

Previously, coronavirus nsp8 proteins were suggested to have template-dependent RNA polymerase activities resembling those of RNA primases or even canonical RNA-dependent RNA polymerases, while more recent studies have suggested an essential cofactor function of nsp8 (plus nsp7) for nsp12-mediated RNA-dependent RNA polymerase activity. In an effort to reconcile conflicting data from earlier studies, the study revisits coronavirus nsp8-associated activities using additional controls and proteins. The data obtained for three coronavirus nsp8 proteins provide evidence that the proteins share metal ion-dependent RNA 3′ polyadenylation activities that are greatly stimulated by a short oligo(U) stretch in the template strand. In contrast, nsp8 was found to be unable to select and incorporate appropriate (matching) nucleotides to produce cRNA products from heteropolymeric and other homooligomeric templates. While confirming the critical role of nsp8 in coronavirus replication, the study amends the list of activities mediated by coronavirus nsp8 proteins in the absence of other proteins.
KEYWORDS: RNA 3′-terminal adenylyltransferase, RNA virus, coronavirus, replication
ABSTRACT
Coronavirus nonstructural protein 8 (nsp8) has been suggested to have diverse activities, including noncanonical template-dependent polymerase activities. Here, we characterized a recombinant form of the human coronavirus 229E (HCoV-229E) nsp8 and found that the protein has metal ion-dependent RNA 3′-terminal adenylyltransferase (TATase) activity, while other nucleotides were not (or very inefficiently) transferred to the 3′ ends of single-stranded and (fully) double-stranded acceptor RNAs. Using partially double-stranded RNAs, very efficient TATase activity was observed if the opposite (template) strand contained a short 5′ oligo(U) sequence, while very little (if any) activity was detected for substrates with other homopolymeric or heteropolymeric sequences in the 5′ overhang. The oligo(U)-assisted/templated TATase activity on partial-duplex RNAs was confirmed for two other coronavirus nsp8 proteins, suggesting that the activity is conserved among coronaviruses. Replacement of a conserved Lys residue with Ala abolished the in vitro RNA-binding and TATase activities of nsp8 and caused a nonviable phenotype when the corresponding mutation was introduced into the HCoV-229E genome, confirming that these activities are mediated by nsp8 and critical for viral replication. In additional experiments, we obtained evidence that nsp8 has a pronounced specificity for adenylate and is unable to incorporate guanylate into RNA products, which strongly argues against the previously proposed template-dependent RNA polymerase activity of this protein. Given the presence of an oligo(U) stretch at the 5′ end of coronavirus minus-strand RNAs, it is tempting to speculate (but remains to be confirmed) that the nsp8-mediated TATase activity is involved in the 3′ polyadenylation of viral plus-strand RNAs.
IMPORTANCE Previously, coronavirus nsp8 proteins were suggested to have template-dependent RNA polymerase activities resembling those of RNA primases or even canonical RNA-dependent RNA polymerases, while more recent studies have suggested an essential cofactor function of nsp8 (plus nsp7) for nsp12-mediated RNA-dependent RNA polymerase activity. In an effort to reconcile conflicting data from earlier studies, the study revisits coronavirus nsp8-associated activities using additional controls and proteins. The data obtained for three coronavirus nsp8 proteins provide evidence that the proteins share metal ion-dependent RNA 3′ polyadenylation activities that are greatly stimulated by a short oligo(U) stretch in the template strand. In contrast, nsp8 was found to be unable to select and incorporate appropriate (matching) nucleotides to produce cRNA products from heteropolymeric and other homooligomeric templates. While confirming the critical role of nsp8 in coronavirus replication, the study amends the list of activities mediated by coronavirus nsp8 proteins in the absence of other proteins.
INTRODUCTION
Coronaviruses include important human and animal pathogens (1, 2). They have very large plus-strand RNA (+RNA) genomes of approximately 30 kb and, compared to most other +RNA viruses, employ strategies of unusual complexity to express and replicate their genomes (3–5). Over the past 2 decades, significant progress has been made in the characterization of key mechanisms and factors involved in the replication and transcription of the coronavirus genome RNA (for a recent review, see reference 6). Although it is now widely accepted that viral RNA synthesis (and modification) is mediated by a large multisubunit protein complex (called the replication-transcription complex [RTC]), our understanding of the proteins involved in individual steps of viral RNA synthesis remains limited. For example, there is little or no information on (i) the viral (or cellular?) factors involved in 5′ capping and 3′ polyadenylation of viral plus-strand RNAs (6, 7) and (ii) the proteins that control the discontinuous RNA synthesis required to produce subgenome-length negative-strand RNAs carrying a 3′ antileader sequence, which are subsequently used to produce mRNAs with a common 5′ leader sequence that is identical to the 5′ end of the genome (8, 9). It has been proposed that nonstructural protein 12 (nsp12), an ∼105-kDa protein comprised of N-terminal nucleotidyl transferase (NiRAN) (10) and C-terminal RNA-dependent RNA polymerase (RdRp) (11) domains, forms a complex with nsp7 and nsp8 (12, 13) and that this complex is required and sufficient for template-dependent RNA synthesis in vitro (14). There is also evidence that the fidelity of this polymerase complex is enhanced by a 3′-to-5′ exoribonuclease (ExoN) activity associated with the N-terminal domain of nsp14 (15, 16). The proposed role of ExoN in ensuring superior RNA replication fidelity has been supported by reverse genetic studies using a range of coronavirus ExoN knockout mutants for which mutator or nonviable phenotypes have been reported (16–22).
The functional characterization of individual replicase gene-encoded proteins revealed that the severe acute respiratory syndrome coronavirus (SARS-CoV) nsp8 has a second noncanonical RNA polymerase activity in vitro that catalyzes the production of short (≤6-nucleotide [nt]) oligonucleotides in an Mn2+ ion- and template-dependent manner, reminiscent of cellular RNA primase activities (23). The short oligonucleotides synthesized by nsp8 were proposed to serve as primers for the canonical RNA polymerase (nsp12). The study also provided data to suggest that nsp8 synthesizes these oligonucleotides de novo and lacks the ability to extend primer/template substrates. In contrast, a subsequent study using a C-terminally His6-tagged form of SARS-CoV nsp8 suggested that nsp8 (alone or in complex with nsp7) is able to extend primed RNA templates in the presence of Mg2+, thus questioning the primase hypothesis proposed earlier (23, 24). The study also postulated that the nsp7-nsp8 complex is capable of synthesizing substantially longer RNA products in both de novo RNA polymerase and primer extension reactions (24). Similarly, de novo RNA polymerase activities resulting in longer transcripts were suggested to be produced by different N-terminally tagged forms of the feline infectious peritonitis virus (FIPV) nsp8, based on the identification of slowly migrating 32P-labeled products generated in reactions in which the reaction mixtures were supplemented with nsp8 and metal ions (12). To our knowledge, the last two studies did not use 3′-blocked template RNAs to exclude the possibility that the radiolabeled template-length RNA products (transcripts) observed in de novo polymerase assays represented 3′-extended forms of the template(s) used in these assays.
This brief overview of nsp8 in vitro studies performed by different laboratories with different protein constructs shows that our understanding of nsp8-associated polymerase (and, possibly, other) activities is incomplete. Based on its conservation among corona- and toroviruses as well as reverse genetics and biochemical data obtained for SARS-CoV nsp8, this small protein is thought to have an important function in coronavirus and, most probably, torovirus replication. However, some of the main conclusions obtained in earlier studies remained controversial and deserve further investigation. We therefore decided to produce three coronavirus nsp8 proteins with their authentic amino termini using previously established protocols. C-terminally His6-tagged wild-type and mutant forms of human coronavirus 229E (HCoV-229E) nsp8 (the latter of which contain single Lys-to-Ala substitutions corresponding to previously characterized replacements in SARS-CoV nsp8) as well as C-terminally His6-tagged wild-type forms of SARS-CoV and FIPV nsp8 were purified to apparent homogeneity. The characterization of (putative) HCoV-229E nsp8 polymerase activities, RNA substrate and nucleotide preferences, RNA-binding activities, and metal ion requirements leads us to conclude that the wild-type protein (but not the nsp8_K3711A mutant) has RNA 3′-terminal adenylyltransferase (TATase) activity if incubated with single-stranded and completely double-stranded substrates. This activity can be significantly stimulated by providing a short oligo(U) stretch as a template. Unlike previous studies, we failed to obtain evidence for a canonical (i.e., high-fidelity and template-dependent) RNA-dependent RNA polymerase activity for this HCoV-229E homolog, which is in line with the protein's pronounced specificity for adenylate. Using a substrate described in a previous study, HCoV-229E nsp8 was found to generate products that corresponded to those obtained previously with SARS-CoV nsp8 (24). However, based on additional data obtained for 3′-blocked versions of the test substrate, we arrived at different conclusions regarding the identities of some of the products. Taken together, our data lead us to suggest that nsp8 acts as an oligo(U)-templated polyadenylyltransferase but also has robust (mono/oligo)adenylate transferase activities when incubated with single-stranded and blunt-ended double-stranded RNAs (dsRNAs). We think that this activity explains most of the data published previously on coronavirus nsp8 homologs, even though additional experiments and controls would be desirable to further substantiate this hypothesis.
RESULTS
Expression and purification of recombinant HCoV-229E nsp8.
To produce a recombinant form of HCoV-229E nsp8 with its authentic N terminus, an expression strategy introduced by Gohara et al. was used (25) (see Materials and Methods). Briefly, the HCoV-229E nsp8-coding sequence (fused to an N-terminal ubiquitin [Ub] tag and a C-terminal hexahistidine tag, Ub-nsp8-CHis6) was inserted into a pASK3-derived plasmid (26) downstream of a tetracycline-inducible promoter. Coexpression of Ub-nsp8-CHis6 and Ubp1 resulted in proteolytic cleavage at the . . . LRGG↓SVAS . . . site between the Ub moiety and the nsp8 sequence in the Ub-nsp8-CHis6 fusion protein, resulting in the release of a C-terminally His6-tagged form of nsp8 with its correct N terminus (NH2-SVAS . . .) (Fig. 1A). The protein was purified by Ni-nitrilotriacetic acid (NTA) affinity and anion-exchange chromatography as described in Materials and Methods. As shown in Fig. 1B, induction of expression in Escherichia coli TB1(pCG1) cells with AHT gave rise to an extra protein of approximately 22 kDa which corresponded well with the molecular mass of 22 kDa calculated for the nsp8-CHis6 protein (Fig. 1B, lanes 1 and 2). Using a two-step protocol, HCoV-229E nsp8 and the (mutant) nsp8_K3687A and nsp8_K3711 proteins could be purified in sufficient amounts for biochemical studies and proved to be stable upon storage at −20°C (Fig. 1A and B, lanes 3 to 5).
