

ABSTRACT
Renal cell carcinoma (RCC) is a common malignant tumour affecting the urinary system, and multidrug resistance is one of the major reasons why chemotherapy for this type of cancer often fails. Previous studies have shown that loss of the human organic cation transporter OCT2 is the main factor contributing to oxaliplatin resistance in RCC, and that DNA hypermethylation and histone methylation play important roles in the transcriptional repression of OCT2 in RCC. In this study, we found that histone acetylation also regulates OCT2 repression in RCC and elucidated the underlying mechanisms. In normal renal cells, HDAC7 combines with MYC at the OCT2 promoter, resulting in a decrease in free HDAC7, which in turn increases the levels of H3K18ac and H3K27ac at the OCT2 promotor and activates OCT2 expression. In RCC cells, however, the interaction between HDAC7 and MYC does not occur, which leads a high abundance of HDAC7 and low levels of H3K18ac and H3K27ac at the OCT2 promoter, thereby resulting in the inhibition of OCT2 transcription. We found that combined treatment using the DNA methylation inhibitor decitabine and the histone deacetylase inhibitor vorinostat significantly increased the expression of OCT2 in RCC cell lines, which sensitized these cells to oxaliplatin. We accordingly propose that the combination of anticancer agents and epigenetic drugs can provide a novel chemotherapeutic regimen.
KEYWORDS: Renal cell carcinoma, histone acetylaion, combination therapy, epigenetic regulation, OCT2
Introduction
Worldwide, kidney cancer accounts for 2–3% of all cancers [1], and approximately 90% of the kidney cancers are renal cell carcinomas (RCCs) [2]. RCC is also one of the tumours prone to multidrug resistance (MDR). The effective rate of clinical chemotherapy for RCC is only 7–10%. This MDR can be attributed in part to the high expression of certain drug transporters in the kidney. The human organic cation transporter member 2 (OCT2, encoded by SLC22A2), which is the most abundant organic cation transporter in the kidney, is mainly expressed on the basolateral or apical side of renal tubular epithelial cells and participates in drug uptake [3–5]. The functions of this transporter are related to the disposal of many clinically endogenous substances and drugs in the body [6,7].
Previous studies have shown that platinum is the main anti-cancer substrate of OCT2 [8], and that OCT2 has different transportability for different platinum compounds (in the order oxaliplatin > Pt [DACH]Cl2 > ornaplatin > transplatin > cisplatin), whereas it is unable to transport carboplatin [9]. In addition, several studies have shown that an increase in the exogenous expression of OCT2 can significantly increase the accumulation of oxaliplatin and thereby enhance cell cytotoxicity [10,11]. Although some other OCT transporters, such as OCT1 [10] and OCT3 [12], are also involved in the transport of platinum, the expression of these transporters in renal tubular epithelial cells is very low [10]. Therefore, the expression and activity of OCT2 are assumed to play decisive roles in the accumulation and cytotoxicity of platinum anti-cancer drugs, particularly oxaliplatin, in renal cancer cells.
Histone acetylation, which is associated with chromatin assembly, DNA repair, and recombination [13,14], is one of the most extensively studied types of epigenetic modification. Dynamic histone acetylation is critical for correct gene expression and is regulated by a balance of histone acetyltransferases (HATs) and histone deacetylases (HDACs). If this balance is disrupted, there exists the potential for tumour development [15].
HATs include members of the p300/CBP and MYS GNAT families. It has been found that CBP and P300 catalyse the process whereby H3K18ac and H3K27ac activate the transcription of downstream genes [16]. On the basis of structural differences, HDACs can be divided into five classes: Class I, including HDAC1, HDAC2, HDAC3, and HDAC8; Class IIa, including HDAC4, HDAC5, HDAC7, and HDAC9; Class IIb, including HDAC6 and HDAC10; Class III, which are related enzymes of silent information regulator 2 (SirT2), and Class IV, which comprises only HDAC11. HDACs regulate tumour growth by inhibiting histone acetylation, which in turn suppresses the transcription of oncogenes [17]; therefore, HDACs can provide key targets for anti-tumour drugs. Vorinostat (suberoylanilide hydroxamic acid, SAHA) was the first HDAC inhibitor approved by the Federal Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma (CTCL), and thereafter Romidepsin (FK228) was also approved by the FDA for treatment of CTCL and peripheral T-cell lymphoma.
Previous studies have demonstrated that the loss of OCT2 in tumour cells is the major cause of oxaliplatin resistance in RCC. DNA and histone methylations are key factors in the transcriptional repression of OCT2 in RCC [18]. Here, we found that aberrant histone acetylation also occurs in the OCT2 promoter and explore the mechanisms whereby an HDAC7-MYC complex mediates the regulation of OCT2 in RCC. Although epigenetic drugs have poor therapeutic effects on most solid tumours [19,20], the combination of epigenetic drugs with cytotoxic drugs [21] or immunotherapeutics [22] can enhance the anti-cancer effect, indicating that it is possible to design novel combination therapies for RCC. As OCT2 expression is increased by the HDAC inhibitor SAHA and DNA methylation inhibitor decitabine (DAC) in RCC cells, we investigated the synergistic effect of SAHA-DAC-oxaliplatin combination therapy on RCC cells.
Results
Aberrant active histone acetylation by H3K27ac and H3K18ac in RCC
OCT2 transcriptional repression in RCC has previously been shown to be regulated by DNA methylation and H3K4me3 [18]; however, to date, it was not known whether histone acetylation is related to OCT2 transcriptional repression. We initially observed that in three pairs of the tumour-adjacent tissues, H3K18ac and H3K27ac were both enriched around the transcription start site (TSS) of the OCT2 promoter, whereas H3K9ac abundance was very low (Figure 1(a)). As H3K18ac, H3K27ac, and H3K9ac are involved in transcriptionally permissive histone modification, the results indicated that H3K18ac and H3K27ac might be associated with OCT2 transcription. We observed that OCT2 transcription was markedly decreased in selected three pairs of RCC tissues (Figure 1(b)), and thus ChIP-qPCR assays were carried out to identify H3K18ac and H3K27ac occupancy in these three pairs of tissues. In the tumour-adjacent tissues, H3K18ac and H3K27ac were highly enriched around the TSS of OCT2 and were reduced to different degrees in the matched tumour tissues, whereas no significant change was detected for H3K9ac (Figure 1(c–e)), indicating that the loss of H3K18ac and H3K27ac, but not that of H3K9ac, might occur at the OCT2 promoter when OCT2 is repressed in RCC.
Figure 1.
