

Abstract
CellMiner (http://discover.nci.nih.gov/cellminer) and CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/) are web-based applications for mining publicly available genomic, molecular, and pharmacological datasets of human cancer cell lines including the NCI-60, Cancer Cell Line Encyclopedia (CCLE), Genomics of Drug Sensitivity in Cancer (GDSC), Cancer Therapeutics Response Portal (CTRP), NCI/DTP small cell lung cancer (SCLC), and NCI Almanac cell line sets. Here we introduce our RNA sequencing data for the NCI-60 and their access and integration with the other databases. Correlation to transcript microarray expression levels for identical genes and identical cell lines across CellMinerCDB demonstrate the high quality of these new RNA-seq data. We provide composite and isoform transcript expression data and demonstrate diversity in isoform composition for individual cancer- and pharmacologically relevant genes, including HRAS, PTEN, EGFR, RAD51, ALKBH2, BRCA1, ERBB2, TP53, FGFR2, and CTNND1. We reveal cell-specific differences in the overall levels of isoforms and show their linkage to expression of RNA processing and splicing genes as well as resultant alterations in cancer and pharmacological gene sets. Gene-drug pairings linked by pathways or functions show specific correlations to isoforms compared to composite gene expression, including ALKBH2-benzaldehyde, AKT3-vandetanib, BCR-imatinib, CDK1 and 20-palbociclib, CASP1-imexon, and FGFR3-pazopanib. Loss of MUC1 20 amino acid variable number tandem repeats, which is used to elicit immune response, and the presence of the androgen receptor AR-V4 and -V7 isoforms in all NCI-60 tissue of origin types demonstrates translational relevance. In summary, we introduce RNA-seq data to our CellMiner and CellMinerCDB web-applications, allowing their exploration for both research and translational purposes.
Keywords: cancer cell lines, genetic variants, drug resistance, biomarkers, pharmacogenomics
Introduction
Cancer cell line drug and genomic databases, pioneered by the National Cancer Institute in the 1980’s with the NCI-60 cancer cell line panel (1–4), have subsequently been expanded to larger numbers of cell lines in initiatives including the Cancer Cell Line Encyclopedia (CCLE) from the Broad Institute, the Genomics of Drug Sensitivity in Cancer (GDSC) from the Massachusetts General Hospital (MGH)-Sanger Institute, and the Genentech Cell Line Screening Initiative (gCSI) (5–7), expanding both molecular and drug response data for the field. Efforts continue in the areas of accumulating, organizing, making available, interpreting and applying these forms of data for the dual purposes of gaining a better understanding of cancers and selecting prospective treatment options (5–11). The NCI-60 cell line screen, with ~21,000 drug and compound activities (in CellMiner and CellMinerCDB) (9,12), and 5,355 two-drug combinations (in NCI ALMANAC) (9,13), in addition to its more extensive molecular characterization, remains the largest repository of drug versus molecular feature information by logs. Integration of the datasets from the NCI, the Broad Institute, the Massachusetts General Hospital and the Sanger Institute, has now been done within CellMinerCDB, facilitating their assessment and comparison (9). However, the ability to understand and utilize the phenotypic effects of the complex translational changes that may occur in multiple genes, multiple molecular pathways, and multiple gene functional groups simultaneously remains challenging.
The widespread application of RNA sequencing to human diseases has provided both opportunities and challenges due to the informative but complex nature of the resultant data (7,14). Desire for a better understanding of this complexity is motivated by the recognition that disruption of transcript fidelity and increased diversity is common in cancer, resulting in functionally variable end-products and cancer-specific isoform variations that contributes to disease progression (15). Currently, RNA-seq is being used diagnostically, prognostically and therapeutically. Examples include: i) identification of differences in clear cell renal carcinoma patients stratified as being low-risk by clinicopathologic prognostic algorithms, who are actually at high risk of death, ii) providing information regarding the biology, prognosis, and therapy for multiple myeloma, iii) the recognition of radiation-sensitive transcriptional pathways in prostate cancer, iv) assessing the association of miR-204 with survival in cancer, and v) combining RNA-seq with other forms of data for enhanced prediction of therapeutic response (16–21).