FIG 1.

Production of HCoV-229E nsp8 in E. coli. (A) Multiple-sequence alignment of the N-terminal regions of coronavirus nsp8 proteins representing the genera Alpha-, Beta-, Gamma-, and Deltacoronavirus. Sequences were aligned using the Clustal Omega program (62) and converted using the ESPript program (63). The black background indicates invariant residues. The alanine substitutions in HCoV-229E nsp8 generated in this study are indicated by arrows. Numbers indicate the positions in the HCoV-229E polyprotein 1a/1ab sequence. SARS-CoV, severe acute respiratory syndrome coronavirus (GenBank accession number AY291315); MERS-CoV, Middle East respiratory syndrome coronavirus (GenBank accession number JX869059.2); MHV, mouse hepatitis virus A59 (GenBank accession number NC_001846.1); IBV, avian infectious bronchitis virus (GenBank accession number NC_001451.1); BW-CoV, beluga whale coronavirus SW1 (GenBank accession number NC_010646); WiCoV, wigeon coronavirus HKU20 (GenBank accession number JQ065048); WECoV, white-eye coronavirus HKU16 (GenBank accession number JQ065044); FIPV, feline infectious peritonitis virus (GenBank accession number NC_002306); HCoV-NL63, human coronavirus NL63 (GenBank accession number NC_005831); HCoV-229E, human coronavirus 229E (GenBank accession number NC_002645). Secondary structure elements determined by crystal structure analysis of SARS-CoV nsp8 (chain H; PDB accession number 2AHM) (13) are shown above the alignment. (B) Coomassie brilliant blue-stained 12% SDS-polyacrylamide gel showing the production and purification of HCoV-229E nsp8-CHis6. Lanes 1 and 2, total lysates of E. coli TB1(pCG1) cells transformed with pASK-Ub-nsp8-CHis6 plasmid DNA and grown in the absence (–) or presence (+) of anhydrotetracycline (AHT); lanes 3 to 5, purified nsp8-CHis6 proteins (wild type [WT] or variants containing the indicated alanine substitutions of conserved residues). Lane M, marker proteins (molecular masses [in kilodaltons] are indicated to the left).
Terminal adenylyltransferase (TATase) activity of HCoV-229E nsp8.
Preliminary experiments using HCoV-229E nsp8 (Fig. 2 and data not shown) revealed the production of radiolabeled products when the protein was incubated with synthetic oligoribonucleotide substrates in the presence of [α-32P]ATP, confirming that the purified protein was active. To optimize reaction conditions for subsequent studies, we used U18 as a test substrate and incubated the reaction mixtures at 30°C for 60 min under various conditions with respect to the (i) nsp8 concentration, (ii) NaCl concentration, (iii) identities and concentrations of metal ions, (iv) pH, and (v) ribonucleotide concentrations. Based on these experiments (Fig. 2 and data not shown), we decided to use the following optimized (standard) reaction buffer in subsequent experiments: 50 mM Tris-Cl, pH 8.0, 50 mM NaCl, 1 MgCl2, 1% Triton X-100, 1 mM dithiothreitol (DTT), 4.5% glycerol, 1 μM substrate RNA, 100 μM the indicated nucleoside triphosphate(s) [NTP(s)], 0.17 μM the indicated [α-32P]NTP(s) (3,000 Ci/mmol), and 2 μM nsp8. Consistent with previous studies of the SARS-CoV nsp8 (23, 24), the data revealed that HCoV-229E nsp8 activity required the presence (of moderate concentrations) of Mg2+ or Mn2+ ions (Fig. 2B). High concentrations of these ions inhibited activity, while other metal ions failed to support nsp8 activity (Fig. 2C). HCoV-229E nsp8 activity proved to be sensitive to salt concentrations above 50 mM (Fig. 2A).
FIG 2.

RNA 3′-terminal nucleotidyl transferase (TNTase) activity of HCoV-229E nsp8. The effects of increasing salt concentrations and the presence of metal ions on TNTase activity were determined. (A) nsp8 activity assays were performed in reaction buffer supplemented with 2 μM nsp8, 1 μM U18 (substrate RNA), 100 μM ATP, 0.17 μM [α-32P]ATP, 1 mM MgCl2, and various concentrations of NaCl (36 to 150 mM). (B) Activity assays were performed in reaction buffer supplemented with 2 μM nsp8, 50 mM NaCl, 100 μM ATP, 0.17 μM [α-32P]ATP, 1 μM U18 (substrate RNA), and the indicated concentrations of MgCl2 and MnCl2 (0 to 20 mM). (C) Activity assays were performed in reaction buffer supplemented with 2 μM nsp8, 50 mM NaCl, 100 μM ATP, 0.17 μM [α-32P]ATP, and 1 μM U18 (left) or KR07 RNA (right), and the indicated divalent metal ions (each at 1 mM). 5′-32P-labeled RNAs of U18 and KR07 were used as 18-nt and 28-nt markers, respectively, as indicated to the left. The reaction mixtures were incubated at 30°C for 60 min. Products were resolved in a TBE-buffered 12% polyacrylamide–7 M urea gel and visualized by phosphorimaging. Lanes M, 5′-32P-labeled RNAs (U18, KR07) were used as markers with sizes (in nucleotides) indicated to the left.
In the presence of [α-32P]ATP (only), radiolabeled products with sizes that exceeded those of the substrate RNAs were found to be generated. These larger-than-expected products were obtained with both the homopolymeric U18 and the heteropolymeric substrate RNA KR07 (5′-UAAUGGAACGGUUUCGAUAUGGAUACAC-3′, representing the 3′ end of the HCoV-229E genome RNA) (Fig. 2C), suggesting that (i) the protein has adenylyltransferase activity and (ii) the products likely represent 3′-polyadenylated forms (rather than complementary copies) of the substrate RNAs used.
To corroborate the idea of HCoV-229E nsp8 acting as an RNA 3′-terminal ribonucleotidyl transferase (TNTase), we decided to include 3′-biotinylated RNA substrates that lacked a free 3′-hydroxyl group as controls and performed reactions with just a single nucleotide, i.e., (i) ATP and [α-32P]ATP, (ii) CTP and [α-32P]CTP, (iii) UTP and [α-32P]UTP, and (iv) GTP and [α-32P]GTP. As shown in Fig. 3, radiolabeled AMP was efficiently incorporated into products by HCoV-229E nsp8 if the reactions were performed with substrate RNAs carrying unmodified 3′ ends, while no radiolabeled products were generated from 3′-biotinylated substrate RNAs in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP (Fig. 3A and B, lanes 2 and 8). With some (but not all) substrate RNAs, a rather inefficient incorporation of CMP was observed (Fig. 3A, lanes 4 and 10; Fig. 3B, lane 4), while there was (nearly) no incorporation of GMP and UMP into any of the test substrates used (Fig. 3). Taken together, these data provide strong support for the proposed RNA 3′-ribonucleotidyl transferase activity of HCoV-229E nsp8 and reveal a pronounced preference for ATP in the transfer reaction, irrespective of the type of substrate RNA used (homopolymeric or heteropolymeric). In an additional set of experiments using dATP instead of ATP, we found that [α-32P]dAMP was not incorporated into any of the substrate RNAs used (data not shown), confirming that the nsp8-mediated RNA 3′ polyadenylation reaction requires the presence of a ribose-2'-hydroxyl group.
FIG 3.

TATase activity of HCoV-229E nsp8. (A) Reactions in which A18 and U18 were used as substrate RNAs. The suffix b indicates 3′ biotinylation of the respective oligoribonucleotide. (B) Reactions in which oligoribonucleotides KR07, KR07-b, KR12, KR12-b, KR05, and KR13 were used as substrate RNAs. The nucleotides used in the respective reactions are indicated above the autoradiogram. Lanes M, 5′-32P-labeled RNA markers, with the size(s) (in nucleotides) indicated to the left; lanes –, reactions performed in the absence of nsp8. Reactions were performed in reaction buffer supplemented with 50 mM NaCl, 1 mM MgCl2, 2 μM nsp8, 100 μM the indicated NTP (along with 0.17 μM the respective 5′-α-32P-labeled NTP), and 1 μM the indicated oligoribonucleotide. The reaction mixtures were incubated at 30°C for 60 min. RNA products were resolved in TBE-buffered 12% polyacrylamide–7 M urea gels and visualized by phosphorimaging.
Next, we explored the possible preferences of the nsp8 TATase activity for specific substrates and asked the question of whether the presence of specific 3′-terminal nucleotides affects the nsp8 TATase activity. The nucleotide sequence of one of the substrates corresponded to that of the 3′ end of the HCoV-229E genome (KR07). In addition, we used a range of KR07 derivatives carrying replacements of the 3′ cytidylate with adenylate, uridylate, and guanylate (Fig. 4A). Reactions were performed under the optimized conditions described above and in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP. Compared to the TATase activity obtained with KR07, nucleotide replacements of the 3′-terminal cytidylate resulted in increased TATase activities, with KR07_C28A and KR07_C28G representing the best substrates. Similarly, using another substrate, JZR3, replacements of the 3′ cytidylate with adenylate or guanylate resulted in superior activities (Fig. 4C, lanes 2 and 5). These data suggest that, at least in vitro, substrates with a 3′-terminal purine (with A being preferred over G) are polyadenylated more efficiently than substrates carrying a 3′-terminal pyrimidine.
FIG 4.