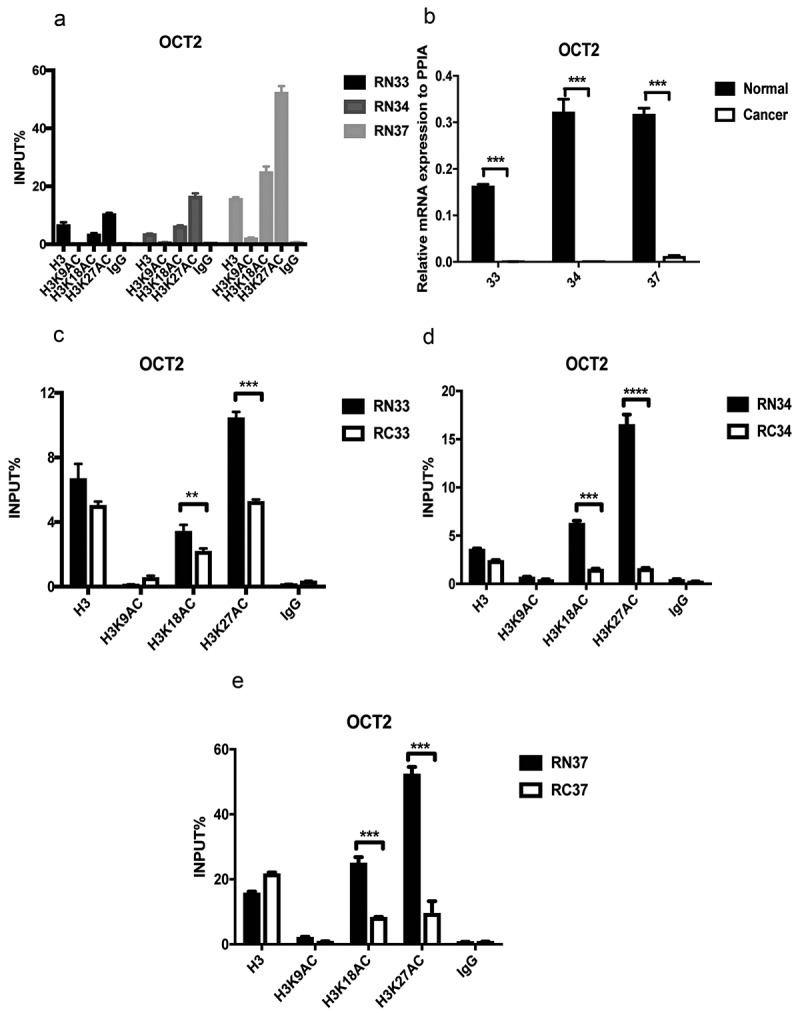
Decreased OCT2 transcriptional expression and aberrant histone acetylation in three pairs of RCC patients tissues (No. 33;34;37). (a) ChIP assay of H3K9ac, H3K18ac and H3K27ac at OCT2 promoter in tumour-adjacent kidney tissues; (b) RT-qPCR analysis of OCT2 mRNA expression; (c, d, e) H3K9ac, H3K18ac and H3K27ac occupancy at OCT2 promoter. The results are expressed as means ± S.D. from technical triplicates in A-E[one-tailed unpaired t-test; **P < 0.01; ***P < 0.001; ****P < 0.0001].
Increased transcriptional expression of HDAC7 and HDAC9 in RCC
Given that the acetylation and deacetylation of histones are catalysed by HATs and HDACs, respectively, we next attempted to determine whether HATs or HDACs mediate OCT2 repression in RCC.
The Oncomine database showed that the expression of CBP and P300, which have been reported to mainly catalyse H3K18ac and H3K27ac, had no significant effect in RCC tumours compared with the tumour-adjacent tissues (Fig. S1). The microarray data indicated that HDACs, but not CBP and P300, are the key factors involved in histone acetylation in RCC. To determine the role of HDACs in the OCT2 regulatory mechanism, the mRNA expression of class I and II HDACs was examined in four randomly selected pairs of RCC tissues with reduced expression of OCT2 (Figure 2(a), Fig. S2) by RT-qPCR. We found that only HDAC7 and HDAC9 were up-regulated in these four RCC tissues (Figure 2(b–e)), suggesting that these two HDACs might mediate histone deacetylation around the TSS of the OCT2 promoter. We then examined the mRNA expression of HDAC7 and HDAC9 in 36 pairs and 30 pairs of RCC tissues, respectively (Figure 2(f–g)); Fig. S3). Due to ethical reasons, the amounts of some of the collected samples were insufficient to enable mRNA expression analysis of both HDAC7 and HDAC9. The results confirmed that higher HDAC7 and HDAC9 expression was detected in RCC tumours than in the paired adjacent tissues. Due to OCT2 repression in RCC, the assembled data further indicated that HDAC7 and HDAC9 regulate the abundance of H3K27ac and H3K18ac at the OCT2 promoter, resulting in a decrease in H3K18ac and H3K27ac in RCC.
Figure 2.
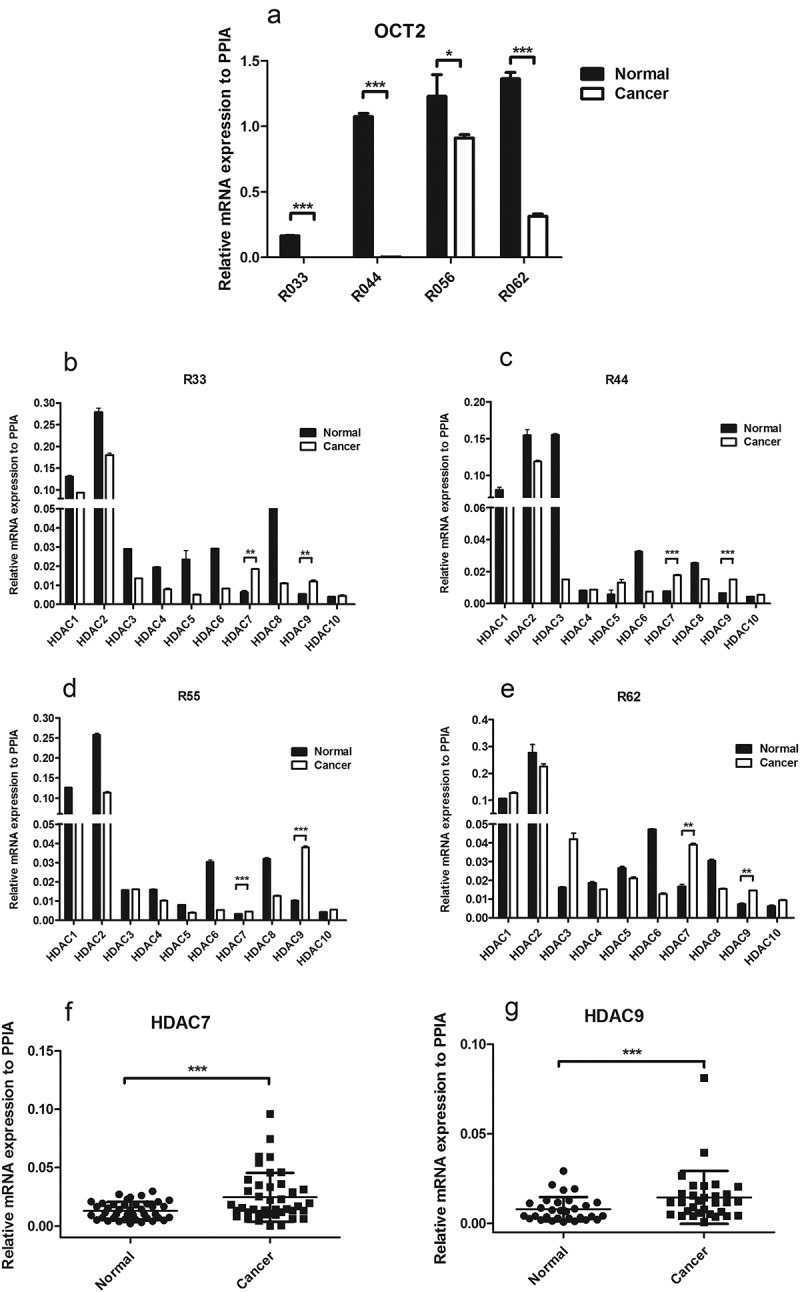
HDACs transcriptional expression in RCC patients tissues. (a) RT-qPCR analysis of OCT2 mRNA expression in four pairs of RCC patient tissues (No. 33,44,55,62); (b, c, d, e) RT-qPCR analysis of HDACs mRNA expression in four pairs of RCC patient tissues (No. 38,44,55,62); (f, g) The two-tailed paired t-test for mRNA expression of HDAC7 (f) and HDAC9 (g) in RCC tissues. The results are expressed as means ± S.D. from technical triplicates in a–f [a–e: one-tailed unpaired t-test: *P<0.1;**P < 0.01; ***P < 0.001; ****P < 0.0001; F-G: two-tailed paired t-test: ***P < 0.001].
