the number of correct consensus sequences/the total number of reads. The copy number for each called tandem repeat was directly extracted from each tool’s result. We aligned each consensus sequence with the ground truth repeat unit sequence and calculated the number of identical bases (the number of equal operations, i.e. ‘=’, in the alignment CIGAR) using the alignment result. Average copy number and identical bases of each dataset were calculated using only correct consensus sequences.
3.1.1 Running speed
TideHunter is approximately 100 times faster than INC-seq on all datasets (Table 1-3), which benefits from the fast repeat unit recognition algorithm and the SIMD acceleration of POA. As an alignment-based method, INC-seq identifies tandem repeat patterns through exhaustive segment alignment, which is expected to be very slow. TRF shows comparable running speed but has clearly lower sensitivity than TideHunter, especially on datasets with higher error rates and longer repeat pattern sizes.
Table 3.
Performance on datasets with five repeat copy numbers (1000 reads for each copy number, error rate is 15%, error distribution is 37:42:21, repeat pattern size is 1000 bp)
| Copy number | Tool | Accuracy (%) | Ave. copy number | Ave. identical base | Run time (CPU min) |
|---|---|---|---|---|---|
| TideHunter | 0.2 | 2.0 | 814.5 | 0.02 | |
| 2 | INC-seq | 0.0 | 0.0 | 0.0 | 18.0 |
| TRF | 0.1 | 1.9 | 902.0 | 0.05 | |
| TideHunter | 89.3 | 2.9 | 887.9 | 0.1 | |
| 3 | INC-seq | 97.3 | 2.0 | 876.7 | 26.5 |
| TRF | 7.6 | 2.3 | 908.6 | 0.2 | |
| TideHunter | 100.0 | 4.8 | 952.5 | 0.3 | |
| 5 | INC-seq | 99.2 | 3.8 | 934.1 | 47.1 |
| TRF | 31.1 | 3.8 | 916.2 | 0.8 | |
| TideHunter | 99.9 | 9.5 | 988.2 | 0.9 | |
| 10 | INC-seq | 100.0 | 7.8 | 971.6 | 91.0 |
| TRF | 60.1 | 6.8 | 921.1 | 3.6 | |
| TideHunter | 100.0 | 18.4 | 996.0 | 2.9 | |
| 20 | INC-seq | 100.0 | 16.9 | 988.3 | 181.5 |
| TRF | 84.6 | 14.6 | 934.0 | 10.3 |
Note: The best performance regarding each specific feature on each dataset.
3.1.2 Performance under varying error rates and distributions
TideHunter shows a higher sensitivity and accuracy than INC-seq and TRF across five datasets with different error rates and error distributions (Table 1). Given a long enough repeat pattern (1000 bp) and a large enough copy number (10), TideHunter and INC-seq are able to detect almost all (>99%) of the tandem repeats regardless of the error rate. TideHunter has the overall highest average copy number and number of identical bases. The strategy of seeding and chaining of short k-mers enables TideHunter to discover the tandem repeat signal embedded in the long-read, even at a very high error rate.
INC-seq shows a slightly lower sensitivity and accuracy than TideHunter, while TRF only detected approximately 70% or fewer of the repeats from the 13%, 15%-a and 16% error rate datasets, and 0 from the 20% dataset. TRF is not expected to be able to process datasets with very high error rates, as it uses a preset error probability model requiring the matching fraction of two adjacent repeats to be at least 80%.
On the 20% error rate dataset, though TideHunter successfully detected tandem repeats from most of the reads, the copy number and number of identical bases dropped substantially as compared to other datasets. This is due to the low sequence identity resulting in more terminations during the tandem repeat searching.
TideHunter shows a higher robustness than TRF and INC-seq across three different error distributions on the 15%-a, 15%-b and 16% error rate datasets. Among the three tools, the error distribution influences TRF’s performance the most in respect to both the total number of correct consensus sequences and the average copy number. TRF favors the dataset with more insertion errors (15%-b, 11:60:29) and identifies the least number of tandem repeats when more deletion errors are in the reads (16%, 28:24:48). The performance of INC-seq also dropped substantially with a high number of deletion errors (16%, 28:24:48).