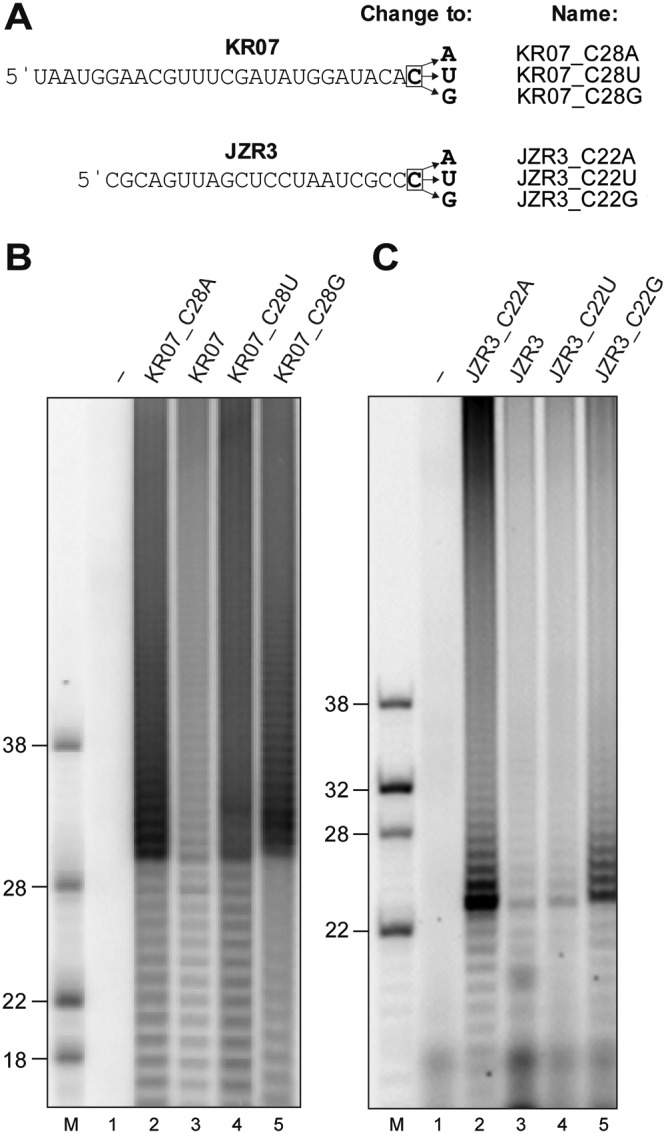
Effects of various 3′-terminal nucleotides on HCoV-229E nsp8 TATase activity. (A) The RNA substrates used in the TATase assays for which the results are shown in panels B and C. (B) TATase assay using 1 μM KR07, KR07_C28A, KR07_C28U, and KR07_C28G. (C) TATase assay using 1 μM JZR3, JZR_C22A, JZR3_C22U, and JZR_C22G, as indicated. 5′-32P-labeled RNAs loaded in lanes M were used as markers. Sizes (in nucleotides) are indicated to the left. Lanes –, reactions performed with RNA KR07 and JZR3 in the absence of nsp8. The reaction mixtures were incubated at 30°C for 60 min. The products were resolved in TBE-buffered 12% polyacrylamide–7 M urea gels and visualized by phosphorimaging.
Activity of HCoV-229E nsp8 on partial-duplex RNA substrates.
The data presented above suggest that HCoV-229E nsp8 is able to 3′ polyadenylate single-stranded RNA (ssRNA) substrates with diverse sequences/structures if these substrates contain a free 3′-hydroxyl group. Apparently, the protein did not require a primer/template hybrid (or a template) to generate radiolabeled RNA products, unlike what was suggested in earlier studies, in which SARS-CoV nsp8 appeared to produce radiolabeled RNA products in a template-dependent manner, indicative of both de novo and primer-dependent RNA polymerase activities (23, 24). In an effort to reconcile the previously published SARS-CoV nsp8 data with data obtained for HCoV-229E nsp8, a series of in vitro activity assays was performed using substrate and reaction conditions corresponding to those described by te Velthuis et al. (24). Of note, the nucleotide sequences of the partially double-stranded RNA substrate (named KR01/JTR1 in the present study) were identical to those of the SAV556/SAV557 substrate used in the previous study. In the presence of 50 μM ATP and 0.17 μM [α-32P]ATP, HCoV-229E nsp8 was revealed to produce multiple radioactively labeled products of ≥40 nt, suggesting efficient 3′ polyadenylation of one or both strands of KR01/JTR1 (Fig. 5B, lane 4). Similar products were obtained in the presence of 50 μM ATP, 50 μM GTP, and 0.17 μM [α-32P]ATP (Fig. 5B, lane 2). In striking contrast, no radiolabeled products were detected if the reaction was performed in the presence of 50 μM ATP, 50 μM GTP, and 0.17 μM [α-32P]GTP (Fig. 5B, lane 3). These data show that nsp8 is unable to incorporate GMP into reaction products, which strongly argues against a true copy process of the C/U sequence present in the primed RNA substrate KR01/JTR1 to generate a G/A sequence. This conclusion is further supported by data obtained in a reaction with a reaction mixture containing 50 μM ATP and 0.17 μM [α-32P]ATP in which prominent products of ≥40 nt that comigrated with the products seen in lane 2 of Fig. 5B were observed, while no labeled product was detected in a reaction containing 50 μM GTP and 0.17 μM [α-32P]GTP, even though the template contained a C at the first position (Fig. 5B; compare lanes 4 and 5). Interestingly, the protein failed to produce radiolabeled products when KR01/JTR1-b hybrid RNA (where the suffix b indicates 3′ biotinylation) was used as a substrate (Fig. 5B, lanes 6 to 9). The fact that 3′ biotinylation of the bottom-strand JTR1-b nearly completely abolished TATase activity led us to conclude that the labeled products seen in lanes 2 and 4 of Fig. 5B represent the 3′-polyadenylated forms of the bottom (JTR1) rather than the top (KR01) strand. Also, the data obtained in this and subsequent experiments (Fig. 6 and data not shown) suggest that, unlike as suggested previously by others (24), the radiolabeled products of the nsp8 activity do not represent 3′-extended primers resulting from an ATP- and GTP-dependent copy process of the (CU)10-containing template sequence. Instead, they represent 3′ mono-, oligo-, and polyadenylated variants of the bottom strand of the partially double-stranded RNA hybrid used in the respective experiment. The data also suggest that, with one important exception (see below), nsp8 TATase activity prefers blunt over recessive 3′ ends in partially double-stranded substrate RNAs.
FIG 5.
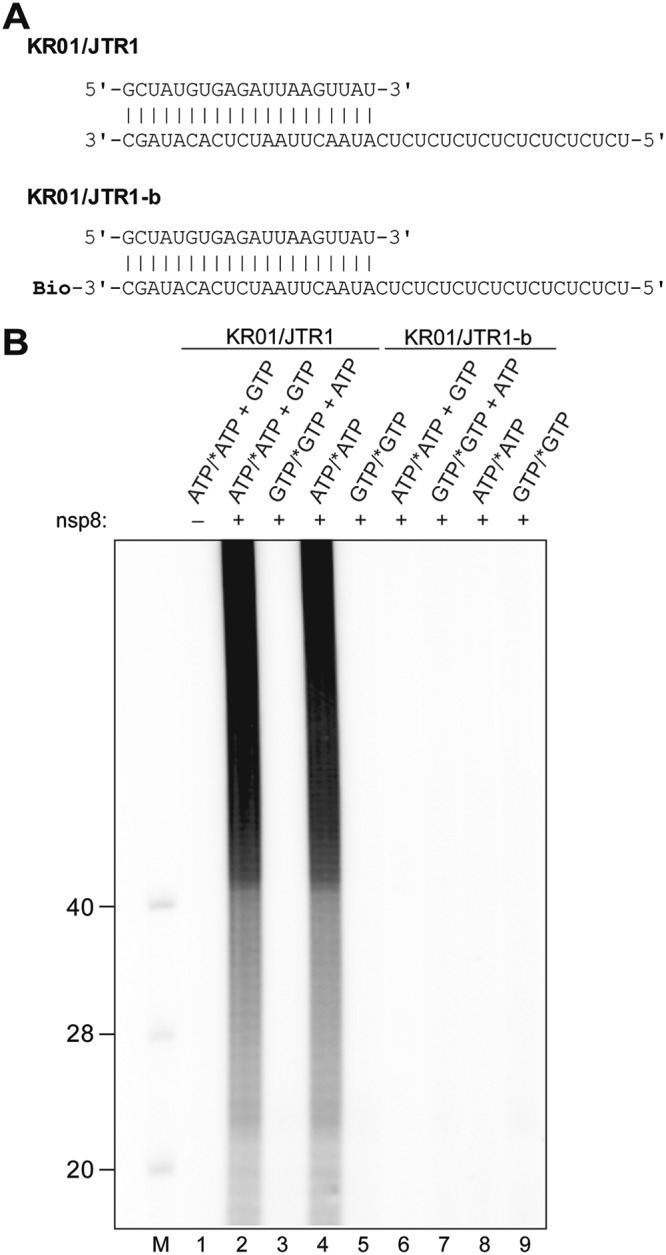
Primer extension assay. nsp8 activity assays were performed using a primer/template substrate (KR01/JTR1) described in an earlier study (24) and a derivative of this substrate in which the bottom strand was 3′ biotinylated (KR01/JTR1-b). (A) Primer/template RNA hybrids used in the experiment. Bio, RNA 3′ biotinylation. (B) Assays were performed in reaction buffer supplemented with 2 μM nsp8, 50 mM NaCl, 4 mM MgCl2, 50 μM ATP and/or GTP, 0.17 μM [α-32P]ATP (*ATP), or [α-32P]GTP (*GTP), as indicated, and 1 μM the indicated RNA substrate. The reaction mixtures were incubated at 30°C for 60 min. Products were resolved in a TBE-buffered 12% polyacrylamide–7 M urea gel and visualized by phosphorimaging. Sizes (in nucleotides) of 5′-32P-labeled marker RNAs (lane M) are indicated to the left. Lane –, activity assay performed with KR01/JTR1 in the absence of nsp8.
FIG 6.

