

Abstract
According to recent research advance, it is interesting to identify new, potent and selective inhibitors of human butyrylcholinesterase (BChE) for therapeutic treatment of both the Alzheimer’s disease (AD) and heroin abuse. In this study, we carried out a structure-based virtual screening followed by in vitro activity assays, with the goal to identify new inhibitors that are selective for BChE over acetylcholinesterase (AChE). As a result, a set of new, selective inhibitors of human BChE were identified from natural products with solanaceous alkaloid scaffolds. The most active one of the natural products (compound 1) identified has an IC50 of 16.8 nM against BChE. It has been demonstrated that the desirable selectivity of these inhibitors for BChE over AChE is mainly controlled by three key residues in the active site cavity, i.e. residues Q119, A277, and A328 in BChE versus the respective residues Y124, W286, and Y337 in AChE. Based on this structural insight, future rational design of new, potent and selective BChE inhibitors may focus on these key structural differences in the active site cavity.
Introduction
Cholinesterases, including both acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), are among the well-known targets for treatment of Alzheimer’s disease (AD), the most serious neurodegenerative disease that has affected about 30 million people [1]. In fact, the first four drugs that have ever been approved by the FDA for the AD treatment are cholinesterase inhibitors that either inhibit both AChE and BChE (tacrine, doneperzil, and rivastigmine) or selectively inhibit AChE (galantamine). However, studies reported in recent years have revealed that BChE might be a better target compared to AChE for the AD treatment [2–4]. In other words, a BChE-selective inhibitor might be more promising than an AChE inhibitor for the AD treatment [2–14].
In addition, in our recently reported study [15], we proposed and validated a new therapeutic strategy for heroin toxicity treatment by using a selective BChE inhibitor to block heroin activation. It has been demonstrated that a selective BChE inhibitor can be used to significantly attenuate the heroin-induced toxicity and physiological effects [15]. Hence, it is interesting to develop potent and selective BChE inhibitors in drug development for therapeutic treatment of both the AD and heroin abuse.
In fact, a variety of cholinesterase inhibitors have been reported in literature, with various scaffolds. Most of reported cholinesterase inhibitors are either not sufficiently potent for BChE [16–27] or almost equally potent for both AChE and BChE [25, 28–41]. For some other BChE inhibitors reported, the selectivity is unknown (not tested at all) [42, 43]. Few types of cholinesterase inhibitors have promising potency and selectivity for BChE over AChE [12, 17, 25, 38, 44–56]. Some BChE inhibitors were also identified through virtual screening methods but either their activity was mediocre [57] or their selectivity over AChE was not reported [58, 59]. It is highly desired to identify new, potent and selective BChE inhibitors as options for further drug development.
Here we report the identification of a set of new, selective BChE inhibitors from natural products with solanaceous alkaloid scaffolds through structure-based virtual screening and in vitro activity assays.
Materials and methods
Structure-based virtual screening.
Our virtual screening was performed on the Development Therapeutics Program (DTP) Release 4 compound library including ~265,000 compounds available at the National Cancer Institute (NCI) (https://cactus.nci.nih.gov/download/nci/) by using the X-ray crystal structures of human BChE (PDB entry 4BDS) [60] and AChE (PDB entry 4EY5) [61]. During the virtual screening, Autodock Vina 1.1.2 software [62] was used to search for the optimum binding conformation for each compound in the NCI compound library. To minimize the searching area, a 15 Å×15 Å×15 Å box containing the active site of BChE or AChE was chosen as the target binding site. The protein was set rigid and all the water molecules in the original crystal structure were removed prior to molecular docking. The default settings of the Autodock Vina was used and no other parameters were modified. The compounds were then ranked by their binding free energies (docking score) with BChE. Within the top-ranked compounds, only the compounds predicted to have a positive binding free energy with AChE will be selected and ordered from the NIH DTP program. All the binding modes were then investigated and visualized using the PyMol [63].
In vitro activity tests.
The top compounds selected from the virtual screening were first ordered from the NCI DTP program and assayed for their inhibitory activity against BChE. The most compromising compounds (1 to 3) were also ordered from Sigma-Aldrich (St Louis, MO) for comparison and verification. Both wild-type AChE and BChE were expressed and purified based on our previous reported protocol [64]. For activity determination, we followed the original Ellman’s protocol, as described in detail in our previous report [64]. The concentration of substrate butyrylthiocholine for BChE (or acetylthiocholine for AChE) was at 10 μM. The tested compounds were first dissolved in DMSO and the final DMSO concentration was at 1%.
