

ABSTRACT
Abnormal accumulation of proteins is a hallmark of a variety of neurological diseases including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Maintenance of protein homeostasis (proteostasis) in neurons via proteasomal and macroautophagy/autophagy-lysosomal degradation is thought to be central for proper neuronal function and survival. We recently reported evolutionarily conserved roles for two ALS-linked proteins, UBQLN2 (ubiquilin 2) and VAPB, in regulation of lysosomal degradation. Ubiquilins are required for v-ATPase-mediated lysosomal acidification, whereas VAPs are required for the PtdIns4P-mediated endo-lysosomal trafficking pathway.
KEYWORDS: ALS, Drosophila, ER stress, lysosomal acidification, MTOR, v-ATPase
Main
Proteins are in a dynamic state of synthesis and degradation. Clearance of damaged or unnecessary proteins and organelles is essential for cellular function and maintenance. The failure to degrade proteins via proteasomes and lysosomes can result in numerous diseases. Neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) most often display protein aggregates, endoplasmic reticulum (ER) stress, proteasomal dysfunction, and autophagosomal or endo-lysosomal defects. Proper lysosomal acidification is critical to maintain normal autophagic flux, and lysosomes contain more than 60 lumenal hydrolases, most of which are activated by low pH in the lysosomal lumen (pH 4.5–5). This acidic pH is generated and maintained by the vacuolar-type H+-translocating ATPase (v-ATPase), a lysosomal proton pump that hydrolyzes ATP to produce a proton gradient across the lysosomal membrane. A growing body of evidence indicates that several ALS-causing genes including those encoding two ubiquilins (UBQLN2 and UBQLN4), VAPB, VCP, and SQSTM1/p62, are required for proper autophagic-lysosomal degradation.
Ubiquilins affect proteostasis via the ubiquitin-proteasome system and autophagy. Our recent study revealed a previously undocumented role for ubiquilins as a potentiator of autophagy by regulating v-ATPase-mediated lysosome acidification both in mutant flies (Ubqn1) and UBQLN1/2/4-depleted human cell lines [1]. Loss of Ubqn in the fly nervous system causes ER stress, an age-dependent neuronal dysfunction, and a slow progressive demise of neurons and glia. Aging Ubqn1 neurons show accumulation of electron-dense aggregates, autophagic vesicles, and aberrant lysosomes. Accumulation of these structures emerges prior to the onset of neurodegeneration and neuronal dysfunction in Ubqn1 mutants. Also, neurons display a severe accumulation of mitochondria in aged flies suggesting that defective autophagy/mitophagy plays a role in neurodegeneration. In addition, glial cells also accumulate aggregates, aberrant lysosomes, and autophagosomes, consistent with previous data showing that glia have an important role in ALS neuropathology progression.
Interestingly, loss of ubiquilins increases autophagic induction by reducing Tor/MTORC1 signaling, yet the autophagic flux is halted due to severely diminished lysosomal acidification. Ubqn1 mutants and UBQLN1/2/4-depleted cells exhibit reduced activity of both MTORC1 and MTORC2. However, it is not obvious if ubiquilins regulate MTORC1 and MTORC2 activity directly or indirectly. Under nutrient-rich conditions, MTORC1 senses lysosomal amino acids through the v-ATPase and localizes to the lysosomal surface where it is activated. In addition, lysosomal MTORC2 is an inhibitor of a specific form of autophagy, chaperone-mediated autophagy, by negatively regulating the assembly of LAMP2A (lysosomal-associated membrane protein 2A). We found that Ubqn protein is enriched in puncta on the surface of lysosomes and therefore, it is possible that ubiquilins regulate the activities of MTOR complexes by its direct association with lysosomal membrane proteins. Mass spectrometry analysis and biochemical assays revealed that ubiquilins interact with multiple v-ATPase subunits. The v-ATPase is a multisubunit complex composed of a peripheral domain (V1) and an integral membrane domain (V0). Loss of ubiquilins causes an accumulation of a fragment of Vha100-1/V0a1, a V0 subunit of the v-ATPase, which is associated with severe acidification defects. Reducing the Vha100-1/V0a1 levels in Ubqn1 mutant flies significantly suppresses the lysosomal acidification and autophagic flux defects. How Ubqn regulates the levels of v-ATPase subunits is not known. However, given that loss of Ubqn causes an accumulation of a Vha100-1/V0a1 fragment (~75 kDa), but does not affect the levels of the full-length protein (~100 kDa), it is likely that ubiquilin is involved in the degradation of misfolded/dysfunctional Vha100-1/V0a1. Although the nature of this accumulated Vha100-1/V0a1 fragment is unknown, subcellular fractionation data show that it incorporates into the lysosomal membrane. We speculate that when the Vha100-1/V0a1 fragment accumulates, the stoichiometric balance between the v-ATPase subunits is impaired, resulting in impaired v-ATPase activity and alkalized lysosomes. Hence, loss of Ubqn interferes with lysosome function and leads to inhibition of autophagic flux even though there is an increase in autophagosome formation. Importantly, feeding fly larvae with acidic nanoparticles alleviates the autophagic degradation defects. Given that long-term feeding of acidic nanoparticles is toxic, therapeutic interventions based on this approach could be considered in future studies if a controlled dosage optimization can be achieved.
