


Abstract
G-quadruplexes play various roles in multiple biological processes, which can be positive when a G4 is involved in the regulation of gene expression or detrimental when the folding of a stable G4 impairs DNA replication promoting genome instability. This duality interrogates the significance of their presence within genomes. To address the potential biased evolution of G4 motifs, we analyzed their occurrence, features and polymorphisms in a large spectrum of species. We found extreme bias of the short-looped G4 motifs, which are the most thermodynamically stable in vitro and thus carry the highest folding potential in vivo. In the human genome, there is an over-representation of single-nucleotide-loop G4 motifs (G4-L1), which are highly conserved among humans and show a striking excess of the thermodynamically least stable G4-L1A (G3AG3AG3AG3) sequences. Functional assays in yeast showed that G4-L1A caused the lowest levels of both spontaneous and G4-ligand-induced instability. Analyses across 600 species revealed the depletion of the most stable G4-L1C/T quadruplexes in most genomes in favor of G4-L1A in vertebrates or G4-L1G in other eukaryotes. We discuss how these trends might be the result of species-specific mutagenic processes associated to a negative selection against the most stable motifs, thus neutralizing their detrimental effects on genome stability while preserving positive G4-associated biological roles.
INTRODUCTION
G-quadruplexes (G4) are four-stranded secondary structures formed by G-rich DNA or RNA sequences. They result from the stacking of multiple stable ‘G quartets’ (a planar arrangement of four guanines) coordinated by cations (1,2). Extensive, biophysical and structural studies revealed an impressive diversity of G4 conformations depending on the number of G quartets, the length of the loops and their sequences as well as strand orientation (3). Compelling evidence clearly implicates G4 motifs in various biological processes, comprising telomere maintenance (4–6), replication (7–9), genome rearrangements (10–12), DNA damage response (13), chromatin structure (14,15) and transcriptional (16–19) or translational regulation (20,21).
Typically, the consensus sequence motif G3+N1-7G3+N1-7G3+N1-7G3+ has been used to identify potential G-quadruplex-forming motif sequences (PQS) (22,23). This led to an estimate of 376 000 motifs in the human hg19 reference genome (updated here to 404 347 in hg38) but other estimates have been proposed. Namely, the number of PQS almost doubles by either considering G4 sequences with varying loop lengths of 1 to 12 nt (24) or by scanning the reference genome with the G4Hunter algorithm (25), reaching 765 400 potential sequences (when setting a score threshold of 1.75). A similar number was observed by using an in vitro replication stop assay performed in the presence of the pyridostatin G4 ligand and allowing the inclusion of ‘non-canonical’ G4s (containing bulges, strand interruptions with snapback guanines or incomplete tetrads) (26). Thus, depending on the algorithms used in silico, the experimental methods used to identify PQS in vitro, as well as the cell lines and their chromatin accessibility for in vivo experiments (26–28), this estimate varies greatly. On the other hand, there are limited reports that incorporate loop base composition as an essential parameter in genome-wide G4 motif identification (29–31). By using natural and mutated human G4-forming minisatellite sequences in wild-type yeast cells treated with a G4 ligand, PhenDC3 or PhenDC6 (32), we previously showed that not all potential G4 motifs induce G4-dependent minisatellite instabilities (33). Indeed, we demonstrated that only G4s with loops of ≤3 nt were able to stimulate the G4-dependent minisatellite instability and that G4s with the consensus G3N1G3N1G3N1G3 (where N is any nucleotide) – herein called G4-L1 – both formed the most stable G4 in vitro and correlatively triggered the highest genetic instability in vivo (33). Furthermore, we showed that the base composition of the loops is important, with the presence of pyrimidine bases being correlated with the most stable G4s, both in vitro and in vivo (33). Here, we report a comprehensive analysis of the G4 PQS, in particular short-looped, and their polymorphisms in humans as well as in a large number of eukaryotes and other branches of the evolutionary tree of life. We found striking biases in motif loop composition, indicating that purine loops are markedly over-represented compared to pyrimidine loops, with a particular enrichment for single A bases in mammals. In contrast, we observed a different trend that favors G bases in distantly-related metazoans and plants. We discuss the biological significance of the G4 motif sequences biases and the potential evolutionary mechanisms that may differentially shape the loop composition of PQS and the length of the [GGGX]n tetra-nucleotide repeats in genomes.
MATERIALS AND METHODS
G4-L1 motif search and annotation
We defined a G4-L1 motif as a 15-nt sequence with four runs of exactly three guanines, separated by loop sequences containing precisely one base (that may itself be a guanine). We searched, by regular expression matching (as first described in the Quadparser method (22)), for the motifs previously defined—([gG]{3}\w{1}){3}[gG]{3}—in the fasta file of the human reference genome hg38. We only counted non-overlapping identical G4-L1 motifs. However, overlapping motifs with different loop sequences were both counted (i.e. GGGAGGGAGGGTGGGAGGG will count for two motifs, one with loops A-A-T and one with loops A-T-A). Motif fold-enrichment in hg38 was calculated by comparing actual G4 sequence counts (for different loop sizes, ranging from 1 to 12 nt) to counts of G4 motifs in a randomized background. To do so, we generated a sub-genome with fixed 5 or 10 Kbp size windows centered at around each identified PQS (slop utility from the BEDtools suite (34)), created fasta files for each interval (bedtools getfasta) and assembled them into a sub-genome fasta file. Then, we performed three independent dinucleotide shuffles in those segments to generate the randomized local background and search for G4 sequences as described for the reference genome. Nucleotide shuffling was performed with a Python implementation of the Altschul-Erikson dinucleotide shuffle algorithm (35). The annotatePeaks.pl Perl script from HOMER software v4.7 (36) was used to annotate the genomic coordinates found, for the overall G4-L1 set as well as for each of the 64 different motifs combinations independently. The inter-motif distances and motif density along chromosomes were calculated in R 3.3.3 for Mac OS X (37).
G4-L1 motif clusters assessment
We assessed the number of G4-L1 and G4-L1,7 motifs found along chromosomes versus sequence size (in base pairs, bp). For G4-L1, we observed two trends in the distribution, with a break point at around 500 bp. For inter-motif distances inferior to 470 bp (in order to fit at least two 15 nt-motifs in a 500-nt span), we calculated the average number of motifs found in 500-bp windows with high G4-L1 density and thus defined a G4-L1 motif cluster as 500-bp sequence containing at least three non-overlapping motifs.
We scanned the hg38 genome for clusters, resulting in 646 G4-L1 motif clusters. We analyzed cluster sizes (i.e. number of G4-L1 motifs found within the 500 bp) and distributions for each of the 23 human chromosomes. Significant differences in proportions of G4-L1 motifs located within clusters between chromosomes were evaluated using the Pearson’s chi-squared test.
Intersection with BG4 ChIP-seq data
We retrieved the latest BG4 ChIP-seq peaks data, specific to the HaCaT cell line, from the GEO database, accession number GSE99205 (38). The exact locations of the BG4 peaks were used to retrieve the corresponding sequences in fasta format using the getfasta command from the bedtools suite, using the hg19 reference genome. We then used the previously described regular expression matching approach to search for G4-L1, G4-L1-7 and G4-L1-12 motifs in these sequences. We also searched for potential G4-forming sequences running the G4Hunter algorithm in command line (with the set of parameters described by Bedrat et al. (25), and particularly with a score threshold of 1.75). Additionally, we split the BG4 peak sequences fasta file into one file per chromosome and ran the Quadron algorithm (39) in command line, with default settings. We counted the number of G4 motifs found, per chromosome, with each of the methods then extracted and analyzed the loop sequences for each motif. The consensus sequences found in the BG4 peaks (with or without PQS detected within the peaks) were found using the RSAT software suite (40).
Intersection with in vitro G4-seq data for multiple species
We retrieved G4-seq maps for 12 species from the GEO database, accession number GSE110582 (41). These species included: Homo sapiens, Mus musculus, Danio rerio, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, Leishmania major, Trypanosoma brucei, Plasmodium falciparum, Arabidopsis thaliana, Escherichia coli and Rhodobacter sphaeroides. The exact coordinates of the mapped G4s were used to retrieve the corresponding sequences in fasta format (bedtools getfasta, using the same reference genomes as described by Marsico et al.). We used the previously described regular expression matching approach to search for G4-L1 motifs and assess G4-L1X (X = {A,T,C,G}) proportions.
G4-L1 motif polymorphism evaluation
The latest variation data from the 1000 Genomes project (42) was retrieved from the dedicated data portal (phase 3 1000 Genomes data, accessed on September 2017) in vcf format. We intersected the positions of each of the reported common SNPs, defined as having a minor allele frequency of ≥0.01 in at least one of the five major human populations [African (AFR), Admixed American (AMR), East Asian (EAS), European (EUR) and South Asian (SAS)] with at least two unrelated individuals having the minor allele. The nucleotide variations were searched within the 15 positions of every G4-L1 motif found in hg38. Pairwise Mann–Whitney u-tests (with P-value adjustment) were applied to evaluate differences in overlapping variant counts in G-runs versus loop positions, and between different G positions. Polymorphism rates were also assessed for six other types of non-B DNA motifs: direct repeats (DR), inverted repeats (IR), mirror repeats (MR), A-phase repeats (APR), short tandem repeats (STR) and Z-DNA, for which coordinates were retrieved from the non-B database (available at http://nonb.abcc.ncifcrf.gov) (43). Proportions of non-polymorphic sequences were compared between the different types of motifs using Chi-squared tests.