Results and discussion
Based on the structure-based virtual screening, we selected a set of 10 compounds (1 to 10 depicted in Figure 1) that were predicted to bind with BChE only, without binding with AChE. All these compounds are natural products with solanaceous alkaloid scaffolds. The binding free energies calculated for AChE binding with compounds 1 to 10 ranged from +4.5 kcal/mol to +17 kcal/mol; the positive binding free energy means that the free energy of the protein-ligand complex is higher than the total free energy of the separated protein and ligand. This is because BChE has a relatively larger active site cavity than AChE such that these larger molecules can only fit the BChE active site cavity, whereas the AChE active site cavity is not large enough to accommodate these molecules (see below for the detailed binding structures).
Figure 1.
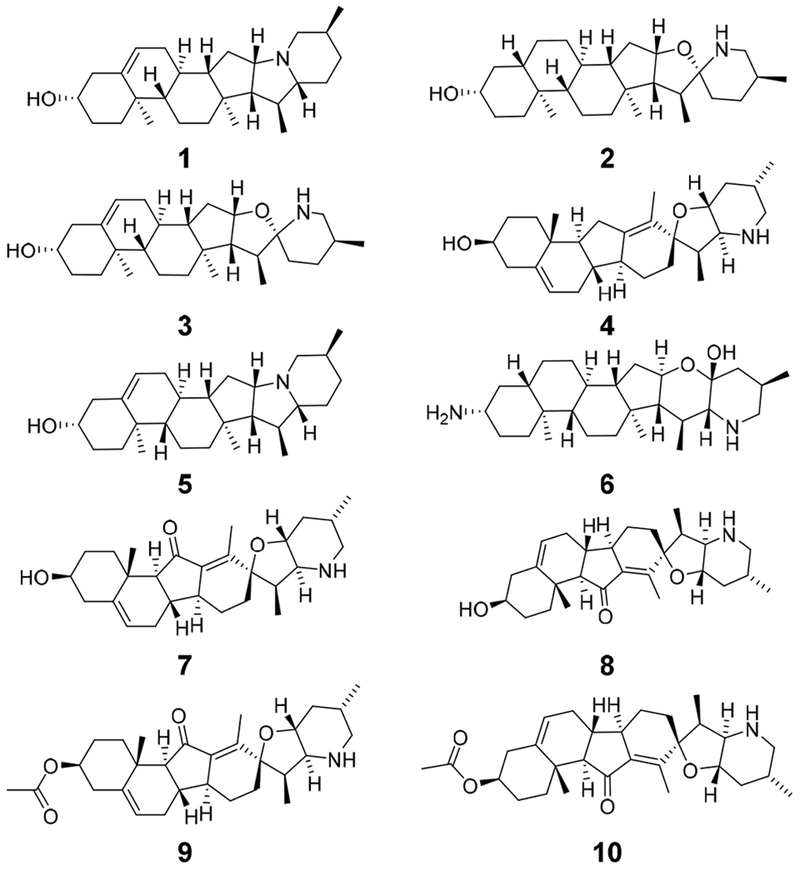
Molecular structures of the 10 compounds with solanaceous alkaloid scaffolds selected through virtual screening
Next, the computationally selected compounds 1 to 10 (ordered from the NCI DTP) were assayed for their inhibitory activity against human BChE. They were assayed first for their inhibitory activity at a concentration of 5 μM. As seen in Table 1, these compounds at 5 μM inhibited the BChE activity by 8-100%. The top-3 compounds (1 to 3) inhibited BChE by at least 95%. The most active compounds (1 to 3) were tested further for the dose-dependent inhibition in order to determine their IC50 values (see Figure 2 and Table 1) against BChE. As seen in Table 1, we obtained IC50 = 16.8 nM, 346 nM, and 391 nM for compounds 1 to 3, respectively. Similar results were also obtained from the use of compounds 1 to 3 ordered from Sigma-Aldrich (St Louis, MO).
Table 1.
Inhibitory activity of computationally selected solanaceous alkaloids (compounds 1 to 3) against human BChE
| Compound ID | NCI ID | Conventional Name | Inhibition (%) of BChE at 5 μM | Inhibition (%) of AChE at 5 μM | IC50 against BChE (nM) |
|---|---|---|---|---|---|
| 1 | 76025 | Solanidine | 100 | 10 | 16.8 |
| 2 | 27592 | Tomatidine | 95 | 19 | 346 |
| 3 | 35543 | Solasodine | 98 | 2 | 391 |
| 4 | 734950 | 44 | |||
| 5 | 76026 | 39 | |||
| 6 | 152144 | 24 | |||
| 7 | 23898 | 22 | |||
| 8 | 7520 | 18 | |||
| 9 | 117607 | 14 | |||
| 10 | 117612 | 8.3 |
Figure 2.