In the current study, we introduce RNA sequencing data for the NCI-60 cell lines as a scientific resource. This form of data is important as it: i) is currently widely used for patient samples, ii) has greater dynamic range than microarrays, and iii) provides information on isoforms. Regarding the latter, here we determine both the composite (of all isoforms) and individual isoform expression levels for all genes. We make that data easily available through our CellMiner and CellMinerCDB web-applications in multiple forms and facilitate its comparison to other forms of molecular and pharmacological data. We enable comparisons of the composite gene expression levels to: i) microarray transcript data, ii) DNA copy number, iii) DNA methylation and iv) protein expression demonstrating significant correlations. We show that the number of isoforms varies across cell lines, and their pattern to have significant correlations to the transcript expression levels of core splicing factor genes at the individual level, as well as being linked at the omic level to RNA processing gene sets by Gene Set Enrichment Analysis. This is logical as transcript splicing and processing factors control the transcriptome. Multiple genes involved in cancers and pharmacological response are also shown to express isoforms that result in proteins with amino acid changes affecting both gene and pathway function. We demonstrate the existence of multiple biologically-linked gene-drug pairs, for which the isoform expression levels are significantly correlated with to drug activity, whereas the same gene composite expression is not. We propose that, when considering candidate biomarkers for pharmacological response, transcript isoforms of known pharmacologically relevant genes deserve consideration, at the same level of importance as mutations.
Materials and methods
Cell line source, culture, and RNA purification
Cell lines were obtained from the National Cancer Institute, Division of Cancer Treatment and Diagnosis (DCTD) tumor cell line repository (https://dtp.cancer.gov/organization/btb/docs/DCTDTumorRepositoryCatalog.pdf). Cell culture was performed as described previously, with harvesting at ~80% confluency (22). Cell line authentication is described at https://discover.nci.nih.gov/cellminer/celllineMetadata.do under the “Fingerprint” header. Mycoplasma testing was done using the Clongen laboratories, LLC Mycoplasma Detection using Polymerase Chain Reaction test. RNA was purified using a RNeasy purification kit (Qiagen, Inc.) using the manufacturer’s instructions.
RNA library preparation, sequencing, alignment, and quality control
RNA was quantified and treated with DNase according to the manufacturers protocol (Qiagen, Inc). RNA was used for generating sequencing libraries using the TotalScript™ RNA-Seq Kit (Epicentre), providing total RNA for sequencing without normalization. The libraries were sequenced at the Center for Cancer Research Sequencing facility using the HiSeq 2000 (Illumina) with paired end 100bp reads using the TruSeq Cluster Kit v3 (Illumina). Data was converted to fastq and aligned back to the human genome assembly 19 with the STAR split-read aligner. RNAeQC was used to analyze data quality (23). The Binary Sequence Alignment Map (BAM) files are deposited and available from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA433861).
Read alignment, and determination of composite gene and isoform transcript expression levels
Reads were aligned to the hg19 genome using the STAR aligner. Aligned reads were used to compute gene and isoform expression using cufflinks (version 2.2.1) (24). Gene and isoform positions were downloaded from the UCSC Table Browser “refGene” table from the “RefSeq Genes” track downloaded on 11 Aug 2016 (https://genome.ucsc.edu/cgi-bin/hgTables). We used the lower confidence limit calculated by cufflinks for expression of each gene in each cell line to detect expressions not significantly above zero. Expression values with lower confidence limit equal to zero were set to zero. Values for both the composite and isoform transcript levels are presented as fragments per kilobase per million reads (FPKM). For 642 genes with multiple locations on the genome, we selected those locations that were present in the NCBI RefSeq GRCh37 annotation. Both the composite and isoform transcript expression levels are available for download at “CellMiner \ Download Data Sets \ Download Processed Data Set \ RNA: RNA-seq”. The CellMiner url is https://discover.nci.nih.gov/cellminer.
Data comparisons and visualizations
For all molecular data comparisons described below, the raw RNA sequencing (RNA-seq) expression levels (Supplemental Table 1) were scaled logarithmically (log2) following addition of 0.1 to each data point, as log2(0) is undefined. For comparison to four other forms of molecular data, the RNA-seq genes were filtered to have a minimum of two cell lines with FPKM values ≥ 1. This molecular data used for comparison may be downloaded from “CellMiner \ Download Data Sets” and includes: i) transcript microarray expression levels from “RNA:5 Platform Gene Transcript \ z scores” used as log2 values, ii) DNA copy numbers from “Combined aCGH \ gene summary”, iii) DNA methylation data from “Illumina 450k methylation \ Gene average”, and iv) protein expression data from “SWATH (Mass spectrometry) \ Protein” (25). The normalizations of each of these data sets has been previously described (12,25–27).