Unbalanced error distributions lead to additional (insertion > deletion) or missing (insertion < deletion) nucleotides in the sequencing reads compared to the original template sequences. In such cases, although the hit distances between two identical k-mers of consecutive copies differ from the true repeat pattern size, TideHunter will still chain these anchors together, as their distances are similar to each other, thus leading to a small chaining cost. As such, the optimal chain will still reflect the true underlying repeat pattern size in an approximate manner. This again illustrates the advantage of TideHunter’s seed-and-chain strategy over other approaches.
3.1.3 Performance under varying repeat pattern sizes
TideHunter identified all the repeats on 500, 1000, 2000 and 3000 bp repeat pattern size datasets (Table 2). On the 100 bp dataset, TideHunter detected tandem repeats from 734 reads, while TRF and INC-seq detected tandem repeats from 702 and 377 reads, respectively. When the repeat pattern is short and the error rate is high, it is more likely that TideHunter will collect a tandem repeat hit having a distance multiple folds larger than the true repeat pattern size, which leads to a false optimal chain.
Table 2.
Performance on datasets with five repeat pattern sizes (1000 reads for each repeat pattern size, error rate is 15%, error distribution is 37:42:21, copy number is 10)
| Repeat pattern size | Tool | Accuracy (%) | Ave. copy number | Ave. identical base | Run time (CPU min) |
|---|---|---|---|---|---|
| TideHunter | 73.4 | 9.5 | 95.9 | 0.03 | |
| 100 bp | INC-seq | 37.7 | 2.6 | 36.5 | 12.3 |
| TRF | 70.2 | 8.4 | 67.3 | 0.2 | |
| TideHunter | 100.0 | 9.5 | 494.0 | 0.3 | |
| 500 bp | INC-seq | 87.4 | 4.2 | 453.8 | 50.8 |
| TRF | 71.9 | 7.6 | 463.0 | 1.8 | |
| TideHunter | 100.0 | 9.5 | 988.5 | 0.9 | |
| 1000 bp | INC-seq | 100.0 | 7.9 | 974.0 | 96.4 |
| TRF | 61.5 | 7.4 | 928.7 | 3.2 | |
| TideHunter | 100.0 | 9.4 | 1976.6 | 4.3 | |
| 2000 bp | INC-seq | 99.8 | 9.0 | 1955.5 | 163.4 |
| TRF | 30.0 | 5.4 | 1819.0 | 1.5 | |
| TideHunter | 100.0 | 9.4 | 2965.1 | 11.6 | |
| 3000 bp | INC-seq | 98.8 | 9.0 | 2934.2 | 300.4 |
| TRF | 0.0 | 0.0 | 0.0 | 0.8 |
Note: The best performance regarding each specific feature on each dataset.
INC-seq is less sensitive on datasets with repeat pattern sizes of 500 bp or shorter. TRF shows lower performance when the repeat pattern size increases and does not function with repeat pattern size of 2000 bp or longer.
3.1.4 Performance under varying copy numbers
All three tools failed with the 2 copy number dataset as the repeat signal is insufficient to be detected (Table 3). For the 3 copy number dataset, INC-seq identified a higher number of repeats than TideHunter. For datasets with the copy number larger than 5, TideHunter and INC-seq both correctly identified almost all repeats. TRF still failed with approximately 15% of the reads even on the 20 copy number dataset.
Given a higher copy number of the repeats, all three tools are able to generate a more accurate consensus sequence. TideHunter always provides more identical bases than INC-seq and TRF, as more copies of repeats are collapsed to call the consensus sequence.
3.2 Real data evaluation
In the real data evaluation, we first evaluated TideHunter along with TRF, INC-seq and C3POa on a synthetic Spike-In RNA Variant (SIRV) E2 dataset (Volden et al., 2018). In total, 603 906 Nanopore 1D reads having at least one splint sequence were used to perform the evaluation. These reads were selected from 828 684 raw reads using the C3POa preprocessing script.