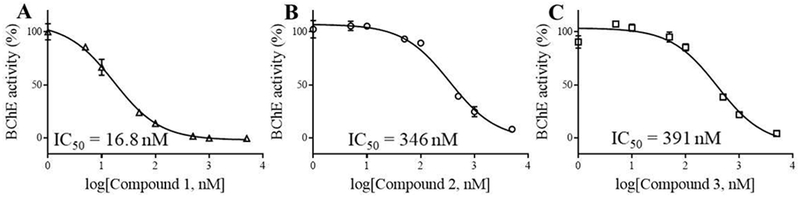
Dose-dependent inhibition of human BChE by compounds 1 (A), 2 (B), and 3 (C).
Interestingly, according to our detailed literature search, compounds 1 to 3 were tested for their inhibitory activity against AChE by Roddick et al. [65], and they demonstrated that none of these compounds (1 to 3) at a high concentration of 100 μM had significant inhibition against AChE. According to our own assays in this study, at a concentration of 5 μM, compounds 1 to 3 from Sigma-Aldrich (St Louis, MO) inhibited the AChE activity by only about 10%, 19%, and 2%, respectively (see Table 1). To the best of our knowledge, we have not found any report of testing these compounds against BChE. Taking all of these experimental data together, we can conclude that compounds 1 to 3 are indeed selective inhibitors of BChE, which is consistent with the aforementioned prediction from computational screening.
Depicted in Figure 3 are the docked binding structures of BChE with the most active compounds (1 to 3) identified. According to the docked binding structures with BChE, the inhibitor (compound 1 or 2 or 3) stays in a mainly hydrophobic environment, but having a favorable hydrogen bond (HB) between the hydroxyl group of the inhibitor and the backbone oxygen of amino-acid residue H438. The HB with compound 1 is the strongest (with the shortest O…H distance of 1.956 Å), explaining why compound 1 is the most potent inhibitor of BChE with IC50 = 16.8 nM within these three inhibitors. With these three inhibitors, their order of the HB strengths (see Figure 2) is consistent with their order of the IC50 values.
Figure 3.
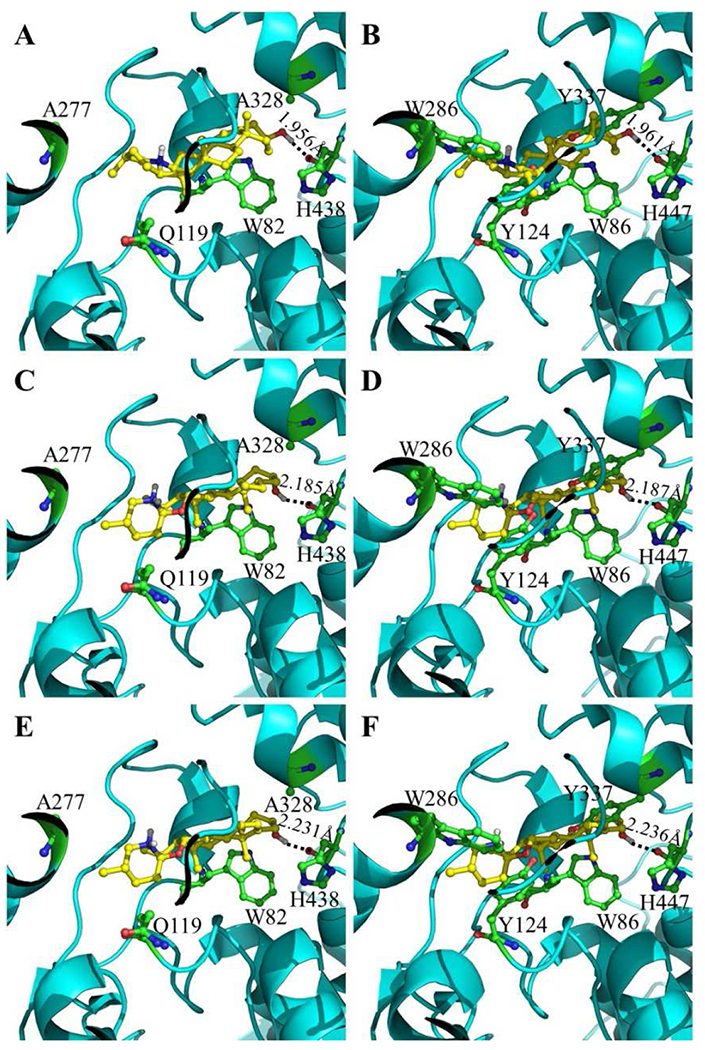
Modeled interactions of BChE and AChE with compounds 1 to 3. (A) Favorable binding structure of compound 1 with BChE. (B) Unfavorable interaction of compound 1 with AChE after BChE in panel A is replaced with AChE. (C) Favorable binding structure of compound 2 with BChE. (D) Unfavorable interaction of compound 2 with AChE after BChE in panel C is replaced with AChE. (E) Favorable binding structure of compound 3 with BChE. (F) Unfavorable interaction of compound 3 with AChE after BChE in panel E is replaced with AChE.
