

Abstract
Degradable poly(amine-co-ester) (PACE) terpolymers hold tremendous promise for siRNA delivery because these materials can be formulated into delivery vehicles with highly efficient siRNA encapsulation, providing effective knockdown with low toxicity. Here, we demonstrate that PACE nanoparticles (NPs) provide substantial protein knockdown in human embryonic kidney 293 cells (HEK293) and hard-to-transfect primary human umbilical vein endothelial cells (HUVECs). After intravenous administration, NPs of solid PACE (sPACE)—synthesized with high monomer content of a hydrophobic lactone—accumulated in the liver and, to a lesser extent, in other tissues. Within the liver, a substantial fraction of sPACE NPs were phagocytosed by liver macrophages, while a smaller fraction of NPs accumulated in hepatic stellate cells and liver sinusoidal endothelial cells, suggesting that sPACE NPs could deliver siRNA to diverse cell populations within the liver. To test this hypothesis, we loaded sPACE NPs with siRNA designed to knockdown Nogo-B, a protein that has been implicated in the progression of alcoholic liver disease and liver fibrosis. These sPACE:siRNA NPs produced up to 60% Nogo-B protein suppression in the liver after systemic administration. We demonstrate that sPACE NPs can effectively deliver siRNA therapeutics to the liver to mediate protein knockdown in vivo.
Keywords: Polymeric nanoparticles, poly(amine-co-ester) (PACE), siRNA, Nogo-B
Graphical Abstract
1. INTRODUCTION
Small interfering RNAs (siRNAs) are short, double stranded nucleic acids that can mediate transient gene knockdown inside a cell. Because siRNAs are unstable and rapidly degraded by serum enzymes and intracellular RNases, challenges remain in the translation of siRNA-based therapies to the clinic. [1–3]. In addition, siRNAs cannot diffuse readily across the cellular membrane due to their large size and negative charge. To address these limitations, a number of delivery platforms have been explored, including liposomes, lipid nanoparticles, polyplexes, and polymeric nanoparticles (NPs) [3–9]. Cationic lipids can transfect cells with high efficiency but are generally toxic to cells and are pro-inflammatory in vivo [1, 2, 10]. Cationic polymers with high charge densities, such as polyethyleneimine (PEI), poly(beta-amino-ester) (PBAE), and poly(L-lysine), can condense siRNA through electrostatic interactions to form polyplexes [11, 12]. Although effective for siRNA delivery in vitro, many of these cationic polymers are not suitable for in vivo delivery due to association with negatively charged serum proteins and high cytotoxicity [1, 11]. Solid polymeric NPs are often used for sustained delivery of drugs, including products that are used clinically [13, 14]. For example, poly(lactic-co-glycolic acid), or PLGA, is often used in NP formulations due to its low toxicity and sustained release capabilities [9, 14–17]. However, the efficacy of PLGA nanoparticles for siRNA is limited by low siRNA encapsulation and transfection efficiency compared to cationic lipids and polymers.
Recently, we synthesized a biodegradable family of poly(amine)-co-ester (PACE) terpolymers with low charge densities through enzymatic copolymerization of 15-pentadecanolide (PDL), diethyl sebacate (DES) and N-methyldiethanolamine [18]. An advantage of the PACE family of polymers is that the cation density is lower than most cationic polymers, leading to lower toxicity; the presence of hydrophobic monomers, such as PDL, in PACE contributes to its ability to efficiently form particles even at low overall cation densities [18]. In our first reports, we found that polymers with low PDL content (10 to 20%) resulted in the formation of polyplexes that were efficient for transfection of plasmid DNA in cultured cells and tumors in animals [18]. More recently, we discovered that solid NPs with slow-release properties could be produced from PACE polymers composed of 50% to 90% PDL [19]: we call these polymers solid PACE (sPACE). These formulations offer stable and efficient loading of nucleic acids, such as pDNA, mRNA, and siRNA [19, 20]. Further, we showed the utility of these solid NPs for ex vivo transfection of cells and tissues, producing extended periods of knockdown that persisted for weeks after transplantation of transfected tissues into animal hosts [19]. In this report, we examine the effect of polymer composition on transfection efficiency, toxicity, and biodistribution after intravenous injection of sPACE NPs.
Reticulon 4B, also known as Nogo-B, is an endoplasmic reticulum (ER) resident protein that is implicated in a number of liver pathologies, such as hepatic fibrosis and alcoholic liver disease [21–23]. In the liver, Nogo-B is highly expressed in non-parenchymal cells [21], such as liver sinusoidal endothelial cells (LSECs), Kupffer cells, and hepatic stellate cells (HSCs). Nogo-B plays a crucial role in the progression of alcoholic liver disease. Specifically, it promotes M1 polarization and inhibits M2 polarization in Kupffer cells to accentuate liver injury in alcoholic liver disease in humans and mice [23]. In addition, Nogo-B also promotes the progression of hepatic fibrosis through facilitating TGFβ signaling and inhibiting apoptosis of activated hepatic stellate cells [21, 22]. For example, Nogo-A/B knockout mice demonstrated slower progression of hepatic fibrosis and development of portal hypertension 4 weeks after bile duct ligation, suggesting that this protein is an important contributor to the development and progression of fibrosis [21, 22]. Based on these studies, suppression of Nogo-B expression using siRNA therapeutics may be an effective strategy for treating hepatic fibrosis and alcoholic liver disease. In this report, we demonstrate, using siRNA-loaded sPACE NPs, that we can achieve greater than 60% Nogo-B suppression in the liver for at least 1 week after a single intravenous dose.