HCoV-229E nsp8 TATase activities on partially double-stranded substrate RNAs with different 10-nt 5′ overhangs. (A) RNA substrates used in the experiment. Bio, RNA 3′ biotinylation. (B) nsp8 activity assays were performed in standard reaction buffer in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP using the RNA substrates indicated above the autoradiograms. (C) nsp8 activity assays were performed in standard reaction buffer using the RNA substrates indicated above the autoradiograms. Reactions were performed in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP (lanes 1 to 4), 100 μM UTP and 0.17 μM [α-32P]UTP (lane 5), and 100 μM GTP and 0.17 μM [α-32P]GTP (lane 6). The reaction mixtures were incubated at 30°C for 60 min. Products were resolved in a TBE-buffered 12% polyacrylamide–7 M urea gel and visualized by phosphorimaging. In lane M, 5′-32P-labeled oligoribonucleotides were loaded as markers. Sizes (in nucleotides) are indicated.
HCoV-229E nsp8 has oligo(U)-assisted TATase activity.
Although we failed to obtain evidence for generic RNA template-dependent RNA polymerase activities, a number of preliminary experiments (Fig. 5 and data not shown) indicated that the 3′-adenylation activity of nsp8 on double-stranded RNA may be affected by the presence or absence of specific 5′ single-stranded regions in the bottom strand. To explore more directly the role of single-stranded RNA regions with various sequences located opposite the strand with ongoing 3′ adenylation, partially double-stranded RNAs were designed in which the top strand (KR07) was hybridized to a cRNA called KR07comp-b. In this case, the bottom strand was 3′ biotinylated to block any potential nsp8-mediated 3′ adenylation of this strand. In addition to this basic construct, a range of 3′-biotinylated bottom strands with different 5′ overhangs, including U10, C5U5, U5C5, C10, and A10, was synthesized and hybridized to KR07 (Fig. 6A). As controls, a number of dsRNAs in which both the top and the bottom strands were 3′ biotinylated were included in these experiments (Fig. 6A).
In line with previous experiments, HCoV-229E nsp8 was found to have robust TATase activity using the single-stranded RNA KR07 as a substrate (Fig. 6B, lane 3). A similar TATase activity was observed for the KR07/KR07comp-b RNA hybrid, suggesting that (fully) double-stranded RNA can be equally efficiently 3′ polyadenylated by nsp8 (Fig. 6B, lane 4). As shown before, 3′-biotinylated forms of single- or double-stranded RNA substrates were not polyadenylated (Fig. 6B, lanes 2, 5, and 6). Maximum 3′ polyadenylation of the top strand was observed for an RNA hybrid in which the bottom strand had an unpaired 5′ U10 overhang (Fig. 6B, lane 8), and only slightly lower activities were observed for 5′ overhangs with U5C5 and C5U5 sequences (Fig. 6B, lanes 7 and 9). In contrast, we failed to detect TATase activity for dsRNA substrates containing 5′ A10 and C10 overhang regions (KR07/A10KR07comp-b and KR07/C10KR07comp-b, respectively) (Fig. 6B, lanes 10 and 11). For the last two substrates, labeled products could also not be detected if the matching nucleotides, UTP and GTP, respectively, were included in the reaction mixture (Fig. 6C). Taken together, the data show that HCoV-229E nsp8 TATase activity is strongly stimulated by the presence of a short (≥5-nt) oligo(U) stretch in the opposite strand, suggesting that nsp8 uses the oligo(U) stretch (or a part of it) as a template. To corroborate this hypothesis, we modified the KR01-containing substrates used in the previous experiment (Fig. 6) to include longer 5′ single-stranded regions in the bottom strand, (i) a mixed (UC)10 sequence and (ii) a homooligomeric U20 sequence (Fig. 7A). If the KR01 3′ end was blocked with biotin, no radiolabeled product was generated, while TATase activity was readily detectable for KR01 with a free 3′-hydroxyl group (Fig. 7B; compare lanes 2 and 3). There was no detectable activity using a substrate containing a mixed (CU)10 sequence in the 5′ single-stranded region of the bottom strand (Fig. 7B, lane 4), while very efficient TATase activity was observed for an RNA hybrid containing an unpaired 5′ U20 sequence in the bottom strand (Fig. 7B; compare lanes 4 and 7), providing additional evidence for the proposed poly(U)-templated TATase activity of nsp8. In line with previous experiments (see above), we failed to detect radiolabeled products in reactions using 3′-biotinylated dsRNA substrates (KR01-b/JTR1-b, KR01-b/U20JTR1comp-b), confirming the requirement of a free 3′ hydroxyl group for nsp8 TATase activity (Fig. 7B, lanes 4 and 5; Fig. 6).
FIG 7.

HCoV-229E nsp8 TATase activity on partially double-stranded substrate RNAs with different 20-nt 5′ overhangs. (A) RNA substrates used in the experiment. Bio, RNA 3′ biotinylation. (B) nsp8 activity assays were performed in standard reaction buffer in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP using the RNA substrates shown in panel A. The reaction mixtures were incubated at 30°C for 60 min. The products were resolved in a TBE-buffered 12% polyacrylamide–7 M urea gel and visualized by phosphorimaging. In lane M, 5′-32P-labeled ribooligonucleotides were loaded as markers. Sizes (in nucleotides) are indicated to the left.
Next, we tested if SARS-CoV nsp8 and FIPV nsp8 display the same poly(U) template-assisted TATase activity described above for the HCoV-229E homolog. To this end, recombinant forms of SARS-CoV and FIPV nsp8 were produced in E. coli (see Materials and Methods) and used in reactions in which the reaction mixtures were supplemented with 2 μM the appropriate nsp8, 50 mM NaCl, 1 mM MgCl2, 100 μM ATP, 0.17 μM [α-32P]ATP, and 1 μM substrate RNA (Fig. 8A). Using a partially double-stranded RNA substrate with an unpaired 5′ U20 sequence in the bottom strand, we found that all three nsp8s displayed robust TATase activity resulting in a product of ≥40 nt (Fig. 8B, lanes 3, 5, and 7). Among the three coronavirus nsp8s used in this experiment, SARS-CoV nsp8 displayed the most efficient TATase activity with this particular substrate (Fig. 8B, lane 7). Together, these data suggest that oligo(U) template-assisted TATase activity is a conserved feature of coronavirus nsp8 proteins.
FIG 8.
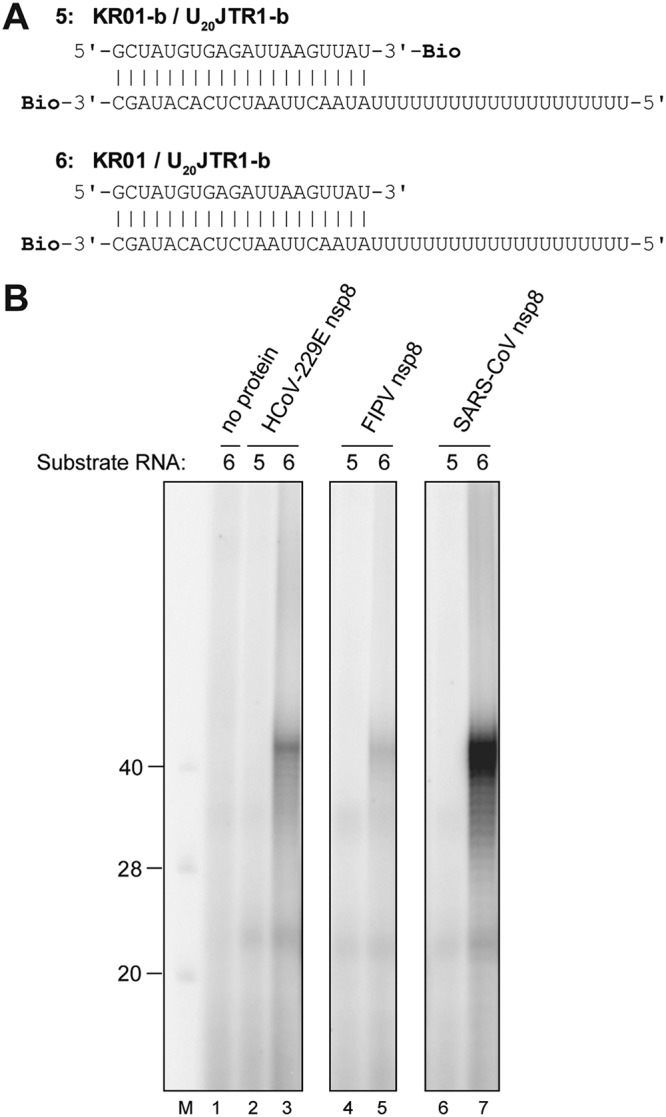
Oligo(U)-templated TATase activities of representative coronavirus nsp8 proteins. (A) RNA substrates used in the experiment. Bio, RNA 3′ biotinylation. (B) Activity assays were performed in standard reaction buffer with the indicated recombinant proteins in the presence of 100 μM ATP and 0.17 μM [α-32P]ATP using the substrate RNAs shown in panel A. The reaction mixtures were incubated at 30°C for 60 min. The products were resolved in a TBE-buffered 12% polyacrylamide–7 M urea gel and visualized by phosphorimaging (selected lanes from the same autoradiogram are shown). 5′-32P-labeled oligonucleotides were loaded as markers (lane M). Sizes (in nucleotides) are indicated to the left.
Role of conserved Lys residues in HCoV-229E nsp8-mediated RNA-binding and TATase activities and viral replication.
Previous structural and biochemical studies identified a number of residues to be essential for nsp8 polymerase/primase activity and protein-protein and protein-RNA interactions (12–14, 23, 24). To assess the potential role in TATase activity of two HCoV-229E nsp8 residues equivalent to conserved Lys residues shown previously to be important for SARS-CoV nsp8 primase activity and SARS-CoV replication (14, 23), we produced mutant forms of HCoV-229E nsp8 in which polyprotein 1a (pp1a) residues Lys-3687 and Lys-3711 were replaced with Ala (Fig. 1). The proteins were produced in E. coli and purified to apparent homogeneity using metal ion affinity and anion-exchange chromatography (Fig. 1B). The TATase activities of the mutant proteins were determined using the reaction conditions established for the wild-type enzyme, and KR07 and U18 were used as test substrates in these assays. As shown in Fig. 9, no TATase activity was detected for the nsp8_K3711A protein, while nsp8_K3687A retained its TATase activity on the two RNA substrates used in this experiment (Fig. 9A and B). Next, we determined the RNA-binding activities of the wild-type and mutant nsp8 proteins using a range of RNAs (A18, U18, C18, and KR07) (see Materials and Methods). As shown in Fig. 9C and D, nsp8_K3687A bound RNA less efficiently than the wild-type protein. The reduced RNA-binding activity of nsp8_K3687A was particularly evident using A18 and C18 (Fig. 9C), while the protein retained RNA-binding activity to U18 and KR07 (Fig. 9C and D). In contrast, the K3711A substitution abolished the RNA-binding activity of the protein completely (Fig. 9C and D), suggesting that the lack of TATase activity was probably caused by the insufficient RNA-binding capacity of this protein.