Panels B, D, and F of Figure 3 also show why the AChE active site cavity cannot accommodate any of these compounds. Specifically, panels A, C, and E show the docked favorable binding structures of BChE with compounds 1, 2, and 3, respectively. The corresponding unfavorable interactions of the compounds with AChE (after BChE is replaced with AChE) are depicted in panels B, D, and F, respectively. For the major differences between BChE and AChE in the protein-ligand interactions, residues Q119, A277, and A328 in BChE are replaced with Y124, W286, and Y337, respectively, in AChE. In the X-ray crystal structures of AChE-inhibitor complexes [66, 67], these residues (Y124, W286, and Y337) also played important roles in AChE binding with the inhibitors through hydrophobic interactions. Possible clash with any of these residues is expected to greatly impair the binding of the compound with AChE. According to our molecular modeling studies (see Figure 3), the compound (1 or 2 or 3) has clash with the side chains of Y124, W286, and Y337 in AChE, explaining why these compounds are selective for BChE over AChE for their inhibitory activities.
Conclusion
Through combined structure-based virtual screening and in vitro activity assays, we have successfully identified a set of new, selective inhibitors of human BChE from natural products with solanaceous alkaloid scaffolds. The most potent BChE inhibitor (compound 1) identified has an IC50 of 16.8 nM against BChE. These interesting outcomes suggest that natural products may be used as a promising resource in our future search of potent and selective inhibitors of BChE for BChE-based drug discovery.
Notably, the selectivity of these compounds for BChE over AChE is mainly controlled by three key residues in the active site cavity, i.e. residues Q119, A277, and A328 in BChE versus the respective residues Y124, W286, and Y337 in AChE (all with relatively larger side chains). In light of this structural insight, it is interesting to focus on these key structural differences in future rational design of new, potent and selective BChE inhibitors.
Supplementary Material
Research Highlights.
Virtual screening using 3D structures of both BChE and AChE is effective.
Highly selective BChE inhibitors have been identified from natural products.
The compounds with a solanaceous alkaloid scaffold can bind with BChE only.
Three key residues are mainly responsible for the extremely high selectivity.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH grants UH2/UH3 DA041115, R01 DA035552, R01 DA032910, R01 DA013930, and R01 DA025100) and the National Science Foundation (NSF grant CHE-1111761).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional Information
Competing Financial Interests statement: The authors declare that there is no conflict of interest for this work.
References
- [1].Dolgin E, How to defeat dementia, Nature News, 539 (2016) 156. [DOI] [PubMed] [Google Scholar]
- [2].Chatonnet A, Lockridge O, Comparison of butyrylcholinesterase and acetylcholinesterase, Biochem J, 260 (1989) 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li B, Duysen EG, Carlson M, Lockridge O, The butyrylcholinesterase knockout mouse as a model for human butyrylcholinesterase deficiency, Journal of Pharmacology and Experimental Therapeutics, 324 (2008) 1146–1154. [DOI] [PubMed] [Google Scholar]
- [4].Manoharan I, Boopathy R, Darvesh S, Lockridge O, A medical health report on individuals with silent butyrylcholinesterase in the Vysya community of India, Clin Chim Acta, 378 (2007) 128–135. [DOI] [PubMed] [Google Scholar]
- [5].Greig NH, Utsuki T, Yu Q, Zhu X, Holloway HW, Perry T, Lee B, Ingram DK, Lahiri DK, A new therapeutic target in Alzheimer’s disease treatment: attention to butyrylcholinesterase, Curr Med Res Opin, 17 (2001) 159–165. [DOI] [PubMed] [Google Scholar]