Increased HDAC7 occupancy upon SAHA treatment and myc-mediated activation of OCT2
In order to examine the influence of histone acetylation on OCT2 expression, 786-O cells were treated with the histone deacetylase inhibitor SAHA. We accordingly observed that OCT2 expression was activated in 786-O cells in response to SAHA treatment (Figure 3(a)). As SAHA inhibits all class I and II HDACs, to further investigate how HDAC7 and HDAC9 affected OCT2 expression, 786-O cells were transfected with nontargeting control siRNA (si-NC), or siRNA targeting HDAC7 (siHDAC7-s1 and siHDAC7-s2) and HDAC9 (siHDAC9-s1 and siHDAC9-s2). The silencing effect of siRNA was assessed by RT-qPCR (Figure 3(c–d)). After transient knockdown expression of HDAC7 and HDAC9, OCT2 expression was significantly increased in 786-O cells (Figure 3(b)), and OCT2 protein expression was increased after HDAC7 and HDAC9 knockdown in 786-O cells (Figure 3(e–f)). Collectively, these results provide evidence that both HDAC7 and HDAC9 regulate OCT2 expression in RCC. In addition, the expression of OCT2 was found to be positively correlated with the efficiency of HDAC inhibition.
Figure 3.
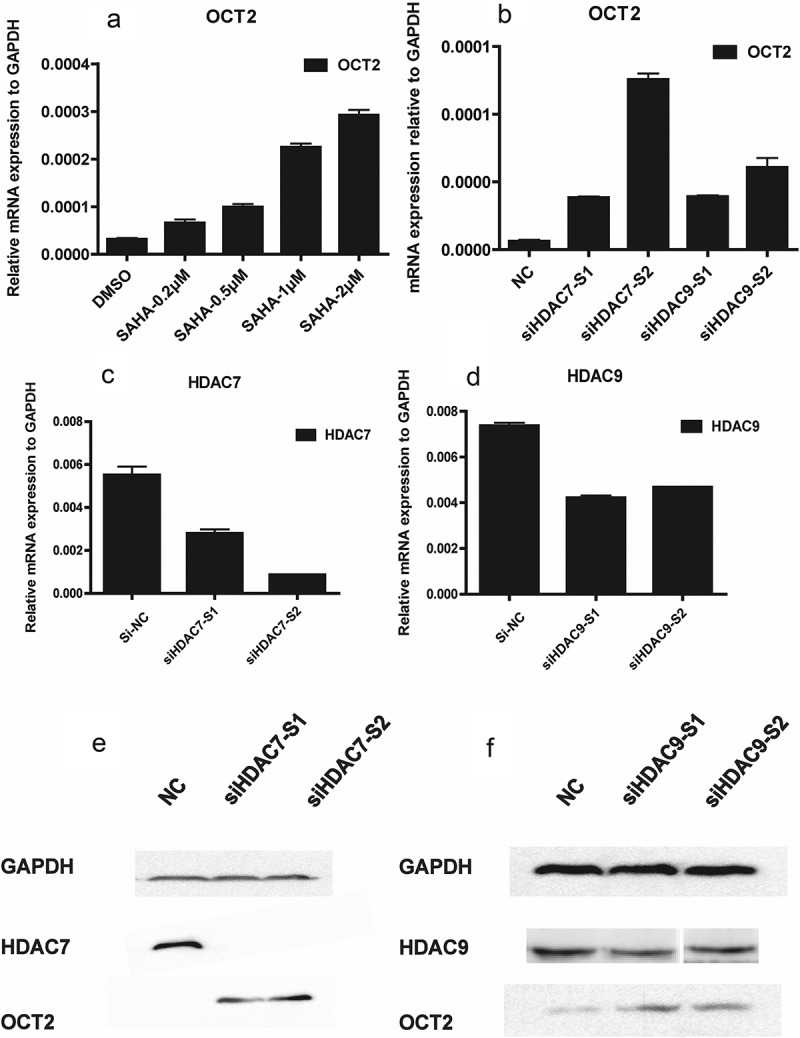
Loss of HDAC7/HDAC9 up-regulated the expression of OCT2 in RCC cell line 786-O. (a) RT-qPCR analysis of OCT2 mRNA expression in 786-O after treated with SAHA at 0.2 µM, 0.5 µM, 1.0 µM, 2.0 µM for 48 h; (b) RT-qPCR analysis of OCT2 mRNA expression after HDAC7 or HDAC9 knocked down in 786-O cells; (c, d) RT-qPCR analysis of HDAC7 (c) or HDAC9 (d) mRNA expression after targeted siRNA was transfected into 786-O cells. (e) HDAC7 and OCT2 protein expression after HDAC7 knockdown in 786-O cells. (f) HDAC9 and OCT2 protein expression after HDAC9 knockdown in 786-O cells. The results are expressed as means ± S.D. from technical triplicates in A-D.