FIG 9.

TATase and RNA-binding activities of wild-type and mutant forms of HCoV-229E nsp8. (A and B) Results of TATase assays performed in standard reaction buffer using the indicated wild-type and mutant HCoV-229E nsp8 proteins and substrate RNAs (U18 and KR07). The products were resolved in TBE-buffered 12% polyacrylamide–7 M urea gels and visualized by phosphorimaging. (C and D) RNA-binding activities of wild-type and mutant HCoV-229E nsp8 proteins with the indicated 5′-32P-labeled homopolymeric substrate RNAs (C) and the heteropolymeric substrate RNA KR07 (D). Products were separated in nondenaturing 10% polyacrylamide gels and visualized by phosphorimaging. The positions of free and protein-bound 32P-labeled RNAs are indicated to the right.
In a final set of experiments, we examined the possible effects of the pp1a/pp1ab K3687A and K3711A substitutions in the context of viral replication in cell culture. Full-length HCoV-229E genome RNAs containing the desired mutations were generated using the reverse genetics system developed by Thiel et al. (27). Following cotransfection of Huh-7 cells with 1.25 μg of in vitro-transcribed 5′-capped full-length HCoV-229E RNA (wild-type and nsp8 mutants) along with 0.75 μg N mRNA, the cells were incubated at 33°C. Determination of the virus titers in the cell culture supernatants collected at 72 h posttransfection revealed that wild-type HCoV-229E was readily recovered with titers of >106 PFU/ml, while no viable viruses could be rescued after transfection of mutant RNAs (HCoV-229E_K3687A and HCoV-229E_K3711A), suggesting critical roles of nsp8-mediated activities in viral replication.
DISCUSSION
Although previous studies on coronavirus nsp8 proteins (including their complexes with nsp7) were focused on recombinant forms of nsp8 of only two viruses, SARS-CoV and FIPV, partly inconsistent or controversial information was obtained in several instances (6, 12, 14, 23, 24). With regard to enzymatic activities and possible roles in viral RNA synthesis, at least four functions have been suggested for nsp8. The protein was shown to be an essential cofactor (together with nsp7) for RdRp activity in vitro. Additional functions were suggested to include (i) a function as a processivity factor for the viral RdRp (in a hexadecameric complex with nsp7), (ii) noncanonical RdRp (primase) activity, and (iii) primer-dependent and de novo RdRp activities (alone or in complex with nsp7) (12–14, 23, 24). Possibly, some of the discrepant findings of these studies may be explained by the presence of a few extra residues in the respective protein constructs (6). Indeed, there is evidence that the presence and identities of N-terminal tags may affect the activities of SARS-CoV and FIPV nsp8 (12, 24). In an effort to revisit the diverse polymerase activities reported previously for two coronavirus nsp8 proteins, we decided to focus on the isolated nsp8 protein by characterizing the activities of the HCoV-229E nsp8 (representing the genus Alphacoronavirus) and, subsequently, extended major findings arising from this work to previously characterized nsp8 homologs from SARS-CoV (genus Betacoronavirus) and FIPV (genus Alphacoronavirus). To produce and purify these proteins in an active form, we took advantage of a previously established system suitable to generate C-terminally His6-tagged forms of nsp8 with their authentic N-terminal residues in a T7 polymerase-free system, thereby eliminating the risk of T7 RNA polymerase contamination, which may interfere with subsequent analyses of the viral RdRp activities produced in E. coli (24, 25). The proteins were purified from E. coli by metal ion affinity and anion-exchange chromatography and characterized in vitro using protocols optimized in this study.
Using SARS-CoV and HCoV-229E nsp8, we failed to obtain evidence for primase activity for these proteins (that is, template-dependent de novo polymerase activity resulting in short oligonucleotides) on any of the substrates used, which is in agreement with the findings of a study by Subissi et al. (14) but contradicts an earlier report (23). We, however, found that HCoV-229E nsp8 transfers (ribo)nucleotides to the 3′ end of substrate RNAs, with ATP being the clearly preferred nucleotide. Importantly, this activity was not detectable if substrates in which the 3′-hydroxyl group was blocked were used, thus essentially excluding the possibility that template-dependent de novo polymerase activities are involved in the production of the radiolabeled products obtained in our assays. Also, the vast majority of products were larger than the substrates used, and virtually identical products were generated from a partially double-stranded RNA containing a single-stranded heteropolymeric (C/U) sequence in reactions in which the reaction mixtures contained either ATP alone or ATP in combination with GTP, arguing against a template-dependent copy process being involved in the production of the labeled RNAs seen in the autoradiograms (Fig. 5). The latter hypothesis is supported by the lack of radiolabeled RNAs being produced if the 3′ end of the bottom strand was blocked with biotin (KR01/JTR1-b), indicating that essentially all radiolabeled products seen for KR01/JTR1 in Fig. 5 (lanes 2 and 4) were produced by 3′ polyadenylation of the bottom strand, JTR1 (i.e., at the substrate's blunt end), rather than a copy process of the template, resulting in the 3′ extension of the top strand, KR01 (to fill up the 3′ recessive end of the substrate).
A very weak 3′ extension of single-stranded substrate RNAs was also observed in the presence of CTP and UTP, while there was no detectable incorporation of GTP in any of the substrates used (Fig. 3 and 5). The nucleotide selectivity of the nsp8-mediated terminal transferase activity resembles that of eukaryotic poly(A) polymerases (PAPs), which are known to be highly specific for ATP (28, 29). The efficient 3′-terminal polynucleotidylation of RNA substrates by PAPs in the presence of ATP (but not CTP, UTP, and GTP) has been suggested to involve nucleotide base stacking (see below). PAPs have been reported to exhibit 800-fold higher catalytic efficiencies with ATP than with GTP (30). Both the different electrostatic properties and shapes of the two purine nucleotides were suggested to be involved in the distinct binding and use of ATP by PAPs (31, 32). Based on the data obtained in this study, it seems reasonable to suggest that coronavirus nsp8 homologs are RNA-specific TATases that probably act in a distributive manner.
Similar to cellular PAPs, many viral RNA polymerases, including picornavirus, calicivirus, flavivirus, nodavirus, alphavirus, and bacteriophage ϕ6 polymerases, have been reported to add nontemplated nucleotides to the 3′ ends of RNA products, and it has been suggested that the addition of a few extra nucleotides reflects an intrinsic terminal transferase activity of (some of) these RdRps (33–41). These studies also showed that the TNTase activity requires the presence of a 3′-hydroxyl group, as confirmed by experiments using blocked 3′ ends, which abolished this activity (40–42). Similarly, although it is not apparently related to viral RdRp, the HCoV-229E nsp8 was shown to have TATase activity on homopolymeric and heteropolymeric RNA substrates, unless their 3′ ends were blocked by 3′ biotinylation (Fig. 3, 6, 7, and 8). The nsp8-associated RNA 3′ nucleotidyl terminal transferase activity displayed a strong preference for ATP (over CTP and UTP), while GTP and dATP were not incorporated.
In previous studies, metal ions were demonstrated to affect the nucleotide selectivity of poliovirus 3Dpol in vitro (33). The use of Mn2+ was found to relax nucleotide selectivity, resulting in decreased RdRp fidelity (33, 43, 44). Nucleotide misincorporations and pronounced primer cleavage occurred in the presence of Mn2+ but not (or much less so) in the presence of Mg2+ ions (33). Mn2+ ions were also reported to affect alphavirus nsP4 (RdRp) TATase activity (39). Because of these special effects of Mn2+ ions when used at (nonphysiologically) high concentrations, we decided to use Mg2+ ions in (almost all) the HCoV-229E nsp8 activity assays included in this study, even though Mn2+ was confirmed to be equally effective in supporting HCoV-229E nsp8-mediated TATase activity (Fig. 2B). Our decision to use Mg2+ ions instead of Mn2+ ions was also prompted by data obtained in a previous study in which a sequence-specific (yet low-fidelity) RdRp activity mediated by SARS-CoV nsp8 was observed in reactions with reaction mixtures containing Mn2+ ions (23), while this particular nsp8-associated activity was no longer observed in a more recent study using Mg2+ ions as a metal ion cofactor in RdRp assays including nsp8 (14).
Although this remains to be formally proven, it is tempting to suggest that the TATase activity established in this study for HCoV-229E nsp8 may have a role in the 3′ polyadenylation of viral RNAs. It has been confirmed for bovine coronavirus (BCoV) that viral plus-strand RNAs are 3′ polyadenylated, while the 5′ ends of negative strands contain a poly(U) track of 9 to 26 nt (45). The efficient nsp8-mediated 3′ polyadenylation of substrate RNAs in the presence of a 5′-oligouridylated template supports the idea that nsp8 may have functions that resemble those of cellular PAPs (see below). Even though the crystal structures of SARS-CoV and FIPV nsp8 have been reported (12, 13), the structural basis for the selective use of ATP for the nsp8-associated terminal transferase activity remains to be established, for example, by further structural studies of nsp8 in the presence of ATP analogs. Based on previous studies on cellular PAPs (which, however, are not evidently related to coronavirus nsp8 proteins), it seems safe to predict that multiple interactions are required for NTP selectivity, RNA substrate binding, and catalysis (30).
