

Abstract
Cancer has long been viewed as a disease of altered metabolism. Although it has long been recognized that the majority of cancer cells display increased dependence on glycolysis, the metabolism of “cancer stem-like cells” (CSCs) that drive tumor growth and metastasis is less well characterized. In this chapter, we review the current state of knowledge of CSC metabolism with an emphasis on the development of therapeutic strategies to exploit the metabolic vulnerabilities of these cells. We outline emerging evidence indicating distinct metabolic pathways active in the proliferative, epithelial- (E) and quiescent, mesenchymal-like (M) CSC states in triple negative breast cancer (TNBC). These CSC states are characterized by their different redox potentials and divergent sensitivities to inhibitors of glycolysis and redox metabolism. We highlight the roles of two redox-regulated signaling pathways, HIF1α and NRF2, in regulating CSC epithelial-mesenchymal plasticity during metabolic/oxidative stress, and discuss clinical strategies using combinations of pro-oxidant based therapeutics simultaneously targeting E- and M-like CSCs. By specifically targeting CSCs of both states, these strategies have the potential to increase the therapeutic efficacy of traditional chemotherapy and radiation therapy.
Keywords: Cancer Stem Cells (CSCs), Triple Negative Breast Cancer, Reactive Oxygen Species (ROS), Glycolysis, Oxidative phosphorylation (OXPHOS), HIF1α, NRF2, Thioredoxin and Glutathione Antioxidant Pathways
1. Introduction
There is substantial evidence that many cancers, including breast cancer, are hierarchically organized and driven by a small population of tumor cells displaying stem cell properties [1–3]. These “cancer stem-like cells (CSCs)” or “tumor initiating cells (TICs)”, which reside at the apex of tumor heterogeneity, have the capacity for self-renewal, tumor initiation and generation of differentiated tumor progeny constituting the tumor bulk. In addition to their tumorigenic potential, CSCs are inherently resistant to traditional cancer therapies including chemotherapy and ionizing radiation, leading to treatment failure, metastases, and cancer relapse [4–10].
As discussed in several recent reviews, tumor cells including the small population of CSCs display remarkable genetic/epigenetic heterogeneity and cellular plasticity [11–13]. Adding another layer of complexity, recent studies demonstrate that BCSCs maintain the plasticity to transition between proliferative, epithelial-like (E) and quiescent, mesenchymal-like (M) states [14]. These distinct E- and M-like BCSCs exhibit discrete patterns of marker expression characterized by elevated aldehyde dehydrogenase (ALDH) activity and CD24−CD44+ expression respectively [14]. The transition of CSCs from the E- to M-like state closely resembles the epithelial-to-mesenchymal transition (EMT), which is associated with the acquisition of stem cell properties [15]. The equilibrium of these dynamic CSC states is regulated by the tumor microenvironment through mechanisms including, but not limited to, cytokine/chemokine signaling, genetic/epigenetic regulation of key transcription factors as well as growth factor receptors and microRNA/LncRNAs [13, 16, 17]. Most recently, we demonstrated that the dynamics of BCSCs in distinct E- and M-like states is tightly controlled by changing reduction-oxidation (redox) states induced by metabolic stressors [18]. In this chapter, we highlight the emerging knowledge regarding redox-regulated CSC plasticity, the underlying signaling mechanisms governing redox regulation of CSC state dynamics, and the different metabolic pathways contributing to their differential redox potentials as well as divergent responses to inhibitors of glycolysis and redox metabolism. We emphasize the implications of these findings for the development of novel combinatory therapeutic strategies to effectively target CSCs of distinct phenotypic states, and discuss how these strategies may enhance tumor responses to the conventional treatment approaches including chemo- and radiation therapies.
2. CSC plasticity as a mechanism conferring treatment resistance and metastatic relapse
Current cancer therapies are mainly aimed at reducing tumor mass by targeting rapidly proliferating bulk tumor cells. Although these therapeutic strategies are effective in reducing the size of tumors, they frequently fail to eradicate advanced tumors and are associated with metastatic relapse. The CSC hypothesis suggests that conventional antitumor strategies targeting rapidly proliferating cells may fail to target CSCs, which divide infrequently and are endowed with multiple mechanisms accounting for their therapeutic resistance[19]. Indeed, a large body of studies has indicated that BCSCs are resistant to ionizing radiation and chemotherapy [9, 20–22]. This has been demonstrated in cultured cancer cell lines, in primary mammary tumor cells [7, 23, 24], in patient derived tumor xenografts [9, 25, 26] as well as in the clinical setting of breast cancer patients undergoing neoadjuvant chemotherapy [8, 25].
The demonstration that BCSCs exist in a dynamic equilibrium of E- and M-like states [14] suggests that CSC epithelial-mesenchymal plasticity may contribute to therapeutic resistance and metastatic relapse. The plasticity of CSCs allowing them to transition from a proliferating epithelial state to an invasive, mesenchymal state facilitates these cells escaping from traditional therapies and disseminating into the circulation and distant organs. Conversely, the plasticity allowing them to transition from an invasive, mesenchymal state to a proliferating epithelial state facilitates metastatic colonization. This suggests that CSCs are not a fixed population but rather a dynamic, metaplastic phenotypic state that is regulated by the tumor microenvironment (i.e., growth factor/inflammatory signaling, stromal-tumor interactions and metabolic reprograming). In agreement with this notion, published studies have shown that HER2 overexpression drives the self-renewal of ALDH+ E-BCSCs that are sensitive to the HER2 antibody trastuzumab [27]. Conversely, resistance to HER2 blockade is associated with an increase in CD24−CD44+ M-BCSCs resulting from the activation of an IL6 driven inflammatory loop [28]. Moreover, in trastuzumab-resistant HER2+ breast cancer, a combinatory approach targeting IL6 receptor by tocilizumab and HER2 by trastuzumab synergistically abrogated tumor growth and metastases by eliminating both M- and E-BCSCs [28], providing a proof-of-concept that simultaneously targeting distinct CSC states may eradicate metastatic/drug-resistant breast cancer by eliminating the refractory CSC population. These studies also imply that therapeutic approaches targeting either state alone may not be sufficient to eliminate CSCs, since the targeted cell population could be rapidly regenerated by cells in alternative states. Thus, multiple CSC therapies attacking distinct forms of CSCs may be required to effectively eliminate these lethal seeds of cancer, leading to more effective therapies.
3. Cell redox as a key regulator of CSC plasticity
Cancer cells are characterized by increased levels of oxidative stress, generated by increased metabolic demands and oncogenic signaling [29, 30]. Many chemotherapeutic agents or ionizing radiation, by inducing oxidative stress, are able to elicit ROS-mediated cytotoxicity in bulk tumor cells which are more susceptible to second oxidative insults compared to their normal counterparts [31, 32]. However, ROS levels in the quiescent CSCs are significantly different as compared to the bulk of tumor cells [7]. Recently, we demonstrated that modulation of redox potential through co-inhibition of glycolysis and NRF2-mediated antioxidant responses is able to disrupt the state equilibrium of BCSCs, leading to terminal differentiation and apoptosis of both M- and E-BCSCs [18]. This provided a novel framework using pro-oxidant based therapeutics targeting metabolism of distinct CSC states to effectively eliminate the refractory CSC populations.
As is the case for their normal tissue counterparts, CSCs obtain energy principally through two different metabolic pathways: mitochondrial respiration, which depends on oxygen; and fermentation of glucose through aerobic glycolysis, which is oxygen-independent. CSCs appear to be capable of deriving energy from both sources. In the quiescent M state, they primarily rely on glycolysis; in the proliferative E state, they utilize oxygen to fuel mitochondrial oxidative phosphorylation (OXPHOS). Such dynamic utilization of distinct metabolic pathways depending on the tumor microenvironment is critical for cells in these CSC states to meet redox homeostasis and bioenergetics needs. Thus, the reactive oxygen species (ROS) generated from the mitochondrial OXPHOS is not merely a by-product of cancer metabolism, but rather serves as a key mediator regulating CSC plasticity.
3.1. ROS as a secondary messenger in cell signaling
Metabolic reactions utilizing molecular oxygen (O2) produce reactive oxygen species (ROS) including superoxide (O2−, hydrogen peroxide (H2O2) and hydroxyl radical (HO·). Although ROS are generated in various cellular compartments such as the peroxisomes and endoplasmic reticulum (ER), it is well recognized that mitochondrial OXPHOS serves the major source of ROS production, as ~2% of the oxygen consumed by the mitochondria is estimated to be reduced to form superoxide anion [29, 33, 34]. Mitochondrial ROS are not stable and rapidly dismutated by the manganese superoxide dismutase (MnSOD, SOD2) at the mitochondria or Cu/Zn-containing SOD (SOD1) in the cytosol into H2O2, which is permeable to the cell/nuclear membranes and further converted to HO· [35].