Our previous data have indicated that MYC, acting as a transcription factor, regulates OCT2 expression by binding to the OCT2 promoter [18]. In our study, we found SAHA induces the mRNA expression of MYC in 786-O (Figure 4(a)). We therefore generated 786-O cells that stably expressed shRNA against MYC as described in a previous study [18]. MYC was efficiently knocked down when shRNA against MYC was turned on by doxycycline treatment (Figure 4(b)). The knockdown of MYC was found to block the transcriptional activation of OCT2 by SAHA in both mRNA and protein level (Figure 4(c–d)). We then performed Co-IP analysis to examine the direct interaction between MYC and HDAC7 in 786-O cells following treatment with SAHA. We found that MYC and HDAC7 bind to each other after SAHA treatment, and when MYC was knocked out, the protein complex disappeared, but there was still free HDAC7 in the cell (Figure 4(e)). However, HDAC9 did not bind to MYC in SAHA-treated 786-O cells (Fig. S4). A subsequent CHIP-qPCR assay showed that both MYC and HDAC7 occupancy at the E-box of the OCT2 promoter were enriched in SAHA-treated 786-O cells (Fig. S5), resulting in the reducing of free HDAC7 at the acetylation site of OCT2 promoter (Figure 4(f)). When HDAC7 was knocked down, H3K18ac and H3K27ac enriched at OCT2 promoter in 786-O cells (Figure 4(g)). We accordingly proposed our assumption that the combination of MYC and HDAC7 at the E-box of OCT2 promoter in normal renal cells results in a decrease in free HDAC7, which may increase the expression H3K18ac and H3K27ac and activate OCT2 transcription. In contrast to normal kidney cells, MYC cannot bind to and interfere with HDAC7 in RCC cells, which increases the amounts of free HDAC7 at the OCT2 promoter and decreases H3K18ac and H3K27ac levels, eventually resulting in OCT2 transcriptional inhibition. However, it remains unclear why the recruitment of HDAC7 by MYC at the OCT2 promoter in RCC cells is disrupted.
Figure 4.
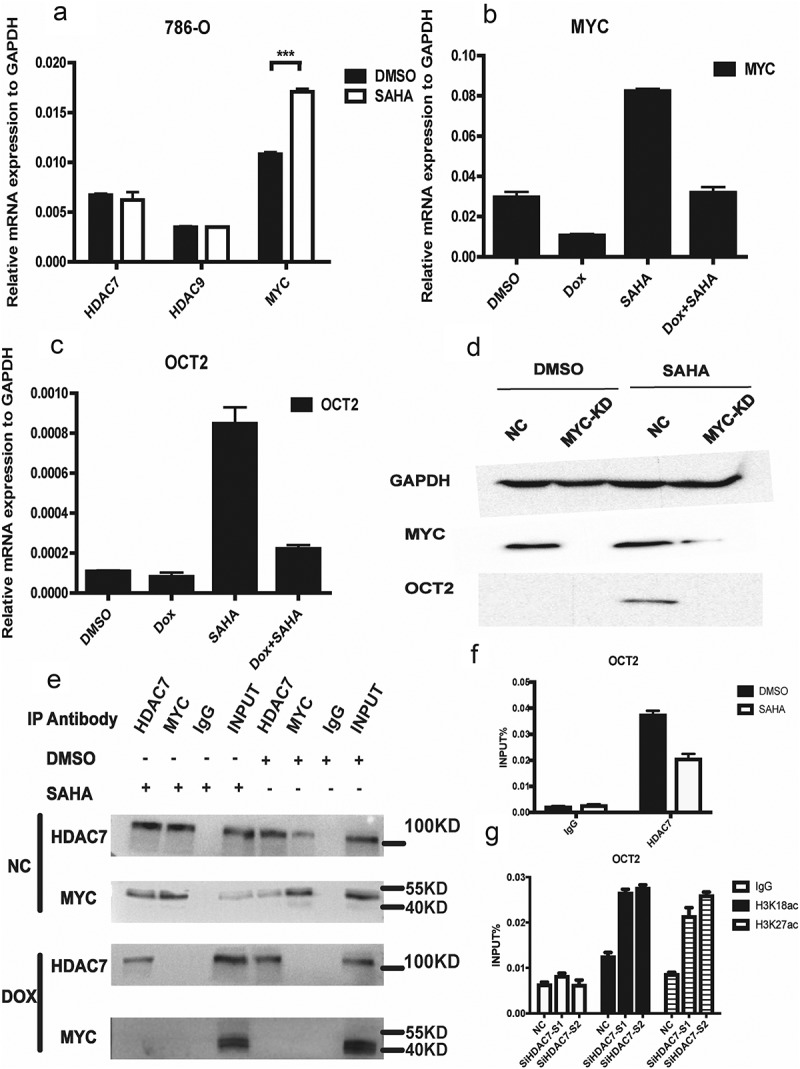
MYC combined with HDAC7 at OCT2 promoter to regulate the OCT2 expression in RCC cell line 786-O. (a) RT-qPCR analysis of HDAC7, HDAC9 and MYC mRNA expression in 786-O after treated with 2 µM SAHA for 48 h; (b, c) RT-qPCR analysis of MYC (b) and OCT2 (c) mRNA expression in 786-O cells after knockdown MYC and treated with 2µM SAHA for 48 h; (d) MYC and OCT2 protein expression in 786-O cells after knockdown MYC and treated with 2 µM SAHA for 48 h. (e) Co-IP analysis of HDAC7-MYC complex in 786-O cells after knockdown MYC and treated with 2µM SAHA for 48 h; (f) ChIP-qPCR analysis of HDAC7 enrichment at OCT2 promoter in 786-O cells after treated with 2 µM SAHA for 48 h; (G) ChIP-qPCR analysis of H3K18ac and H3K27ac enrichment at OCT2 promoter in 786-O cells after HDAC7 knockdown. The results are expressed as means ± S.D. from technical triplicates in a, b, c, f, g [a: one-tailed unpaired t-test: P = 0.0008].