- [6].Mushtaq G, Greig NH, Khan JA, Kamal MA, Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus, CNS Neurol Disord Drug Targets, 13 (2014) 1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Giacobini E, Cholinergic function and Alzheimer’s disease, International Journal of Geriatric Psychiatry, 18 (2003) S1–S5. [DOI] [PubMed] [Google Scholar]
- [8].Ciro A, Park J, Burkhard G, Yan N, Geula C, Biochemical differentiation of cholinesterases from normal and Alzheimer’s disease cortex, Current Alzheimer Research, 9 (2012) 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, Yu Q-S, Mamczarz J, Holloway HW, Giordano T, Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent, Proceedings of the National Academy of Sciences of the United States of America, 102 (2005) 17213–17218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].PERRY EK, Perry R, Blessed G, Tomlinson B, Changes in brain cholinesterases in senile dementia of Alzheimer type, Neuropathology and applied neurobiology, 4 (1978) 273–277. [DOI] [PubMed] [Google Scholar]
- [11].Davies P, Maloney A, Selective loss of central cholinergic neurons in Alzheimer’s disease, The Lancet, 308 (1976) 1403. [DOI] [PubMed] [Google Scholar]
- [12].Kosak U, Brus B, Knez D, Sink R, Zakelj S, Trontelj J, Pislar A, Slenc J, Gobec M, Zivin M, Tratnjek L, Perse M, Salat K, Podkowa A, Filipek B, Nachon F, Brazzolotto X, Wieckowska A, Malawska B, Stojan J, Rascan IM, Kos J, Coquelle N, Colletier JP, Gobec S, Development of an in-vivo active reversible butyrylcholinesterase inhibitor, Sci Rep, 6 (2016) 39495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Furukawa-Hibi Y, Alkam T, Nitta A, Matsuyama A, Mizoguchi H, Suzuki K, Moussaoui S, Yu Q-S, Greig NH, Nagai T, Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice, Behavioural brain research, 225 (2011) 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hartmann J, Kiewert C, Duysen EG, Lockridge O, Greig NH, Klein J, Excessive hippocampal acetylcholine levels in acetylcholinesterase □ deficient mice are moderated by butyrylcholinesterase activity, Journal of neurochemistry, 100 (2007) 1421–1429. [DOI] [PubMed] [Google Scholar]
- [15].Zhang T, Zheng X, Kim K, Zheng F, Zhan C-G, Attenuate heroin’s toxicity and physiological effects by blocking its activation, Cell Chem. Biol (submitted), (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schott Y, Decker M, Rommelspacher H, Lehmann J, 6-Hydroxy- and 6-methoxy-beta-carbolines as acetyl- and butyrylcholinesterase inhibitors, Bioorganic & medicinal chemistry letters, 16 (2006) 5840–5843. [DOI] [PubMed] [Google Scholar]
- [17].Decker M, Novel inhibitors of acetyl- and butyrylcholinesterase derived from the alkaloids dehydroevodiamine and rutaecarpine, European journal of medicinal chemistry, 40 (2005) 305–313. [DOI] [PubMed] [Google Scholar]
- [18].Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, Morton CL, Obenauer JC, Damodaran K, Beroza P, Danks MK, Potter PM, Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases, J Med Chem, 48 (2005) 2906–2915. [DOI] [PubMed] [Google Scholar]
- [19].Hatfield MJ, Tsurkan LG, Hyatt JL, Edwards CC, Lemoff A, Jeffries C, Yan B, Potter PM, Modulation of esterified drug metabolism by tanshinones from Salvia miltiorrhiza (“Danshen”), J Nat Prod, 76 (2013) 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leader H, Smejkal RM, Payne CS, Padilla FN, Doctor BP, Gordon RK, Chiang PK, Binary antidotes for organophosphate poisoning: aprophen analogues that are both antimuscarinics and carbamates, J Med Chem, 32 (1989) 1522–1528. [DOI] [PubMed] [Google Scholar]
- [21].Hicks LD, Hyatt JL, Moak T, Edwards CC, Tsurkan L, Wierdl M, Ferreira AM, Wadkins RM, Potter PM, Analysis of the inhibition of mammalian carboxylesterases by novel fluorobenzoins and fluorobenzils, Bioorg Med Chem, 15 (2007) 3801–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zaheer-Ul-Haq ZU, Wellenzohn B, Liedl KR, Rode BM, Molecular docking studies of natural cholinesterase-inhibiting steroidal alkaloids from Sarcococca saligna, J Med Chem, 46 (2003) 5087–5090. [DOI] [PubMed] [Google Scholar]