At high levels, ROS induce damage to various cellular components including DNA, proteins and lipids, leading to cell senescence, death or oncogenic transformation[29]. The extensive production of ROS during normal metabolism requires the activation of adequate antioxidant defenses to maintain redox homeostasis. Recent evidence has suggested that, although high levels of ROS are toxic, low-to-intermediate levels of ROS are crucial for cell development and homeostasis [36]. ROS act as important secondary massager to modulate a wide variety of signaling molecules including kinases, phosphatases, and transcription factors including JAK-STAT[37, 38], p38 MAPK/ERK/JNK[39–41 ], PI3K-AKT[42, 43], NF-kB[44–46], PKA[47], PKC[48, 49], NRF2/Keap1[50], Hippo-FOXO[51], etc., which in turn stimulate diverse cellular responses including cell survival, proliferation and differentiation[52, 53] and cell migration, adhesion and invasion[54, 55].
3.2. CSC radio-resistance is associated with low reactive oxygen species
The antitumor effects of both radiation and chemotherapy are mediated, at least partially, by the generation of ROS, which results in DNA damage and subsequent single-stranded and double-stranded breaks [56–58]. In neural stem cells (NSCs) and hematopoietic stem cells (HSCs), protection from oxidative stress is critical for the maintenance of their self-renewal [59–61], and mice deficient in ATM kinase or FoxO1, FoxO3, and FoxO4 transcription factors exhibit elevated ROS levels in the HSC compartment, leading to rapid extinction of HSCs [60, 62, 63]. In parallel studies, Diehn et al. documented that human and mouse BCSCs, similar to their normal tissue counterparts, are characterized by low levels of ROS associated with increased expression of free radical scavenging systems that account for the radio-resistance of these cells[7]. This study suggests that strategies that block these antioxidant defenses may successfully target CSCs. Indeed, pharmacological depletion of ROS scavengers in BCSCs markedly decreases their clonogenicity resulting in radio-sensitization [7]. Accordingly, treatment of human acute myeloid leukemia (AML) with parthenolide, a naturally occurring compound that induces ROS, effectively targets AML stem and progenitor cells for apoptosis [64].
3.3. E- and M-like CSCs intrinsically differ in their redox states
As summarized above, there is compelling evidence that low levels of intracellular ROS are required for maintaining CSCs including hematopoietic stem cells (HSCs) [60–62] and mammary stem cells (MaSCs) [7]. Like their normal counterparts, the CD44+/CD24− mesenchymal BCSCs are characterized by low ROS [7]. Epigenetic silencing of the gluconeogenic enzyme fructose-1,6-biphosphatase FBP1 by Snail facilitates increased glycolysis while suppressing OXPHOS to maintain low ROS levels in M-like BCSCs, promoting their self-renewal and an EMT-like CSC phenotype in TNBC [65].
The mammary epithelium is comprised of an outer layer of basal and an inner layer of luminal epithelial cells maintained by distinct basal and luminal stem cells that give rise to cells restricted to the basal and luminal lineages respectively during mammary gland development [66]. Interestingly, MaSCs located in the basal compartment (EpCAM−/lowMUC1−CD49f+CD90+) are characterized by lower ROS levels while the luminal epithelial cells including the primitive luminal stem and/or progenitor cells (EpCAM+MUC1+CD49f+CD90−) are characterized by higher levels of ROS [67]. Moreover, this study also demonstrated that the primitive luminal stem cells/progenitor cells display increased mitochondria mass and greater rates of oxygen consumption, and that glutathione-dependent and -independent antioxidant defense mechanisms are active in the basal and luminal mammary epithelial compartments respectively [67]. These distinct luminal and basal stem cell populations identified in mammary epithelium are reflective of the ALDH+ and CD24−CD44+ CSC states isolated in different subtypes of breast cancers [14].
The finding that basal and luminal MaSCs maintain different ROS levels and lineage-specific mechanisms of ROS control suggest that the E- and M-like BCSCs reflective of their respective luminal and basal stem cell lineages may inherit distinct ROS levels and metabolic pathways to control their respective redox states. Indeed, our recent studies in TNBC revealed that E-BCSCs and M-BCSCs display significantly different ROS levels and divergent responses to metabolic/oxidant stressors [18]. We further demonstrated that metabolic/oxidative stress generated by 2DG, H2O2 or hypoxia promotes the transition of ROSlo M-BCSCs to a ROShi E-state, and this transition was reversed by N-acetylcysteine and mediated by activation of the AMPK-HIF1α axis [18]. These differential responses of E- and M-BCSCs to metabolic/oxidative stressors are linked to their distinct bioenergetics and redox metabolism, which is discussed in the following section.
4. Metabolic pathways of CSCs
For most slowly proliferating normal tissue cells, mitochondria serve as the main source of energy production through the tricarboxylic acid (TCA) cycle coupled with OXPHOS in the mitochondria membrane, which generates 36 ATP per molecule of glucose. Cancer cells, however, are characterized by high rate of proliferation and thus need to adapt their cellular metabolism to support increased proliferation by providing rapid ATP generation as well as biosynthesis of nucleotides, proteins and lipids to support tumor growth. Almost 100 years ago, Otto Warburg reported that cancer cells preferentially utilize glycolysis (which generates 2 ATP per unit of glucose) to generate copious amounts of lactate, regardless of the presence of oxygen[68]. Such an inefficient way of energy production, however, is necessary for generation of various metabolic intermediates important for maintaining tumor redox homeostasis and anabolic metabolism (macromolecular biosynthesis). Despite a glycolytic phenotype in the bulk of tumor cells, the metabolic pathways utilized by the small subset of CSCs are less well characterized. Currently there is no consensus on the metabolism of CSCs and a growing body of evidence in different cancer types indicates that the metabolism of CSCs is context-dependent and reliant on glycolysis and/or OXPHOS.
4.1. Metabolic heterogeneity and plasticity of CSCs
It was originally hypothesized that CSCs display a glycolytic phenotype with similarities to the quiescent adult stem cells [69]. This hypothesis is supported by the findings that transition from somatic oxidative metabolism into pluripotency-dependent glycolysis facilitates nuclear reprogramming and formation of induced pluripotent stem cells (iPSCs) [70, 71]. In contrast, bioenergetics transition from glycolysis to OXPHOS occurs during differentiation of human embryonic stem cells (hESCs) [72]. Thus, metabolic reprograming is not merely associated with sternness, but also acts as a regulator of cell differentiation.
A number of studies in breast cancer supported the above notion by demonstrating that BCSCs enriched by sphere formation [73], CD24−CD44+[65, 73] or CD49fhighEpCAMlow [74] preferentially utilize glycolysis over mitochondrial OXPHOS compared to the differentiated cells from the same tumor. This glycolytic phenotype is also found in the CD133+ hepatocellular CSCs [75] and the radio-resistant sphere-forming nasopharyngeal carcinoma cells [76]. Despite the studies suggesting a dependence of CSCs on glycolysis, other studies demonstrate that CSCs might also rely on oxidative metabolism. For example, quantitative proteomics studies of breast cancer cells revealed that key mitochondrial-related proteins involving in fatty acid β-oxidation and ketone metabolism as well as mitochondrial biogenesis were significantly upregulated in the mammospheres relative to the epithelial monolayers, suggesting that clonal expansion of BCSCs requires mitochondrial OXPHOS [77]. Indeed, XCT790, a well-established inhibitor of estrogen-related receptor a (ERRa), which functions as an essential cofactor of PGC-1α required for mitochondrial biogenesis [78, 79], suppresses BCSC activity by blocking several independent signaling pathways normally required for the self-renewal of CSCs, including Sonic hedgehog, TGFβ-SMAD, STAT3 and Wnt signaling [80]. This PGC-1α mediated mitochondrial biogenesis and OXPHOS is essential for the functional motility of breast cancer cells and metastasis [81]. The requirement of mitochondrial biogenesis for the self-renewal of CSCs is also supported by the findings that a number of antibiotics (i.e., Azithromycin and Tetracycline) targeting mitochondrial ribosomes effectively eradicate CSCs across multiple tumor types [82]. This functional link of mitochondria to the CSC phenotype was subsequently confirmed by the findings that proliferative ALDH+ BCSCs display increased mitochondrial mass and activity, and that high mitochondrial mass identifies a sub-population of BCSCs that are chemo-resistant [83, 84]. A recent study further demonstrated that MYC and MCL1, two genes frequently amplified in TNBC, cooperatively promote mitochondrial OXPHOS, leading to ROS-mediated HIF1α stabilization and enrichment of ALDH+ BCSCs [85].