Combination therapy using epigenetic drugs and oxaliplatin enhances the chemosensitivity of RCC cells
Previous studies have shown that DAC can effectively induce the expression of OCT2 and that sequential combination of DAC and oxaliplatin sensitizes RCC cells to oxaliplatin both in vitro and in xenografts [18]. In our study, we observed a 28.4-fold increase in epigenetic activation of OCT2 in RCC cells following DAC+SAHA treatment (Figure 5(a)). The rank order of induction was as follows: DMSO > SAHA > DAC > DAC+SAHA in both mRNA and protein level (Figure 5(a–b)). The platinum cellular uptake in 786-O has the same trend as OCT2 up-regulated expression (Figure 5(c)). We therefore examined whether pretreatment with DAC and SAHA synergistically promoted stronger cytotoxicity compared with DAC or SAHA alone. Cells of the 786-O line were initially treated sequentially with increasing concentrations of SAHA, DAC, or oxaliplatin (Fig. S6A). The median inhibitory concentration (IC50) values of these three drugs were calculated as follows: IC50 (DAC) = 12.01 µM, IC50 (SAHA) = 4.69 µM, and IC50 (oxaliplatin) = 22.39 µM (Fig. S6B-D). Finally, the 786-O cells were pretreated sequentially with increasing concentrations of SAHA and/or DAC, followed by application of various concentrations of oxaliplatin at 48 h (Supplemental Table 4) to evaluate the combination index (CI). According to the Chou-Talalay method, the gradient concentrations of drugs are determined by their IC50 values [23]. As shown in Figure 5(d), DAC and oxaliplatin alone induced an approximate 50% reduction in the survival rate of 786-O cells at maximal concentrations of 50 µM and 150 µM, respectively, whereas SAHA had a 70–80% cytotoxic effect at the maximal concentration of 20 µM. Given that it is essentially unproductive to examine the effects of drug combinations when the cell growth inhibition is extremely low, we only investigated the effect of such combinations when the cell growth inhibition rate was greater than 50%. Whereas a synergistic or additive effect was observed after treatment of 786-O cells with SAHA+oxaliplatin (CI = 0.29–0.89), only synergistic effects were observed after 786-O cells had been treated with DAC+oxaliplatin and DAC+SAHA+oxaliplatin (CIDAC+OXA = 0.12–0.47, CIDAC+SAHA+OXA = 0.11–0.63). In order to compare the synergistic effect of DAC+oxaliplatin and DAC+SAHA+oxaliplatin in 786-O cells, we calculated the IC50 value and dose-reduction index (DRI) of oxaliplatin in 786-O cells (Figure 5(e)), where DRI is the ratio of the dose of oxaliplatin(alone) to the dose of oxaliplatin(combo), which represents the fold-dose decrease in oxaliplatin in combination. As shown in Figure 5(e), the DRI(oxaliplatin) of DAC+SAHA+oxaliplatin was clearly higher than that of the other combinations, indicating that pretreatment of 786-O cells with DAC+SAHA followed by treatment with oxaliplatin had the strongest synergetic cytotoxicity compared with any other combination.
Figure 5.
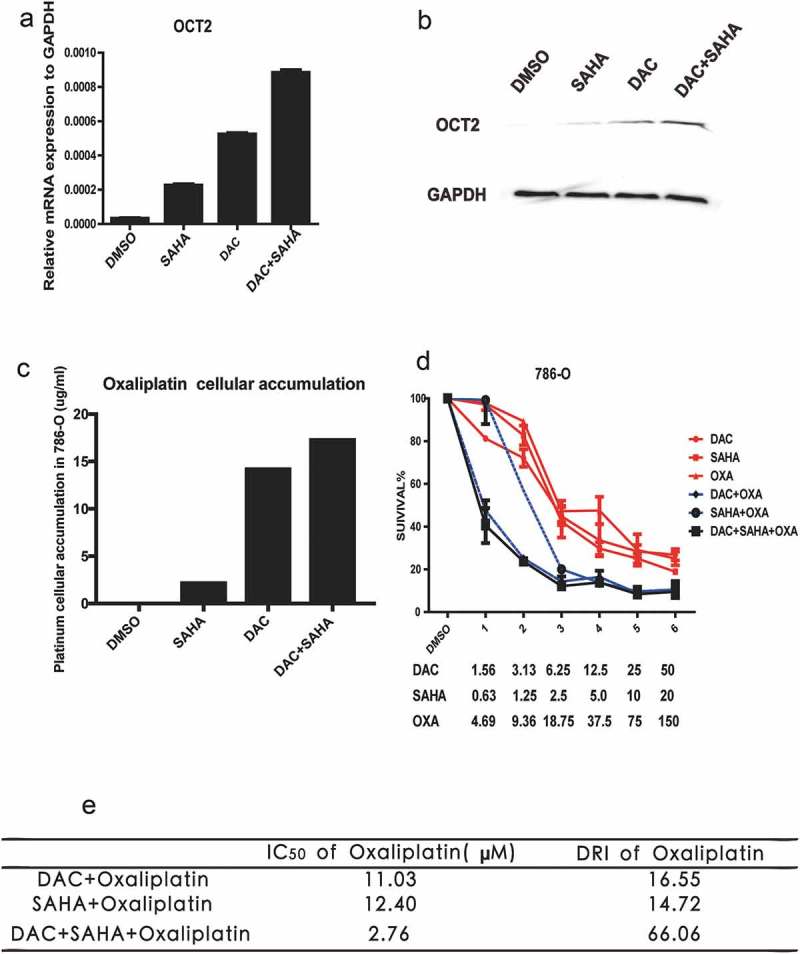
DAC, SAHA and oxaliplatin combinations. (a) RT-qPCR analysis of OCT2 mRNA expression after treated with DAC, SAHA and DAC+SAHA in 786-O cells, respectively; (b) OCT2 protein expression of DAC, SAHA, DAC+SAHA treatment in 786-O cells; (c) Platinum cellular accumulation after treated with DAC, SAHA and DAC+SAHA in 786-O; (d) Dose-Survival Curve of DAC, SAHA, OXA, DAC+SAHA, DAC+OXA, SAHA+OXA, DAC-SAHA-OXA in 786-O cells; (e) The IC50 and dose-reduction index (DRI) of OXA. DRI is the ratio of dose of OXA (alone) to dose of OXA (combo), which represents the fold-dose decrease of OXA in combination. CI < 0.8, 0.8 < CI<1, and CI > 1 indicate synergism, additive effect, and antagonism, respectively. The results are expressed as means ± S.D. from technical triplicates in a–d.
Discussion
Many renal masses remain asymptomatic or nonpalpable until the advanced stages of RCC [1]. The 5-year survival rate for patients with metastatic RCC is less than 10% [24], with the low survival rate being attributable to the high resistance of RCC to both chemotherapy and radiation therapy [25]. Such resistance to chemotherapy can often be ascribed to MDR mediated by drug transporters [26,27].
OCT2 is an uptake transporter expressed in kidney tubules. In our previous studies, we detected a correlation between the loss of OCT2 expression and tumour cell resistance to platinum, which showed that the aberrant OCT2 promoter in RCC is characterized by a hypermethylated CpG island and low H3K4 tri-methylation occupancy. In the present study, we further investigated other epigenetic factors related to OCT2 repression. We accordingly detected significant decreases in H3K18ac and H3K27ac at the OCT2 promoter in RCC tumour tissue compared with the matched adjacent tissue (Figure 1(c–e)). H3K18ac and H3K27ac are involved in a variety of physiological processes and the relative balance between histone acetylation and deacetylation is essential for normal cell growth. The findings of a previous study have indicated that loss of H3K18ac causes suppression of the nonreceptor tyrosine kinase EtK, which plays an essential role in prostate cancer cell survival and growth [28], whereas disruption of the association between BRD4 and H3K27ac has been shown to downregulate MYC and promote antitumour activity in preclinical animal models of human cancers [29]. We therefore hypothesize that OCT2 repression in RCC may be attributable to the loss of H3K18ac and H3K27ac. However, on the basis of our present results, we cannot exclude the possibility that other HATs are involved in the transcriptional regulation of OCT2, and this accordingly warrants further research.