G4-L1 motifs in other species
Besides the human genome, we searched for G4-L1 motifs in 77 additional vertebrate genomes: 55 mammals (including 16 primates), 22 other jawed vertebrates and the lamprey genome. In addition, we also analyzed 25 invertebrate genomes (accessed from the UCSC Genomes portal (44)), 189 protist genomes (accessed from the EnsemblProtists database (45)) as well as 217 genome assemblies for bacterial genomes (accessed from the EnsemblBacteria database (45)). The total number of G4-L1 motifs was evaluated along with loop composition, total GC content (defined as the sum of G and C nucleotides in the respective assembly) and genome size for each of the assemblies. In order to account for the heterogeneity of GC content in higher mammals, we retrieved chromosome size information for 20 of the mammalian genome assemblies. With this information, we binned each reference genome into non-overlapping 10 Kbp (kilo base pairs) or 1 Mbp (mega base pairs) fixed-size windows (using the makewindows utility from the bedtools suite). Then, we counted the number of G4-L1 motifs present in each of these bins (bedtools intersect utility, -c option to report the number of hits). At last, correlations between GC content and G4-L1 counts were computed for each bin. Spearman’s correlation coefficients were used to test for correlations among the different variables.
G4-L1 motifs in other species
Principal component analysis (PCA) was performed using the FactoMineR and factoextra packages in the R environment, initially for eukaryotes only and then for all species. The analysis was performed on loop composition information (active variables: A-A-A, T-T-T, G-G-G, C-C-C and mixed loops content), after having normalized the data matrixes (variables were centered and reduced), as described by Lê et al. (46). Confidence level ellipses were assessed using the fviz_pca_ind function of the factoextra package. Correlation between A-A-A (T-T-T, C-C-C or mixed) loop content and G-G-G loop content in eukaryotes was estimated by calculating Spearman’s correlation coefficients. Estimated divergence times between different eukaryotic species and the human genome (in Mya, millions of years ago) were retrieved from the TimeTree database (47). The correlations between divergence time and A-A-A, G-G-G, T-T-T, C-C-C or mixed loops content were assessed using R scripts.
Phylogenetic tree of vertebrates
We retrieved branch lengths for 100 vertebrate genomes in Newick format from the UCSC Genome Browser database and reconstructed the phylogenetic tree using FigTree [available for download at: http://tree.bio.ed.ac.uk/software/figtree/]. Information about the phylogeny of the AID/APOBEC genes was retrieved from published work (48,49).
[GGGX]n microsatellite motifs
We interrogated, by regular expression matching, the human reference genome for [NNNX]2+ repeats, where ‘2+’ denotes at least two consecutive repeats of the subunit and X = {A,T,C,G}. We obtained the overall number and genomic locations for each of the possible subunit repeats, comprising specifically the [GGGX]2+ repeats. Then, [GGGX]2+ microsatellite polymorphism was assessed by intersecting the motif positions with known common SNPs from phase3 1000 Genomes variation data. Data describing the distribution of other [NNNX]2+ motifs in the human genome is available from the corresponding authors.
Circular dichroism
CD experiments were carried out at 20°C with a JASCO J-1500 spectropolarimeter equipped with a Peltier temperature controller (Jasco PTC-348WI) interfaced to a PC, by using 0.5 cm path rectangular quartz cells (1 ml reaction volume). Scans were recorded from 210 to 330 nm with the following parameters: 100 mdeg sensitivity, 1 nm data pitch, 200 nm min−1 scan speed, 1 s response, 1 nm band width and 4 accumulations. CD spectra of solutions containing 3 μM of (GGGX)4 were recorded in lithium cacodylate (10 mM) supplemented with KCl (100 mM) at pH 7.2. The signal was further smoothened through the Savitzky-Golay method (2 order, 20 points window). The CD data were blank-subtracted and normalized to molar dichroic absorption (Δϵ) on the basis of concentration using Eq. Δϵ = θ/ (32980 x c x l), with θ the ellipticity in millidegrees, c concentration in mol L−1 and l the path length in cm. Sequences used for the experiment:
[GGGA]4: TTGGGAGGGAGGGAGGGATT,
[GGGC]4: TTGGGCGGGCGGGCGGGCTT,
[GGGG]4: TTGGGGGGGGGGGGGGGGTT,
[GGGT]4: TTGGGTGGGTGGGTGGGTTT.
FRET-melting experiments
Quadruplex-structure stabilization in the presence of PhenDC3 was monitored via Fluorescence Resonance Energy Transfer (FRET)-melting assay performed in 96-well plates on real time polymerase chain reaction (PCR) apparatus 7900HT Fast Real-Time PCR System as follow: 5 min at 25°C, then increase of 0.5°C every minute until 95°C. Each experimental condition was tested in duplicated in a volume of 25 μl for each sample. FRET-melting assay was performed with oligonucleotides equipped with FRET partners (here: FAM and TAMRA) at each extremity in the presence or absence of PhenDC3. The oligonucleotides were prepared at 0.2 μM and the ligands at 1 μM final concentrations. Measurements were made with excitation at 492 nm and detection at 516 nm in the so-called K+1 buffer containing lithium cacodylate (10 mM, pH 7.2), KCl (1 mM) completed by LiCl (99 mM). The stabilization induced by PhenDC3 on the G-quadruplex structures was measured by identifying the temperature at half denaturation of the G4 in the presence or absence of the ligand. The experiments were carried out on pre-folded G-quadruplex structures: the sequences were heated at 90°C for 5 min and left to cool down at 4°C overnight. Sequences used for the experiment:
F[GGGA]4T: 6FAM-(TTGGGAGGGAGGGAGGGATT)-3′TAMRA,
F[GGGC]4T: 6FAM-(TTGGGCGGGCGGGCGGGCTT)-3′TAMRA,
F[GGGG]4T: 6FAM-(TTGGGGGGGGGGGGGGGGTT)-3′TAMRA,
F[GGGT]4T: 6FAM-(TTGGGTGGGTGGGTGGGTTT)-3′TAMRA,
with 6FAM: 6-carboxyfluorescein and TAMRA: 6-carboxy-tetramethylrhodamine).
[GGGX]4 repeat instability in yeast cells
The synthetic [GGGA]4, [GGGT]4, [GGGC]4 and [CAGT]4 DNA microsatellite repeat clones were designed and purchased from GenScript and then integrated by the Gibson cloning assembly method at the same cloning site into the CEN11 TRP1 ARS replicative URA3 reporter pSH44 plasmid, as previously described (50). The resulting plasmids were introduced into the haploid MGD131-102A strain (MATa arg4Δ-2060 leu2-3,112 trpl-289 ura3-52 cyhRstrain (51), upon selection for tryptophan prototroph transformants. As expected, yeast cells carrying the plasmid also became uracil prototrophic, indicating that the in-frame [GGGX]4 inserts did not inactivate the expression of the URA3 gene phenotype. To test the G18 array, we used the ORT 5604 strain (same background as MGD131-102A) (10). To measure the instability of the four [GGGX]4 repeats, in each case, 12 parallel cultures were grown for 8 generations and appropriate dilutions were plated on SC-TRP and 5-FOA-TRP containing media, in order to count the viable cells that retain the reporter plasmid and select for ura3 auxotrophic cells (5-FOA resistant cells retaining the plasmid), respectively. The mutation rates per cell and per division were calculated with the bz-rates tool (52). Briefly, the tool's estimator calculates the mean number of mutations and mutation rate, taking into account differential growth rates and plating efficiency.
Media
Saccharomyces cerevisiae single colonies were cultured in liquid synthetic complete SC-URA and then for eight generations in SC-TRP in the presence of dimethyl sulfoxide (DMSO) and the presence or absence of PhenDC3 (10 μM). Cells were diluted and plated on SD-TRP and SD +5FOA-TRP. 5FOA-TRP plates were prepared according to standard protocols (53). SC liquid media containing PhenDC3 (10 μM) have been prepared as previously described (11).
Statistics
All statistical analyses were performed in R 3.3.3 for Mac OS X (37).
RESULTS
Short loop G-quadruplexes (G4-L1) in the human genome
To identify and annotate G4 motifs in the latest human reference genome (hg38), we first examined their prevalence in silico, while varying the loop size between 1 (G4-L1) and 12 (G4-L1-12) nucleotides, irrespective of the nucleotide loop composition (Table 1). Then, to evaluate their enrichment in the genome, we calculated their expected frequencies in a locally randomized nucleotide background (see ‘Materials and Methods’ section). We found that the G4 sequences with small loop lengths are markedly enriched in the human genome since their number largely exceeds random expectation (Table 1). The observed/expected ratios are 37, 9 and 5 for the G4-L1, G4-L1-7 and G4-L1-12, respectively, (comparisons between all ratios, ANOVA P < 2.2 × 10−16; from 37 to 9 and from 9 to 5, t-test adjP < 0.001; Table 1). Recently, an in vitro study of DNA G4 maps has also shown that quadruplexes carrying long loops are less enriched than shorter ones (fold enrichment of G3+L1–7 and G3+L8–12 sequences are 47.3 and 15.4, respectively) (41). These results strongly suggests that the G4 motifs in general, and particularly short-looped sequences, have evolved under certain selective pressures in the human genome.