The roles of mitochondrial oxidative metabolism in conferring therapeutic resistance by promoting the maintenance of CSCs have also been described in other malignancies. For example, the oncofetal insulin-like growth factor 2 mRNA-binding protein IMP2 in glioblastoma has been shown to control OXPHOS, which is crucial for preserving CD133+ glioblastoma CSCs [86]. In acute myelogenous leukemia (AMF), the quiescent leukemia stem cells (FSCs) express high levels of BCF-2 and have a selective dependency on oxidative respiration [87]. Furthermore, the BCF-2 inhibitors ABT-737 and ABT-263 selectively inhibit the quiescent FSCs by targeting OXPHOS [87]. Similarly, in chronic myeloid leukemia (CMF), the primitive CD34+ leukemic stem cells (FSCs) rely on oxidative metabolism for their survival. Treatment with the combination of imatinib and tigecycline, an antibiotic inhibiting mitochondrial protein translation, selectively eradicates CML LSCs in vitro and in a xenograft model of human CML [88]. The antidiabetic drug metformin, which blocks mitochondrial function via inhibition of the ETC complex I, inhibits cellular transformation and selectively kills BCSCs by depleting the TCA cycle and glycolytic intermediates and suppressing nucleotide biosynthesis [89]. In pancreatic ductal adenocarcinoma (PDAC), metformin specifically targets CD133+ CSCs, which display elevated expression of PGC-1α essential for mitochondrial biogenesis, OXPHOS functionality, and the self-renewal and tumorigenic potential of pancreatic CSCs [90]. Interestingly, metformin-resistant CSC clones in PDAC emerge due to an intermediate glycolytic/OXPHOS phenotype driven by increased expression of MYC, and genetic/pharmacological suppression of MYC restores metformin sensitivity in metformin-resistant pancreatic CSCs [90]. Thus, the balance of MYC/PGC-1α expression appears to determine the dynamic metabolic states (glycolysis versus OXPHOS) of pancreatic CSCs. This metabolic heterogeneity and plasticity of CSCs identified in various tumor types highlight that CSCs display metabolic plasticity enabling them to adapt to changing tumor microenvironments and nutrient availability.
4.2. CSCs in distinct E and M states depend upon different metabolic pathways
As discussed in the previous section, the Warburg effect characterized by increased aerobic glycolysis in the bulk of tumor cells represents a striking metabolic difference between cancer and its normal tissue cells. However, despite the dependence of cancers on glycolysis, glycolytic inhibitors such as 2-Deoxyglucose (2DG) and Lonidamine exhibit little effect on solid tumor growth [91, 92]. Although the mechanisms accounting for the lack of sensitivity of cancer cells to glycolytic inhibition remain to be fully characterized, a recent study suggested that mTORCl-dependent metabolic rewiring underlies the escape of cancer cells to glycolytic addiction [93].
The phenotypic plasticity of BCSCs enabling them to transition between a quiescent mesenchymal state and proliferative epithelial state facilitates the capacity of BCSCs to initiate and grow primary tumors, to invade the basement membrane and tissue vasculature, and ultimately colonize distant organs to form clinically significant metastases [17, 19]. Despite the functional significance of this CSC plasticity in tumor development, treatment resistance and metastatic relapse, the cellular and molecular mechanisms regulating BCSC plasticity by the complex tumor microenvironment remain largely elusive. Currently, little is known about the metabolic pathways active in distinct BCSC states and how these CSC states evolve under various metabolic stressors such as glycolytic inhibition, nutrient deprivation, hypoxia, and xenobiotic/oxidant stress. These metabolic differences of M- and E-like CSCs may confer resistance to glycolytic inhibition and play critical roles for tumor cell survival and progression under stress. As CSCs exhibit a dynamic equilibrium of proliferative E- and quiescent M-like states coordinately driving tumor growth, metastasis and treatment resistance, elucidation of these metabolic differences will help to define and exploit metabolic vulnerabilities of each CSC state, providing a conceptual framework to effectively target this critical tumor cell population.
To address these questions, we investigated how metabolic/oxidative stress modulates the state dynamics of BCSCs and identified markedly different responses of M- and E-BCSCs to oxidant stress that are closely linked to their distinct redox potentials. Specifically, metabolic/oxidative stress generated by 2DG, H2O2 or hypoxia, by activating AMPK-dependent HIF1α stabilization and NRF2-mediatted antioxidant responses, promotes the transition of BCSCs from a quiescent, ROSlow M-like state to a ROShigh proliferative E-state [18]. Interestingly, these divergent responses of M- and E-BCSCs to oxidative stress are closely associated with their different metabolic pathways. As depicted in Figure 1, the CD24−CD44+M-BCSCs and ALDH+ E-BCSCs significantly differ in many aspects of cellular metabolism. Although both CSC states exhibit elevated glycolysis regulatory genes, the relatively quiescent M-BCSCs exhibit higher glycolytic rate in glucose-rich culturing conditions. In contrast, the proliferative E-BCSCs are endowed with robust expression of mitochondrial OXPHOS regulatory genes and exhibit highest metabolic plasticity for OXPHOS under glycolytic inhibited conditions. These proliferative E-BCSCs are also endowed with robust NRF2-mediated antioxidant responses, exemplified by highly elevated expression of a wide variety of antioxidant genes involved in drug transport and detoxification, NADPH production, TXN and GSH antioxidant pathways, etc. These metabolic differences of M- and E-BCSCs suggest that while glycolysis plays an important role in supporting the low redox states of M-BCSCs, both mitochondrial respiration and NRF2-mediated antioxidant defenses are important for E-BCSCs to maintain their proliferation and to prevent the accumulation of oxidative stress generated from mitochondrial OXPHOS.
Figure 1. Metabolic differences of E- and M-BCSCs versus the bulk tumor cells.
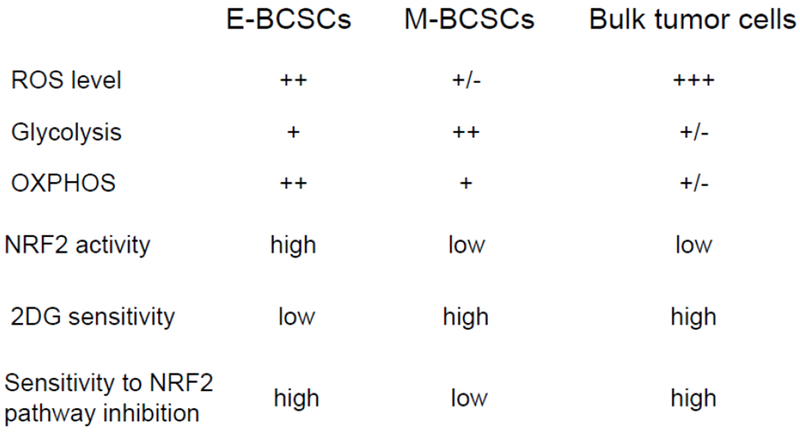
The CD24−CD44+ M-BCSCs and ALDH+E-BCSCs significant differ in many aspects of cellular metabolism as compared to the bulk tumor cells identified as ALDH−CD24+CD44−.
As metabolic/oxidative stress produced by 2DG-mediated glycolytic inhibition facilitates the transition of quiescent, ROSlow M-BCSCs to a proliferative, ROShigh E-state susceptible to the blockade of NRF2 or its downstream thioredoxin (TXN) and glutathione (GSH) antioxidant pathways, we validated a combinatory treatment approach utilizing 2DG together with inhibitors of TXN (by AUR) and GSH (by BSO) antioxidant pathways. This combination treatment approach significantly suppressed tumor growth, tumor-initiating potential and metastasis by abrogating both M- and E-BCSCs in patient-derived xenograft (PDX) and systemic metastasis models of TNBC [18]. As 2DG is not an FDA-approved cancer therapeutic, our preclinical studies suggest a pro-oxidant based therapeutic approach utilizing standard-of-care radio-chemotherapy and antiangiogenic therapies (all of which generate oxidative stress) in combination with inhibitors of NRF2-mediated antioxidant responses to treat metastatic cancer (i.e., TNBC). As illustrated in Figure 2, this combinatory approach, by elevating oxidant stress in the ROSlow M-like CSCs, promotes the transition of quiescent M-BCSCs to a ROShigh E-state that is more susceptible to inhibition of NRF-mediated antioxidant pathways (i.e., NRF2 inhibitor Trig or inhibitors of TXN/GSH pathways such as AUR/BSO), leading to terminal differentiation and subsequent apoptosis of the both M- and E-like BCSCs. Thus, elucidating metabolic differences of distinct CSC states may provide a conceptual framework to effectively target CSCs in various tumor types, leading to enhanced treatment responses for traditional cancer therapies.
Figure 2. A pro-oxidant based therapeutic approach with potential to increase treatment responses for traditional cancer therapies.
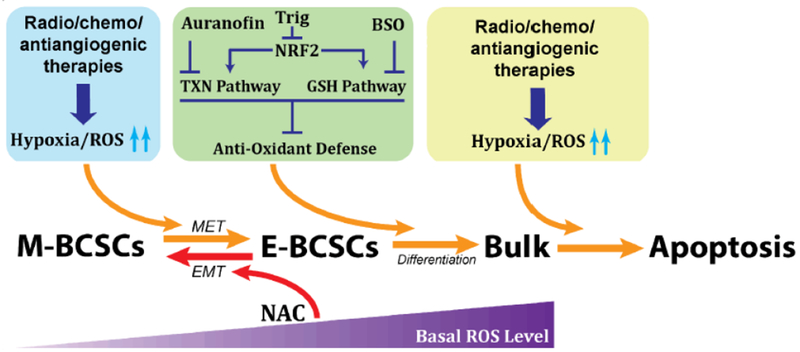
Combination strategies utilizing traditional cancer therapies (i.e., standard radio-chemotherapies or anti-angiogenic therapies) in combination with inhibitors of NRF2-mediated antioxidant responses (i.e., NRF2 inhibitor Trig or inhibitors of TXN/GSH antioxidant pathways including AUR/BSO) enhance treatment responses by simultaneously targeting CSCs of distinct states.