Histone acetylation is co-regulated by HATs and HDACs. HATs promote acetylation to generate less condensed chromatin structures, thereby enabling the activation of transcription, whereas HDACs mediate chromatin aggregation [30]. In the present study, we found that, compared with paired adjacent tissues, the expression of HDAC7 and HDAC9 increased in most of the RCC tissues, whereas the expression of other HDACs varied. These abnormal increases occurred during the early stage of RCC pathological grading, suggesting that HDAC7 and HDAC9 may be potential targets and early diagnostic markers for the treatment of RCC. Furthermore, we also speculated that the loss of H3K18ac and H3K27ac at the OCT2 promoter is caused by HDAC7/9. It has been reported that the expression of class I HDACs in tumours is abnormally increased at both the mRNA and protein levels when compared with paired adjacent tissues. In contrast to class I HDACs, expression of class II HDACs has been found to decrease in tumours, and a higher expression of class II HDACs predicts a better prognosis in patients [31]. HDAC7 and HDAC9 belong to class II HDACs and Osada et al. [32] found that HDAC7 mRNA expression was higher in node-negative, low-grade lymph tumours compared with late-stage lymph node metastases. Furthermore, in pancreatic cancer, both the mRNA and protein expression levels of HDAC7 are significantly up-regulated in tumours [33]. These studies therefore indicate that the expression of HDACs differs among different cancer types.
Histone deacetylation, and particularly the inhibitors of HDACs, has been a hot topic in anti-cancer drug research in recent years [34]. The SAHA we used in this study is a hydroxamic acid and has the characteristics of a longer half-life, lower toxicity, and greater stability compared with other hydroxamic acids such as TSA [31]. In 2006, the FDA gave approval for the use of SAHA in the treatment of cutaneous T-cell lymphomas. However, although it can induce the expression of OCT2 mRNA in 786-O cells (Figure 3(a)), it does not induce such expression in other RCC cell lines, 769-P or CAKI (Fig. S7), which indicated the limitation of our study: the SAHA induction mechanism we studied may only work in certain part of RCC patients. Although the three cell lines are derived from primary renal clear cell carcinomas, they were obtained from different patients, which suggests different factors such as the oxygen content in the tumour microenvironment affecting the regulatory mechanism.
In our study, SAHA slightly up-regulated MYC in 786-O cells, whereas MYC itself can recruit both HATs and HDACs. Recent studies have shown that the recruitment of HATs by MYC may be mediated by TRRAP [35], which is not a HAT but is present in a different macro-molecular complex containing HAT sub-units, including GCN5, PCAF, and TIP60 [36]. MYC also recruits HDACs to gene promoters, and it has previously been demonstrated that MYC recruits HDAC3 to form a transcriptional suppressor complex, co-localizes on the miR-451 promoter, and finally induces its downregulation in acute myeloid leukaemia [37]. However, on the basis of the findings of the present study, we speculate that MYC performed an opposite function in complex recruitment in normal renal cells. As epigenetic regulation invariably involves the functioning of several different pathways, there are likely to be complex interactions relating to DNA methylation and HDACs. Previous studies have shown that synergy between HDAC activity and DNA methylation is operative for a key epigenetic abnormality in cancer cells, namely, transcriptional silencing of tumour suppressor genes [38].
Based on the above results, we propose a hypothesis that the histone acetylation mechanism underlying OCT2 repression in RCC is regulated by the enrichment of free HDAC7 at OCT2 promoter (Figure 6). In normal renal cells, HDAC7 combines with MYC at the E-box site on OCT2 promoter, resulting in a decrease of free HDAC7s at the promoter, which increases H3K18ac and H3K27ac at the promoter and activates OCT2 expression. However, in RCC cells, the HDAC7-MYC complex is not established, leading to an increase in free HDAC7s at the OCT2 promoter and a decrease in the levels of H3K18ac and H3K27ac, thereby resulting in OCT2 transcriptional inhibition.
Figure 6.
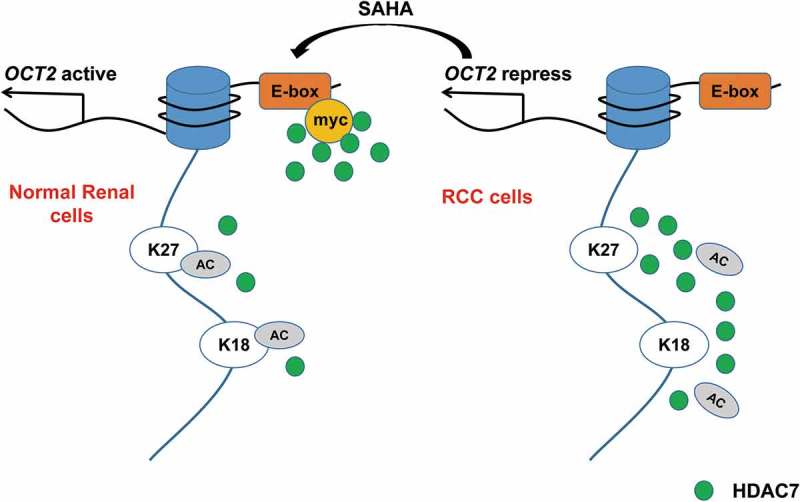
Histone acetylation mechanism of OCT2 repression in RCC. In normal renal cells, HDAC7 combines with MYC at the E-box site on OCT2 promoter, resulting in a decrease of free HDAC7s at the promoter, which increases H3K18ac and H3K27ac at the promoter and activates OCT2 expression. However, in RCC cells, the HDAC7-MYC complex is not established, leading to an increase in free HDAC7s at the OCT2 promoter and a decrease in the levels of H3K18ac and H3K27ac, thereby resulting in OCT2 transcriptional inhibition.
It has previously been shown that the cellular accumulation of oxaliplatin is the key determinant of the cytotoxicity of this drug [39]. In renal cells, oxaliplatin is mainly bound by OCT2 and effluxed by MATE-2K [40]. Both OCT2 and MATE-2K are repressed in RCC, whereas treatment with DAC or SAHA can induce the expression of OCT2 but not MATE-2K, resulting in the high accumulation and cytotoxicity of OXA in RCC cells [18]. TSA, which is similar to SAHA, can up-regulate both OCT2 and MATE-2K [41], and is hence not suitable for use in combination therapy.