The array comparative genomic hybridization (aCGH) data with total ranges greater than or equal to 1.15 (ie. ‘max copy number’ – ‘min copy number’ ≥ 1.15) were used, as this to removes genes without copy number change. Genes without copy number change will not have an influence on transcript level. Throughout the manuscript, Pearson’s correlation coefficients and p values were calculated, and the density plots and bar graphs generated using R computing unless otherwise designated (http://www.r-project.org).
CellMinerCDB databases
The cell line sets included in CellMiner Cross-Data-Base (CDB) currently are the National Cancer Institute 60 (NCI-60), Cancer Cell Line Encyclopedia (CCLE), Genomics and Drug Sensitivity in Cancer (GDSC), Cancer Therapeutics Response Portal (CTRP), Developmental Therapeutics Program Small Cell Lung Cancer Project (DTP SCLC), and the NCI Almanac. The urls for each of these are accessible through CellMinerCDB within Metadata by clicking “Select here to learn more about…” for each Cell Line Set (9). The CellMinerCDB url is https://discover.nci.nih.gov/cellminercdb/.
Gene Set Enrichment Analysis
A pre-ranked Gene Set Enrichment Analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp) was run based on a gene correlation score using the classic enrichment statistic with 1000 permutations. For each gene, we calculated the correlation value and p-value between the total number of isoforms and the composite transcript levels across the same cell lines. The gene correlation score was defined as 1/p-value (inverse of p-value) for positive correlation genes and −1/p-value otherwise.
Quantitation of drug and compound activity levels
Drug activity levels expressed as 50% growth-inhibitory levels (GI50) were determined by the DTP at 48 hours using the sulforhodamine B assay (2). That data is passed through quality control for repeat experiment consistency, a minimum range ≥1.2 log10, and a minimum of 36 cell lines with activity values as described previously, and as accessible within CellMiner (26).
MUC1 clinical trials and patient treatment based on specific cancer molecular alterations
The MUC1 clinical trials url is https://clinicaltrials.gov/ct2/results?cond=&term=muc1&cntry=&state=&city=&dist=.
Patient treatment based on specific cancer molecular alterations urls https://dctd.cancer.gov/majorinitiatives/NCI-sponsored_trials_in_precision_medicine.htm, https://clinicaltrials.gov/ct2/show/NCT01771458 and https://clinicaltrials.gov/ct2/show/NCT01306045.
Results
RNA-seq comparison to microarray transcript expression in the NCI-60
Composite transcript expression levels were determined genome-wide by RNA-seq in the NCI-60 cell lines (23,826 genes filtered after removal of duplicate genes; Supplemental Table 1). For the purpose of global comparison to other data types, genes were filtered to remove those with low levels of expression (Materials and Methods). The resultant composite transcript expression levels were compared to our microarray transcript expression data to determine consistency between platforms and assess reliability. The 14,572 selected genes in both forms of data had strong correlations between RNA-seq and microarrays gene expression, demonstrating both overall consistency between platforms and reliability of the NCI-60 RNA-seq data (Supplemental Figure 1A). The mean of the corresponding Pearson’s correlations was 0.64 (0.59 using Spearman’s), including 13,014 genes (89.3%) with significant correlations (r ≥ 0.33, p ≤ 0.01; n = 60 cell lines). Supplemental Figure 2 shows microarray versus RNA-seq expression scatter plots for nine representative genes (TP53, CDKN2A, RB1, CCNE1, SLFN11, TOP1, BRCA1, TDP1 and ERBB2). Similar and additional plots for any chosen gene can be readily obtained at the CellMinerCDB.
Comparison of RNA-seq and microarray transcript expression to other molecular data
Supplemental Figures 1B, C and D compare both the composite RNA-seq and microarray measurements of transcript expression to other forms of biologically-linked molecular data to both compare and assess. Supplemental Figure 1B compares the transcript measurements to aCGH measurements of DNA copy number change. Correlations are expected to be most positive for those genes in which DNA copy number alterations are dominant, and less for those for which aCGH variation is superseded by other regulatory influences (27). This occurs, with mean Pearson’s correlations of 0.28 and 0.32 (0.27 and 0.28 using Spearman’s) for the RNA-seq-aCGH, and transcript microarray-aCGH data, respectively.