With respect to substrate specificity, we were interested to examine if (and to what extent) the identities of the 3′-terminal nucleotides affect the HCoV-229E nsp8-mediated TATase activity. We found that replacement of the 3′-terminal cytidylate present in KR07 and JZR3 with adenylate resulted in increased nsp8 TATase activities (Fig. 4B and C). Also, in the presence of a 3′-terminal guanylate, the nsp8 TATase activity was enhanced, suggesting that purines are preferred over pyrimidines at the 3′-terminal position. Similar observations were previously made for the TNTase activity of the HCV RdRp (35), where a C-to-G substitution at the 3′-terminal position resulted in enhanced 3′ adenylation. Early studies performed in the 1960s proposed differential stacking interactions between different types of nucleobases to be responsible for this preference: purine-purine > purine-pyrimidine > pyrimidine-pyrimidine (46–48). Theoretical studies indicated that A-A stacking (purine-purine) is more favorable than A-C stacking (purine-pyrimidine) (49, 50). As mentioned above, nsp8 TATase activity was found to be increased if the 3′-terminal pyrimidine (cytidylate) present in two oligonucleotide substrates was replaced with purine (A or G) (Fig. 4B and C), while a moderate (or no) stimulation of activity was observed when the cytidylate was replaced with another pyrimidine (uridylate). The effects observed for 3′-terminal nucleotide substitutions support the idea that a differential base-stacking interaction may have a role in the initiation of 3′ polyadenylation. Similar to the observations made in our study, a 10-fold lower catalytic efficiency was reported for a cellular PAP using an A18 substrate in which the 3′-terminal A was replaced with C (A17C) (51). In line with this interpretation, we found that replacements of the penultimate nucleotide had no detectable effect on nsp8 TATase activity (data not shown).
In the context of an earlier study addressing possible primer-dependent and de novo RdRp activities of the SARS-CoV nsp8 and an nsp7-nsp8 complex, evidence was presented that (if a primer/template complex is provided) nsp8 is able to generate radiolabeled RNAs with sizes that exceed those of the template, suggesting nontemplated 3′ TNTase activity (24). This observation is consistent with what we propose here for the HCoV-229E nsp8 homolog. In contrast, however, our data do not support the template-dependent copy process described in that earlier study. Our data rather suggest that the near-substrate-length labeled products observed in these experiments represent 3′ mono- or oligoadenylated forms of the template strand. To test this hypothesis, we used the primed template described previously (24) and analyzed the radiolabeled products generated by nsp8 in the presence of different (combinations of) nucleotides. Labeled products could be detected only if [32P]ATP was included in the reaction mixture, while [32P]GTP was not incorporated into any of the products, thereby ruling out the possibility that the labeled RNA product represented a copy of the (CU)10 template (Fig. 5 and 6 and data not shown). We also observed that, at low ATP concentrations, only one major product (of approximately substrate length and probably representing a 3′-monoadenylated RNA) was produced, while higher ATP concentrations (50 to 100 μM) resulted in polyadenylated products. Importantly, no such products were observed in the presence of [α-32P]GTP (Fig. 5 and data not shown), once again supporting the adenylate-specific 3′-terminal ribonucleotidyl transferase activity established in this study.
To address the possible roles of specific RNA structures and sequences in supporting the TNTase activity of nsp8, a series of partial-duplex RNAs with one strand representing the 3′ end of the HCoV-229E genome and the other representing the complementary 5′ end of the viral antigenome (with and without 5′ single-stranded tails) was used in activity assays (Fig. 6A). Using an RNA hybrid containing a 5′ overhang with a short oligo(U) stretch (U10, U5C5, and C5U5), a very efficient polyadenylation of KR07 was observed, while nearly no TATase activity was observed if the partially double-stranded RNAs contained 5′ tails with A10 or C10 sequences in the bottom strand (Fig. 6B). The same result was obtained in reactions in which the reaction mixtures included appropriate complementary nucleotides (UTP and GTP) (Fig. 6C), again confirming the nucleotide selectivity of nsp8. Furthermore, the data show that HCoV-229E nsp8 TATase is equally active on single-stranded and (the blunt ends of) double-stranded RNA substrates (Fig. 6B, lanes 3 and 4), whereas recessive 3′ ends in partially double-stranded RNAs are clearly disfavored (Fig. 5 and 6), unless the bottom strand contains a stretch of uridylates. While it is clear that the nsp8 TATase does not strictly require an oligo(U) template, the activity was greatly stimulated by the presence of a single-stranded oligo(U) sequence opposite the 3′ polyadenylation site, with U5–10 stretches stimulating polyadenylation activity most efficiently. Interestingly, the position of the U5 sequence in the mixed sequence (C5U5 or U5C5) was not critical, possibly indicating low processivity and repeated use of the oligo(U) template. The situation appeared to be different if the 5′ oligo(U) tail was extended up to 20 nucleotides. In this case, a rather defined product which was not efficiently extended beyond template length (42 nt) was observed. This was possibly caused by the lack of a suitable oligo(U) template once an extended (and, thus, stable) A-U base-paired structure had been formed by the 3′ polyadenylation reaction.
Based on the data presented in this study, it seems reasonable to suggest a model in which the nsp8-mediated TATase (in conjunction with other RTC components) acts to 3′ polyadenylate genomic RNA and subgenomic mRNAs in an oligo(U)-assisted manner. If confirmed, this mechanism would resemble the mechanisms (including polyadenylation signals) employed by negative-strand RNA viruses to produce polyadenylated mRNAs (52–55) and the mechanisms proposed for the poliovirus polymerase (56–58), while cellular PAPs generally add poly(A) tails in a template-independent manner and depend on specific polyadenylation signals (59). The factors and mechanisms that drive and regulate 3′ poly(A) tailing of coronavirus plus-strand RNAs are currently unclear and deserve further investigation.
In a previous study, the SARS-CoV nsp8 Lys-58 residue was implicated in the dsRNA-binding activity of the nsp7-nsp8 complex (13). We replaced the homologous Lys residue in the HCoV-229E nsp8 (K3687A) and examined the RNA-binding and TATase activities of this protein. Consistent with data obtained for the corresponding SARS-CoV nsp8 mutant nsp8_K58A (13, 14, 24), the RNA-binding activity of HCoV-229E nsp8_K3687A was found to be reduced by approximately 50% (Fig. 9C and D). Despite this reduced RNA-binding affinity, the TATase activity of nsp8_K3687A was not affected or was even higher than that of the wild-type protein (Fig. 7A and B). Earlier studies reported a complete abolishment of nsp8 primase and polymerase activities for the equivalent substitution in the SARS-CoV nsp8 (K58A) (23, 24). Subsequently, the same replacement was shown to abolish de novo initiation, while it retained significant primer extension activity in polymerase assays of the nsp7-nsp8-nsp12 complex (14). Although the basis of these differential effects is currently unclear, the data obtained in these different virus systems and assays lead us to suggest that this particular substitution retains (some) in vitro activities. The remaining ∼50% RNA-binding affinity of HCoV-229E nsp8_K3687A was probably sufficient to retain TATase activity, while the SARS-CoV equivalent retained some of its capacity to support the polymerase activity of the nsp7-nsp8-nsp12 complex (14). However, these functional defects resulted in nonviable viruses in both cases (no RNA accumulation or virus reproduction) (14), reinforcing the important role of a fully functional nsp8 in viral replication.
Replacement of another highly conserved lysine residue in nsp8, HCoV-229E pp1a Lys-3711, with Ala resulted in a >90% reduction of RNA-binding activity (Fig. 9B and D), while the TATase activity of this protein was completely abolished using two different substrates (Fig. 7A and C). A mutant HCoV-229E full-length RNA containing this particular mutation did not give rise to viable virus following transfection into Huh-7 cells. The HCoV-229E nsp8 mutagenesis data are thus in agreement with biochemical data reported for the equivalent substitution in SARS-CoV nsp8 (23) as well as reverse genetics data for this virus. A SARS-CoV mutant containing the equivalent mutation in nsp8 was shown to be crippled and rapidly evolved compensatory mutations to restore nearly wild-type growth kinetics in cell culture (14). It should also be mentioned that the complete loss of TATase activity in the HCoV-229E_K3711A protein represents an important control because it excludes that potential contaminations with bacterial RNA 3′ polyadenylation activities were responsible for the TATase activity obtained with reaction mixtures containing the recombinant HCoV-229E nsp8 wild-type protein produced and purified under identical conditions.
In summary, our study is in line with previous reports (12, 23, 24) by showing that coronavirus nsp8 proteins act as nucleotidyl transferases. However, it does not support previous suggestions on nsp8-associated primase and RdRp activities. Based on our findings, we consider it possible that, in those earlier studies, coronavirus nsp8 polymerase activity data were overinterpreted because two important controls were omitted: (i) there was no proof to show that [α-32P]GMP (in addition to [α-32P]AMP) was actually incorporated into the product during primed RNA synthesis from a CU template and (ii) no 3′-blocked template RNA was used in de novo RdRp assays to exclude the possibility that the presumed full-length transcripts identified in these assays represented 3′-mono- or oligonucleotidylated forms of the template RNA (12, 24). Despite these considerations, we think that the combined data from this and earlier studies provide sufficient evidence to suggest divalent metal ion-dependent RNA 3′ TATase activities for coronavirus nsp8 proteins. This activity is strongly enhanced by the presence of an oligo(U) template strand and can be blocked by replacing a single conserved Lys residue with Ala. While it is clear from this and a previous study that nsp8 is critically involved in coronavirus replication and acts as a cofactor for nsp12-mediated RdRp activity in vitro (14), the diverse functions of nsp8 (both alone and in complex with other replicase subunits) remain to be investigated in more detail, including a possible involvement of the TATase activity in the production of 3′-polyadenylated plus-strand RNAs.
MATERIALS AND METHODS
Cloning, mutagenesis, and protein production.