2. RESULTS AND DISCUSSION
2.1. Formulation and characterization of PACE nanoparticles
In prior studies, we demonstrated that PACE (with 10–20% PDL) can condense plasmid DNA into nanosized polyplexes that provide more efficient transfection of HEK293 cells than Lipofectamine or PEI [18]. In addition, PACE polyplexes resulted in substantially lower toxicity to cells than other cationic polymers such as PEI. However, PACE polymers composed of 10–20% PDL are liquid at room temperature [20]. When complexed with nucleic acids in solution, these low-PDL content polymers form polyplexes that only provide short-term stability in solution. In addition, due to the lack of stability of these polyplexes in solution, release of the encapsulated material can be difficult to control. To produce a more stable formulation, we synthesized PACE polymers with a higher fraction of PDL and obtained sPACE polymers that were solid at room temperature (Figure 1A) [20]. The molecular weights of the sPACE polymers synthesized are as follows: 37600, 46800, 31600, 35400, and 41500 Da for PACE-50, 60, 60, 80, and 90, respectively [19]. Solid PACE polymers, composed of 50, 60, 70, 80, and 90% PDL, showed varying rates of degradation in Tris-EDTA buffer: for example, sPACE composed of 60% PDL decreased in molecular weight by 80% after 4 weeks, whereas sPACE composed of 90% PDL was not substantially degraded over the same timeframe (Supplemental Figure 1). We formulated siRNA-loaded sPACE NPs using polymers composed of 50, 60, 70, 80, or 90% PDL (PACE-50, PACE-60, PACE-70, PACE-80, and PACE-90) (Figure 1B,C) and quantified NP size, surface charge, and siRNA encapsulation efficiency (Figure 1D,E,F; Supplemental Figure 2). All NPs were similar in size (240–300 nm), consistent with our previous studies [19, 20]. NPs produced from sPACE with lower PDL content (i.e. PACE-50) had a higher surface charge (i.e. +23 mV), whereas NPs formulated using sPACE with higher PDL content (i.e. PACE-90) had lower surface charge. On the other hand, NPs composed of higher lactone % (i.e. PACE-90) had the lowest surface charge (i.e. 1.3 mV). These results suggest that NP surface charge correlates with the density of protonizable amines in the PACE polymer. Next, all NPs were loaded with 300 to 400 pmol/mg of siRNA (representing 60– 80% encapsulation efficiency). These results are similar to our previous findings [19, 20]; here, we used a different siRNA molecule, but achieved similar NP loading, size, and surface charge over the range of sPACE compositions. We have also previously reported that these formulations result in sustained release of siRNA over a period of day to weeks, with polymer degradation occurring slowly over a period of weeks (Supplemental Figure 1) [19, 20].
Figure 1. Characterization of siRNA-loaded PACE NPs.

(A) PACE polymers were synthesized by enzyme-catalyzed copolymerization of 15-pentadecanolide (PDL), diethyl sebacate (DES) and N-methyldiethanolamine (MDEA), as described previously [18]. (B) Cationic amines on PACE NPs condense siRNA molecules into the core of NPs and also facilitate NP uptake into cells. (C,D) Scanning electron microscopy (SEM) images of siRNA-loaded PACE NPs synthesized with 50% to 90% PDL. NP diameter was quantified using image J. Scale bar: 1 μm. (E) NP zeta potential was measured using a Zetasizer (Malvern Instruments). (F) NPs were dissolved in methylene chloride for 2 hours. siRNA was extracted into Tris-EDTA buffer and siRNA was quantified using QuantIT PicoGreen assay (Invitrogen). The siRNA amount for all formulations was 500 pmol siRNA/mg of polymer, and encapsulation efficiency was calculated as: encapsulated siRNA/total feed siRNA x 100%. In panels D, E, and F, n = 4 for all formulations.
3.2. Cytotoxicity and protein knockdown in vitro
We measured the transfection efficiency of sPACE NPs (PACE-50, 60, 70, 80, 90) in human embryonic kidney cells (HEK293) and primary human umbilical vein endothelial cells (HUVECs). HEK293 cells have been used in the past as a reporter cell-line for the study of siRNA transfection and HUVECs are hard-to-transfect primary cells. Luciferase-HEK293 cells were treated with siRNA-loaded NPs for 6 hours and cultured in fresh medium for 72 hours. Negative control groups were treated with control (CTL) siRNA-loaded NPs and positive control groups were treated with Lipofectamine. HEK293 cells treated with Lipofectamine demonstrated 60% suppression of luciferase expression after 72 hours. In comparison, HEK293 cells treated with PACE-50, 60, and 70 NPs demonstrated > 80% suppression of luciferase expression (Figure 2A). Confluent HUVECs were treated with siRNA-loaded NPs targeting survivin for 6 hours, cultured for 48 hours, and survivin mRNA expression was quantified using qRT-PCR (Figure 2B). HUVECs treated with either PACE-50, 60, 70 NPs or Lipofectamine demonstrated > 60% knockdown of mRNA expression. The degree of protein suppression in both HEK293 cells and HUVECs is comparable to previously published studies in HUVECs using a different siRNA [19]. Our findings demonstrate that PACE-50, 60, and 70 NPs are effective for siRNA transfection in both HEK293 cells and HUVECs in vitro.