Table 1.
Number of G4-L1-a motifs in the human genome (a = (2,12))
| Number of PQS in hg38 | Number of PQS in local background | Ratio(observed/expected) 10 Kbp bins around PQS | ||||
|---|---|---|---|---|---|---|
| Motif | G3 | G3+ | G3 | G3+ | G3 | G3+ |
| G4-L1 | 18 153 | 39 634 | 487 (±3) | 695 (± 9) | 37 (±0.22) | 57 (±0.76) |
| G4-L1-2 | 51 008 | 79 831 | 2196 (±17) | 2548 (± 19) | 23 (±0.18) | 31 (±0.24) |
| G4-L1-3 | 116 628 | 131 266 | 5612 (±47) | 6053 (± 54) | 21 (±0.18) | 22 (±0.20) |
| G4-L1-4 | 195 501 | 201 984 | 11 300 (±91) | 11 813 (± 74) | 17 (±0.14) | 17 (±0.11) |
| G4-L1-5 | 264 284 | 268 562 | 19 593 (±164) | 20 182 (± 159) | 13 (±0.11) | 13 (±0.10) |
| G4-L1-6 | 331 699 | 334 529 | 30 762 (±176) | 31 389 (± 170) | 11 (±0.06) | 11 (±0.06) |
| G4-L1-7 | 404 347 | 406 531 | 44 983 (±164) | 45 636 (± 199) | 9 (±0.03) | 9 (±0.04) |
| G4-L1-8 | 483 818 | 485 601 | 62 345 (±289) | 63 028 (± 309) | 8 (±0.04) | 8 (±0.04) |
| G4-L1-9 | 563 173 | 564 198 | 82 848 (±334) | 83 593 (± 326) | 7 (±0.03) | 7 (±0.03) |
| G4-L1-10 | 640 520 | 641 386 | 106 862 (±217) | 107 609 (± 173) | 6 (±0.01) | 6 (±0.01) |
| G4-L1-11 | 717 046 | 717 600 | 134 115 (±82) | 134 834 (± 61) | 5 (±0.00) | 5 (±0.00) |
| G4-L1-12 | 796 765 | 797 358 | 164 834 (±21) | 165 584 (± 24) | 5 (±0.00) | 5 (±0.00) |
PQS, putative quadruplex sequences; G3, runs of exactly 3 guanines; G3+, runs of 3 or more guanines. Numbers between parentheses show standard deviation values (from counts in n = 3 independent shuffles).
Then, we examined the G4 loop length and base compositions among the subset of 10 560 sites identified by ChIP-seq using the BG4 antibody (27) and the HaCaT cell line (28). We used three different search algorithms: regular expression matching for G4-L1, G4-L1-7 and G4-L1-12 motifs, G4Hunter (25) and the machine learning model Quadron (39) (see ‘Materials and Methods’ section). By regular expression matching, we identified 171 G4-L1 and 14 703 G4-L1-12 motifs, and up to 18 876 PQS with the G4Hunter algorithm (Supplementary Figure S1A). Outstandingly, short loops are the most abundant (1–3 nt), with single-nucleotide loops representing the most frequent ones (Supplementary Figure S1A), particularly C-rich loops (Supplementary Figure S1B).
To study the potential singularities of these quadruplexes, we examined the sequence and the coordinates of the 18 153 G4-L1 (G3N1G3N1G3N1G3) motifs present in the hg38 reference (Supplementary Table S1). Genome annotations of G4-L1 motifs show relative distributions that are very similar to previous annotations of the ∼376 000 G4-L1-7 dataset (54,55) (Supplementary Figure S2A). G4-L1s are present in ribosomal DNA (23 motifs), with two motifs found in the promoter of the RNA5-8S5 gene (RNA, 5.8S ribosomal 5) and 21 motifs found in the gene bodies of the RNA28S5 (RNA, 28S ribosomal 5), RNA5-8S5, RNA5S15 (RNA, 5S ribosomal 15) and RNA18S5 (RNA, 18S ribosomal 5) genes. G4-L1 sequences are also enriched in gene promoters, 5′UTR regions (Supplementary Figure S2A) and within the first introns of genic regions (Supplementary Figure S2A). In contrast, G4-L1 sequences are strongly depleted in exon regions (Supplementary Figure S2A). As described previously for the consensus PQS (55), there is a strand bias toward the presence of G4-L1 motifs in the non-template strand of gene first introns (Supplementary Figure S2B). Conversely, we see an opposite bias in UTR regions, where G4-L1 sequences tend to accumulate in the template strand of transcription (Supplementary Figure S2B), suggesting two different mechanisms by which the formation of G4 could impact transcription. In conclusion, the enriched G4-L1 subset is representative of the G4-L1-7 PQS in terms of associations with these overall genome features.
The G4-L1A motif (G3AG3AG3AG3) is largely predominant in the human genome
Next, we examined the base composition of the 64 possible G4-L1 sequences. All combinations of L1 loop sequences exist (Figure 1A and Supplementary Table S1) but remarkably, the motifs carrying three A loops (G3AG3AG3AG3, or G4-L1A) represent 49% of the total G4-L1 set (Figure 1B), while the other homogenous motifs G4-L1T, G4-L1G or G4-L1C are considerably less frequent, representing only 4.4, 4.2 and 1.0% of all G4-L1, respectively. The remaining G4-L1 motifs (41.5%) are mixed, with a combination of A, T, C or G loops. Thus, the G4-L1 loops are strongly biased both in their base composition (A >> T ∼ G > C) and in their propensity to bear identical nucleotides within the loops (58.5% of all motifs, the G4-L1A representing 84% of these). These observations cannot be viewed solely as a consequence of an intrinsic bias in the human genome nucleotide richness (∼58% A:T versus ∼42% G:C), since the observed loop composition distribution is significantly different from that observed in the shuffled reference genome (Figure 1C), where G4-L1A, G4-L1T, G4-L1C and mixed motif proportions are close to the expected values (1 combination out of 64 possible ones:  ). To determine whether this remarkable bias of base composition could be specific to the G4-L1 motifs, we also examined the G4 PQS with loops of 2–4 nt. The G4-L2 (51 008 occurrences) containing three AA loops are also frequent (6.1%) and over-represented compared to the other G4-L2 carrying homogeneous CC, TT, GG or mixed loop compositions as compared to the expectations based on random local shuffling of the genome sequence (two-proportions z-tests P = 1.1 × 10−7; Supplementary Figure S3). Similarly, the G4 motifs bearing 3-nt loops are enriched in homogenous A loops, albeit to a much lesser extent (two-proportions z-tests P = 0.046; Supplementary Figure S3). Further, we found that G4-L1C motifs are more strongly correlated with various functional features, such as promoters and 5′UTR regions (Supplementary Figure S4A) whereas G4-L1A motifs are particularly enriched in annotated repetitive regions, mainly present in low-complexity regions (simple repeats, G-rich regions) and in transposable elements (Supplementary Figure S4B). G4 formation within transposable elements has been previously reported (56,57), and shown to stimulate retrotransposition (58) and gene transcription (59).
). To determine whether this remarkable bias of base composition could be specific to the G4-L1 motifs, we also examined the G4 PQS with loops of 2–4 nt. The G4-L2 (51 008 occurrences) containing three AA loops are also frequent (6.1%) and over-represented compared to the other G4-L2 carrying homogeneous CC, TT, GG or mixed loop compositions as compared to the expectations based on random local shuffling of the genome sequence (two-proportions z-tests P = 1.1 × 10−7; Supplementary Figure S3). Similarly, the G4 motifs bearing 3-nt loops are enriched in homogenous A loops, albeit to a much lesser extent (two-proportions z-tests P = 0.046; Supplementary Figure S3). Further, we found that G4-L1C motifs are more strongly correlated with various functional features, such as promoters and 5′UTR regions (Supplementary Figure S4A) whereas G4-L1A motifs are particularly enriched in annotated repetitive regions, mainly present in low-complexity regions (simple repeats, G-rich regions) and in transposable elements (Supplementary Figure S4B). G4 formation within transposable elements has been previously reported (56,57), and shown to stimulate retrotransposition (58) and gene transcription (59).
Figure 1.

Mapping the G4-L1 motifs across the human genome. (A) From the outermost to the innermost circle: chromosome cytobands for the hg38 reference genome; rainfall plots (showing the motif locations on the x-axis versus the distance between consecutive motifs on the y-axis); and density plots (distribution shape over chromosomes) of G4-L1 motif (G3N1G3N1G3N1G3) distribution across the human genome. Red, N = A; green, N = T; blue, N = G; orange, N = C and purple, N = {A,T,G,C}. Inset: linear representation for chromosome X, all loop combinations (gray). (B) Distribution of the different loop compositions across the human genome. Red, N = A; green, N = T; blue, N = G; orange, N = C and purple, N = {A,T,G,C}. (C) Distribution of the different loop compositions across the background (random expectation). Red, N = A; green, N = T; blue, N = G; and purple, N = {A,T,G,C}.