5. Redox-regulated signaling pathways modulating CSC phenotypic/metabolic plasticity and therapeutic responses
Although a plethora of ROS-dependent signaling pathways including PTEN/PI3K/AKT/mTOR [94, 95], JAK/STAT[37, 96], Notch [97–99], Hippo [51], ATM/p53 [100–102], Wnt [65, 103, 104] and NF-kB [46, 105–107] cascades have been implicated in regulating CSCs through modulation of cancer metabolism and redox homeostasis, our recent studies demonstrate a prominent role of ROS-induced HIF1α and NRF2, two redox-sensitive transcription factors, in regulation of BCSC plasticity during metabolic stress[18]. In this section, we focus on the roles of HIF1α and NRF2 in regulating CSC metabolism and phenotypic plasticity as well as the clinical implications of targeting these redox-sensitive signaling pathways to overcome therapeutic resistance.
5.1. Roles of HIF1α in regulation of redox and intermediary metabolism
Advanced cancers often display intra-tumor hypoxia, which promotes tumor metastasis and therapeutic resistance largely mediated by induction/stabilization of hypoxia-inducible factors (HIFs) [108, 109]. HIFs including HIF1 and HIF2 are transcription factors mediating hypoxic responses and HIF1 functions as a master regulator of glycolysis [110]. HIF1 has two subunits: HIF1α and HI FI β. HIF1 protein levels increase under hypoxic stress and decrease under normoxic conditions due to oxygen-dependent prolyl hydroxylation of HIF1α, which targets HIF1α for proteasomal degradation via the von Hippel-Lindau (VHL) ubiquitin ligase complex [111]. HIF1 promotes glycolysis through transcriptional activation of a number of glycolytic genes including glucose transporters (i.e., GLUT1 and GLUT3), hexokinases (i.e., HK1 and HK2), pyruvate kinase (PKM) and lactate dehydrogenase (LDHA). HIF1α also promotes the transcription of the PDK1 gene encoding pyruvate dehydrogenase kinase I, which phosphorylates and inactivates pyruvate dehydrogenase (PDH), the first enzymatic entry into the TCA cycle [112, 113]. This HIF1α-mediated metabolic rewiring by increased influx to the glycolytic pathway and reduced influx to the TCA cycle helps to overcome elevated oxidant stress associated with hypoxia by blunting the production of mitochondrial ROS.
In addition to modifying metabolic influx to glycolytic pathway versus TCA cycle, HIF1α also mediates increased metabolic influx to the serine biosynthetic pathway and mitochondrial one carbon (folate cycle) metabolism. This facilitates NADPH and glutathione synthesis to reduce mitochondrial oxidant stress to promote BCSC maintenance and lung metastasis [114, 115]. This role of HIF1α in maintaining ROS homeostasis including redox balance at the mitochondria is critical for the maintenance of CSCs under metabolic stressful conditions (i.e., chemotherapy or antiangiogenic therapy). Indeed, when TNBCs were treated with paclitaxel or gemcitabine, induction of HIF activity including HIF1α and HIF2α led to increased ALDH+ BCSCs and chemotherapy resistance, associated with increased secretion of IL-6 and IL-8 [116], two prominent inflammatory cytokines promoting BCSC activity [117, 118]. Subsequent studies identified a redox-mediated mechanism of HIF1α promoting a chemotherapy-induced BCSC phenotype [119].
The development of antiangiogenic agents was thought to be a major advance in cancer treatment. Several therapeutic agents targeting tumor neovascularization have been developed, including antibodies (i.e., bevacizumab) targeting vascular endothelial growth factor (VEGF) and receptor tyrosine kinase inhibitors (i.e., Sunitinib and Nintedanib) that block signaling mediated by the VEGF receptor. Despite the encouraging responses of some tumor types including colorectal [120], renal cell [121] and pancreatic neuroendocrine [122] carcinomas to antiangiogenic therapies, numerous other tumor types, particularly breast cancer [123–125], are poorly responsive to antiangiogenic regimens. Previous studies in our laboratory showed that that tumor hypoxia generated by anti-angiogenic agents enriches ALDH+ E-BCSCs within the tumor hypoxic zones in a HIF1α dependent manner [126], suggesting that the induction of ALDH+ E-BCSCs by standard antiangiogenic therapy may contribute to antiangiogenic resistance in breast cancer. Thus, targeting HIF1α-dependent induction of ALDH+ BCSCs by chemo or antiangiogenic therapy may overcome chemotherapy or antiangiogenic resistance. In support of this idea, co-administration of HIF inhibitors such as digoxin together with standard chemotherapeutic (paclitaxel and gemcitabine) overcome chemo resistance and prevented tumor relapse in TNBC xenograft mice [116]. Similarly, concurrent administration of a novel HIF1α inhibitor CRLX101 (an investigational nanoparticle-drug conjugated with camptothecin) and bevacizumab led to decreased induction of both HIF1α and ALDH+ CSCs in breast tumors leading to tumor regression and delayed tumor recurrence in preclinical mouse models [127].
5.2. Roles of NRF2 in regulation of redox and intermediary metabolism
The nuclear factor erythroid 2-related factor 2 (NRF2) is one of the most important transcription factors orchestrating intrinsic resistance to oxidative stress and adaptive antioxidant responses to various environmental stressors [50, 128]. NRF2 has been traditionally regarded as a tumor suppressor due to its roles as the main defense mechanism of the cell and a major regulator of cell survival. Yet, recent studies demonstrate that hyperactivation of the NRF2 pathway creates an environment that not only supports the survival of the normal cells, but also the growth and progression of malignant cells, protecting them against oxidative/xenobiotic stress, chemotherapeutic agents, and radiotherapy[129]. Accumulating evidence is emerging to demonstrate that NRF2 profoundly influences the metabolism of glucose, lipids, amino acids and nucleotides [128, 130]. Through regulation of redox and intermediary metabolism of tumor cells, NRF2 pathway serves as a driver of cancer progression, metastasis, and resistance to therapy.
5.2.1. Regulation of NRF2 activity
NRF2 belongs to the cap ‘n’ collar (CNC) subfamily of basic leucine zipper (bZIP) transcription factors equipped with 7 modular NRF2-ECH homology domains (Nehl-7), each of which preforming distinct functions to regulate NRF2 activity [128]. In unstressed conditions, NRF2 protein levels remain low in the cytoplasm, where it forms a complex through dimerization of its Neh2 domain with the Kelch-like ECH-associated protein 1 (Keap1), a NRF2 negative regulator that targets NRF2 for ubiquitination and proteasomal degradation [131, 132]. Electrophiles and oxidants inhibit Keap1-mediated proteasomal degradation of NRF2, thereby enabling NRF2 accumulation and nuclear translocation to initiate the transcription of a wide variety of antioxidant genes that facilitate cellular adaptation to stress[128]. Once activated, NRF2 hetero-dimerizes with the small MAF proteins to control the basal and inducible expression of over 200 genes that contain antioxidant response elements (AREs) in their regulatory regions [133]. Although NRF2 is principally regulated by Keap1, the Neh6 domain of NRF2 confers additional mechanism of stability control by recruiting β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for the S-phase kinase-associated protein 1 (Skpl)–Cul1–Rbx1 core E3 complex. This negative regulation of NRF2 by β-TrCP is enhanced by glycogen synthase kinase 3 (GSK3) activities [134–136].
In addition to ubiquitination, NRF2 protein expression is subject to other forms of post-translational modifications including phosphorylation and acetylation, which allow modulation of NRF2 activity by fine-tuning its subcellular localization in the cytoplasm/nucleus. For example, activation of the AMP-activated protein kinase (AMPK), a master regulator of metabolism to maintain cellular bioenergetics during metabolic stress, increases NRF2 activity through phosphorylation of GSK3β, leading to the inhibition of GSK3β function in promoting NRF2 nuclear exclusion and degradation [137, 138]. AMPK is also capable of directly phosphorylating NRF2 at Ser550 to facilitate its nuclear localization [139]. In addition to the phosphorylation of NRF2, multiple lysine residues in the Nehl domain of NRF2 were shown to be acetylated by p300/CBP, which augments promoter-specific DNA binding of NRF2 to ARE sequences and induction of antioxidant responsive gene expression during stress [140].