Figure 2. Transfection and toxicity in HEK293s and HUVECs.

(A) Luciferase HEK293s were treated with luciferase siRNA-loaded PACE NPs (0.1 mg/mL, 30 nM siRNA or Lipofectamine (30 nM siRNA) for 6 hours. After incubation, cells were washed 3 times with PBS and cultured in fresh medium for 72 hours. Luciferase expression was measured on a luminometer. (B) HUVECs were treated with survivin siRNA-loaded PACE NPs (0.1 mg/mL, 30 nM siRNA) or Lipofectamine (30 nM) for 6 hours. After incubation, cells were washed 3x with PBS and cultured in fresh medium for 72 hours. Survivin mRNA expression was quantified using qRT-PCR and normalized to GAPDH mRNA expression. Data shown represents the mean ± SD (n = 4). ** p < 0.01, *** p < 0.001, **** p < 0.0001 by one-way ANOVA with Bonferroni’s post-test for multiple comparisons. (C, D) HEK293s and HUVECs were treated with siRNA-loaded PACE NPs for 24 hours. Cell viability was measured using the CellTiter Blue assay, with n = 4 for all formulations.
We further evaluated the toxicity of PACE NPs loaded with siRNA against luciferase or survivin in both HEK293 cells and HUVECs. Both cultured cells were seeded into 96 well plates and incubated with PACE-50, 60, 70, 80, and 90 NPs for 24 hours. In HEK293 cells, we observed minimal toxicity (20% cell death) for PACE-50 and 60 formulations, even at the highest NP concentration (5000 μg/mL NPs or 1500 pmol/mL and 1750 pmol/mL siRNA for PACE-50 and PACE-60, respectively) (Figure 2C). In comparison, cationic polymers with a high charge density, such as polyethyleneimine (PEI) caused substantial toxicity; for example, HEK293 cells treated with PEI-plasmid DNA demonstrated > 80% cell death at 100 μg/mL [18]. Our findings suggest that lower polymer charge density in PACE reduces the degree of toxicity in cells. Further, we observed that PACE NPs resulted in a range of toxicity when incubated with HUVECs for 24 hours. An increase in cytotoxicity was correlated to a decrease in PACE PDL composition; for example, PACE-90 particles demonstrated very low toxicity (20% cell death at 2500 μg /mL NPs or 750 pmol/mL siRNA) while PACE-50 demonstrated the greatest toxicity (60–70% cell death at 2500 μg /mL NPs or 875 pmol/mL siRNA) (Figure 2D). Our observed sPACE NP toxicity in HUVECs is comparable to toxicity reported in a previously published study using sPACE NPs to treat HUVECs using a different siRNA [19].
2.3. Biodistribution of PACE-70 nanoparticles
Out of all PACE formulations, PACE-60, 70, and 80 NPs demonstrated both effective protein knockdown and low cytotoxicity. To test their potential for in vivo transfection, we examined the biodistribution of PACE-60, 70, and 80 NP formulations after tail vein intravenous (IV) injection. Three mg of DiD-loaded NPs in 150 μL of PBS was injected into C57BL/6 mice and allowed to circulate for 24 hours. After 24 hours, the brain, lung, heart, stomach, pancreas, liver, spleen, and kidneys were harvested and imaged using Carestream In-Vivo Imaging System. For all sPACE formulations, the majority of NPs accumulated in the lungs, liver, and spleen, but to varying degrees (Figure 3A,B). The route of IV administration did not, however, affect biodistribution. For the same NP type, similar biodistribution profiles were observed following IV retro-orbital or tail-vein IV administration (Supplemental Figure 3). These observed differences suggest that biodistribution of these NPs is governed, to some extent, by PACE polymer composition, perhaps because of the differences in NP surface charge (Figure 1E). We also examined the kinetics of NP clearance from the blood after intravenous administration via retro-orbital IV injection (Figure 3C). these data were obtained from small (2 μL) volumes of blood collected over a 24-hour period, with the concentration of DiD-loaded NPs quantified via microscopy, using a method we described previously [24]. We observed a rapid decline in NP concentration in blood within 30 minutes after IV injection for all sPACE formulations. Retention in the circulation at 24 hours was significantly different for various sPACE formulations; 0% of initial PACE-60 NPs, 10% of initial PACE-70 NPs, and 20% of initial PACE-80 NPs remained in circulation after 24 hours. Other studies have demonstrated that positively charged NPs adsorb serum proteins, which promote opsonization and phagocytosis by the mononuclear phagocyte system in the liver and spleen, leading to rapid clearance from the blood stream [25, 26]. Consistent with these previous observations, we observed that NPs with a higher surface charge (i.e. PACE-60) were cleared from the blood completely within 24 hours whereas 20% of nanoparticles with a lower surface charge (i.e. PACE-80) remained in circulation after 24 hours. Our results suggest that sPACE NPs are capable of effective circulation in the blood and PDL composition of the polymer can substantially affect circulation kinetics of these formulations.