HDACs have a potent effect in haematological diseases or lymphoid malignancies alone, but often fail to achieve satisfactory therapeutic effects in the case of solid tumours. In our study, we also found that synergistic effects of the SAHA-oxaliplatin combination were the lowest among the three combination groups examined, indicating that SAHA does not work well in combination with oxaliplatin. This may be related to the fact that SAHA does not cause a sufficiently high induction of OCT2. Therefore, a combination of DAC and SAHA has been used for tumour treatment. Chen et al. [42], for example, found that the combination of DAC and SAHA inhibited ovarian cancer growth by inducing apoptosis, G2/M arrest, autophagy, and re-expression of imprinted tumour suppressor genes, whereas Dong et al. [43] demonstrated that combined treatment with DAC and SAHA enhanced the antiproliferative activity of glioma cells. In our study, we designed a clinical protocol based on the combined use of DAC, SAHA, and the anti-cancer drug oxaliplatin, and we demonstrated that the toxicity of DAC+SAHA+oxaliplatin to 786-O cells was higher than that of DAC+oxaliplatin, and that the synergistic effect of this combination was stronger than that of DAC+oxaliplatin at high dose. Nowadays, there is a heightened focus on multidrug combination research in an effort to solve the pervasive problem of MDR. In this study, we found that DAC and SAHA significantly induced the expression of OCT2 in RCC cells, enhanced the cellular accumulation of oxaliplatin, and significantly reversed drug resistance, thereby providing new insights for the revision of clinical oxaliplatin guidelines for RCC.
Materials and methods
Tissues and cell culture
Thirty-six paired tissue samples of RCC patients were provided by the Specimen Bank of Zhejiang Cancer Hospital (Hangzhou, China) and were approved by the Institutional Review Board of Zhejiang Cancer Hospital [No. (2014)-08-76; for detailed patient information, see Supplemental Table 1].
HEK293T cells and cells of the RCC cell line 786-O were purchased from the Chinese Academy of Science Committee on Type Culture Collection Cell Libraries. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, and those of the 786-O line were cultured in RPMI-1640 medium containing 10% fetal bovine serum. All cells were maintained at 37°C in 5% CO2. For epigenetic drug treatment, the 786-O cells were cultured in medium containing SAHA (Selleck Chemical) for 48 h or DAC (Sigma-Aldrich) for 72 h, with the medium being refreshed every 24 h.
Real-time quantitative polymerase chain reaction analysis
Total RNA of tissues was isolated using a total RNA mini-prep kit (Tiangen, Beijing, China), whereas total RNA from cell lines was isolated using a total RNA mini-prep kit (Axygen, Suzhou, China). The isolated RNA was subsequently reverse transcribed to cDNA using PrimeScript RT Master Mix (Takara, Tokyo, Japan). We performed real-time quantitative polymerase chain reactions (RT-qPCR) using a StepOnePlus System (Applied Biosystems) and SYBR Premix EX Taq (Takara, Tokyo, Japan). Peptidylprolyl isomerase A (PPIA) in tissues and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cell lines served as the respective internal controls. The relative mRNA expression levels were normalized to PPIA or GAPDH. The sequences of the primers used for amplification are listed in Supplemental Table 2.
Short hairpin RNA transfection of the RCC cell line 786-O
Short hairpin RNAs (shRNAs) were cloned as described by Paddison et al. [44], and inserted into pTRIPZ lentiviral vectors (Thermo Scientific, Waltham, MA). To package virus, HEK293T cells were transfected with shRNA against MYC (shRNA-MYC) using Lipofectamine 2000 (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Cells of the 786-O line were infected by the supernatant virus of HEK293T cells. The sequences of the shRNAs used are listed in Supplemental Table 3. Cells of the 786-O line expressed shRNA following treatment with 1 μg/mL doxycycline for 48 h.
Small interfering RNA transfection
The small interfering RNAs (siRNAs) were synthesized by GenePharma (Shanghai, China) and transfected into cells for 48 h using Lipofectamine 3000 (Life Technologies, Waltham, MA) according to the manufacturer’s instructions. The target sequences of the siRNAs used are listed in Supplemental Table 3.
Chromatin immunoprecipitation assay (ChIP)
The procedures used for ChIP analysis have been described previously [45]. RT-qPCR was performed using the specific primers listed in Supplemental Table 2. The antibodies used for ChIP were as follows: anti-H3 (Abcam, Cambridge, MA; Ab1791), anti-H3K9ac (Abcam, Cambridge, MA; Ab4441), anti-H3K18ac (Abcam, Cambridge, MA; Ab1191), anti-H3K27ac (Abcam, Cambridge, MA; ab4729), anti-HDAC7 (Sigma, H2662), anti-MYC (Abcam, Cambridge, MA; ab32072), anti-HDAC9 (Abcam, Cambridge, MA; ab59718), and normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA; sc-2027) as a control. The IP beads used were Anti-Rabbit Ig IP Agarose Beads (Rockland, 00-8800-25).
Co-immunoprecipitation (Co-IP)
For co-immunoprecipitation, we used previously described procedures [46] with some modifications. Briefly, a lysis buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.5 mM EDTA, 1% Nonidet P-40, 1% Triton X-100) containing protease inhibitors was prepared freshly to lyze cells for immunoprecipitation. The lysate, together with the aforementioned antibodies and IP beads were incubated overnight at 4°C. The beads were then washed three times with IP lysis buffer and boiled in 2 × SDS loading buffer to elute the bound complexes. Western blotting was performed to determine the Co-IP results.
MTT assay
Cells of the 786-O line were seeded in 96-well plates at a density of 1000 cells/well, cultured for 24 h before drug treatment (see in Supplemental Table 4), and maintained in drug-free medium for 48 h. Thereafter, 100 µL of fresh medium containing 0.5 mg/mL MTT was added to each well of the 96-well plate as described previously [47]. The DAC, SAHA, and oxaliplatin contents in 786-O cells were maintained at a constant molar ratio of 2.5:1:7.5 based on the potency (IC50) of each drug. Drug synergy quantification was analysed using Compusyn software (ComboSyn, Inc.) according to the Chou-Talalay method [23]. The combined drug effects were quantified using the combination index (CI), where CI < 0.8, 0.8 < CI < 1.2, and CI > 1.2 indicated synergism, additive effect, and antagonism, respectively.
Platinum cellular uptake in 786-O
Cells of the 786-O line were seeded in 6 cm cee dishes. After OCT2 expression was induced by DAC, SAHA or DAC+SAHA, 100 µM oxaliplatin was administrated for 24 h. The platinum cellular uptake was detected by ICP-MS, completed by Hangzhou Leading Pharmatech Co., Ltd.
Statistical analysis
A two-tailed paired t-test was used to analyse differences in HDAC7/HDAC9 expression between RCC tissues and tumour-adjacent tissues. Multiple t-tests were used to evaluate differences in the expressions of MYC, HDAC7, and HDAC9 determined by RT-qPCR and histone acetylation enrichment in ChIP assays. A P value of less than 0.05 was considered significant. All statistical analyses and drug IC50 calculations were performed using Prism version 6.0.
Funding Statement
This project was supported by National Natural Science Foundation of China [81773817], National Key R&D Program of China [No. 2017YFC0908600, 2017YFE0102200], and the National Natural Science Foundation of China [No. 81702801].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Materials
Supplemental data for this article can be accessed here.