NRF2 activity is also controlled by transcriptional regulation. For example, functional AREs in mouse NRF2 promoter region have been identified, which mediate the autoregulation of NRF2 through transcriptional activation, providing a positive feedback mechanism [141]. NRF2 activity can also be regulated by various polymorphisms in its promoter and protein coding regions and a single nucleotide polymorphism in the ARE-like sequences of human NRF2 promoter is associated with diminished NRF2 expression a single nucleotide polymorphism in the ARE-like sequences of human NRF2 promoter is associated with diminished NRF2 expression and increased susceptibility to lung cancer[142]. The NRF2 promoter also contains a NF-kB binding site, thereby enabling its transcriptional activation by inflammatory stimuli such as lipopolysaccharide [143]. This constitutively high NRF2 expression driven by NF-kB in human acute myeloid leukemia underlies its chemo-resistance [144].
5.2.2. NRF2 in metabolic reprograming of cancer
Beyond its function as a master regulator of antioxidant responses, NRF2 has recently been recognized as a key transcription factor mediating metabolic reprogramming in cancer cells [128, 130, 145]. Through direct regulation of key metabolic genes or indirectly through crosstalk with other transcription factors, NRF2 serves as a major hub to modulate numerous enzymes in major metabolic pathways of carbohydrates, nucleic acids, lipids, and amino acids.
NRF2 and glucose metabolism
Metabolic reprogramming refers to a process that cancer cells utilize in order to rewire their metabolic pathways and energy production network to support their proliferative and/or invasive characteristics. This phenomenon was first demonstrated in cancers by the Warburg effect of aerobic glycolysis observed in the 1920s [68]. Although glycolysis represents one of the remarkable features of cancer metabolic alternations, the underlying mechanisms driving this metabolic alteration and its influences on cancer development and progression are still not fully understood. Activation of NRF2 increases glucose uptake and diverts it to the pentose phosphate pathway (PPP) by promoting enzyme expression in the oxidative (including glucose-6-phosphate dehydrogenase, G6PD and 6-phosphogluconate dehydrogenase, PGD) and non-oxidative (including transketolase, TKT and transaldolase, TALDO1) arms of PPP [130, 146, 147]. In addition to G6PD and PGD, NRF2 also promotes the expression of two other NADPH synthetic enzymes ME1 and IDH1[146], suggesting a role of NRF2 in replenishing NADPH, the main reducing equivalent required for redox hemostasis and biosynthesis of lipids and nucleotides. This role of NRF2 in promoting nucleotide synthesis is supported by the findings that the expression of de novo purine synthesis enzymes, phosphoribosyl pyrophosphate amidotransferase (PPAT) and methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), was significantly decreased following knockdown of NRF2 in A549 cells (which is deficient of Keap1) [146]. The role of NRF2 in redirecting glycolytic intermediates into anabolic pathways to support tumor growth was further demonstrated in a recent study by showing that activation of NRF2 upregulates the expression of key serine biosynthesis enzymes including PHGDH, PS ATI and SHMT and diverts glycolytic intermediate 3-phosphoglycerate (3PG) to serine/glycine synthetic pathway, which augments glutathione and nucleotide production and confers poor prognosis in human non-small cell lung cancer (NSCLC) [148].
The folate cycle, through transfer of one carbon unit between tetrahydrofolate and its derivatives in the cytoplasm or mitochondria, produces metabolites essential for cell growth, including nucleotides, methionine and NADPH [149]. Interestingly, MTHFD1L, an enzyme critical for folate cycle maintenance[150], is transcriptionally activated by NRF2, which confers metabolic advantages in hepatocellular carcinoma [151]. Together with the study showing that HIF1α induced by chemotherapeutic agents promotes BCSCs and lung metastasis by redirecting glycolytic intermediates to serine synthesis and mitochondrial one carbon metabolism [115], these studies provide evidence for a role of NRF2 and HIF1α to confer metabolic advantages to cancer cells including CSCs through metabolic reprograming.
NRF2 and glutamine metabolism
In addition to glycolysis, many tumors fuel their cellular bioenergetics by degradation of glutamine through glutaminolysis, which is catalyzed by a NRF2 target gene glutaminase (GLS), thereby providing cancer cells with nitrogen for the biosynthesis of nucleotides and nonessential amino acids [128, 146]. The glutamate generated by GLS can be deaminated into α-ketoglutarate to fuel the TCA cycle and this step is enhanced by the NRF2 target gene ME1[152]. Alternatively, glutamate can be used to fuel glutathione biosynthesis, which is catalyzed by glutamate-cysteine ligase (with catalytic GCLC and regulatory GCLM subunits) and glutathione synthetase (GS), two enzymes also regulated NRF2[152]. Upregulated expression of GCLC and GCLM and ME1 by NRF2 has been shown to direct increased carbon flux from glutamine toward GSH biosynthesis and TCA cycle [146]. illustrating a role of NRF2 in rewiring glutamine metabolism in cancer. Of note, the ability of NRF2 to promote cell proliferation through metabolic reprogramming (i.e., PPP shunting, NADPH production, nucleotide synthesis and glutamine metabolism) is augmented in the presence of active PI3K-Akt signaling [146]. suggesting a positive feedback mechanism between oncogenic pathways and NRF2 in driving the malignant phenotype.
NRF2 and lipid metabolism
As summarized in two recent reviews [128, 130]. NRF2 negatively regulates a variety of genes involved in fatty acid biosynthesis, desaturation and transport. In contrast, a number of lipases involved in the degradation of triglycerides/phospholipids and enzymes involved in mitochondrial fatty acid β-oxidation (FAO) are positively regulated by NRF2. The FAO products including NADH and FADH2 counteract ROS accumulation to reduce oxidative stress and increase ATP production by serving as the electron carriers of the ETC complex [153]. Several studies have reported a function of FAO in the maintenance of hematopoietic stem cells (HSCs) and CD8+ memory T cells, which display some stem like characteristics [154–156].
Recent studies also implicate a role of FAO in driving a CSC-like phenotype and chemo-resistance. For instances, the promyelocytic leukemia (PML) gene expressed in breast cancer acts as both a negative regulator of PGC-1α acetylation and a potent activator of PPAR signaling and fatty acid oxidation, which promotes ATP production and inhibits anoikis, a property associated with CSCs and metastasis[157]. The expression of the oncogenic transcription factor MYC is elevated in many TNBCs, which drives dysregulated FAO activity [158]. Pharmacologic inhibition of FAO catastrophically decreased energy metabolism in MYC-overexpressing TNBC cells and blocked tumor growth in a MYC-driven transgenic TNBC model and a MYC-overexpressing TNBC patient-derived xenograft model [158]. The activation of JAK/STAT3 promotes M-like BCSC phenotype and tumor growth [159]. Inhibiting JAK/STAT3 blocks the self-renewal of M-like BCSCs and the expression of diverse lipid metabolic genes, including carnitine palmitoyltransferase IB (CPT1B), a rate-limiting enzyme of FAO pathway; and blocking FAO re-sensitize breast cancer cells to chemotherapy while reducing cancer sternness in vivo [160].
5.3. Roles of NRF2 and HIF1α in promoting CSC plasticity
Our recent studies demonstrated that NRF2 and HIF1α are critical for promoting an E-BCSC phenotype during metabolic/oxidative stress and suppression of NRF2 or HIF1α impedes the maintenance and clonal expansion of E-BCSCs [18]. As shown in Figure 3, three different mechanisms by which metabolic/oxidant stress induces an enhanced E-BCSC phenotype are identified: 1) conversion of M- to E-BCSCs, which is mediated by AMPK-dependent HIF1α stabilization, 2) activation of HIF1α-NOTCH self-renewal pathway, 3) activation of NRF2-mediated ALDH1A1/3 expression, which is independent of HIF1α.
Figure 3. NRF2 acts as a central hub to facilitate the transition of BCSCs from the M to E state as well as the maintenance/proliferation of E-BCSCs under metabolic/oxidant stress.
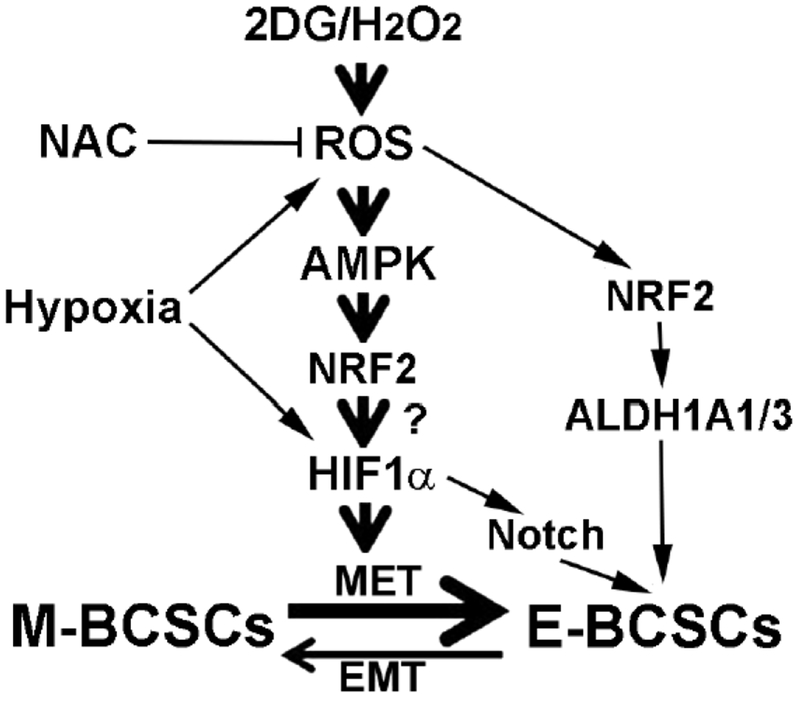
The activation of NRF2 during metabolic/oxidant stress promotes CSC plasticity by mediating AMPK-dependent HIF1α stabilization, which is required for the conversion of M- to E-BCSCs. NRF2 activation also mediates the activation of HIF1α-NOTCH self-renewal pathway and the expression of NRF2 target genes ALDH1A1/3 to facilitate the propagation of E-BCSCs.