Figure 3.

Biodistribution of PACE NPs. (A,B) DiD-loaded PACE NPs in PBS were injected intravenously into mice. After 24 hours, organs were harvested and imaged using the Bruker In-Vivo MS FX PRO. Organ mean fluorescence intensity (MFI) (brain, heart, lungs, liver, pancreas, spleen, kidneys) was measured and reported. * p < 0.05, ** p < 0.01. (C) DiD-loaded NPs in PBS were injected retro-orbitally into mice and 2 μL of blood were collected via tail nick at each time point. Circulating NP concentrations in the blood were quantified using microscopy as previously described [24]. Data shown is mean ± SD (n = 4).
2.4. Cellular distribution of nanoparticles in the mouse liver
Although many studies have examined delivery of siRNA therapeutics to the liver, only a few studies have examined the cellular distribution of nanocarriers after intravenous administration [27]. To better understand the cellular fate of NPs in the liver, we administered DiD-loaded PACE-70 NPs via IV tail vein injection and studied liver distribution after 24 hours. We choose PACE-70 NPs for this analysis because treatment with these NPs results in effective protein knockdown, low cytotoxicity, and robust accumulation in the liver. Overall, PACE-70 NPs were effectively internalized by liver non-parenchymal cells (i.e. kupffer cells, hematopoietic stem cells (HSCs), and liver sinusoidal endothelial cells (LSECs)); for example, 95% of non-parenchymal cells were associated with the DiD-loaded NPs, but only 68% of hepatocytes were associated with the DiD-loaded NPs (Figure 4B,C). To estimate the strength of the fluorescence signal – and therefore the total number of NPs associated with each cell population – the mean fluorescence intensity (MFI) was determined for each cell population (Figure 4D). As expected, Kupffer cells demonstrated the highest degree of NP association per cell. In comparison, HSCs and LSECs showed 4-fold lower NP association and hepatocytes showed 15-fold lower NP association compared to Kupffer cells. To confirm our observations, and to determine whether the NPs associated with cells were internalized, we sectioned and imaged the livers 24 hours after PACE-70 NP injection (Figure 4E). The liver sections were fixed in 4% PFA, stained using antibodies against CD31 (LSECs), CD68 (Kupffer cells), or desmin (HSCs), and imaged using confocal microscopy. Consistent with our flow cytometry findings, large quantities of sPACE NPs (red) co-localized with Kupffer cells. LSECs (green) and HSCs (green) showed clear NP (red) co-localization, although less than Kupffer cells, and hepatocytes showed obvious but sparse NP (red) co-localization. Together, these data suggest that liver macrophages sequestered a large fraction of systemically administered PACE-70 NPs. We also observed substantial accumulation of NPs in HSCs, and LSECs 24 hours after injection, suggesting that PACE-70 NPs can reliably deliver agents, such as Nogo-B siRNA, to liver macrophages, HSCs and LSECs.
Figure 4. Cellular biodistribution of PACE-70 NPs in the liver.

(A, B) Mice were injected with PACE-70 NPs. After 24 hours, isolated livers were incubated in collagenase for 4 h ours. Cells were centrifuged at 50g for 3 minutes to isolate hepatocytes. Non-parenchymal cells were stained for CD68 (KCs), CD31 (LSECs), and CD38 (HSCs) and analyzed by flow cytometry. Hepatocytes were isolated separately. (C) NP-positive cell populations in the liver were quantified using flow cytometry. (D) LSEC, HSC, KC, and hepatocyte NP MFI. Data shown is mean ± SD (n = 4). (E) 24 hours after NP injection, livers were fixed in 4% PFA, sectioned, and stained for CD68 (KCs), CD31 (LSECs), and desmin (HSCs) and imaged under the confocal microscope. Hepatocytes were imaged via autofluorescence. Scale bar: 25 μm.