References
- [1].Ljungberg B, Cowan NC, Hanbury DC, et al. EAU guidelines on renal cell carcinoma: the 2010 update. Eur Urol. 2010;58(3):398–406. [DOI] [PubMed] [Google Scholar]
- [2].Baldewijns MM, van Vlodrop IJ, Schouten LJ, et al. Genetics and epigenetics of renal cell cancer. Biochim Biophys Acta. 2008;1785(2):133–155. [DOI] [PubMed] [Google Scholar]
- [3].Gorboulev V, Ulzheimer JC, Akhoundova A, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16(7):871–881. [DOI] [PubMed] [Google Scholar]
- [4].Motohashi H, Sakurai Y, Saito H, et al. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13(4):866–874. [DOI] [PubMed] [Google Scholar]
- [5].Prasad B, Johnson K, Billington S, et al. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos. 2016;44(12):1920–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Koepsell H, Gorboulev V, Arndt P.. Molecular pharmacology of organic cation transporters in kidney. J Membr Biol. 1999;167(2):103–117. [DOI] [PubMed] [Google Scholar]
- [7].Pritchard JB, Miller DS. Renal secretion of organic anions and cations. Kidney Int. 1996;49(6):1649–1654. [DOI] [PubMed] [Google Scholar]
- [8].Budiman T, Bamberg E, Koepsell H, et al. Mechanism of electrogenic cation transport by the cloned organic cation transporter 2 from rat. J Biol Chem. 2000;275(38):29413–29420. [DOI] [PubMed] [Google Scholar]
- [9].Burger H, Zoumaro-Djayoon A, Boersma AW, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br J Pharmacol. 2010;159(4):898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang S, Lovejoy KS, Shima JE, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66(17):8847–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yao Z, Guo H, Yuan Y, et al. Retrospective analysis of docetaxel, oxaliplatin plus fluorouracil compared with epirubicin, cisplatin and fluorouracil as first-line therapy for advanced gastric cancer. J Chemother. 2014;26(2):117–121. [DOI] [PubMed] [Google Scholar]
- [12].Yokoo S, Masuda S, Yonezawa A, et al. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab Dispos. 2008;36(11):2299–2306. [DOI] [PubMed] [Google Scholar]
- [13].McMurry MT, Krangel MS. Pillars article: a role for histone acetylation in the developmental regulation of V(D)J recombination. Science. 2000;287:495–498. [PubMed] [Google Scholar]
- [14].Wurtele H, Li Q, Zhou H, et al. Histone acetylation and chromatin assembly. Med Sci. 2009;25(2):121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dedes KJ, Dedes I, Imesch P, et al. Acquired vorinostat resistance shows partial cross-resistance to ‘second-generation’ HDAC inhibitors and correlates with loss of histone acetylation and apoptosis but not with altered HDAC and HAT activities. Anticancer Drugs. 2009;20(5):321–333. [DOI] [PubMed] [Google Scholar]
- [16].Jin Q, Yu LR, Wang L, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. Embo J. 2011;30(2):249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu Y, Zheng X, Yu Q, et al. Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin. Sci Transl Med. 2016;8(348):348r–97r. [DOI] [PubMed] [Google Scholar]
- [19].Stewart DJ, Issa JP, Kurzrock R, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15(11):3881–3888. [DOI] [PubMed] [Google Scholar]
- [20].Ljungberg B, Bensalah K, Canfield S, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol. 2015;67(5):913–924. [DOI] [PubMed] [Google Scholar]
- [21].Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116(17):4043–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chiappinelli KB, Zahnow CA, Ahuja N, et al. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016;76(7):1683–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. [DOI] [PubMed] [Google Scholar]
- [24].Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. New Engl J Med. 1996;335(12):865. [DOI] [PubMed] [Google Scholar]
- [25].Cohen HT, McGovern FJ. Renal-cell carcinoma. New Engl J Med. 2005;353(23):2477. [DOI] [PubMed] [Google Scholar]
- [26].Fletcher JI, Haber M, Henderson MJ, et al. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10(2):147–156. [DOI] [PubMed] [Google Scholar]
- [27].Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. [DOI] [PubMed] [Google Scholar]
- [28].Dai B, Kim O, Xie Y, et al. Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 2006;66(16):8058–8064. [DOI] [PubMed] [Google Scholar]
- [29].Sengupta D, Kannan A, Kern M, et al. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics. 2015;10(6):460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lehrmann H, Pritchard LL, Harel-Bellan A. Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation. Adv Cancer Res. 2002;86:41–65. [DOI] [PubMed] [Google Scholar]
- [31].Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280(2):168–176. [DOI] [PubMed] [Google Scholar]
- [32].Osada H, Tatematsu Y, Saito H, et al. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004;112(1):26–32. [DOI] [PubMed] [Google Scholar]
- [33].Ouaissi M, Sielezneff I, Silvestre R, et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol. 2008;15(8):2318–2328. [DOI] [PubMed] [Google Scholar]
- [34].Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6(6):579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McMahon SB, Van Buskirk HA, Dugan KA, et al. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94(3):363–374. [DOI] [PubMed] [Google Scholar]
- [36].Frank SR, Parisi T, Taubert S, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4(6):575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Su R, Gong JN, Chen MT, et al. c-Myc suppresses miR-451 YWTAZ/AKT axis via recruiting HDAC3 in acute myeloid leukemia. Oncotarget. 2016;7(47):77430–77443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rountree MR, Bachman KE, Herman JG, et al. DNA methylation, chromatin inheritance, and cancer. Oncogene. 2001;20(24):3156–3165. [DOI] [PubMed] [Google Scholar]
- [39].Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. [DOI] [PubMed] [Google Scholar]
- [40].Efferth T. Resistance to targeted ABC transporters in cancer. Springer International Publishing Switzerland; 2015. [Google Scholar]
- [41].Yu Q, Liu Y, Zheng X, et al. Histone H3 lysine 4 trimethylation, lysine 27 trimethylation, and lysine 27 acetylation contribute to the transcriptional repression of solute carrier family 47 member 2 in renal cell carcinoma. Drug Metab Dispos. 2017;45(1):109–117. [DOI] [PubMed] [Google Scholar]
- [42].Chen MY, Liao WS, Lu Z, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer. 2011;117(19):4424–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dong Y, Desmond JC, Ong JM, et al. Suberoylanilide hydroxamic acid (SAHA) crosses the blood-brain barrier and inhibits growth of human multiforme glioblastomas in the brains of immunodeficient mice. Cancer Res. 2005;65:S771. [Google Scholar]
- [44].Paddison PJ, Cleary M, Silva JM, et al. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat Methods. 2004;1(2):163–167. [DOI] [PubMed] [Google Scholar]
- [45].Richon VM, Webb Y, Merger R, et al. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc Natl Acad Sci USA. 1996;93(12):5705–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Foltman M, Sanchez-Diaz A. Studying protein-protein interactions in budding yeast using co-immunoprecipitation. Methods Mol Biol. 2016;1369:239–256. [DOI] [PubMed] [Google Scholar]
- [47].Plumb JA. Cell sensitivity assays: the MTT assay. Methods Mol Med. 1999;28:25–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.