Although our studies identify ROS-AMPK-HIF1α and NRF2 pathways in regulation of BCSC state dynamics, additional studies are necessary to determine how activation of AMPK regulates HIF1α stability during metabolic/oxidative stress. Interestingly, previous studies showed that NRF2 knockdown reduces HIF1α protein levels and, consequently, the expression of VEGF, PDGF, and angiopoietin in glioblastoma [161], suggesting that NRF2 mediates HIF1α stabilization induced by hypoxic stress. Therefore, it is likely that that enhanced NRF2 expression not only promotes the self-renewal/proliferation of E-BCSCs, but also mediates AMPK-dependent HIF1α stabilization required for facilitating M to E state transition of BCSCs under metabolic/oxidative stress. If so, NRF2 would serve as an important hub in facilitating BCSC M to E state transition as well as maintenance and proliferation of E-BCSCs under metabolic/oxidant stress (Figure 3). In addition, as the tumor microenvironment including nutrient/oxygen availability and tumor stroma constantly impacts tumor behavior, future studies to assess how these environmental factors affect BCSC metabolism and phenotypic plasticity in vivo will be critical.
One plausible mechanism of NRF2 and HIF1α mediating treatment resistance is through metabolic reprograming, which confers metabolic advantages in CSCs, especially E-like CSCs that express elevated levels of NRF2 and HIF1α. This idea is supported by our recent gene profiling analyses of E- and M-BCSCs isolated from two patient derived xenograft (PDX) models of TNBC, which identified differential expression patterns of glycolysis and PPP pathway genes in M- and E-BCSCs[18]. Specifically, a wide variety of glycolytic enzymes ranging from HK1 to LDHD were highly elevated in M- but less robustly in E-BCSCs. In contrast, various PPP enzyme genes, including G6PD, were highly upregulated in E- but less robustly in M-BCSCs [18]. This robust elevation of PPP genes in E- but not M-BCSCs suggests that elevated expression of NRF2 and/or HIF1α in E-BCSCs redirects glycolytic intermediates to PPP pathway, promoting NADPH production and nucleotide biosynthesis to support the propagation of E-BCSCs.
6. Concluding remarks
Recent findings that E- and M-like CSCs significantly differ in their redox states, metabolic pathways and sensitivities to metabolic/oxidant stress have identified a novel framework exploiting metabolic vulnerabilities of distinct CSC states [18]. As increased oxidant stress, through activation of HIF1α and NRF2 signaling, promotes the transition of quiescent M-CSCs to an E-like state that is more susceptible to the inhibition of redox metabolism especially NRF2-mediated antioxidant pathways [18], future pro-oxidant-based therapeutic strategies utilizing conventional cancer therapies (i.e., standard chemotherapeutic/antiangiogenic agents, radiation therapy) together with agents targeting NRF2-mediated antioxidant responses may prove to be efficient in overcoming resistance associated with traditional cancer therapies by targeting both M- and E-like CSCs.
In addition to traditional cancer therapies that are associated with toxicity to normal cells, pharmacological ascorbate (high doses of Vitamin C given via intravenous injection) has re-emerged as a non-toxic and easily implementable pro-oxidant which has the potential to increase treatment efficacy when combined with standard-of-care radio-chemotherapy [162–164]. Recent studies have demonstrated that oxidant-mediated disruption of iron metabolism causes the selective susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate [165], supporting a generalized mechanism for the use of pharmacological ascorbate in cancer therapy. As iron-dependent accumulation of lipid hydroperoxides can trigger ferroptosis, a regulated cell death nexus linking metabolism, redox biology and diseases [166], future studies are necessary to determine if pharmacological ascorbate disrupts NRF2-regulated iron metabolism in CSCs to induce ferroptosis.
Considering the central roles of NRF2 in regulating antioxidant defenses and metabolism, future metabolic tracing studies dissecting how activation of NRF2 rewires the metabolism of CSCs to promote their phenotypic/metabolic plasticity and proliferation under metabolic/oxidative stress are also required, which will not only validate NRF2 as a critical target to abrogate CSC plasticity and proliferation, but also identify potential new targets downstream of NRF2 in driving CSC self-renewal/proliferation and treatment resistance. These mechanistic studies may reveal novel therapeutic strategies to target the metabolic vulnerabilities of CSCs, thereby improving treatment efficacy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wicha MS, Liu S, Dontu G: Cancer stem cells: an old idea--a paradigm shift. Cancer research 2006, 66(4):1883–1890; discussion 1895–1886. [DOI] [PubMed] [Google Scholar]
- 2.Liu S, Wicha MS: Targeting breast cancer stem cells. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010, 28(25):4006–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS: Cancer stem cells in breast: current opinion and future challenges. Pathobiology 2008, 75(2):75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ: Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clinical cancer research : an official journal of the American Association for Cancer Research 2006, 12(19):5615–5621. [DOI] [PubMed] [Google Scholar]
- 5.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI et al. : Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America 2009, 106(33):13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean M, Fojo T, Bates S: Tumour stem cells and drug resistance. Nature reviews Cancer 2005, 5(4):275–284. [DOI] [PubMed] [Google Scholar]
- 7.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M et al. : Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458(7239):780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC et al. : Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008, 100(9):672–679. [DOI] [PubMed] [Google Scholar]
- 9.Phillips TM, McBride WH, Pajonk F: The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006, 98(24):1777–1785. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Atkinson RL, Rosen JM: Selective targeting of radiation-resistant tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(8):3522–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien-Ball C, Biddle A: Reprogramming to developmental plasticity in cancer stem cells. Developmental biology 2017, 430(2):266–274. [DOI] [PubMed] [Google Scholar]
- 12.Wainwright EN, Scaffidi P: Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends in cancer 2017, 3(5):372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks MD, Burness ML, Wicha MS: Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell stem cell 2015, 17(3):260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD et al. : Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Reports 2014, 2(1):78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al. : The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133(4):704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Luo M, Brooks M, Clouthier SG, Wicha MS: Biological and clinical significance of cancer stem cell plasticity. Clin Transl Med 2014, 3(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo M, Brooks M, Wicha MS: Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des 2015, 21(10):1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, Conley S, Fath MA, Davis A, Gheordunescu E et al. : Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell metabolism 2018, 28(1):69–86 e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo M, Clouthier SG, Deol Y, Liu S, Nagrath S, Azizi E, Wicha MS: Breast cancer stem cells: current advances and clinical implications. Methods Mol Biol 2015, 1293:1–49. [DOI] [PubMed] [Google Scholar]
- 20.Lagadec C, Vlashi E, Della Donna L, Meng Y, Dekmezian C, Kim K, Pajonk F: Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast cancer research : BCR 2010, 12(1):R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karimi-Busheri F, Rasouli-Nia A, Mackey JR, Weinfeld M: Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast cancer research : BCR 2010, 12(3):R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fillmore CM, Kuperwasser C: Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast cancer research : BCR 2008, 10(2):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY: Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer research 2008, 68(9):3243–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM: WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proceedings of the National Academy of Sciences of the United States of America 2007, 104(2):618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J et al. : let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131(6):1109–1123. [DOI] [PubMed] [Google Scholar]
- 26.Zielske SP, Spalding AC, Wicha MS, Lawrence TS: Ablation of breast cancer stem cells with radiation. Transl Oncol 2011, 4(4):227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A et al. : HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer research 2013, 73(5):1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK et al. : Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Molecular cell 2012, 47(4):570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorrini C, Harris IS, Mak TW: Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery 2013, 12(12):931–947. [DOI] [PubMed] [Google Scholar]
- 30.Szatrowski TP, Nathan CF: Production of large amounts of hydrogen peroxide by human tumor cells. Cancer research 1991, 51(3):794–798. [PubMed] [Google Scholar]
- 31.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF et al. : Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475(7355):231–234. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Trachootham D, Alexandre J, Huang P: Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature reviews Drug discovery 2009, 8(7):579–591. [DOI] [PubMed] [Google Scholar]
- 33.Finkel T: Signal transduction by mitochondrial oxidants. The Journal of biological chemistry 2012, 287(7):4434–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handy DE, Loscalzo J: Redox regulation of mitochondrial function. Antioxidants & redox signaling 2012, 16(11):1323–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hancock JT, Desikan R, Neill SJ: Role of reactive oxygen species in cell signalling pathways. Biochemical Society transactions 2001, 29(Pt 2):345–350. [DOI] [PubMed] [Google Scholar]
- 36.Raman D, Foo CH, Clement MV, Pervaiz S: Breast Cancer: A Molecular and Redox Snapshot. Antioxidants & redox signaling 2016, 25(6):337–370. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Cheung SH, Evans EL, Shaw PE: Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer research 2010, 70(20):8222–8232. [DOI] [PubMed] [Google Scholar]
- 38.Simon AR, Rai U, Fanburg BL, Cochran BH: Activation of the JAK-STAT pathway by reactive oxygen species. The American journal of physiology 1998, 275(6 Pt 1):C1640–1652. [DOI] [PubMed] [Google Scholar]
- 39.Son Y, Kim S, Chung HT, Pae HO: Reactive oxygen species in the activation of MAP kinases. Methods in enzymology 2013, 528:27–48. [DOI] [PubMed] [Google Scholar]
- 40.Martindale JL, Holbrook NJ: Cellular response to oxidative stress: signaling for suicide and survival. Journal of cellular physiology 2002, 192(1):1–15. [DOI] [PubMed] [Google Scholar]