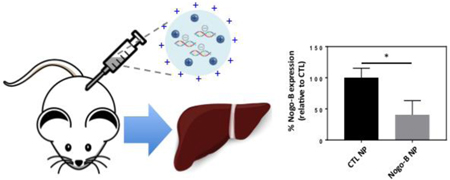
2.5. Nogo-B knockdown
We next examined if PACE-70 NPs could effect knockdown of Nogo-B expression. We first treated cultured mouse macrophages (RAW264.7) using siRNA-loaded NPs (10 μg/mL, 3.5 nM siRNA) for 24 hours; PACE-70 NPs were abundantly internalized (Figure 5A). Macrophages treated with Nogo-B siRNA-loaded NPs showed > 95% protein knockdown. In comparison, cells treated with a commercial transfection reagent, ScreenFect A, demonstrated only 60% protein knockdown (Figure 5B, C). Next, given that sPACE NPs resulted in preferential biodistribution to the liver, we determined whether siRNA-loaded PACE-70 NPs could suppress Nogo-B in the liver after injection into the spleen (Figure 5D). We have previously reported that biodistribution within the liver is comparable between IV administration and direct injection into the spleen [27]. One week after injection, mice were sacrificed and Nogo-B expression in the liver was quantified by western blot. We observed more than 60% suppression of liver Nogo-B levels in mice treated with Nogo-B siRNA-loaded NPs (Figure 5E,F). In comparison, mice treated with CTL siRNA-loaded NPs or siRNA alone did not demonstrate Nogo-B knockdown. Our findings suggest that a single, low dose (1 mg/kg) NP injection can result in effective Nogo-B suppression in the liver for 1 week. Moreover, PACE NP treatment is non-toxic in vivo; mice treated with 1 mg of PLGA NPs (known to have low toxicity) or 1 mg of PACE NPs had similar blood chemistry profiles compared to untreated mice (Supplemental Figure 4) [9, 14–17]. To the best of our knowledge, this is the first report of in vivo knockdown of Nogo-B expression in the liver using Nogo-B siRNA-loaded NPs. This study demonstrates the feasibility of PACE-based nanotechnology to regulate expression of Nogo-B protein in the liver, which has the potential to be translated into a therapeutic to improve the outcomes of alcoholic liver disease and liver fibrosis.
Figure 5. Nogo-B knockdown.

(A) RAW264.7 cells were treated with PACE NPs for 24 h ours. Cells were stained using phalloidin and DAPI and imaged on a confocal microscope. Scale bar: 50 μm. (B, C) RAW264.7 cells were treated with PACE-70 NPs for 24 hours. After incubation, cells were washed 3 times and cells were cultured in fresh medium for another 72 h ours. Protein expression was quantified by Western Blot. (D) Mice were injected with PACE NPs (1 mg/kg) through the spleen. (E, F) After 1 week, livers were harvested and Nogo-B protein expression was quantified by Western Blot. Data shown is mean ± SD (n = 4). * p < 0.05, ** p < 0.01 by student’s t test.
3. MATERIALS AND METHODS
3.1. Cell lines.
Human embryonic kidney 293 (HEK293) cells and murine macrophage RAW264.7 cells were obtained from American Type Culture Collection (Manassas, VA), and were stably transfected with luciferase, as described previously [28], and cultured in DMEM medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 ug/mL streptomycin. Human umbilical vein endothelial cells (HUVECs) were pur chased from the Vascular Biology and Therapeutics Program (Yale University). HUVECs were cultured in M199 medium supplemented with 20% FBS (Invitrogen), 2mM L-glutamine (Invitrogen), 100 U/mL penicillin, 100 ug/mL streptomycin, and 0.1% endothelial cell growth supplement (BD Bioscience, Franklin Lakes, NJ) on 0.1% gelatin coated tissue culture plates. RAW264.7 cells and mouse MF-HSCs (myofibroblasts) were cultured in DMEM medium (ThermoFisher) supplemented with 10% FBS and 5% penicillin/streptomycin, as described previously [21].
3.2. Synthesis of poly(amine-co-ester) terpolymers.
Poly(amine-co-ester) (PACE) polymers were synthesized via enzyme-catalyzed copolymerization of 15-pentadecanolide (PDL), diethyl sebacate (DES) and N-methyldiethanolamine (MDEA), as described previously, with minor modifications to the composition of PDL (50 – 90%) [18]. 15-pentadecanolide (PDL), diethyl sebacate (DES), N-methyldiethanolamine (MDEA), Novozym 435 catalyst, and diphenyl ether solvent were stirred at 90°C and under 1 atm of argon gas for 24 hours. The pressure was reduced to 1.6 mmHg at 90°C and the reaction was continued for another 72 hours. The resulting polymer was purified in hexane to remove residual solvent, dissolved in methylene chloride, and filtered to remove the enzyme catalyst. The added methylene chloride was removed at 40 °C and 1.0mmHg. The filtrates were analyzed by GPC using polystyrene standards to determine polymer molecular weights.
3.3. Formulation of nanoparticles.
Nanoparticles were formulated using a modified water-in-oil-in-water (w/o/w) double-emulsion solvent evaporation technique, as described previously [9, 29]. Briefly, 25 nmols (500 pmol/mg of polymer) of Nogo-B siRNA (5’-GAAGCGCAAAGCAGAAUGAUU-3’) (GE Dharmacon), Luciferase siRNA (5’-GCUAUGAAGCGCUAUGGGC-3’), Survivin siRNA (5’-GUCCGGUUGCGCUUUCCUUUC-3’) or nontargeting control siRNA (GE Dharmacon) was dissolved in pH 5.2 sodium acetate buffer containing 1mM EDTA was added dropwise under vortex to 50 mg of PACE in methylene chloride, and sonicated to form the first water-in-oil emulsion. Next, the emulsion was added dropwise under vortex to a 5% PVA solution and sonicated to form the second water-in-oil-in-water emulsion. The particles were hardened in 0.3% PVA solution for 3 hours, and washed in water three times to remove excess PVA. Nanoparticles were lyophilized for 48 hours and stored in −20 °C prior to use.