- [23].Hyatt JL, Moak T, Hatfield MJ, Tsurkan L, Edwards CC, Wierdl M, Danks MK, Wadkins RM, Potter PM, Selective inhibition of carboxylesterases by isatins, indole-2,3-diones, J Med Chem, 50 (2007) 1876–1885. [DOI] [PubMed] [Google Scholar]
- [24].Decker M, Krauth F, Lehmann J, Novel tricyclic quinazolinimines and related tetracyclic nitrogen bridgehead compounds as cholinesterase inhibitors with selectivity towards butyrylcholinesterase, Bioorg Med Chem, 14 (2006) 1966–1977. [DOI] [PubMed] [Google Scholar]
- [25].Savini L, Campiani G, Gaeta A, Pellerano C, Fattorusso C, Chiasserini L, Fedorko JM, Saxena A, Novel and potent tacrine-related hetero- and homobivalent ligands for acetylcholinesterase and butyrylcholinesterase, Bioorganic & medicinal chemistry letters, 11 (2001) 1779–1782. [DOI] [PubMed] [Google Scholar]
- [26].Darvesh S, Pottie IR, Darvesh KV, McDonald RS, Walsh R, Conrad S, Penwell A, Mataija D, Martin E, Differential binding of phenothiazine urea derivatives to wild-type human cholinesterases and butyrylcholinesterase mutants, Bioorg Med Chem, 18 (2010) 2232–2244. [DOI] [PubMed] [Google Scholar]
- [27].Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, Ross CR, 2nd, S.A. Cantalupo, G. Crundwell, M.K. Danks, R.K. Guy, P.M. Potter, Planarity and constraint of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1, J Med Chem, 50 (2007) 5727–5734. [DOI] [PubMed] [Google Scholar]
- [28].Savini L, Gaeta A, Fattorusso C, Catalanotti B, Campiani G, Chiasserini L, Pellerano C, Novellino E, McKissic D, Saxena A, Specific targeting of acetylcholinesterase and butyrylcholinesterase recognition sites. Rational design of novel, selective, and highly potent cholinesterase inhibitors, J Med Chem, 46 (2003) 1–4. [DOI] [PubMed] [Google Scholar]
- [29].Fang L, Appenroth D, Decker M, Kiehntopf M, Roegler C, Deufel T, Fleck C, Peng S, Zhang Y, Lehmann J, Synthesis and biological evaluation of NO-donor-tacrine hybrids as hepatoprotective anti-Alzheimer drug candidates, J Med Chem, 51 (2008) 713–716. [DOI] [PubMed] [Google Scholar]
- [30].Decker M, Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterases with dirigible selectivity toward butyrylcholinesterase, J Med Chem, 49 (2006) 5411–5413. [DOI] [PubMed] [Google Scholar]
- [31].Butini S, Guarino E, Campiani G, Brindisi M, Coccone SS, Fiorini I, Novellino E, Belinskaya T, Saxena A, Gemma S, Tacrine based human cholinesterase inhibitors: synthesis of peptidic-tethered derivatives and their effect on potency and selectivity, Bioorganic & medicinal chemistry letters, 18 (2008) 5213–5216. [DOI] [PubMed] [Google Scholar]
- [32].Gemma S, Gabellieri E, Huleatt P, Fattorusso C, Borriello M, Catalanotti B, Butini S, De Angelis M, Novellino E, Nacci V, Belinskaya T, Saxena A, Campiani G, Discovery of huperzine A-tacrine hybrids as potent inhibitors of human cholinesterases targeting their midgorge recognition sites, J Med Chem, 49 (2006) 3421–3425. [DOI] [PubMed] [Google Scholar]
- [33].Chen WS, Cocolas GH, Cavallito CJ, Chai KJ, Potent reversible anticholinesterase agents. Bis- and mono-N-substituted benzoquinolinium halides, J Med Chem, 20 (1977) 1617–1623. [DOI] [PubMed] [Google Scholar]
- [34].Sun Q, Peng DY, Yang SG, Zhu XL, Yang WC, Yang GF, Syntheses of coumarin-tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity against butylcholinesterase, Abeta aggregation, and beta-secretase, Bioorg Med Chem, 22 (2014) 4784–4791. [DOI] [PubMed] [Google Scholar]
- [35].Peng DY, Sun Q, Zhu XL, Lin HY, Chen Q, Yu NX, Yang WC, Yang GF, Design, synthesis, and bioevaluation of benzamides: novel acetylcholinesterase inhibitors with multifunctions on butylcholinesterase, Abeta aggregation, and beta-secretase, Bioorg Med Chem, 20 (2012) 6739–6750. [DOI] [PubMed] [Google Scholar]
- [36].Lin G, Tsai HJ, Tsai YH, Cage amines as the stopper inhibitors of cholinesterases, Bioorganic & medicinal chemistry letters, 13 (2003) 2887–2890. [DOI] [PubMed] [Google Scholar]