- 41.Gutierrez-Uzquiza A, Arechederra M, Bragado P, Aguirre-Ghiso JA, Porras A: p38alpha mediates cell survival in response to oxidative stress via induction of antioxidant genes: effect on the p70S6K pathway. The Journal of biological chemistry 2012, 287(4):2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP: Redox regulation of PI 3-kinase signalling via inactivation of PTEN. The EMBO journal 2003, 22(20):5501–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo L, Kaur Kumar J, Clement MV: Redox control of cytosolic Akt phosphorylation in PTEN null cells. Free radical biology & medicine 2012, 53(9):1697–1707. [DOI] [PubMed] [Google Scholar]
- 44.Cichon MA, Radisky DC: ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of Snail. Oncotarget 2014, 5(9):2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gloire G, Legrand-Poels S, Piette J: NF-kappaB activation by reactive oxygen species: fifteen years later. Biochemical pharmacology 2006, 72(11):1493–1505. [DOI] [PubMed] [Google Scholar]
- 46.Morgan MJ, Liu ZG: Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell research 2011, 21(1):103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P: Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. The Journal of biological chemistry 2006, 281(31):21827–21836. [DOI] [PubMed] [Google Scholar]
- 48.Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A et al. : Redox control of protein kinase C: cell- and disease-specific aspects. Antioxidants & redox signaling 2010, 13(7):1051–1085. [DOI] [PubMed] [Google Scholar]
- 49.Perez LM, Milkiewicz P, Ahmed-Choudhury J, Elias E, Ochoa JE, Sanchez Pozzi EJ, Coleman R, Roma MG: Oxidative stress induces actin-cytoskeletal and tight-junctional alterations in hepatocytes by a Ca2+ - dependent, PKC-mediated mechanism: protective effect of PKA. Free radical biology & medicine 2006, 40(11):2005–2017. [DOI] [PubMed] [Google Scholar]
- 50.Tonelli C, Chio IIC, Tuveson DA: Transcriptional Regulation by Nrf2. Antioxidants & redox signaling 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashraf A, Pervaiz S: Hippo circuitry and the redox modulation of hippo components in cancer cell fate decisions. The international journal of biochemistry & cell biology 2015, 69:20–28. [DOI] [PubMed] [Google Scholar]
- 52.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A: Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free radical biology & medicine 2008, 45(1): 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhee SG: Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312(5782):1882–1883. [DOI] [PubMed] [Google Scholar]
- 54.Hurd TR, DeGennaro M, Lehmann R: Redox regulation of cell migration and adhesion. Trends in cell biology 2012, 22(2):107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tochhawng L, Deng S, Pervaiz S, Yap CT: Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13(3):246–253. [DOI] [PubMed] [Google Scholar]
- 56.Ward JF: Biochemistry of DNA lesions. Radiat Res Suppl 1985, 8:S103–111. [PubMed] [Google Scholar]
- 57.Powell S, McMillan TJ: DNA damage and repair following treatment with ionizing radiation. Radiother Oncol 1990, 19(2):95–108. [DOI] [PubMed] [Google Scholar]
- 58.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG: Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res 2011, 711(1–2):193–201. [DOI] [PubMed] [Google Scholar]
- 59.Smith J, Ladi E, Mayer-Proschel M, Noble M: Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proceedings of the National Academy of Sciences of the United States of America 2000, 97(18):10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N et al. : Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431(7011):997–1002. [DOI] [PubMed] [Google Scholar]
- 61.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y et al. : Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature medicine 2006, 12(4):446–451. [DOI] [PubMed] [Google Scholar]
- 62.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C et al. : FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128(2):325–339. [DOI] [PubMed] [Google Scholar]
- 63.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M et al. : Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell stem cell 2007, 1(1):101–112. [DOI] [PubMed] [Google Scholar]
- 64.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT: The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005, 105(11):4163–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T et al. : Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer cell 2013, 23(3):316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, Sharma N, Dekoninck S, Blanpain C: Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479(7372):189–193. [DOI] [PubMed] [Google Scholar]
- 67.Kannan N, Nguyen LV, Makarem M, Dong Y, Shih K, Eirew P, Raouf A, Emerman JT, Eaves CJ: Glutathione-dependent and -independent oxidative stress-control mechanisms distinguish normal human mammary epithelial cell subsets. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(21):7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warburg O, Wind F, Negelein E: The Metabolism of Tumors in the Body. J Gen Physiol 1927, 8(6):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Folmes CD, Dzeja PP, Nelson TJ, Terzic A: Metabolic plasticity in stem cell homeostasis and differentiation. Cell stem cell 2012, 11(5):596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A: Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell metabolism 2011, 14(2):264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerias A, Batchelder EM, Plongthongkum N, Lutz M et al. : The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell research 2012, 22(1):168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park DJ, Park KS, Lee HK: Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochemical and biophysical research communications 2006, 348(4):1472–1478. [DOI] [PubMed] [Google Scholar]
- 73.Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, De Cola A, Scavo E, Carollo R, D’Agostino D et al. : Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell death & disease 2014, 5:e1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, Cai S, Scheeren FA, Kuo AH, Diehn M: Targeting unique metabolic properties of breast tumor initiating cells. Stem cells 2014, 32(7):1734–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song K, Kwon H, Han C, Zhang J, Dash S, Lim K, Wu T: Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: regulation by MIR-122. Oncotarget 2015, 6(38):40822–40835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH: Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell cycle 2015, 14(1):86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F: Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT½ inhibition. Oncotarget 2014, 5(22):11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deblois G, Giguere V: Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochimica et biophysica acta 2011, 1812(8):1032–1040. [DOI] [PubMed] [Google Scholar]
- 79.Deblois G, St-Pierre J, Giguere V: The PGC-1/ERR signaling axis in cancer. Oncogene 2013, (30):3483–3490. [DOI] [PubMed] [Google Scholar]
- 80.De Luca A, Fiorillo M, Peiris-Pages M, Ozsvari B, Smith DL, Sanchez-Alvarez R, Martinez-Outschoorn UE, Cappello AR, Pezzi V, Lisanti MP et al. : Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6(17):14777–14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM et al. : PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature cell biology 2014, 16(10):992–1003, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP: Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015, 6(7):4569–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, Bevilacqua G, Mazzanti CM, McDonnell LA, Naccarato AG et al. : Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6(31):30453–30471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Farnie G, Sotgia F, Lisanti MP: High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6(31):30472–30486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sanchez V, Sanders ME et al. : MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell metabolism 2017, 26(4):633–647 e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I: Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes & development 2012, 26(17):1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM et al. : BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell stem cell 2013, 12(3):329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, Helgason GV, Gottlieb E: Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nature medicine 2017, 23(10):1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K: Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(29):10574–10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O et al. : MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell metabolism 2015, 22(4):590–605. [DOI] [PubMed] [Google Scholar]
- 91.Papaldo P, Lopez M, Cortesi E, Cammilluzzi E, Antimi M, Terzoli E, Lepidini G, Vici P, Barone C, Ferretti G et al. : Addition of either lonidamine or granulocyte colony-stimulating factor does not improve survival in early breast cancer patients treated with high-dose epirubicin and cyclophosphamide. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2003, 21(18):3462–3468. [DOI] [PubMed] [Google Scholar]
- 92.Prasanna VK, Venkataramana NK, Dwarakanath BS, Santhosh V: Differential responses of tumors and normal brain to the combined treatment of 2-DG and radiation in glioablastoma. Journal of cancer research and therapeutics 2009, 5 Suppl 1:S44–47. [DOI] [PubMed] [Google Scholar]
- 93.Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M, Settleman J: mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer cell 2016, 29(4):548–562. [DOI] [PubMed] [Google Scholar]
- 94.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM et al. : Akt stimulates aerobic glycolysis in cancer cells. Cancer research 2004, 64(11):3892–3899. [DOI] [PubMed] [Google Scholar]
- 95.Guertin DA, Sabatini DM: Defining the role of mTOR in cancer. Cancer cell 2007, 12(1):9–22. [DOI] [PubMed] [Google Scholar]
- 96.Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E, 3rd, Zhang Y: Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proceedings of the National Academy of Sciences of the United States of America 2007, 104(41):16158–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC: Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell stem cell 2010, 6(2):141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH et al. : Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proceedings of the National Academy of Sciences of the United States of America 2012, 109(43):E2939–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS: Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast cancer research : BCR 2004, 6(6):R605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cosentino C, Grieco D, Costanzo V: ATM activates the pentose phosphate pathway promoting anti oxidant defence and DNA repair. The EMBO journal 2011, 30(3):546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM: The antioxidant function of the p53 tumor suppressor. Nature medicine 2005, 11(12):1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gandhi UH, Kaushal N, Hegde S, Finch ER, Kudva AK, Kennett MJ, Jordan CT, Paulson RF, Prabhu KS: Selenium suppresses leukemia through the action of endogenous eicosanoids. Cancer research 2014, 74(14):3890–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Funato Y, Michiue T, Asashima M, Miki H: The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nature cell biology 2006, 8(5):501–508. [DOI] [PubMed] [Google Scholar]