3.4. Characterization of nanoparticles.
Nanoparticle size and morphology were characterized using the scanning electron microscope (FEI, Hillsboro, Oregon) and quantified using ImageJ. Zeta potential was measured using the zetasizer (Malvern Instruments). siRNA loading was measured by dissolving nanoparticles in methylene chloride for 2 hours, followed by siRNA extraction into pH 7.4 TE buffer (10mM Tris-HCl, 1mM EDTA) containing 5000 U/mL heparin twice overnight. siRNA loading was quantified using QuantIT PicoGreen assay (Invitrogen) per manufacturer’s instructions. Encapsulation efficiency of nanoparticles was calculated as: encapsulated siRNA/total feed siRNA x 100%. siRNA concentration in nanoparticles was calculated as: % encapsulation efficiency x total feed siRNA x NP concentration.
3.5. Polymer degradation studies.
PACE polymers (50% to 90%) were incubated in pH 7.4 Tris-EDTA buffer on a 37°C on a shaker for 1 to 4 weeks. After incubation, polymers were centrifuged, washed 3x with water, and lyophilized for 48 h ours. Polymer molecular weight before and after incubation was measured using Gel Permeation Chromatography (GPC).
3.6. In vitro nanoparticle transfection.
siRNA transfection studies were performed in the HEK293 reporter cell-line stably expressing luciferase. Briefly, HEK293 cells stably expressing luciferase were incubated with nanoparticles or Lipofectamine for 24 hours. After incubation, cells were cultured in fresh medium for another 48 hours. After incubation, cells were harvested using trypsin. For the luciferase assay, cells were lysed using Reporter Lysis Buffer (Promega), frozen at −80 °C for 1 hour, and defrosted at room temperature. Once the cells have thawed, the cell lysates were centrifuged. The supernatant was added to the Luciferase Assay Reagent (Promega) and luminescence was measured on the luminometer (Glomax). To normalize luminescence to total protein, protein concentrations in each well was measured using BCA Assay (ThermoFisher) according to manufacturer instructions. To measure expression levels of survivin, total RNA was isolated from trypsinized cells using the Qiagen RNeasy Mini Kit (Qiagen). Isolated RNA was used to synthesize cDNA using a High-Capacity cDNA Reverse Transcription Kit (ThermoFisher). Transcript levels were measured by quantitative real-time PCR using a BioRad Mini-Opticon instrument and the iTaq Universal SYBR Green Supermix (BioRad). All transcript levels were normalized to that of GAPDH in the same sample. To evaluate Nogo-B knockdown, RAW264.7 cells were incubated with siRNA-loaded PACE-70 nanoparticles for 24 hours. After incubation, nanoparticles were removed and cells were washed 3 times with PBS and cultured in fresh medium for an additional 72 hours. Cells were then harvested using trypsin and protein levels were quantified with western blot.
3.7. Nogo-B western blot analysis.
Cells were homogenized in lysis buffer containing 50 mM Tris HCl, 0.1 mM EGTA, 0.1 mM EDTA, 0.1% SDS, 0.1% deoxycholic acid, 1% Nonidet P-40, 5 mM sodium fluoride, 1 mM sodium pyrophosphate, 1 mM activated sodium vanadate, 0.32% protease inhibitor cocktail (Roche), and 0.027% Pefabloc. Lysates were centrifuged at 14,000 rpm for 10 minutes at 4°C and the supernatants were collected. Protein concentration was measured using the Lowry assay. Protein (50–80 μg) from each sample was loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and resolved by gel electrophoresis at 180 V for ~1 hour using a Tris/Glycine/SDS electrophoresis buffer. Samples were then transferred to nitrocellulose membranes in a transfer cell at 70V for 2 hours using a Tris/Glycine transfer buffer. Following transfer, membranes were blocked with 5% nonfat milk in Tris-buffered saline with Tween 20 (TBST). Blocked membranes were probed with antibodies against rabbit anti-Nogo serum (1761A, 1:5000, a gift from Dr. William C. Sessa, Yale University), heat shock protein 90 (hsp90, 1:1000, BD Biosciences), and β-actin (1:5000, Sigma-Aldrich). Next, membranes were incubated with fluorophore-conjugated secondary antibodies (680 nm or 800 nm emission) and signals were visualized using the Odyssey Infrared Imaging System (Li-Cor Biotechnology). Bands were quantified using ImageJ software.
3.8. Biodistribution.
PACE NPs (0.2% DiD fluorescent dye) were resuspended in PBS at a concentration of 3 mg per 150μL and sonicated for 10 seconds using a water sonicator (Branson 2510). The nanoparticle solution (150μL) was injected into the tail veins of 8 −10-week-old C57BL/6 mice (Charles River Laboratories). After 24 hours, animals were sacrificed and their organs (brain, heart, lungs, liver, pancreas, spleen, and kidneys) were harvested. DiD NP accumulation in the organs was quantified using Bruker In-Vivo MS FX PRO.