Supplemental Figure 1C compares the transcript measurements to DNA methylation levels. Correlations are expected to be negative for those genes in which DNA methylation is influential (12). This occurs, with mean Pearson’s correlations of −0.18 and −0.17 (−0.18 and −0.17 using Spearman’s) for the RNA-seq-DNA methylation and transcript microarray-DNA methylation data, respectively.
Supplemental Figure 1D compares transcript measurements to protein expression levels (28). Correlations are expected to be positive for the subset of genes for which these parameters coincide. This occurs, with mean Pearson’s correlations of 0.27 and 0.34 (0.29 and 0.35 using Spearman’s) for the RNA-seq-protein expression, and transcript microarray-protein expression data, respectively.
Isoforms expression levels and characterization
Structural variability of transcripts resulting in multiple isoforms for a given gene can occur for multiple reasons including variable start or stop sites, splicing variation, gene fusions, nucleotide insertions and deletions, or genes that appear in multiple locations. Further assessment of the same genes presented in Supplemental Table 1 yielded transcript expression levels for a total of 46,834 gene isoforms. Expression levels for all cell lines are presented in Supplemental Table 2. In this table, the “Transcript” reference sequences define the transcripts. Also included are the “Mean abundance ratio”, the proportion of that gene transcript expressed as that isoform, and the transcript start and stop sites. The protein reference sequences, number of amino acids per isoform, and chromosome number are also indicated.
Data access using the CellMiner and CellMinerCDB web-based applications
The RNA-seq data are available both through CellMiner and CellMinerCDB, each of which have complementary content and functions. Details are provided within the respective web-applications and outlined in Figure 1.
Figure 1.
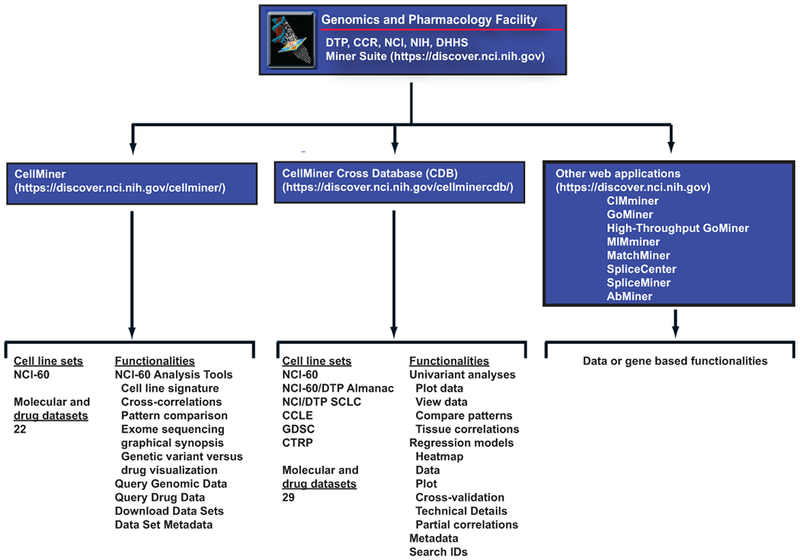
Schematic of the Genomics and Pharmacology web-applications, including their urls, cell line sets, and functionalities.
CellMiner1 provides the RNA-seq data for the NCI-60 cancer cell lines in multiple formats (Figure 2). The “Download Data Sets” tab illustrated in Figure 2A allows one to download the entire composite gene (Supplemental Table 1) or isoform (Supplemental Table 2) expression data as tables. The “Query Genomic Data” tab in Figure 2B provides access to subsets of that same data, queryable by gene name, Entrez Gene identifier, mRNA or protein RefSeq, chromosome, or genomic location. The “NCI-60 Analysis Tools” tab in Figure 2C provides three additional outputs. “Cell line signature \ RNA-seq gene expression values” provides the composite transcript expression levels for a gene (Supplemental Table 1), as well as isoform transcript expression levels for that gene (Supplemental Table 2). “Pattern comparison” compares any input pattern to the patterns of ~21,766 compound activities, ~103,881 molecular parameters (including the composite RNA-seq transcript expression levels) with ~49 phenotypic indicators and provides their correlations and p values. “Drug vs. gene variant/isoforms” is an expansion of the pre-existing “Drug vs. gene variant” tool that provides a comparison between the activity of a chosen compound and the DNA variants of a chosen gene, now updated to include isoform transcript expression level comparisons (to the same drug activity). Figure 2D exemplifies cell line signature snapshots from the CellMiner website for AR (androgen receptor) transcript levels as measured by both RNA-seq and microarray.