- 104.Korswagen HC: Regulation of the Wnt/beta-catenin pathway by redox signaling. Developmental cell 2006, 10(6):687–688. [DOI] [PubMed] [Google Scholar]
- 105.Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D et al. : ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell stem cell 2013, 12(6):761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, Sun X, Wu Y, Zhou J, Pan J: Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer research 2010, 70(6):2516–2527. [DOI] [PubMed] [Google Scholar]
- 107.Bubici C, Papa S, Dean K, Franzoso G: Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 2006, 25(51):6731–6748. [DOI] [PubMed] [Google Scholar]
- 108.Wilson WR, Hay MP: Targeting hypoxia in cancer therapy. Nature reviews Cancer 2011, 11(6):393–410. [DOI] [PubMed] [Google Scholar]
- 109.Semenza GL: HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation 2013, 123(9):3664–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Denko NC: Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature reviews Cancer 2008, 8(9):705–713. [DOI] [PubMed] [Google Scholar]
- 111.Semenza GL: Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148(3): 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim JW, Tchernyshyov I, Semenza GL, Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metabolism 2006, 3(3):177–185. [DOI] [PubMed] [Google Scholar]
- 113.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC: HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell metabolism 2006, 3(3):187–197. [DOI] [PubMed] [Google Scholar]
- 114.Semenza GL: Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. The EMBO journal 2017, 36(3):252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL: PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer research 2016, 76(15):4430–4442. [DOI] [PubMed] [Google Scholar]
- 116.Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL: Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(50):E5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, Korkaya H, Heath A, Dutcher J, Kleer CG et al. : Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer research 2011, 71(2):614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D et al. : CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. The Journal of clinical investigation 2010, 120(2):485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lu H, Samanta D, Xiang L, Zhang H, Hu H, Chen I, Bullen JW, Semenza GL: Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proceedings of the National Academy of Sciences of the United States of America 2015, 112(33):E4600–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E et al. : Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. The New England journal of medicine 2004, 350(23):2335–2342. [DOI] [PubMed] [Google Scholar]
- 121.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST et al. : Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. The New England journal of medicine 2007, 356(2):115–124. [DOI] [PubMed] [Google Scholar]
- 122.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A et al. : Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. The New England journal of medicine 2011, 364(6):501–513. [DOI] [PubMed] [Google Scholar]
- 123.Kerbel RS: Issues regarding improving the impact of antiangiogenic drugs for the treatment of breast cancer. Breast 2009, 18 Suppl 3:S41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE: Paclitaxel plus bevacizumab versus paclitaxel alone for metasta tic breast cancer. The New England journal of medicine 2007, 357(26):2666–2676. [DOI] [PubMed] [Google Scholar]
- 125.Rose S: FDA pulls approval for avastin in breast cancer. Cancer discovery 2011, 1(7):OF1–2. [DOI] [PubMed] [Google Scholar]
- 126.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS: Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences of the United States of America 2012, 109(8):2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Conley SJ, Baker TL, Burnett JP, Theisen RL, Lazarus D, Peters CG, Clouthier SG, Eliasof S, Wicha MS: CRLX101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast cancer research and treatment 2015, 150(3):559–567. [DOI] [PubMed] [Google Scholar]
- 128.Hayes JD, Dinkova-Kostova AT: The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in biochemical sciences 2014, 39(4):199–218. [DOI] [PubMed] [Google Scholar]
- 129.Menegon S, Columbano A, Giordano S: The Dual Roles of NRF2 in Cancer. Trends in molecular medicine 2016, 22(7):578–593. [DOI] [PubMed] [Google Scholar]
- 130.Rojo de la Vega M, Chapman E, Zhang DD: NRF2 and the Hallmarks of Cancer. Cancer cell 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD: Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. The Journal of biological chemistry 2006, 281(34):24756–24768. [DOI] [PubMed] [Google Scholar]
- 132.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M: Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Molecular and cellular biology 2006, 26(8):2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhu M, Fahl WE: Functional characterization of transcription regulators that interact with the electrophile response element. Biochemical and biophysical research communications 2001, 289(1):212–219. [DOI] [PubMed] [Google Scholar]
- 134.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD: Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32(32):3765–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A: SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Molecular and cellular biology 2011, 31(6):1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F et al. : Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Molecular and cellular biology 2012, 32(17):3486–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jain AK, Jaiswal AK: GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. The Journal of biological chemistry 2007, 282(22):16502–16510. [DOI] [PubMed] [Google Scholar]
- 138.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A: Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. The Journal of biological chemistry 2006, 281(21):14841–14851. [DOI] [PubMed] [Google Scholar]
- 139.Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG: AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Molecular and cellular biology 2016, 36(14):1931–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun Z, Chin YE, Zhang DD: Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Molecular and cellular biology 2009, 29(10):2658–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kwak MK, Itoh K, Yamamoto M, Kensler TW: Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Molecular and cellular biology 2002, 22(9):2883–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Okano Y, Nezu U, Enokida Y, Lee MT, Kinoshita H, Lezhava A, Hayashizaki Y, Morita S, Taguri M, Ichikawa Y et al. : SNP (−617C>A) in ARE-like loci of the NRF2 gene: a new biomarker for prognosis of lung adenocarcinoma in Japanese non-smoking women. PloS one 2013, 8(9):e73794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rushworth SA, MacEwan DJ, O’Connell MA: Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. Journal of immunology 2008, 181(10):6730–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ: The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120(26):5188–5198. [DOI] [PubMed] [Google Scholar]
- 145.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD: Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free radical biology & medicine 2015, 88(Pt B):108–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H: Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer cell 2012, 22(1):66–79. [DOI] [PubMed] [Google Scholar]
- 147.Heiss EH, Schachner D, Zimmermann K, Dirsch VM: Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox biology 2013, 1:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE et al. : NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nature genetics 2015, 47(12):1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD: Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510(7504):298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tibbetts AS, Appling DR: Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annual review of nutrition 2010, 30:57–81. [DOI] [PubMed] [Google Scholar]
- 151.Lee D, Xu IM, Chiu DK, Lai RK, Tse AP, Lan Li L, Law CT, Tsang FH, Wei LL, Chan CY et al. : Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. The Journal of clinical investigation 2017, 127(5):1856–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Altman BJ, Stine ZE, Dang CV: From Krebs to clinic: glutamine metabolism to cancer therapy. Nature reviews Cancer 2016, 16(10):619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Carracedo A, Cantley LC, Pandolfi PP: Cancer metabolism: fatty acid oxidation in the limelight. Nature reviews Cancer 2013, 13(4):227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH et al. : A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nature medicine 2012, 18(9):1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C et al. : A human memory T cell subset with stem cell-like properties. Nature medicine 2011, 17(10):1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, Klatt NR, Brenchley JM, Vaccari M, Gostick E et al. : Superior T memory stem cell persistence supports long-lived T cell memory. The Journal of clinical investigation 2013, 123(2):594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K et al. : A metabolic prosurvival role for PML in breast cancer. The Journal of clinical investigation 2012, 122(9):3088–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G et al. : Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nature medicine 2016, 22(4):427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R et al. : The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation 2011, 121(7):2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y et al. : JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell metabolism 2018, 27(1):136–150 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W, Zhou Y, Cong Z, Yan F, Chen S: Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. International journal of cancer 2014, 135(3):574–584. [DOI] [PubMed] [Google Scholar]
- 162.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q: High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Science translational medicine 2014, 6(222):222ra218. [DOI] [PubMed] [Google Scholar]
- 163.Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, Pillai MV, Newberg AB, Deshmukh S, Levine M: Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PloS one 2012, 7(1):e29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, Yee NS, Bodeker KL, Du J, Roberts LJ, 2nd et al. : Pharmacological ascorbate with gemcitabine for the control of metastatic and node positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer chemotherapy and pharmacology 2013, 71(3):765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T et al. : O2(−) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer cell 2017, 31(4):487–500 e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE et al. : Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]