G4-L1 motif clusters: high G4-L1 motif density regions
To assess the relative density of the G4-L1 motifs along the human genome, we examined the genome-wide inter-motif distances (inner circles, Figure 1A and Supplementary Figure S5A). We identified several chromosome regions with small inter-motif distances and thus high G4-L1 density—notably within subtelomeric and pericentromeric regions, a trend previously observed for G4-L1-7 motifs (60). Figure 2A reports the number of motifs found with increasing inter-motif distances, limited to 50 Kbp for clarity (all log-scaled distances are reported in Supplementary Figure S5B). It reveals two distinct trends in the distribution of the G4 sequences: first, for both G4-L1 and G4-L1-7 motifs, a large set of sequences exhibit short inter-motif distances (500 bp or less); second, there is a strong inflexion in the increase of the number of motifs only for G4-L1 at ∼500 bp. The average density of G4-L1 found to have inter-motif distances of 500 bp or less is 50-fold higher than the PQS average density found for the whole genome (Figure 2B and Supplementary Table S2). Thus, a G4-L1 cluster can be defined as a 500 bp region where at least three non-overlapping motifs occur (Figure 2B). Using this threshold, there are 646 G4-L1 clusters in the hg38 reference genome (Supplementary Table S3), comprising 2428 motifs (13.4% of the G4-L1 genome set). Once again, the clustered G4-L1 sequences display an over-representation of identical loop motifs (78.4%), of which 63.9% were G4-L1A, the other types being much less frequent (G4-L1G: 8.4%, G4-L1T: 5.3% and G4-L1C: 0.8%) (Figure 2C). In terms of localization, the G4-L1 clusters are present in every chromosome (Supplementary Figure S6A) with no global strand bias; 325 clusters are located on the template strand and 321 on the non-template strand. Nevertheless, the G4-L1 clusters were significantly more prevalent in chromosomes 2, 14, 21, X and Y (Supplementary Figure S6A), where they also displayed a higher density of motifs within the 500 bp window (we calculated an average of 8.8 motifs/Kbp in clusters within these chromosomes and particularly 10 motifs/Kbp for chromosome X). Particularly, there are two identical clusters within the PAR1 region of chromosome X/Y (starting at 2 227 721 and 2 228 257), carrying 16 G4-L1A and 8 G4-L1A motifs, respectively, in a 500 bp window. In contrast, we only found one region with a cluster bearing ≥3 consecutive G4-L1G, located on chromosome 2 (chr2: 32,916,229-32,916,625). In terms of annotation, G4-L1 clusters are particularly enriched within gene promoters and TTS regions (Supplementary Figure S6B). As an extreme, G4-L1 motifs with identical loops are part of tetra-nucleotide microsatellites arrays [GGGX]n, where X is any of the four nucleotides {A,T,C,G} and n (n ≥ 2) the number of repeats of the core four-letter pattern. To test whether these microsatellites display a similar evolutionary trend as G4-L1 motifs, we analyzed the composition of the >400 000 [GGGX]n motifs present in the human genome (see ‘Materials and Methods’ section). Again, there is a clear bias toward [GGGA]n (chi-squared goodness of fit tests P < 0.05 for n ≥ 2 when comparing against a homogenous representation of each base; Figure 2D) with an almost absolute predominance of A nucleotides in large (n > 4) [GGGX]n motifs. This predominance is specific to [GGGX]n since, for instance, in [AAAX]n the X = A prevalence rapidly decreases after n = 7 repeats while in [GGX]n this decrease is seen after n = 3 repeats (Supplementary Figure S7). With regard to polymorphisms, we identified variants in each class of [GGGX]n microsatellites (using, as described below, common SNP sites from the 1000 Genomes project (40)). The [GGGG]≥3 sequence is the most polymorphic at the population level, in the cluster comprised of short repeat sequences (5 ≥ n ≥ 2) obtained by hierarchical clustering (Figure 2E). Conversely, [GGGC]4-5 motifs are particularly conserved (Figure 2E), which could be correlated with the enrichment for such motifs in promoters. Together, these observations demonstrate that the loop composition within [GGGX]n tetra-nucleotide microsatellites recapitulates the overall G4-L1 biases.
Figure 2.

G4-L1 motif clusters. (A) Number of G4 motifs, G4-L1-7 in red (primary y-axis) and G4-L1 in blue (secondary y-axis), found within increasing sequence distances (in base pairs, bp). (B) Schematic representation of a G4-L1 cluster, defined as a 500 bp region containing at least three non-overlapping G4-L1 motifs (i.e. 3 motifs/0.5 Kbp or 6 motifs/Kbp). (C) Distribution of the different loop compositions of G4-L1 motifs found within clusters across the human genome. Red, N = A; green, N = T; blue, N = G; orange, N = C and purple, N = {A,T,G,C}. (D) Microsatellite motifs were defined as n ≥ 2 repeats of the [GGGX] subunit. To perform the search, positions −3 to −1 were set to G and position 0 was variable (X). The number of repeats (n) of the subunit is shown on the x-axis, whilst the y-axis represents the nucleotide composition of the X position for each of the n values. The number of occurrences, for each value of n, is shown over the corresponding bar. Red, X = A; green, X = T; blue, X = G; orange and X = C. *, Chi-squared goodness of fit tests P < 0.05 (comparison of the observed distributions to an expected homogenous distribution). (E) Variability of the [GGGX]n micro-satellites in the human genome. The heatmap shows log2 fold-change differences between [GGGX]n motif counts in hg38 and in a genome where common SNPs (from the 1000 Genomes Project database) were masked, for different micro-satellite motif sizes. >0: motif less polymorphic than expected; <0: motif more polymorphic than expected. Dendrograms created through by-column and by-row hierarchical clustering were added on top and on the side of the heatmap, respectively.
Polymorphisms in the human G4-L1 motifs are restricted
Next, to examine the polymorphism of G4-L1 sequences in the human population, we analyzed the occurrence and the sequence variability of the G4-L1 motifs within the 1000 Genomes project data (42). We found 2845 (16% of the genome set) polymorphic G4-L1 motifs, i.e. overlapping with common SNPs (Single Nucleotide Polymorphisms, see ‘Materials and Methods’ section). The nature of the base changes and their positions are summarized in Figure 3 and the complete list and coordinates of the variants are reported in Supplementary Table S4. For comparison, the rate of polymorphism of six other types of motifs prone to form non-canonical secondary structures (referred to as non-B DNA motifs) is reported in Supplementary Figure S6A. These comprise direct repeats (DR), inverted repeats (IR), mirror repeats (MR), A-phase repeats (APR), short tandem repeats (STR) and Z-DNA. Coordinates were retrieved from the non-B database (available at http://nonb.abcc.ncifcrf.gov) (41). The G4-L1 rate of polymorphism is similar to the one of Z-DNA (17%), slightly higher than those of IR, STR and APR (13, 10 and 14%, respectively), and lower than those of DR (22%) and MR (29%) (Supplementary Figure S8A). Particularly, G4-L1 motifs are slightly less polymorphic than the consensus PQS G4-L1-7 (22%), showing that the most stable in vitro structures are also best conserved within the human population. Interestingly, we observed that the genomic regions carrying G4-L1 sequences, similar to any other non-B DNA motif, appear to be more polymorphic than regions devoid of such motifs (Supplementary Figure S8A).
Figure 3.

Polymorphism of G4-L1 sequences in the human genome. (A) Base composition of the motif loops in polymorphic G4-L1 sequences. Red, N = A; green, N = T; blue, N = G; and orange, N = C. P-values were calculated using Chi-squared goodness of fit tests. Loop 1, P = 0.029; Loop 2, P = 0.0019; Loop 3, P = 0.054. Inset represents the most frequent motif found within polymorphic G4-L1 sequences. (B) The number of point common variants (SNPs) overlapping with each of the positions of G4-L1 motifs was estimated genome-wide. The number polymorphic positions is shown on the y-axis, and the positions in the G4-L1 sequence are reported on the x-axis. Gray, loop position; black, G-run. (C) Comparison of the amount of polymorphic nucleotides found by position in the G4-L1 15nt-motif. ‘G’ refers to positions {1,3,5,7,9,11,10,12}; ‘middle G’ to positions {2,6,10,14} and ‘N’ to positions {4,8,12}. Adjusted P-values were calculated using pairwise t-tests. *, P < 0.01 (one-way ANOVA P-value = 0.00067). (D) Variant composition by position in the G4-L1 motif and by DNA strand. ‘G’ refers to positions {1,3,5,7,9,11,10,12}; ‘middle G’ to positions {2,6,10,14} and ‘N’ to positions {4,8,12}. Red, N = A; green, N = T; blue, N = G; and orange, N = C. (+), non-template strand; (−), template strand.