Another three synthetic 16S ribosomal RNA (rRNA) datasets (Li et al., 2016) were used to evaluate the first three tools excluding C3POa, as it requires additional splint sequence information as input. The first 16S rRNA dataset is a simple synthetic community with only three bacteria, while the other two datasets include two independent replicates having ten bacteria. For all three datasets, only Nanopore 2D reads were used.
Three of the tools were run with default or recommended settings on all the real datasets. INC-seq was run on the SIRV E2 dataset with the same settings as in the simulation study, as the SIRV E2 dataset has template sequences shorter than 500 bp, which is the default anchor length for INC-seq.
To focus on high-quality consensus results, we filtered out tandem repeats with less than six copies. The threshold six was chosen based on the default setting of INC-seq.
Moreover, for the SIRV E2 dataset, all consensus sequences were trimmed to full-length transcript reads using the C3POa post-processing script. Full-length reads and consensus sequences were mapped to the artificial SIRVome sequences (Volden et al., 2018) and a customized 16S rRNA reference database (Li et al., 2016), respectively, using minimap2 (Li, 2018).
3.2.1 Performance on the SIRV E2 dataset
On the SIRV E2 dataset, TideHunter is approximately 40 times and 120 times faster than C3POa and INC-seq respectively, and 2.6 times faster than TRF (Table 4). It is more sensitive in terms of generating consensus sequences and full-length transcripts. Over 25% of the reads were detected to have six or more copies of tandem repeats. C3POa has the highest mappable ratio, but overall, TideHunter provided the most mappable full-length consensus sequences.
Table 4.
Performance on the SIRV E2 dataset (603 906 Nanopore 1D reads with at least one splint sequence were used for evaluation)
| Tool | # consensus | # full-length reads | # mappable reads (mappable ratio %) | Error rate (%) | Run time (CPU hour) |
|---|---|---|---|---|---|
| TideHunter | 155 261 | 148 126 | 142 208 (96.0) | 5.1 | 5.2 |
| C3POa | 136 243 | 119 503 | 119 267 (99.8) | 4.2 | 204.5 |
| INC-seq | 115 963 | 110 645 | 107 159 (96.8) | 6.5 | 630.6 |
| TRF | 118 040 | 110 079 | 105 145 (95.5) | 6.7 | 13.8 |
To evaluate the accuracy of the consensus sequences, we calculated the error rate based on the minimap2 alignments. Consensus sequences generated by TideHunter are more accurate than INC-seq and TRF, but are 0.9% less accurate than C3POa. This is likely due to the sophisticated consensus calling strategy that C3POa adopts. Unlike TideHunter, which directly generates the final consensus sequence through POA, C3POa considers the POA output as a preliminary consensus sequence. It then aligns all the partitioned segments back to the preliminary consensus sequence. These alignments are used as input to further error-correct the consensus sequence by racon (Vaser et al., 2017). Racon splits the consensus sequence and partitioned segments into several chunks using non-overlapping windows, then independently performs another round of POA within each window. We may implement a similar strategy in TideHunter in the future in order to further improve the consensus accuracy.
3.2.2 Performance on synthetic 16S rRNA datasets
On three 16S rRNA datasets, TideHunter is over 25 times faster than INC-seq and three times faster than TRF (Table 5). It also has the most identified tandem repeats and mappable consensus sequences. TRF shows a slightly higher sensitivity than INC-seq, which is likely due to Nanopore 2D reads having a relatively higher sequencing accuracy.
Table 5.
Performance on synthetic 16S rRNA datasets (only Nanopore 2D reads were used for evaluation)
| Dataset (# reads) | Tool | # consensus | # mappable reads (mappable ratio %) | Error rate (%) | Run time (CPU min) |
|---|---|---|---|---|---|
| TideHunter | 3860 | 3853 (99.8) | 4.4 | 4.7 | |
| Simplea (14 580) | INC-seq | 2178 | 2174 (99.8) | 4.8 | 119.0 |
| TRF | 2542 | 2540 (99.9) | 4.5 | 14.5 | |
| TideHunter | 1596 | 1590 (99.6) | 3.1 | 1.5 | |
| Rep.1b (7 444) | INC-seq | 1076 | 1074 (99.8) | 4.7 | 50.4 |
| TRF | 1360 | 1356 (99.7) | 3.5 | 5.5 | |
| TideHunter | 1564 | 1558 (99.6) | 3.0 | 1.5 | |
| Rep.2c (2 904) | INC-seq | 1183 | 1178 (99.6) | 4.3 | 43.9 |
| TRF | 1330 | 1326 (99.7) | 3.4 | 4.8 |
Simple: simple community dataset with 3 bacteria.