- [37].Atta ur R, Khalid A, Sultana N, Ghayur MN, Mesaik MA, Khan MR, Gilani AH, Choudhary MI, New natural cholinesterase inhibiting and calcium channel blocking quinoline alkaloids, J Enzyme Inhib Med Chem, 21 (2006) 703–710. [DOI] [PubMed] [Google Scholar]
- [38].Darvesh S, Darvesh KV, McDonald RS, Mataija D, Walsh R, Mothana S, Lockridge O, Martin E, Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase, J Med Chem, 51 (2008) 4200–4212. [DOI] [PubMed] [Google Scholar]
- [39].Raza R, Saeed A, Arif M, Mahmood S, Muddassar M, Raza A, Iqbal J, Synthesis and biological evaluation of 3-thiazolocoumarinyl Schiff-base derivatives as cholinesterase inhibitors, Chem Biol Drug Des, 80 (2012) 605–615. [DOI] [PubMed] [Google Scholar]
- [40].Musilek K, Roder J, Komloova M, Holas O, Hrabinova M, Pohanka M, Dohnal V, Opletalova V, Kuca K, Jung YS, Preparation, in vitro screening and molecular modelling of symmetrical 4-tert-butylpyridinium cholinesterase inhibitors--analogues of SAD-128, Bioorganic & medicinal chemistry letters, 21 (2011) 150–154. [DOI] [PubMed] [Google Scholar]
- [41].Butini S, Campiani G, Borriello M, Gemma S, Panico A, Persico M, Catalanotti B, Ros S, Brindisi M, Agnusdei M, Fiorini I, Nacci V, Novellino E, Belinskaya T, Saxena A, Fattorusso C, Exploiting protein fluctuations at the active-site gorge of human cholinesterases: further optimization of the design strategy to develop extremely potent inhibitors, J Med Chem, 51 (2008) 3154–3170. [DOI] [PubMed] [Google Scholar]
- [42].Lin G, Chen GH, Ho HC, Conformationally restricted carbamate inhibitors of horse serum butyrylcholinesterase, Bioorganic & medicinal chemistry letters, 8 (1998) 2747–2750. [DOI] [PubMed] [Google Scholar]
- [43].Kalir A, Teomy S, Amir A, Fuchs P, Lee SA, Holsztynska EJ, Rocki W, Domino EF, N-allyl analogues of phencyclidine: chemical synthesis and pharmacological properties, J Med Chem, 27 (1984) 1267–1271. [DOI] [PubMed] [Google Scholar]
- [44].Luo W, Yu QS, Zhan M, Parrish D, Deschamps JR, Kulkarni SS, Holloway HW, Alley GM, Lahiri DK, Brossi A, Greig NH, Novel anticholinesterases based on the molecular skeletons of furobenzofuran and methanobenzodioxepine, J Med Chem, 48 (2005) 986–994. [DOI] [PubMed] [Google Scholar]
- [45].Sawatzky E, Wehle S, Kling B, Wendrich J, Bringmann G, Sotriffer CA, Heilmann J, Decker M, Discovery of Highly Selective and Nanomolar Carbamate-Based Butyrylcholinesterase Inhibitors by Rational Investigation into Their Inhibition Mode, J Med Chem, 59 (2016) 2067–2082. [DOI] [PubMed] [Google Scholar]
- [46].Wang J, Wang ZM, Li XM, Li F, Wu JJ, Kong LY, Wang XB, Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer’s disease based on the fusion of donepezil and melatonin, Bioorg Med Chem, 24 (2016) 4324–4338. [DOI] [PubMed] [Google Scholar]
- [47].Arbiser JL, Kraeft SK, van Leeuwen R, Hurwitz SJ, Selig M, Dickersin GR, Flint A, Byers R, Chen LB, Clioquinol-zinc chelate: a candidate causative agent of subacute myelo-optic neuropathy, Mol Med, 4 (1998) 665–670. [PMC free article] [PubMed] [Google Scholar]
- [48].Prati F, Bergamini C, Fato R, Soukup O, Korabecny J, Andrisano V, Bartolini M, Bolognesi ML, Novel 8-Hydroxyquinoline Derivatives as Multitarget Compounds for the Treatment of Alzheimer’s Disease, ChemMedChem, 11 (2016) 1284–1295. [DOI] [PubMed] [Google Scholar]
- [49].Makhaeva GF, Boltneva NP, Lushchekina SV, Serebryakova OG, Stupina TS, Terentiev AA, Serkov IV, Proshin AN, Bachurin SO, Richardson RJ, Synthesis, molecular docking and biological evaluation of N,N-disubstituted 2-aminothiazolines as a new class of butyrylcholinesterase and carboxylesterase inhibitors, Bioorg Med Chem, 24 (2016) 1050–1062. [DOI] [PubMed] [Google Scholar]
- [50].Carlier PR, Han YF, Chow ES, Li CP, Wang H, Lieu TX, Wong HS, Pang YP, Evaluation of short-tether bis-THA AChE inhibitors. A further test of the dual binding site hypothesis, Bioorg Med Chem, 7 (1999) 351–357. [DOI] [PubMed] [Google Scholar]