With respect to base composition, the motifs carrying all T or all A loops are significantly less polymorphic than those carrying all G or mixed loops, while those bearing all C loops are intermediate (Supplementary Figure S8B). Likewise, motifs within clusters are also less polymorphic (Supplementary Figure S8B). However, when inspecting individually each of the base changes in polymorphic motif loops, subtle differences emerge. Namely, in the first loop, C or T bases varied more frequently than A or G while in the second loop, T or G bases were the most variable (Figure 3A). Moreover, the most frequent polymorphic motif found carries preferentially C or T in loop 1 (N1), C or G in loop 2 (N2) and C or G in loop 3 (N3) (inset Figure 3A). In addition, the loop nucleotides (located at positions 4, 8 and 12 in a 15-nt G4-L1 motif) are less polymorphic than the G tracts; in average, every loop position varied 213 times, while every G position varied 281 times (two-sample t-test, P = 0.03) (Figure 3B). The degree of polymorphism also differs within G tracts, where the flanking Gs (positions {1, 3, 5, 7, 9,11, 13, 15}) are significantly more polymorphic than the central Gs (positions {2, 6, 10, 14}) (two-sample t-test, adj.P = 0.0027) (Figure 3C). Although all types of variations are observed for the loop nucleotides, {A,T,C}>G is the most frequent (Figure 3D). On the other hand, within the G-runs, the most frequent change is G>A, irrespective of its position within the G tract (Figure 3D). Interestingly, we did not observe differential variability among the 15 nt positions of the minimal G4-L1G motif (Supplementary Figure S9A) and the most frequent variant was G>T (Supplementary Figure S9B), suggesting that G15 homopolymers behave differently than other G4-L1 sequences.
In conclusion, at the population level, the pyrimidine and G nucleotides in the loops are more polymorphic than the A loops. The remarkable variations in the degree of polymorphism of the various G4-L1 motifs and the large scale divergence of G4-L1 loop composition between species described hereafter raise the question of the molecular origin of this selective variation, occurring at the nucleotide level resolution (see ‘Discussion’ section).
G4-L1A is the least stable quadruplex in vitro and the least prone to trigger genetic instability in vivo
As previously shown by UV/CD spectroscopy, G4-L1A, T or C containing oligonucleotides (18 nt length) can form stable G-quadruplex structures in vitro, but with a melting temperature (T1/2) highly dependent on the residue in the loop (G4-L1T ∼ C > G4-L1A) (33). Upon FRET-melting analysis of the four [GGGX]4 oligonucleotides (16 nt G-rich core + TT in 3′ and 5′), the G4-L1C, the G4-L1T sequences and the G4-L1G homopolymer exhibit the highest melting temperatures (64.5, 63.2 and 63.9°C, respectively) and G4-L1A the lowest (47.9°C) (Table 2 and Supplementary Figure S10). In the presence of the G4 ligand PhenDC3 (1, 2 and 5 molar equiv.), we observed largely enhanced T1/2 values in all cases (e.g. 14.4 < ΔT1/2 < 25.2°C at 2 molar equiv.) which increase in a dose-dependent manner. This indicates strong binding of the ligand on each preformed G4 secondary structure irrespective of the sequence (Table 2). In each case, the circular dichroism signature was typical of parallel G-quadruplexes (Figure 4A). However, the G4-L1G ([GGGG]4) homopolymer (with a 16 nt G-rich core similar to the dG16 oligonucleotide described by Masiero et al. (61)) showed no clear transition in the FRET-melting curves, suggesting the presence of several species (intra- and intermolecular G4s). Thus, all data indicates that each G4-L1 motif readily folds into a G-quadruplex structure but, remarkably, the G4-L1A motif that is significantly over-represented in the human genome, is the least stable structure in vitro.
Table 2.
Melting temperatures (T1/2) measured by FRET-melting in K+ 1 buffer (Li cacodylate 10 mM, KCl 1mM, LiCl 99 mM)
| Sequence | T1/2 (°C) | T1/2 (°C) 1eq PhenDC3 | ΔT1/2 (°C) 1eq PhenDC3 | T1/2 (°C) 2eq PhenDC3 | ΔT1/2 (°C) 2eq PhenDC3 | T1/2 (°C) 5eq PhenDC3 | ΔT1/2 (°C) 5eq PhenDC3 |
|---|---|---|---|---|---|---|---|
| F-(GGGA)4-T | 47.9 ± 2.4** | 64.8 ± 1.0 | 17.9 | 72.1 ± 0.6 | 25.2 | >83.0 ± 0.4 | >36.1 |
| F-(GGGC)4-T | 64.5 ± 0.5 | 76.7 ± 1.0 | 12.2 | 82.6 ± 1.4 | 18.1 | >88.6 ± 0.3 | >24.1 |
| F-(GGGG)4-T | 63.9 ± 0.6* | 72.7 ± 1.2 | 8.8 | 78.3 ± 0.6 | 14.4 | >85.9 ± 1.0 | >22.0 |
| F-(GGGT)4-T | 63.7 ± 0.5 | 80.1 ± 0.3 | 16.4 | 84.5 ± 1.1 | 20.8 | >89.6 ± 0.2 | >25.9 |
*indicated approximate value as the curves show no clear transition, indicating the presence of several species (intra- and intermolecular G-quadruplexes).
**T1/2 values with high standard deviation (likely due to the existence of several species in equilibrium as suggested by the drift of the melting curve at low temperatures).
Figure 4.

In vitro and in vivo stability of G4-L1 quadruplexes. (A) Circular dichroism (CD) spectra of G4-L1 sequences. All spectra exhibit the characteristic features of parallel G4 structure i.e. negative peak at ∼240 nm and positive peak at ∼260 nm. Black, G4-L1A; red, G4-L1C; blue, G4-L1G and magenta, G4-L1T. (B) Mutation rates per cell per division, corrected by the plating efficiency. Black, culture in DMSO; gray, culture +PhenDC3. Significant fold changes between DMSO and +PhenDC3 conditions are shown. Bars, upper 95% confidence interval bound.
Next, to examine the behavior of the different G4-L1 motifs in vivo, we exploited a highly sensitive plasmid assay, developed in S. cerevisiae (62). For this purpose, each [GGGX]4 sequence, as well as a control [CAGT]4 motif, was inserted in frame within the coding sequence of the plasmid-borne URA3 gene. This assay allows to phenotypically measure the occurrence of out-of-frame mutations that yield 5FOA resistant (uracil auxotroph) colonies (see ‘Materials and Methods’ section). The mutation rates per cell per division for each sequence, along with their 95% confidence intervals, are reported in Supplementary Table S5. In untreated conditions (DMSO), the [GGGA]4 and the control [CAGT]4 arrays exhibited the lowest level of instability, followed by G18 (3-fold increase, no overlap of the 95% confidence limits) and then by [GGGT]4 and [GGGC]4 which exhibited a 5- to 6-fold increase of instability compared to the [GGGA]4 array (Figure 4B). Treatment of the cells with PhenDC3 led to significant increases in the mutation rates of the [GGGX]4 arrays but not in the control sequence. The increase reached 7-fold for G4-L1A, 14-fold for G4-L1T and 34-fold for G4-L1C (Figure 4B). These results, and our previous assay using non-transcribed human minisatellite arrays (33), demonstrate the differential capacity of G4-L1s, either spontaneously or when targeted by small molecules, to trigger genetic instability in vivo. The correlation with the thermal stability of every G4-L1 emphasizes the dramatic differential consequence of the loop base composition and their capacity to form quadruplexes in vivo. To note, the stability of the final complex [(GGGA)4 + PhenDC3] observed (80°C) may contrast with the still lower genetic instability of this sequence as compared to the others. However, it is also likely that the probability of formation of this G4 in vivo is much lower (and/or its lifetime shorter) due to its low T1/2, thereby resulting in a weaker ability of PhenDC3 to target it in vivo.
These results raise the question of the molecular mechanisms that drive G4-L1 sequence maintenance and dynamic evolution, being either isolated, clustered in sub-chromosomal regions or organized in microsatellite repeats.
Sequence specificity of the G4-L1 motifs across species
Given the over-representation of G4-L1A motifs in the human genome, we assessed whether similar biases in loop composition exist in other species. Altogether, we mined 78 vertebrates, 25 other metazoans (invertebrates), 189 protists, 20 plants, 70 yeasts as well as 217 bacterial genomes for G4-L1 sequences. Our analyses showed that G4-L1 motifs are present in all eukaryotes (Supplementary Table S6), exhibiting different motif densities (G4-L1 motifs per mega base pair, Mbp) by phylogenetic group (Figure 5A). Indeed, primate genomes (17 assemblies) contain significantly less G4-L1 sequences than other placental mammals (Wilcoxon rank sum test with P-value adjustment for multiple comparisons, P = 0.005; 34 assemblies) and particularly, the yeast genomes carry significantly less (pairwise comparisons using Wilcoxon rank sum tests with P-value adjustment for multiple comparisons, each P < 0.05) G4-L1 sequences than any other eukaryote examined (Figure 5A). Our analysis across 193 eukaryotic genomes revealed that the absolute number of G4-L1 motifs within a genome is positively correlated with genome size (Spearman's rho = 0.87; Figure 5B) and also to a large extent, to %GC content (Spearman's rho = 0.47) although the significance of this last relationship is highly dependent on the phylogenetic group (Supplementary Figure S11A). Indeed, when using global GC genome content, the positive correlation between G4-L1 and %GC content is mostly explained by non-primate vertebrate genomes (Spearman's rho = 0.31; 61 assemblies) and yeast genomes (Spearman's rho = 0.37), as the relationship is not significant for primate, invertebrate metazoans and plant genomes (Supplementary Figure S11A). However, the connection between these two variables was lost when using overall genomic %GC in higher mammals, given the heterogeneity of GC content in these genomes. Although the correlations are moderate, they are significant when proceeding by 10 Kbp or 1 Mbp fixed-size windows for correlation assessment in mammals (see ‘Materials and Methods’ section; Supplementary Figure S11B). Intriguingly, there is no overall significant relationship between G4-L1G (polyG15) content and GC content (Figure 5B), with only non-primate vertebrate genomes (Spearman's rho = 0.37, P = 0.0349) showing a significant positive correlation between these two variables (Supplementary Figure S12).