To produce HCoV-229E nsp8 in Escherichia coli, the coding sequence of HCoV-229E pp1a residues 3630 to 3824 was amplified by reverse transcription-PCR (RT-PCR) from viral RNA isolated from HCoV-229E-infected Huh-7 cells and inserted into pASK3-Ub-CHis6 using restriction- and ligation-free cloning methods (60, 61). The resulting plasmid encoded the full-length HCoV-229E nsp8 fused to an N-terminal ubiquitin tag and a C-terminal His6 tag (25, 26). Similarly, the coding sequence of FIPV nsp8 (strain 79/1146; GenBank accession number DQ010921; pp1a residues 3506 to 3700) was inserted into pASK3-Ub-CHis6 plasmid DNA and used to produce an N-terminally ubiquitin-tagged and C-terminally hexahistidine-tagged form of FIPV nsp8. For the production of SARS-CoV nsp8, plasmid pASK3-Ub-nsp8-CHis was used (24). The protein expressed from this plasmid comprised residues 3920 to 4117 of the pp1a of SARS-CoV (strain Frankfurt-1; GenBank accession number AY291315) fused to an N-terminal ubiquitin tag and a short C-terminal sequence, GSSGHHHHHH, including a hexahistidine tag. For all plasmid constructs, expression was under the control of a tetracycline-inducible promoter. Site-specific mutagenesis of the plasmid constructs was done using a PCR-based approach (60, 61). The primers used for cloning and mutagenesis are available upon request.
Heterologous expression of nsp8 in E. coli and protein purification.
For the production of wild-type and mutant coronavirus nsp8 proteins, E. coli TB1 cells were used. Bacteria were cotransformed with the appropriate pASK3-Ub expression construct and pCGI plasmid DNA, the latter of which encodes the ubiquitin-specific carboxyl-terminal hydrolase 1 (Ubp1) (25). LB medium containing ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) was inoculated with an overnight culture of E. coli TB1 cells carrying the appropriate plasmids, and the culture was incubated in a shaking incubator at 37°C. At an optical density at 600 nm of 0.4, protein production was induced with anhydrotetracycline (AHT; 200 ng/ml; IBA Lifesciences) and the cells were incubated for another 17 h at 17°C under vigorous shaking. Next, the cells were harvested by centrifugation (3,000 × g for 10 min) and resuspended in ice-cold NP7a buffer (20 mM Tris-Cl, pH 8.0, 250 mM NaCl, 10 mM imidazole, 15 mM β-mercaptoethanol). After addition of lysozyme (1.5 mg/ml) and EDTA-free protease inhibitor cocktail (Roche) and incubation for 30 min on ice, the cells were sonicated (20 10-s pulses) and insoluble material was removed by centrifugation for 30 min at 40,000 × g at 4°C. The supernatant was incubated with preequilibrated Ni-NTA agarose beads (Macherey-Nagel) for 2 h at 4°C under gentle agitation. The suspension was then loaded onto a disposable filter column and, after washing with 20 ml NP7a buffer and 20 ml buffer NP8a (20 mM Tris-HCl, pH 8.0, 250 mM NaCl, 20 mM imidazole, 15 mM β-mercaptoethanol), the protein was eluted with NP9a buffer (20 mM Tris-Cl, pH 8.0, 250 mM NaCl, 200 mM imidazole, 15 mM β-mercaptoethanol). Eluate fractions were analyzed by SDS-PAGE, and fractions containing nsp8 were pooled and subjected to anion-exchange chromatography using an ÄKTAprime plus instrument (GE Healthcare). To this end, eluate fractions from the Ni-NTA chromatography were diluted 10-fold with buffer A (20 mM Tris-Cl, pH 8.0, 5% [vol/vol] glycerol, 10 mM β-mercaptoethanol) and loaded onto a HiTrap Q column (1 ml; GE Healthcare). Nonspecifically bound proteins were removed by washing with 30 ml of A30 buffer (20 mM Tris-Cl, pH 8.0, 30 mM NaCl, 5% glycerol, 10 mM β-mercaptoethanol), and the recombinant protein was eluted using a continuous NaCl gradient (30 mM to 1 M) in buffer containing 20 mM Tris-Cl, pH 8.0, 5% glycerol, and 10 mM β-mercaptoethanol. Peak fractions containing the desired protein were identified by SDS-PAGE, pooled, dialyzed against storage buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 45% glycerol, 15 mM β-mercaptoethanol), and stored at −20°C until further use.
nsp8 activity assay.
For in vitro activity assays, the various forms of coronavirus nsp8 produced in this study were incubated with single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and partial-duplex substrate RNAs. In addition, a range of 3′-biotinylated substrate RNAs was used. Unless stated otherwise in the text, the reaction mixtures contained 50 mM Tris-Cl, pH 8.0, 50 mM NaCl, 1 MgCl2, 1% Triton X-100, 1 mM DTT, 1.5 mM β-mercaptoethanol, 4.5% glycerol, 1 μM substrate RNA, 100 μM the indicated NTP(s), 0.17 μM the indicated [α-32P]NTP(s), and 2 μM nsp8. The reaction mixtures were incubated for 60 min at 30°C, and the reactions were terminated by the addition of sodium acetate (pH 5.2; final concentration, 300 mM) and 10 volumes of ice-cold ethanol. Following centrifugation, the air-dried pellets were resuspended in PCR-grade proteinase K solution (final concentration, 1 mg/ml; Invitrogen) and incubated at 55°C for 15 min. Reactions were stopped by adding Fu-mix (6 M urea, 80% deionized formamide, 1× TBE [Tris-borate-EDTA], 0.1% [wt/vol] bromophenol blue, 0.1% [wt/vol] xylene cyanol). Following denaturation for 10 min at 65°C, the reaction products were separated in 1× TBE-buffered 12% polyacrylamide gels containing 7 M urea and analyzed by phosphorimaging using a Typhoon 9200 imager (GE Healthcare) and Quantity One software (Bio-Rad).
Electrophoretic mobility shift assay (EMSA).
For RNA-binding assays, denatured/renatured RNAs were used. First, 5′-32P-labeled RNAs (600 nM) were denatured in STE buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA) at 95°C for 2 min and then put on ice for 3 min. RNA refolding was done at room temperature for 10 min in buffer containing 50 mM Tris-HCl, pH 8.0, 7.5 mM NaCl, 1 mM MgCl2, 1% Triton X-100, 1 mM DTT. Next, the refolded RNA was mixed with nsp8 and incubated on ice for 1 h. As a control, a reaction was performed in the absence of nsp8. Typical reaction mixtures (10 μl total volume) contained 60 nM RNA, 2 μM nsp8, 55 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM MgCl2, 1% Triton X-100, 1 mM DTT, 1.5 mM β-mercaptoethanol, 4.5% glycerol, and 0.1 mM EDTA. After 1 h, the reaction mixtures were loaded onto 0.5× TBE-buffered, nondenaturing 10% polyacrylamide gels containing 5% glycerol and separated at 4°C by electrophoresis at a constant voltage (200 V) for 6 h. The dried gels were exposed to phosphorimaging screens, and the products were analyzed using a Typhoon 9200 imager and Quantity One software (Bio-Rad).
Substrate RNAs.
The synthetic RNAs used in this study were purchased from Integrated DNA Technologies (IDT). RNA hybrids (10 μM) were prepared by annealing two RNA oligonucleotides with (fully or partially) complementary sequences in STE buffer in a final volume of 100 μl. The mixture was incubated at 95°C for 5 min and then slowly cooled to room temperature. Annealed RNA hybrids were stored at −20°C until further use. For the sequences of the ribooligonucleotides used in this study, see Fig. 4, 5, 6, 8, and 9. In addition, the following RNAs were used: KR12, 5′-ACUUAAGUACCUUAUCUAUCUACAGAUA-3′; KR12-b, 5′-ACUUAAGUACCUUAUCUAUCUACAGAUA-3′-biotin; KR05, 5′-UAUCUGUAGAUAGAUAAGGUACUUAAGU-3; and KR13, 5′-CUUCCGUCUUAUGGC-CAGUCCAAAUAGU-3′.
ACKNOWLEDGMENTS
We thank Craig E. Cameron, Pennsylvania State University, for providing plasmid pCG1 and Eric J. Snijder, Leiden University Medical Center, for providing plasmid pASK3-Ub-SARS-CoV-nsp8-CHis6. We also thank Konstantin Ivanov for his contributions to experiments performed at an early stage of this work.
The work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 1021, A01; IRTG 1384) and the BBSRC (BB/G012067/1).