We recently showed that additional ALS-causing proteins, VAPA (VAMP associated protein A) and VAPB (VAMP associated protein B and C) in human (Vap33 in flies), is also required for autophagic-lysosomal degradation [2]. Similar to loss of ubiquilins, loss of Vaps leads to accumulation of lysosomes with aberrant acidity. Vap33Δ20 mutants display a dramatic accumulation of autophagic vesicles, especially autolysosomes. However, Ubqn and Vap regulate lysosomal activity through different mechanisms. Vap proteins are localized to the ER membrane, tether ER-Golgi membrane, and directly interact with lipid transfer proteins. This tethering facilitates Golgi to ER transfer of phosphatidylinositol-4-phosphate (PtdIns4P). Hence, loss of Vaps leads to accumulation of PtdIns4P in the Golgi, which strongly promotes the production of endosomes. Those endosomes are not properly acidified to become functional lysosomes. Therefore, the degradation capacity of autolysosomes is impaired. Reducing the PtdIns4P levels with a drug or removing one copy of an endosomal protein required for proper trafficking suppresses the lysosomal degradation defects as well as behavioral defects observed in Vap33Δ20 mutants.
Interestingly, both Ubqn1 and Vap33Δ20 mutants display ER quality control defects, ER stress, and unfolded protein response activation through the PEK/PERK (pancreatic eIF-2α kinase) pathway. It is likely that PEK/PERK activation is responsible for upregulation of chaperones and increased autophagy induction to recruit autophagy as a compensatory degradation mechanism. Indeed, several lines of evidence argue that low levels of ER stress can be beneficial and act as a protective mechanism in ALS pathogenesis.
Together, our findings in two ALS-linked protein families, ubiquilins and Vaps, show that maintenance of normal lysosome activity is highly important for prevention of neuronal demise (Figure 1). Identifying the underlying molecular mechanisms that lead to ALS and other neurological diseases is an important step for potential therapeutic interventions. Given that we found an intimate relation between lysosome activity and ALS-linked proteins, it may be possible to target proteins in the autophagy-lysosome pathway or promote the acidification of lysosomes to slow down the progression of the disease.
Figure 1.
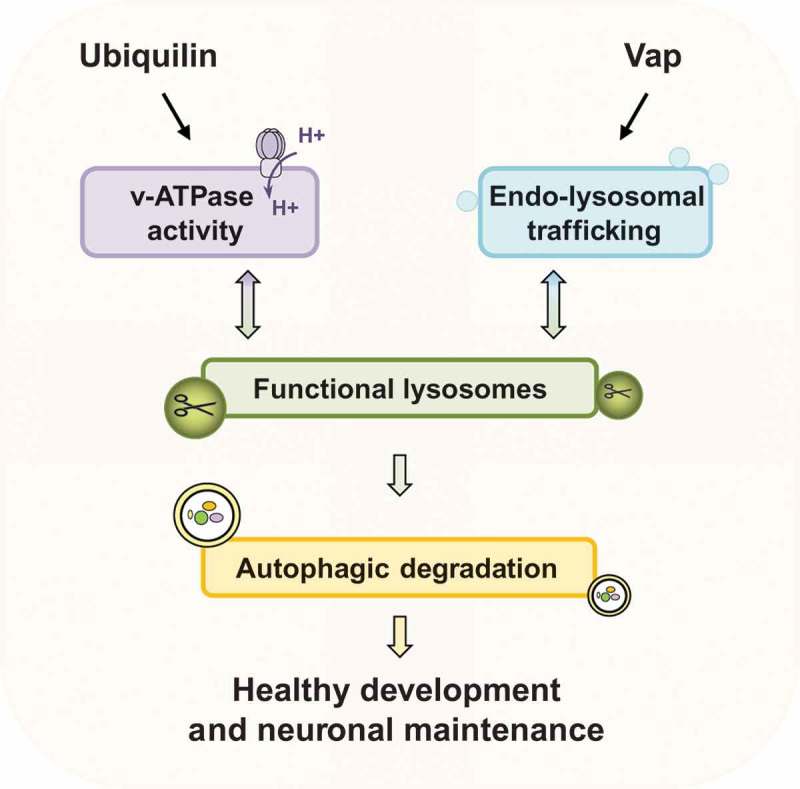
Regulation of lysosomal activity is critical for proper autophagic flux. Ubiquilins regulate lysosomal acidification through v-ATPase activity. Vaps regulate endo-lysosomal trafficking.
Funding Statement
This work was supported by the NIH Office of the Director [R24OD022005]; The Huffington Foundation; Robert A. and Renee E. Belfer Family Foundation; Target ALS
Acknowledgments
H.J.B. is supported by the Robert A. and Renee E. Belfer Family Foundation, the Huffington Foundation, Target ALS, and the NIH Office of the Director (R24OD022005). H.J.B is an Investigator of the Howard Hughes Medical Institute.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Senturk M, Lin G, Zuo Z, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21:384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mao D, Lin G, Tepe B, et al. VAMP associated proteins are required for autophagic and lysosomal degradation by promoting a PtdIns4P-mediated endosomal pathway. Autophagy. 2019;125:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]