Rep.1: replicate 1 of 10 bacteria community.
Rep.2: replicate 2 of 10 bacteria community.
95.4% and 97.4% of TRF and INC-seq’s mappable consensus sequences are detected by TideHunter, while the overlapping ratio is 90.7% for TRF and INC-seq. For all three tools, over 99.6% of the consensus sequences are mappable, and they all have a length of 500–1000 bp, consistent with the size of 16S rRNA.
We calculated the error rate using the primary alignment record for each consensus sequence. All three tools are able to produce a high-quality (error rate <5%) consensus sequence given six or more copies of the tandem repeat. Again, TideHunter slightly outperforms the other two tools.
4 Discussion
The recently proposed RCA-based long-read sequencing workflow provides a new strategy for producing high-accuracy long-read data. With additional computational processing steps, the error rate of the resulting consensus sequences can be lower than 5%, which is much lower than the raw error rate [over 13% (Weirather et al., 2017)].
TideHunter is an efficient and sensitive tandem repeat detection and consensus calling tool specifically designed for RCA-based long-read data. It works with noisy long-reads (PacBio and ONT) at error rates of up to 20% and does not have any limitation of the maximal repeat pattern size as for traditional tandem repeat detection tools (Benson et al., 1999). TideHunter is 20 to 100 times faster than existing alignment-based methods (Li et al., 2016; Volden et al., 2018) and shows a higher sensitivity on all evaluation datasets.
The high efficiency and sensitivity of TideHunter come from its specifically designed seed-and-chain-based repeat unit recognition algorithm and the SIMD acceleration of POA. The seed-and-chain algorithm enables TideHunter to make full use of the identical k-mers between tandem copies, then efficiently identify the repeat pattern size through the search for the optimal chain. The proper chaining score and cost functions used during the chaining step enhance the robustness of TideHunter across datasets with different error rates and unbalanced error distributions. To the best of our knowledge, TideHunter is the first tandem repeat detection tool that adopts the seed-and-chain strategy. Moreover, the overall speed is further accelerated by the SIMD-based implementation of POA.
The configuration of the k-mer length and step size (k and s) is crucial for the performance of TideHunter. Larger k-mer length may reduce the number of tandem repeat hits as the probability of sequencing error showing up in the k-mer becomes higher. On the other hand, if the k-mer is too short, the chance of two random k-mers being identical increases greatly, which likely leads to a false optimal chain. For the step size, exhaustively collecting all k-mers will ensure TideHunter does not miss any useful information in the determination of the repeat pattern size. Our evaluation suggests that the default setting of k = 8 and s = 1 enables TideHunter to tolerate sequencing errors and achieve the highest sensitivity.
Instead of using a larger step size, other long-read aligners utilize MinHash (Berlin et al., 2015) or minimizer (Li, 2016) to sample sequences in a reduced representation. Although we have implemented minimizer seeding in TideHunter, it is not recommended to enable this function as the speed improvement is insignificant (1.2 times faster with parameter window-size = 5), but the sensitivity is slightly lower.
TideHunter is not designed to detect satellite repeats [repeat pattern size: 5–170 bp (Tyler-Smith and Brown, 1987)] from long-read data without RCA. Although alpha satellites having a pattern size of 170 bp can be detected in our simulation, TideHunter shows a poor sensitivity on other very short repeat pattern (<100 bp) datasets (simulated data not shown). Satellite repeat discovery is the main intended usage of TRF. However, its performance is significantly reduced by the high sequencing error rate. We anticipate that new tools need to be developed for discovering satellite repeats from noisy long-read data.