3.9. Circulating NP concentration.
Mice were anesthetized via bell-jar induction using a 7:3 mixture of isofluorane to propylene glycol. Once respirations reduced to 1 breath/second, 150uL of 20mg/mL DiD PACE particles were injected retro-orbitally using a 27-gauge needle and 1 mL syringe. Once animals were awake, 2 μL blood samples were collected via tail nick at 2 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 10 hours, and 24 hours. Blood volumes were promptly mixed 1:1 with a reference DiO nanoparticle in heparinized tubes via pipette. Samples were then snap frozen using liquid nitrogen and stored at −80°C until analysis. After thawing the samples, 2 μL of the collected blood/reference NP solution was spotted on the untreated side of a glad slide and covered with a 12mm diameter circular cover slip. Images were collected using standard GFP and FITC filters at 500ms and 100ms exposures respectively with an air immersion apochromatic 20x objective (NA 1.4, Olympus) on an Olympus Z71 inverted microscope illuminated by an LED light source (Olympus L300) and captured by an Olympus Retiga R6 CCD camera. For intensity quantification, the mean intensity was calculated in ImageJ and then the background (from an identically prepared sample with blood only) was subtracted. Statistical analyses were performed in Prism.
3.10. Liver flow cytometry.
To digest the liver, 0.4% type II collagenase (Worthington) was perfused through the portal vein for 10 min, as described previously [30, 31]. The inferior vena cava was clamped and additional collagenase was infused for an additional 3 minutes. After perfusion, the liver was removed, and incubated in 10 mL of collagenase solution in a 37 °C shaker for 20 minutes. The digested liver was passed through a 100 μm filter. Cell suspensions were centrifuged at 50 g for 3 minutes at 4 °C to isolate hepatocytes. The supernatant containing non-parenchymal cells (HSCs, LSECs, and KCs) were transferred to a new tube and centrifuged at 500 g for 5 minutes at 4 °C. Cell suspensions were stained using Alexa Fluor 488 anti-mouse CD68 (Biolegend), Alexa Fluor 48 anti-mouse CD31 (Biolegend), Alexa Fluor anti-mouse CD38 (Biolegend). Cells were incubated in antibodies for 30 min at 4 °C, and washed 3 times for 20 min each. Cell fluorescence was quantified using Attune NxT flow cytometer.
3.11. Immunofluorescence.
Livers were fixed in 4% PFA solution for 2 hours at room temperature and incubated in 30% sucrose solution for 24 hours. After fixation, livers were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek) on dry ice and cut into 10 μm sections using a cryostat (Leica). Liver sections were fixed using 4% PFA for 10 min and incubated in block solution (10% BSA in PBS) for 1 hour. Sections were permeabilized using 0.2% Triton-X 100 for 30 min and incubated in primary antibodies for 24 hours at 4 °C (Rat anti-mouse CD31 (Biolegend), rat anti-mouse CD68 (Biolegend), rabbit anti-mouse desmin (Cell Signaling)). After 24 hours, sections were washed (PBS containing 2% BSA) 3 times and incubated in secondary antibodies for 30 min (goat anti-rat secondary antibody, Alexa Fluor 488 conjugate (Thermo Fischer Scientific)). Coverslips were mounted using VECTASHIELD anti-fade mounting medium with DAPI (Vector Labs).
3.12. In vivo Nogo-B knockdown.
All animal experiments were performed in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committees of Yale University and the Veterans Affairs Connecticut Healthcare System. siRNA-loaded PACE-70 nanoparticles (1 mg/kg in 200 μL of saline) were administered through the spleen into Male C57BL/6 mice, as described previously [27]. Briefly, mice were anesthetized using ketamine (100mg/kg) and xy lazine (10mg/kg). An abdominal incision is performed and NPs were injected slowly into the spleen at a dose of 1 mg/kg in 200 μL of saline. After 1 week, mice were sacrificed and livers were collected and snap-frozen. Livers were then homogenized with a mortar and pestle and then lysed with lysis buffer as described above and sonicated. Protein concentration was measured using the Lowry assay and protein expression was quantified by western blot as described above.
3.13. Statistical analysis.
Data were reported as mean ± standard deviation. Student’s t-test and One-way ANOVA were performed to determine statistical significance. P values < 0.05 were considered statistically significant.
4. CONCLUSION
We have demonstrated that sPACE NPs densely loaded with siRNA mediate robust protein knockdown in HEK293 cells and HUVECs in vitro. Following IV injection, the majority of sPACE NPs accumulated in the liver, presenting an opportunity to deliver siRNA therapy to the non-parenchymal liver cells involved in alcoholic liver disease and liver fibrosis. Further, we demonstrated, using siRNA designed to knockdown Nogo-B, a protein implicated in the progression of alcoholic liver disease and liver fibrosis, up to 60% Nogo-B protein suppression in the liver following a single systemic IV NP administration. sPACE NPs can effectively deliver siRNA therapeutics to the liver to mediate protein knockdown in vivo.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (P30 DK045735, R21 AA023599, R21 AI126166, and U01 AI32895) and Connecticut DPH grant #2015-0901 (Y.I.). J.C. was supported by an NIH predoctoral fellowship (F31 HL132469), A.S.P. was supported by NIH National Research Service Awards (NRSAs): T32 (GM86287) training grant and F32 (HL142144) individual postdoctoral fellowship. G.T.T. was supported by an NIH postdoctoral fellowship (F32 HL131270).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS STATEMENT
A.S.P. and W.M.S. are inventors on a patent application on PACE polymers for the delivery of gene editing agents. A.S.P. and W.M.S. are consultants for Trucode Gene Repair, Inc. W.M.S. also has equity interests in Trucode Gene Repair, Inc.