REFERENCES
- 1.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2012. Family Coronaviridae, p 806–828. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 2.Zumla A, Hui DS, Perlman S. 2015. Middle East respiratory syndrome. Lancet 386:995–1007. doi: 10.1016/S0140-6736(15)60454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res 117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziebuhr J. 2008. Coronavirus replicative proteins, p 65–81. In Perlman S, Gallagher T, Snijder EJ (ed), Nidoviruses. ASM Press, Washington, DC. [Google Scholar]
- 5.Masters PS, Perlman S. 2013. Coronaviridae, p 825–858. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Snijder EJ, Decroly E, Ziebuhr J. 2016. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res 96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevajol M, Subissi L, Decroly E, Canard B, Imbert I. 2014. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res 194:90–99. doi: 10.1016/j.virusres.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sola I, Mateos-Gomez PA, Almazan F, Zuñiga S, Enjuanes L. 2011. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol 8:237–248. doi: 10.4161/rna.8.2.14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sola I, Almazan F, Zuniga S, Enjuanes L. 2015. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol 2:265–288. doi: 10.1146/annurev-virology-100114-055218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehmann KC, Gulyaeva A, Zevenhoven-Dobbe JC, Janssen GM, Ruben M, Overkleeft HS, van Veelen PA, Samborskiy DV, Kravchenko AA, Leontovich AM, Sidorov IA, Snijder EJ, Posthuma CC, Gorbalenya AE. 2015. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res 43:8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. 1989. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res 17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao Y, Ma Q, Restle T, Shang W, Svergun DI, Ponnusamy R, Sczakiel G, Hilgenfeld R. 2012. Nonstructural proteins 7 and 8 of feline coronavirus form a 2:1 heterotrimer that exhibits primer-independent RNA polymerase activity. J Virol 86:4444–4454. doi: 10.1128/JVI.06635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhai Y, Sun F, Li X, Pang H, Xu X, Bartlam M, Rao Z. 2005. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat Struct Mol Biol 12:980–986. doi: 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subissi L, Posthuma CC, Collet A, Zevenhoven-Dobbe JC, Gorbalenya AE, Decroly E, Snijder EJ, Canard B, Imbert I. 2014. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc Natl Acad Sci U S A 111:E3900–E3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferron F, Subissi L, Silveira De Morais AT, Le NTT, Sevajol M, Gluais L, Decroly E, Vonrhein C, Bricogne G, Canard B, Imbert I. 2018. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc Natl Acad Sci U S A 115:E162–E171. doi: 10.1073/pnas.1718806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, Ziebuhr J. 2006. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci U S A 103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith EC, Blanc H, Surdel MC, Vignuzzi M, Denison MR. 2013. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog 9:e1003565. doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith EC, Sexton NR, Denison MR. 2014. Thinking outside the triangle: replication fidelity of the largest RNA viruses. Annu Rev Virol 1:111–132. doi: 10.1146/annurev-virology-031413-085507. [DOI] [PubMed] [Google Scholar]
- 19.Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell TB, Spiro DJ, Denison MR. 2010. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog 6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckerle LD, Lu X, Sperry SM, Choi L, Denison MR. 2007. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J Virol 81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ulferts R, Ziebuhr J. 2011. Nidovirus ribonucleases: structures and functions in viral replication. RNA Biol 8:295–304. doi: 10.4161/rna.8.2.15196. [DOI] [PubMed] [Google Scholar]
- 22.Becares M, Pascual-Iglesias A, Nogales A, Sola I, Enjuanes L, Zuñiga S. 2016. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the innate immune response. J Virol 90:5399–5414. doi: 10.1128/JVI.03259-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imbert I, Guillemot JC, Bourhis JM, Bussetta C, Coutard B, Egloff MP, Ferron F, Gorbalenya AE, Canard B. 2006. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J 25:4933–4942. doi: 10.1038/sj.emboj.7601368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.te Velthuis AJ, van den Worm SH, Snijder EJ. 2012. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res 40:1737–1747. doi: 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. 1999. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr Purif 17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 26.Te Velthuis AJ, Arnold JJ, Cameron CE, van den Worm SH, Snijder EJ. 2010. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res 38:203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiel V, Herold J, Schelle B, Siddell SG. 2001. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J Gen Virol 82:1273–1281. doi: 10.1099/0022-1317-82-6-1273. [DOI] [PubMed] [Google Scholar]
- 28.Hafe LA, Keller EB. 1975. The polyadenylate polymerases from yeast. J Biol Chem 250:1838–1846. [PubMed] [Google Scholar]
- 29.Tsiapalis CM, Dorson JW, De Sante DM, Bollum FJ. 1973. Terminal riboadenylate transferase: a polyadenylate polymerase from calf thymus gland. Biochem Biophys Res Commun 50:737–743. doi: 10.1016/0006-291X(73)91306-5. [DOI] [PubMed] [Google Scholar]
- 30.Balbo PB, Bohm A. 2007. Mechanism of poly(A) polymerase: structure of the enzyme-MgATP-RNA ternary complex and kinetic analysis. Structure 15:1117–1131. doi: 10.1016/j.str.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moodie SL, Mitchell JB, Thornton JM. 1996. Protein recognition of adenylate: an example of a fuzzy recognition template. J Mol Biol 263:486–500. doi: 10.1006/jmbi.1996.0591. [DOI] [PubMed] [Google Scholar]
- 32.Nobeli I, Laskowski RA, Valdar WS, Thornton JM. 2001. On the molecular discrimination between adenine and guanine by proteins. Nucleic Acids Res 29:4294–4309. doi: 10.1093/nar/29.21.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnold JJ, Cameron CE. 1999. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J Biol Chem 274:2706–2716. doi: 10.1074/jbc.274.5.2706. [DOI] [PubMed] [Google Scholar]
- 34.Neufeld KL, Galarza JM, Richards OC, Summers DF, Ehrenfeld E. 1994. Identification of terminal adenylyl transferase activity of the poliovirus polymerase 3Dpol. J Virol 68:5811–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ranjith-Kumar CT, Gajewski J, Gutshall L, Maley D, Sarisky RT, Kao CC. 2001. Terminal nucleotidyl transferase activity of recombinant Flaviviridae RNA-dependent RNA polymerases: implication for viral RNA synthesis. J Virol 75:8615–8623. doi: 10.1128/JVI.75.18.8615-8623.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poranen MM, Koivunen MR, Bamford DH. 2008. Nontemplated terminal nucleotidyltransferase activity of double-stranded RNA bacteriophage phi6 RNA-dependent RNA polymerase. J Virol 82:9254–9264. doi: 10.1128/JVI.01044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fullerton SW, Blaschke M, Coutard B, Gebhardt J, Gorbalenya A, Canard B, Tucker PA, Rohayem J. 2007. Structural and functional characterization of sapovirus RNA-dependent RNA polymerase. J Virol 81:1858–1871. doi: 10.1128/JVI.01462-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rohayem J, Jager K, Robel I, Scheffler U, Temme A, Rudolph W. 2006. Characterization of norovirus 3Dpol RNA-dependent RNA polymerase activity and initiation of RNA synthesis. J Gen Virol 87:2621–2630. doi: 10.1099/vir.0.81802-0. [DOI] [PubMed] [Google Scholar]
- 39.Tomar S, Hardy RW, Smith JL, Kuhn RJ. 2006. Catalytic core of alphavirus nonstructural protein nsP4 possesses terminal adenylyltransferase activity. J Virol 80:9962–9969. doi: 10.1128/JVI.01067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu W, Wang Z, Xia H, Liu Y, Qiu Y, Liu Y, Hu Y, Zhou X. 2014. Flock house virus RNA polymerase initiates RNA synthesis de novo and possesses a terminal nucleotidyl transferase activity. PLoS One 9:e86876. doi: 10.1371/journal.pone.0086876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Qiu Y, Liu Y, Qi N, Si J, Xia X, Wu D, Hu Y, Zhou X. 2013. Characterization of a nodavirus replicase revealed a de novo initiation mechanism of RNA synthesis and terminal nucleotidyltransferase activity. J Biol Chem 288:30785–30801. doi: 10.1074/jbc.M113.492728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ranjith-Kumar CT, Gutshall L, Kim MJ, Sarisky RT, Kao CC. 2002. Requirements for de novo initiation of RNA synthesis by recombinant flaviviral RNA-dependent RNA polymerases. J Virol 76:12526–12536. doi: 10.1128/JVI.76.24.12526-12536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang SG, Klingenberg M. 1996. Two-stage nucleotide binding mechanism and its implications to H+ transport inhibition of the uncoupling protein from brown adipose tissue mitochondria. Biochemistry 35:7846–7854. doi: 10.1021/bi960244p. [DOI] [PubMed] [Google Scholar]
- 44.Tabor S, Richardson CC. 1989. Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc Natl Acad Sci U S A 86:4076–4080. doi: 10.1073/pnas.86.11.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmann MA, Brian DA. 1991. The 5′ end of coronavirus minus-strand RNAs contains a short poly(U) tract. J Virol 65:6331–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schweizer MP, Chan SI, Ts'o PO. 1965. Interaction and association of bases and nucleosides in aqueous solutions. IV. Proton magnetic resonance studies of the association of pyrimidine nucleosides and their interactions with purine. J Am Chem Soc 87:5241–5247. doi: 10.1021/ja00950a045. [DOI] [PubMed] [Google Scholar]
- 47.Ts'O PO, Helmkamp GK, Sander C. 1962. Interaction of nucleosides and related compounds with nucleic acids as indicated by the change of helix-coil transition temperature. Proc Natl Acad Sci U S A 48:686–698. doi: 10.1073/pnas.48.4.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ts'o PO, Kondo NS, Schweizer MP, Hollis DP. 1969. Studies of the conformation and interaction in dinucleoside mono- and diphosphates by proton magnetic resonance. Biochemistry 8:997–1029. doi: 10.1021/bi00831a033. [DOI] [PubMed] [Google Scholar]
- 49.Friedman RA, Honig B. 1995. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys J 69:1528–1535. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Norberg J, Nilsson L. 1995. Stacking free energy profiles for all 16 natural ribodinucleoside monophosphates in aqueous solution. J Am Chem Soc 117:10832–10840. doi: 10.1021/ja00149a006. [DOI] [Google Scholar]
- 51.Balbo PB, Meinke G, Bohm A. 2005. Kinetic studies of yeast polyA polymerase indicate an induced fit mechanism for nucleotide specificity. Biochemistry 44:7777–7786. doi: 10.1021/bi050089r. [DOI] [PubMed] [Google Scholar]
- 52.Ortin J, Martin-Benito J. 2015. The RNA synthesis machinery of negative-stranded RNA viruses. Virology 479-480:532–544. doi: 10.1016/j.virol.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 53.Hwang LN, Englund N, Pattnaik AK. 1998. Polyadenylation of vesicular stomatitis virus mRNA dictates efficient transcription termination at the intercistronic gene junctions. J Virol 72:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robertson JS, Schubert M, Lazzarini RA. 1981. Polyadenylation sites for influenza virus mRNA. J Virol 38:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK. 1996. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci U S A 93:11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorsch-Hasler K, Yogo Y, Wimmer E. 1975. Replication of picornaviruses. I. Evidence from in vitro RNA synthesis that poly(A) of the poliovirus genome is genetically coded. J Virol 16:1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spector DH, Villa-Komaroff L, Baltimore D. 1975. Studies on the function of polyadenylic acid on poliovirus RNA. Cell 6:41–44. doi: 10.1016/0092-8674(75)90071-9. [DOI] [PubMed] [Google Scholar]
- 58.Larsen GR, Dorner AJ, Harris TJ, Wimmer E. 1980. The structure of poliovirus replicative form. Nucleic Acids Res 8:1217–1229. doi: 10.1093/nar/8.6.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheets MD, Wickens M. 1989. Two phases in the addition of a poly(A) tail. Genes Dev 3:1401–1412. doi: 10.1101/gad.3.9.1401. [DOI] [PubMed] [Google Scholar]
- 60.Yao Z, Jones DH, Grose C. 1992. Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl 1:205–207. doi: 10.1101/gr.1.3.205. [DOI] [PubMed] [Google Scholar]
- 61.Jones DH, Howard BH. 1991. A rapid method for recombination and site-specific mutagenesis by placing homologous ends on DNA using polymerase chain reaction. Biotechniques 10:62–66. [PubMed] [Google Scholar]
- 62.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]