Figure 5.

Genome metrics and G4-L1 motif content of various eukaryotic genomes. (A) Genome size (in mega base pairs, Mbp), GC content and G4-L1 motif density (number of motifs found per Mbp) for different groups of eukaryotes. (B) Relationship between genome size (in mega base pairs, Mbp) and G4-L1 motif counts, GC content and G4-L1 motif counts and GC content and G4-L1G (polyG15) motif content. Spearman correlation coefficients (rho) and their statistical significance are provided at the top of each panel. Regression lines are shown in blue (Preg, linear regression significance).
As observed in the human genome, G4-L1 sequences found in the other eukaryotic genomes are also enriched but exhibit different loop sequence biases. As illustrated in Figure 6A, unsupervised learning (the phylogenetic group belonging was excluded from the analysis) allows to detect two trends in the compositional bias of the G4-L1 loops: in placental mammals—primates in particular—there is a bias toward G4-L1A motifs while in amphibians, fish and invertebrate genomes, as well as in plants and protists (Supplementary Figure S13A) there is an excess of G4-L1G motifs. Intriguingly, the proportions of these two types of G4-L1 motifs exhibit the strongest negative correlation (Spearman's rho = -0.81, P < 2.2e-16) (inset Figure 6A and Supplementary Figure S14). Detailed analysis of G4-L1 loop compositions for each of the phylogenetic groups confirms the trends showing a significantly higher proportion of G4-L1A motifs in primates than in any other group (pairwise comparisons using Wilcoxon rank sum tests with P-value adjustment for multiple comparisons, each P < 0.05) and, conversely, significantly lower proportions of G4-L1G motifs (Figure 6B). Moreover, the lack of significant correlation between %GC content and G4-L1G content, indicates that this trend is not a mere consequence of genome nucleotide composition. At last, it is also striking that G4-L1T and G4-L1C always represent a small fraction of G4-L1 motifs (Figure 6B), with respective mean values of ∼3% and ∼1%, albeit a slightly higher proportion of G4-L1T sequences in vertebrates than in other eukaryotes (Figure 6B).
Figure 6.

Two distinct loop composition trends exhibited by G4-L1 motifs in various eukaryotic genomes. (A) Unsupervised learning by PCA was performed using the five principal loop compositions (G4-L1A, G4-L1T, G4-L1G, G4-L1AC and mixed loops G4-L1) as variables. Principal components 1 and 2 are plotted on the x- and y-axes, respectively, and show the similarity (distance) between each species based on their quadruplex sequence content only. Each dot represents an organism and each color represents its phylogenetic group. Ellipses were generated using 70% confidence intervals around the barycenters of each phylogenetic group. PC1 accounts for 46% of the variance in the dataset and largely separates G4-L1A rich species (right) from G4-L1G rich species (left). Inset shows the correlation between A loop content (x-axis) and G loop content (y-axis). rho, Spearman's correlation coefficient; Preg, linear regression significance. (B) G4-L1 loop content of the different phylogenetic groups of eukaryotes. P, Kruskal–Wallis rank sum test P-values. Red asterisks indicate mean values significantly different (pairwise comparisons using Wilcoxon rank sum tests, adj P < 0.05) from those of all other groups.
Furthermore, when evaluating loop content versus estimated divergence times between different eukaryotic species and the human genome (Mya, millions of years ago), we observed more distinctly these two trends in vertebrates and other eukaryotes (Figure 7). Indeed, G4-L1A motifs seem to ‘emerge’ in jawless fish (600 Mya) and their fraction increases progressively in vertebrate genomes, with a proportion of 0.25 G4-L1A sequences in placental mammals (∼200 Mya). Remarkably, the repertoire of G4-L1A expands in Primates (∼55 Mya), reaching up to 67% of all G4-L1 motifs in the Macaca fascicularis genome. Conversely, the occurrence of G4-L1G slowly decreases from plants (over 1500 MYA) to invertebrate metazoans (around 650 MYA) and drops more rapidly within vertebrates up to primates, which carry the lowest proportions of polyG15 (median for 17 genomes, 9.5%).
Figure 7.

Relationship between G4-L1 loop composition and divergence times with the human genome. Loop composition versus divergence with the human genome (Mya, 106 years ago). Upper panels: left, %G-G-G; right, %A-A-A. Lower panels: left, %T-T-T; center, %C-C-C; right, % Mixed loops. Green, primates only; Blue, vertebrates (other than primates); Red, Eukaryotes non-vertebrates. Black dotted lines indicate 55, 200 and 600 Mya, respectively.
At the extremes, on the G-rich side, we found C. elegans with 79.6% G4-L1G motifs (1130 motifs total) and the extremophilic unicellular red alga Galdieria sulphuraria with only two G4-L1G motifs; on the A-rich side, we found 10 Primate species with 50.3–66.9% G4-L1A motifs (from a total of 13 255 motifs in the gibbon, to a total of 14 970 motifs in the crab-eating macaque). Notably, there are only small differences in the observed trends between closely related species (Supplementary Figure S15). To measure intra-species biases, we investigated 11 Drosophila species (including D. melanogaster), 10 Caernorhabditis species (including C. elegans) and 5 Saccharomyces species (including 64 strains of S. cerevisiae) (Supplementary Table S6). The bias toward G4-L1G sequences is conserved within these species, with a median polyG15 content of 57, 70 and 67%, respectively. Curiously, some genera of protists exhibit exceptionally high proportions of G4-L1A and particularly G4-L1T motifs (Leishmania and Trypanosoma genera; Supplementary Figure S13B and Table S7), while most Plasmodium species fit the global observed trend and are G4-L1G-high (Plasmodium, Supplementary Figure S13B and Table S7). Although rare (as observed in any G4-L1-7 motif), the G4-L1 motifs present in bacterial genomes exhibit slight loop biases, favoring G4-L1G motifs (in average, 13% of the G4-L1 motifs found) and mixed loops motifs (in average, 20% of the G4-L1 motifs found) (Supplementary Table S7).
Of note, our examination of the G4-L1X (X = {A,T,C,G}) content in G4 maps obtained in vitro (41) in a subset of 12 species corroborates the existence of the trends that we observed in silico (see ‘Materials and Methods’ section), with the human and mouse G4 maps being G4-L1A high and conversely, the lower eukaryotes and bacteria being G4-L1G high (Supplementary Figure S16). In addition, very similar G4-L1X proportions were found in silico and in folded G4s predicted in vitro.
DISCUSSION
Estimates of G-quadruplex content in the human genome range from 18 000 (G4-L1) to over 790 000 (G4-L1-12) sites, with limited knowledge of which fraction likely carries cellular functions. Compelling evidence already implicates distinct G4 motifs in various biological processes, suggesting that at least some quadruplexes are likely ‘at work’. However, the risk of assuming the reliance of a phenotype on the ability of a large set of extended length sequences to adopt such structure in vitro remains uncertain. As a step-forward, here, we focused on the subset of short loop PQS that by themselves already exhibit differential genomic and function-related features.
Singularities of the G-quadruplex forming sequences
A total of 18 153 single-nucleotide-loop G-quadruplex motifs are widespread in the human genome. They occur as isolated motifs (15 725) or clustered (2428) in sub-chromosomal regions at an average density of 1 motif per Mbp, although there are regions of markedly high G4-L1 density (up to 6 motifs per Kbp). Furthermore, G4-L1 motifs also occur as microsatellite tetra-nucleotide arrays [GGGX]n (N = 8522, n 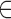 (4,20)), including 1–5 full-length G4-L1 motifs. Intriguingly, in all cases, the G4-L1 sequences show a significant bias in G-quadruplex loop composition, favoring identical adenine loops (G3AG3AG3AG3 or G4-L1A) not only in the human genome but also within the 54 other mammalian genomes examined here. Among the four [GGGX]4 motifs, the [GGGA]4 sequence yields the least thermodynamically stable quadruplex structure in vitro and correspondingly bears the lowest potential to trigger quadruplex-dependent genetic instability, both spontaneously and in the presence of a G-quadruplex ligand (Table 2). Of note, the present G4-dependent assay monitored genetic instability in a coding region (potentially dependent on transcription) while our previous G4-L1A/T/C loop minisatellite assay was independent of transcription but dependent on replication directionality (63). The similar hierarchy of the effect of the loop composition suggests that the G4-dependent instability is dependent on replication directionality in both assays. Alternatively, if a combined replication/transcription-dependent mechanism is involved (perhaps related to R-loop formation), it is similarly sensitive to the G4 loop base composition.
(4,20)), including 1–5 full-length G4-L1 motifs. Intriguingly, in all cases, the G4-L1 sequences show a significant bias in G-quadruplex loop composition, favoring identical adenine loops (G3AG3AG3AG3 or G4-L1A) not only in the human genome but also within the 54 other mammalian genomes examined here. Among the four [GGGX]4 motifs, the [GGGA]4 sequence yields the least thermodynamically stable quadruplex structure in vitro and correspondingly bears the lowest potential to trigger quadruplex-dependent genetic instability, both spontaneously and in the presence of a G-quadruplex ligand (Table 2). Of note, the present G4-dependent assay monitored genetic instability in a coding region (potentially dependent on transcription) while our previous G4-L1A/T/C loop minisatellite assay was independent of transcription but dependent on replication directionality (63). The similar hierarchy of the effect of the loop composition suggests that the G4-dependent instability is dependent on replication directionality in both assays. Alternatively, if a combined replication/transcription-dependent mechanism is involved (perhaps related to R-loop formation), it is similarly sensitive to the G4 loop base composition.