- [51].Lan J-S, Xie S-S, Li S-Y, Pan L-F, Wang X-B, Kong L-Y, Design, synthesis and evaluation of novel tacrine-^-carboline) hybrids as multifunctional agents for the treatment of Alzheimer’s disease, Bioorganic & medicinal chemistry, 22 (2014) 6089–6104. [DOI] [PubMed] [Google Scholar]
- [52].Tunek A, Svensson LA, Bambuterol, a carbamate ester prodrug of terbutaline, as inhibitor of cholinesterases in human blood, Drug Metab Dispos, 16 (1988) 759–764. [PubMed] [Google Scholar]
- [53].Sitar DS, Clinical pharmacokinetics of bambuterol, Clin Pharmacokinet, 31 (1996) 246–256. [DOI] [PubMed] [Google Scholar]
- [54].Pistolozzi M, Du H, Wei H, Tan W, Stereoselective inhibition of human butyrylcholinesterase by the enantiomers of bambuterol and their intermediates, Drug Metab Dispos, 43 (2015) 344–352. [DOI] [PubMed] [Google Scholar]
- [55].Gulçin İ, Abbasova M, Taslimi P, Huyut Z, Safarova L, Sujayev A, Farzaliyev V, Beydemir ş, Alwasel SH, Supuran CT, Synthesis and biological evaluation of aminomethyl and alkoxymethyl derivatives as carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase inhibitors, Journal of enzyme inhibition and medicinal chemistry, 32 (2017) 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kosak U, Brus B, Knez D, Zakelj S, Trontelj J, Pislar A, Sink R, Jukic M, Zivin M, Podkowa A, The magic of crystal structure-based inhibitor optimization: development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity, Journal of medicinal chemistry, 61 (2017) 119–139. [DOI] [PubMed] [Google Scholar]
- [57].Dighe SN, Deora GS, De la Mora E, Nachon F, Chan S, Parat M-O, Brazzolotto X, Ross BP, Discovery and Structure-Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening, Journal of medicinal chemistry, 59 (2016) 7683–7689. [DOI] [PubMed] [Google Scholar]
- [58].Sakkiah S, Lee KW, Pharmacophore-based virtual screening and density functional theory approach to identifying novel butyrylcholinesterase inhibitors, Acta Pharmacologica Sinica, 33 (2012) 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lešnik S, Štular T, Brus B, Knez D, Gobec S, Janežič D.a., Konc J, LiSiCA: a software for ligand-based virtual screening and its application for the discovery of butyrylcholinesterase inhibitors, Journal of chemical information and modeling, 55 (2015) 1521–1528. [DOI] [PubMed] [Google Scholar]
- [60].Nachon F, Carletti E, Ronco C, Trovaslet M, Nicolet Y, Jean L, Renard P-Y, Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl-and butyryl-cholinesterase, Biochemical Journal, 453 (2013) 393–399. [DOI] [PubMed] [Google Scholar]
- [61].Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Franklin MC, Height JJ, Structures of human acetylcholinesterase in complex with pharmacologically important ligands, Journal of medicinal chemistry, 55 (2012) 10282–10286. [DOI] [PubMed] [Google Scholar]
- [62].Trott O, Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J Comput Chem, 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schrödinger L, The PyMOL Molecular Graphics System, Version 2.0, (2018). [Google Scholar]
- [64].Hou S, Xue L, Yang W, Fang L, Zheng F, Zhan C-G, Substrate selectivity of high-activity mutants of human butyrylcholinesterase, Org. Biomol. Chem, 11 (2013) 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Roddick JG, The acetylcholinesterase-inhibitory activity of steroidal glycoalkaloids and their aglycones, Phytochemistry, 28 (1989) 2631–2634. [Google Scholar]
- [66].Harel M, Hyatt JL, Brumshtein B, Morton CL, Yoon KJP, Wadkins RM, Silman I, Sussman JL, Potter PM, The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action, Molecular pharmacology, 67 (2005) 1874–1881. [DOI] [PubMed] [Google Scholar]
- [67].Dvir H, Jiang H, Wong D, Harel M, Chetrit M, He X, Jin G, Yu G, Tang X, Silman I, X-ray structures of Torpedo californica acetylcholinesterase complexed with (+)-huperzine A and (−)-huperzine B: structural evidence for an active site rearrangement, Biochemistry, 41 (2002) 10810–10818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.