REFERENCES
- 1.Whitehead KA, Langer R, and Anderson DG, Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov, 2009. 8(2): p. 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pecot CV, et al. , RNA interference in the clinic: challenges and future directions. Nat Rev Cancer, 2011. 11(1): p. 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kseniya Gavrilov WMS, Therapeutic siRNA: Principles, challenges, and strategies. Yale Journal of Biology and Medicine, 2012. 85(2): p. 187. [PMC free article] [PubMed] [Google Scholar]
- 4.Santel A, et al. , A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther, 2006. 13(16): p. 1222–34. [DOI] [PubMed] [Google Scholar]
- 5.Werth S, et al. , A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. Journal of Controlled Release, 2006. 112(2): p. 257–270. [DOI] [PubMed] [Google Scholar]
- 6.Steinbach JM, Seo YE, and Saltzman WM, Cell penetrating peptide-modified poly(lactic-co-glycolic acid) nanoparticles with enhanced cell internalization. Acta Biomaterialia, 2016. 30: p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wittrup A, et al. , Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol, 2015. 33(8): p. 870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahlman JE, et al. , In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol, 2014. [DOI] [PMC free article] [PubMed]
- 9.Woodrow KA, et al. , Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater, 2009. 8(6): p. 526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lv H, et al. , Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release, 2006. 114(1): p. 100–9. [DOI] [PubMed] [Google Scholar]
- 11.Gary DJ, Puri N, and Won YY, Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release, 2007. 121(1–2): p. 64–73. [DOI] [PubMed] [Google Scholar]
- 12.Patil Y and Panyam J, Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm, 2009. 367(1–2): p. 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang AZ, Langer R, and Farokhzad OC, Nanoparticle delivery of cancer drugs. Annu Rev Med, 2012. 63: p. 185–98. [DOI] [PubMed] [Google Scholar]
- 14.Makadia HK and Siegel SJ, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel), 2011. 3(3): p. 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahara K, et al. , Establishing chitosan coated PLGA nanosphere platform loaded with wide variety of nucleic acid by complexation with cationic compound for gene delivery. Int J Pharm, 2008. 354(1–2): p. 210–6. [DOI] [PubMed] [Google Scholar]
- 16.Patil YB, et al. , The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials, 2010. 31(2): p. 358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danhier F, et al. , PLGA-based nanoparticles: an overview of biomedical applications. J Control Release, 2012. 161(2): p. 505–22. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, et al. , Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat Mater, 2012. 11(1): p. 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui J, Qin L, Zhang J, Abrahimi P, Li H, Li G, Tietjen GT, Tellides G, Pober JS, Saltzman WM, Ex vivo pretreatment of human vessels with siRNA-loaded nanoparticles provides long-lasting protein silencing in endothelial cells. Nature Communications, 2017. 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kauffman AC, et al. , Tunability of Biodegradable Poly(amine-co-ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications. Biomacromolecules, 2018. 19(9): p. 3861–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D, et al. , Reticulon 4B (Nogo-B) is a novel regulator of hepatic fibrosis. Hepatology, 2011. 53(4): p. 1306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tashiro K, et al. , Absence of Nogo-B (reticulon 4B) facilitates hepatic stellate cell apoptosis and diminishes hepatic fibrosis in mice. Am J Pathol, 2013. 182(3): p. 786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JK, et al. , An endoplasmic reticulum protein, Nogo-B, facilitates alcoholic liver disease through regulation of Kupffer cell polarization. Hepatology, 2017. [DOI] [PMC free article] [PubMed]
- 24.Tietjen GT, DiRito J, Pober JS, Saltzman WM, Quantitative microscopy-based measurements of circulating nanoparticle concentration using microliter blood volumes. Nanomedicine: Nanotechnology, Biology, and Medicine, 2017. [DOI] [PMC free article] [PubMed]
- 25.Alexis F, et al. , Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm, 2008. 5(4): p. 505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens DE 3rd and Peppas NA, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm, 2006. 307(1): p. 93–102. [DOI] [PubMed] [Google Scholar]
- 27.Park JK, et al. , Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine, 2016. 12(5): p. 1365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng CJ and Saltzman WM, Enhanced siRNA delivery into cells by exploiting the synergy between targeting ligands and cell-penetrating peptides. Biomaterials, 2011. 32(26): p. 6194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devalliere J, et al. , Sustained delivery of proangiogenic microRNA-132 by nanoparticle transfection improves endothelial cell transplantation. FASEB J, 2014. 28(2): p. 908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Severgnini M, et al. , A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology, 2012. 64(2): p. 187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohar I, B.K., Murray SA, Ebrahimkhani MR, Crispe IN., Isolation of Non-parenchymal Cells from the Mouse Liver. Malaria Vaccines: Methods and Protocols, Methods in Molecular Biology, 2015. 1325: p. 3–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.