By integrating sequence-based prediction with genome annotation, we found that the G4-L1 quadruplex motifs colocalize with functionally significant loci, such as promoters and 5′UTR regions, recapitulating the location of the much larger PQS consensus dataset (G4-L1-7, up to 404 347 sites). On the other hand, we identified several singular features among the G4-L1 motifs that specifically raise the question of their functionality, and therefore whether or not they represent ‘unwanted’ sequences at-risk able to trigger genome instability. Alternatively, but not exclusively, some short loop G4s may have acquired specific functions and thus play important biological roles. These intriguing features are: (i) the over-representation of the G4-L1 motif per se; (ii) the preferred homogeneity of the loop base composition; (iii) the particularly high frequency of the thermodynamically stable G4-L1C sequences targeted by the BG4 antibody and associated with active chromatin; (iv) the depletion of G4-L1C and G4-L1T sequences in most eukaryotes; (v) the overwhelming proportion of G4-L1A in mammals or G4-L1G–motifs in other eukaryotes (discussed hereafter). These differential features strongly suggest that the G4-L1A and G4-L1G motifs and, to a significant but lower extent, the G4-L2 and G4-L3 motifs with purine loops, have been subjected to selective forces that favor their emergence and maintenance in genomes, perhaps in connection with their capacity to form more labile quadruplexes. This selection could have been functionally advantageous, for instance if bound by endogenous regulatory binding proteins involved in transcriptional regulation (18) or adapted to perform post-replicative chromatin-related epigenetic functions (64). Mechanistically, the conjunction of these singular features could sufficiently but not exclusively explain that all species tend to reduce the G4 threat either by mutating or eliminating the most stable G4 secondary structures, carrying Cs and Ts in their loops. Since all G4-L1s adopt parallel G4 structures in vitro, it is attractive to consider that the bases forming the loops are the most exposed to mutagenic processes. However, with the exception of the G4-L1G in which all bases are equally polymorphic, these loop positions are less polymorphic than the G-runs themselves.
Another consideration, intimately linked to the formation of G-quadruplex structure, is the possibility for the PQS complementary strand to form a different C-based i-motif. In particular, recent results indicate that the long (C3T3)nC3 single strand DNA can form an i-motif under physiological conditions (notably at physiological pH) (65,66), although i-motifs are about 100-fold less thermodynamically stable than the corresponding G4 structures and their formation is not kinetically favored (67,68). In comparison, the (C3T)3C3 i-motif, complementary to the G4-L1A motif, was shown to be the least stable i-motif and to form intermolecular, rather than intramolecular structures (66). Although these observations argue against a predominant formation of i-motifs in front of a G4-L1A quadruplex, its competitive or synergistic role on G4-L3A quadruplex formation and evolutionary trend remains to be studied.
Evolutionary trend of G4L1-loop base composition
The human G4-L1 sequences per se are rather conserved across the human population whilst being flanked by regions of higher variability. Also, the G4-L1G sequence (G15) is the most polymorphic of all G4-L1s carrying identical loops and the guanines flanking the loops are more variable than the guanines that form the middle quartet. These observations raise the possibility that guanines that participate to the formation of the top and bottom quartets and to the loops are more prone to mutagenesis, in particular oxidation (69,70), thus pointing to a mutational mechanism linked to DNA damage and repair.
Our comprehensive data mining of more than 500 species genomes uncovered a strong correlation between evolutionary taxonomy and G4-L1 loop composition. It appears that the accumulation of G4-L1A motifs is specific to mammals—and primates in particular—with a marked increase in their proportion among G4-L1 sequences in detriment of G4-L1G motifs. In contrast, G4-L1A motifs are rare in non-vertebrate metazoans, plants, fungi and prokaryotes, whilst G≥15 sequences are widespread. These mutually exclusive biased loop compositions may suggest the existence of putative species-specific mutagenic processes that could drive ancestral PQS sequences toward GA-L1G on one side and to G4-L1A on the other. Another possibility is that the ancestral PQS sequence was a G homopolymer that evolved on the vertebrate branch toward G4-L1A through mechanisms that introduced G>A modifications with certain periodicity. Speculatively, and as suggested by the breaking point observed at around 600 Mya when comparing G>A/A>G trends with species divergence times, this putative mechanism may have appeared with the first vertebrates. When looking at the evolution of DNA processing proteins that originated at the beginning of the vertebrate radiation, the AID/APOBEC family, which comprises several proteins capable of deaminating cytosine into uracil (71), stands out as a potential candidate (48,72) (summarized in Supplementary Figure S17). Indeed, the activity of these enzymes meditates the C > U conversion, which then could lead to G > A on the G-rich strand. Interestingly, the duplication and divergence of the double deaminase domain APOBEC3 genes present in placental mammals, which led to expansion of the family, with the addition of the APOBEC3A-H genes in primates (48,49), could help explain the acceleration of the G>A conversion seen in this phylogenetic branch. On the other hand, the extreme prevalence (98%) of the G-runs versus the other GGGX motifs in the C. elegans genome and the finding that these sequences are eliminated by complete deletion during development and in animals deficient for the dog-1 helicase (12) suggest that different molecular mechanisms can play a role in handling the equilibrium between the maintenance and the inactivation of short-loop G4-L1 motifs. Alternatively, G4-L1 motifs embedded in microsatellite arrays can evolve by replication slippage, ‘killing’ the quadruplex by deletion when less than 4 [GGGX] repeats are retained.
Lastly, G4-induced instability could in some instances be positively selected for, as it may be exploited as a rudimentary inducer of genetic diversity. Beyond the driving forces of the different mutagenic processes in individual species, selection is also an essential driving force that ultimately shapes the genome content. For instance, the only short-loop G4 motif (G4-L1-2-1) with Ts in the genome of the bacteria Neisseria gonorrhoeae is located in the promoter of the pilin expression locus pilE and stimulates its recombination on polymorphic pilS pseudogenes, thus promoting antigenic variation (73). Alongside functional selection, this study raises the key question of the impact of mutagenic processes in shaping the mode of genome evolution in species or, more broadly, taxa.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Sophie Loeillet and Alexandre Serero (Institut Curie) for their assistance with the repeat instability assays, and Claire Beauvineau (Institut Curie) for helping with the FRET-melting experiments. We thank Jean-Louis Mergny for fruitful discussions.