Unlike TRF which allows up to three overlapping repeats to be reported, TideHunter only selects one optimal period size in each read region. Thus, in some rare cases where the template itself is a tandem repeat, TideHunter is likely to generate a consensus sequence containing a single repeat unit of the template. This is because chains with a shorter hit distance always tend to have more anchors and higher chaining scores. However, this issue can be addressed if adaptor sequences are added during the library preparation. Chains of single repeat units will be separated by the adaptor sequence and thus, the true optimal chain will still have the highest score.
A potential future goal is to further improve the consensus quality, as the error rates of the called consensus sequences are still relatively high. Currently, TideHunter simply takes the consensus sequence generated by POA as the final result. A possible solution is to incorporate base quality scores into the consensus calling algorithm, i.e. bases with higher quality have higher weights in the graph. This can be a feasible solution as base quality scores have been used to produce high-quality consensus sequences in other applications, such as genome assembly, read re-alignment and variant calling.
Acknowledgements
We thank Sidney Taiko Sheehan and Eddie Park for reading and editing the manuscript.
Funding
This study was supported by a startup fund from the Children’s Hospital of Philadelphia. This study was also partially supported by the National Key Research and Development Program of China (Nos 2018YFC0910504 and 2017YFC0907503).
Conflict of Interest: none declared.
References
- Benson G. et al. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res., 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K. et al. (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 33, 623.. [DOI] [PubMed] [Google Scholar]
- Calus S.T. et al. (2018) NanoAmpli-Seq: a workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. GigaScience, 7, giy140.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C.-S. et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods, 10, 563.. [DOI] [PubMed] [Google Scholar]
- Chin C.-S. et al. (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods, 13, 1050.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lannoy C. et al. (2017) A sequencer coming of age: de novo genome assembly using MinION reads. F1000Research, 6, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S. et al. (2015) Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res., 25, 1750–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S. et al. (2016) Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genetics, 17, 333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R.S. et al. (2018) Noise-Cancelling Repeat Finder: uncovering tandem repeats in error-prone long-read sequencing data. bioRxiv, doi: 10.1101/475194. [DOI] [PMC free article] [PubMed]
- Koren S. et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol., 30, 693.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. (2003) Generating consensus sequences from partial order multiple sequence alignment graphs. Bioinformatics, 19, 999–1008. [DOI] [PubMed] [Google Scholar]
- Lee C. et al. (2002) Multiple sequence alignment using partial order graphs. Bioinformatics, 18, 452–464. [DOI] [PubMed] [Google Scholar]
- Li C. et al. (2016) INC-Seq: accurate single molecule reads using nanopore sequencing. GigaScience, 5, 34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2016) Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 32, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.G. et al. (2013) Review of tandem repeat search tools: a systematic approach to evaluating algorithmic performance. Brief. Bioinform., 14, 67–81. [DOI] [PubMed] [Google Scholar]
- Liu B. et al. (2017) LAMSA: fast split read alignment with long approximate matches. Bioinformatics, 33, 192–201. [DOI] [PubMed] [Google Scholar]
- Ono Y. et al. (2013) PBSIM: PacBio reads simulator–toward accurate genome assembly. Bioinformatics, 29, 119–121. [DOI] [PubMed] [Google Scholar]
- Pellegrini M. et al. (2010) TRStalker: an efficient heuristic for finding fuzzy tandem repeats. Bioinformatics, 26, i358–i366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmela L., Rivals E. (2014) LoRDEC: accurate and efficient long read error correction. Bioinformatics, 30, 3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmela L. et al. (2017) Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics, 33, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-Smith C., Brown W.R. (1987) Structure of the major block of alphoid satellite dna on the human Y chromosome. J. Mol. Biol., 195, 457–470. [DOI] [PubMed] [Google Scholar]
- Vaser R. et al. (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res., 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volden R. et al. (2018) Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl. Acad. Sci. USA, 115, 9726–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather J.L. et al. (2017) Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research, 6, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A.V. et al. (2013) The MaSuRCA genome assembler. Bioinformatics, 29, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]