Authors' contributions: E.P.L., A.N. and A.L.V. conceived the concepts and analytical framework. E.P.L. designed and performed the in silico data analysis and statistical analyses, and wrote the code for analyzing and presenting the data. D.V. performed the FRET-melting and the circular dichroism experiments. A.H. performed the analysis of repeat instability in yeast. M.P.T.F. provided the PhenDC3 compound for the in vitro and in vivo experiments. E.P.L., A.N. and A.L.V. wrote the manuscript. All authors read and approved the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR 14-CE35-0003-02]; PIC3i Program from Institut Curie [n°91730 ‘Prospects of Anticancer’]; French Ministry of Education, Research and Technology Doctoral Fellowship (to E.P.L.). Funding for open access charge: Research Grants/author’s Host Department.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gellert M., Lipsett M.N., Davies D.R.. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. U.S.A. 1962; 48:2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sen D., Gilbert W.. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988; 334:364–366. [DOI] [PubMed] [Google Scholar]
- 3. Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S.. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006; 34:5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paeschke K., Simonsson T., Postberg J., Lipps H.J.. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Mol. Biol. 2005; 12:847–854. [DOI] [PubMed] [Google Scholar]
- 5. Paeschke K., Juranek S., Simonsson T., Hempel A., Rhodes D., Lipps H.J.. Telomerase recruitment by the telomere end binding protein-beta facilitates G-quadruplex DNA unfolding in ciliates. Nat. Struct. Mol. Biol. 2008; 15:598–604. [DOI] [PubMed] [Google Scholar]
- 6. Smith J.S., Chen Q., Yatsunyk L.A., Nicoludis J.M., Garcia M.S., Kranaster R., Balasubramanian S., Monchaud D., Teulade-Fichou M.P., Abramowitz L. et al.. Rudimentary G-quadruplex-based telomere capping in Saccharomyces cerevisiae. Nat. Struct. Mol. Biol. 2011; 18:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Besnard E., Babled A., Lapasset L., Milhavet O., Parrinello H., Dantec C., Marin J.M., Lemaitre J.M.. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012; 19:837–844. [DOI] [PubMed] [Google Scholar]
- 8. Valton A.L., Hassan-Zadeh V., Lema I., Boggetto N., Alberti P., Saintomé C., Riou J.F., Prioleau M.N.. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014; 33:732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castillo Bosch P., Segura-Bayona S., Koole W., van Heteren J.T., Dewar J.M., Tijsterman M., Knipscheer P.. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014; 33:2521–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ribeyre C., Lopes J., Boulé J.B., Piazza A., Guédin A., Zakian V.A., Mergny J.L., Nicolas A.. The Yeast Pif1 helicase prevents genomic instability caused by G-Quadruplex-Forming CEB1 sequences in vivo. PLos Genet. 2009; 5:e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Piazza A., Boulé J.B., Lopes J., Mingo K., Largy E., Teulade-Fichou M.P., Nicolas A.. Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 2010; 38:4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lemmens B., van Schendel R., Tijsterman M.. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun. 2015; 13:8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S.P.. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012; 8:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sarkies P., Reams C., Simpson L.J., Sale J.E.. Epigenetic instability due to defective replication of structured DNA. Mol. Cell. 2010; 40:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleming A.M., Zhu J., Ding Y., Visser J.A., Zhu J., Burrows C.J.. Human DNA repair genes possess potential G-quadruplex sequences in their promoters and 5′-untranslated regions. Biochemistry. 2018; 57:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H.. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernando H., Sewitz S., Darot J., Tavaré S., Huppert J.L., Balasubramanian S.. Genome-wide analysis of a G-quadruplex-specific single-chain antibody that regulates gene expression. Nucleic Acids Res. 2009; 37:6716–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gray L.T., Vallur A.C., Eddy J., Maizels N.. G-quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014; 10:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fleming A.M., Zhu J., Ding Y., Burrows C.J.. 8-Oxo-7,8-dihydroguanine in the context of a gene promoter G-quadruplex is an on-off switch for transcription. ACS Chem. Biol. 2017; 12:2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wieland M., Hartig J.S.. RNA quadruplex-based modulation of gene expression. Chem. Biol. 2007; 14:757–763. [DOI] [PubMed] [Google Scholar]
- 21. Kumari S., Bugaut A., Huppert J.L., Balasubramanian S.. An RNA G-quadruplex in the 5′UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007; 3:218–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huppert J.L., Balasubramanian S.. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Todd A.K., Johnston M., Neidle S.. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005; 33:2901–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maizels N., Gray L.T.. The G4 genome. PLoS Genet. 2013; 9:e1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bedrat A., Lacroix L., Mergny J.L.. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 27. Biffi G., Tannahill D., McCafferty J., Balasubramanian S.. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hänsel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Kimura H., Narita M. et al.. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 29. Risitano A., Fox K.R.. Influence of loop size on the stability of intramolecular DNA quadruplexes. Nucleic Acids Res. 2004; 32:2598–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rachwal P.A., Brown T., Fox K.R.. Sequence effects of single base loops in intramolecular quadruplex DNA. FEBS Lett. 2007; 581:1657–1660. [DOI] [PubMed] [Google Scholar]
- 31. Guédin A., Gros J., Alberti P., Mergny J.L.. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010; 38:7858–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Cian A., Delemos E., Mergny J.L., Teulade-Fichou M.P., Monchaud D.. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 2007; 129:1856–1857. [DOI] [PubMed] [Google Scholar]
- 33. Piazza A., Adrian M., Samazan F., Heddi B., Hamon F., Serero A., Lopes J., Teulade-Fichou M.P., Phan A.T., Nicolas A.. Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. EMBO J. 2015; 34:1718–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Altschul S.F., Erickson B.W.. Significance of nucleotide sequence alignments: a method for random sequence permutation that preserves dinucleotide and codon usage. Mol. Biol. Evol. 1985; 2:526–538. [DOI] [PubMed] [Google Scholar]
- 36. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of Lineage-Determining transcription factors prime cis-Regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. R Core Team R: A language and environment for statistical computing. 2017; Vienna: R Foundation for Statistical Computing. [Google Scholar]
- 38. Hänsel-Hertsch R., Spiegel J., Marsico G., Tannahill D., Balasubramanian S.. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018; 13:551–564. [DOI] [PubMed] [Google Scholar]
- 39. Sahakyan A.B., Chambers V.S., Marsico G., Santner T., Di Antonio M., Balasubramanian S.. Machine learning model for sequence-driven DNA G-quadruplex formation. Sci. Rep. 2017; 7:14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas-Chollier M., Sand O., Turatsinze J.V., Janky R., Defrance M., Vervisch E., Brohée S., van Helden J.. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 2008; 36:W119–W127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marsico G., Chambers V.S., Sahakyan A.B., McCauley P., Boutell J.M., Di Antonio M., Balasubramanian S.. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 2019; 47:3862–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. The 1000 Genomes Project Consortium Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cer R.Z., Donohue D.E., Mudunuri U.S., Temiz N.A., Loss M.A., Starner N.J., Halusa G.N., Volfovsky N., Yi M., Luke B.T. et al.. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013; 41:D94–D100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kersey P.J., Allen J.E., Allot A., Barba M., Boddu S., Bolt B.J., Carvalho-Silva D., Christensen M., Davis P., Grabmueller C. et al.. Ensembl Genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018; 46:D802–D808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lê S., Josse J., Husson F.. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 2008; 25:1–18. [Google Scholar]
- 47. Kumar S., Stecher G., Suleski M., Hedges S.B.. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017; 34:1812–1819. [DOI] [PubMed] [Google Scholar]
- 48. Conticello S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008; 9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krishnan A., Iyer L.M., Holland S.J., Boehm T., Aravind L.. Diversification of AID/APOBEC-like deaminases in metazoa: multiplicity of clades and widespread roles in immunity. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E3201–E3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kokoska R.J., Stefanovic L., Tran H.T., Resnick M.A., Gordenin D.A., Petes T.D.. Destabilization of Yeast Micro- and Minisatellite DNA sequences by mutations affecting a nuclease involved in okazaki fragment processing (rad27) and DNA polymerase δ (pol3-t). Mol. Cell. Biol. 1998; 18:2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Massy B., Baudat F., Nicolas A.. Initiation of recombination in Saccharomyces cerevisiae haploid meiosis. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11929–11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gillet-Markowska A., Louvel G., Fischer G.. bz-rates: a web tool to estimate mutation rates from fluctuation analysis. G3 (Bethesda). 2015; 5:2323–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Treco D.A., Lundblad V.. Preparation of yeast media. Curr. Protoc. Mol. Biol. 2001; doi:10.1002/0471142727.mb1301s23. [DOI] [PubMed] [Google Scholar]
- 54. Huppert J.L., Balasubramanian S.. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007; 35:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eddy J., Maizels N.. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008; 36:1321–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lexa M., Kejnovský E., Šteflová P., Konvalinová H., Vorlíčková M., Vyskot B.. Quadruplex-forming sequences occupy discrete regions inside plant LTR retrotransposons. Nucleic Acids Res. 2014; 42:968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lexa M., Šteflová P., Martinek T., Vorlíčková M., Vyskot B., Kejnovský E.. Guanine quadruplexes are formed by specific regions of human transposable elements. BMC Genomics. 2014; 15:1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sahakyan A.B., Murat P., Mayer C., Balasubramanian S.. G-quadruplex structures within the 3′ UTR of LINE-1 elements stimulate retrotransposition. Nat. Struct. Mol. Biol. 2017; 24:243–247. [DOI] [PubMed] [Google Scholar]
- 59. Kejnovský E., Lexa M.. Quadruplex-forming DNA sequences spread by retrotransposons may serve as genome regulators. Mob. Genet. Elements. 2014; 4:e28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lam E.Y., Beraldi D., Tannahill D., Balasubramanian S.. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013; 4:1796–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Masiero S., Trotta R., Pieraccini S., de Tito S., Perone R., Randazzo A., Spada G.P.. A non-empirical chromophoric interpretation of CD spectra of DNA G-quadruplex structures. Org. Biomol. Chem. 2010; 8:2683–2692. [DOI] [PubMed] [Google Scholar]
- 62. Henderson S.T., Petes T.D.. Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992; 12:2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M.P., Foiani M., Nicolas A.. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011; 30:4033–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schiavone D., Guilbaud G., Murat P., Papadopoulou C., Sarkies P., Prioleau M.N., Balasubramanian S., Sale J.. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014; 33:2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wright E.P., Huppert J.L., Waller Z.A.E.. Identification of multiple genomic DNA sequences which form i-motif structures at neutral pH. Nucleic Acids Res. 2017; 45:13095–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Školáková P., Renčiuk D., Palacký J., Krafčík D., Dvořáková Z., Kejnovská I., Bednářová K., Vorlíčková M.. Systematic investigation of sequence requirements for DNA i-motif formation. Nucleic Acids Res. 2019; 47:2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Phan A.T., Mergny J.L.. Human telomeric DNA: G-quadruplex, i-motif and Watson-Crick double helix. Nucleic Acids Res. 2002; 30:4618–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lane A.N., Chaires J.B., Gray R.D., Trent J.O.. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008; 36:5482–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Delaney S., Barton J.K.. Charge transport in DNA duplex/quadruplex conjugates. Biochemistry. 2003; 42:14159–14165. [DOI] [PubMed] [Google Scholar]
- 70. Fleming A.M., Ding Y., Burrows C.J.. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Navaratnam N., Morrison J.R., Bhattacharya S., Patel D., Funahashi T., Giannoni F., Teng B.B., Davidson N.O., Scott J.. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J. Biol. Chem. 1993; 268:20709–20712. [PubMed] [Google Scholar]
- 72. Conticello S.G., Thomas C.J., Petersen-Mahrt S.K., Neuberger M.S.. Evolution of the AID/APOBEC family of polynucleoide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005; 22:367–377. [DOI] [PubMed] [Google Scholar]
- 73. Cahoon L., Seifert H.S.. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009; 325:764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.