Figure 2.
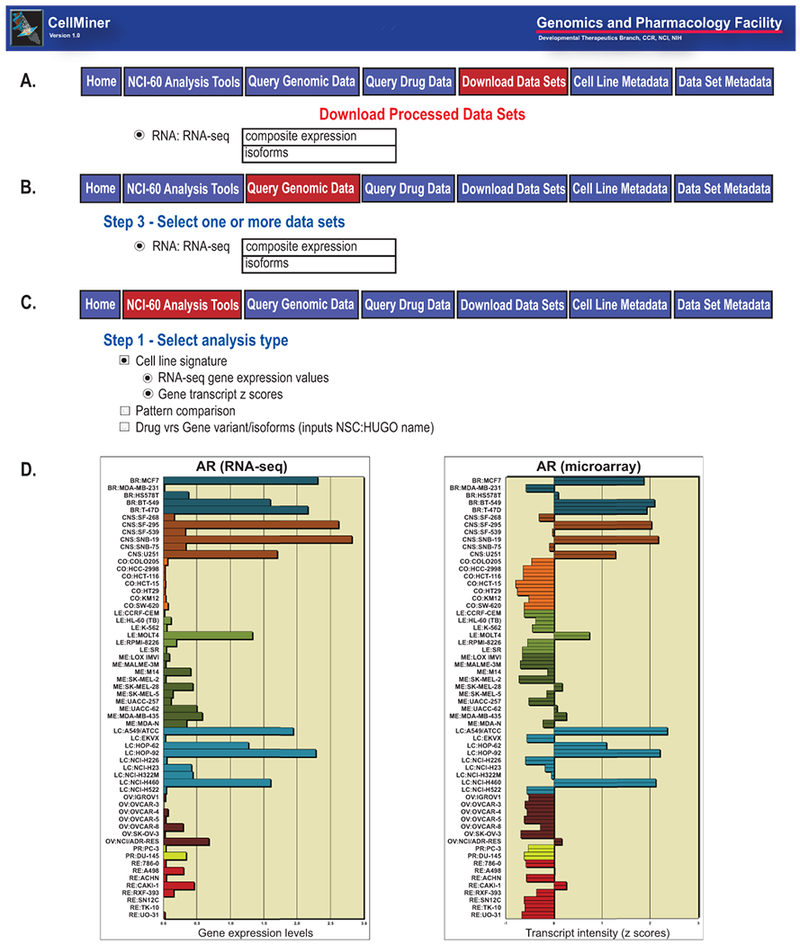
CellMiner output types1. A. The CellMiner Download Data Sets tab and the two forms of RNA-seq data available. B. The CellMiner Query Genomic Data tab and the two forms of RNA-seq data it makes available. C. The CellMiner NCI-60 Analysis Tools tab and the three tools providing RNA-seq data and/or its relationships to other parameters. D. Exemplary CellMiner Cell line signature outputs for AR transcript expression as measured by RNA-seq (log2 FPKM + 0.1) and microarray z score on the x-axis, with cell lines on the y-axis.
The recently introduced CellMinerCDB (cross-database)2 (9) provides both additional functionality and access to additional cell line sets (the CCLE, GDSC, CTRP, SCLC, and the NCI Almanac). The cell line sets and data types included have undergone identifier matching to allow comparisons. The Univariant Analysis \ Plot Data tab generates interactive scatter plots (Figure 3). Figures 3A and B exemplifies the gene AR (androgen receptor) for transcript levels as measured by RNA-seq and microarray from the NCI-60 (A) and the CCLE (B). Figures 3C and D visualize the gene TP53 for transcript levels as measured by RNA-seq from the NCI-60 and the CCLE (C), and RNA-seq from the NCI-60 and microarray for CCLE (D). Figures 3E and F visualize the gene CDH1 (E-cadherin) RNA-seq versus DNA methylation from the NCI-60 (E) and CCLE (F). In all cases the scatter plots demonstrate consistency between platforms, institutions and, most importantly cell lines.
Figure 3.
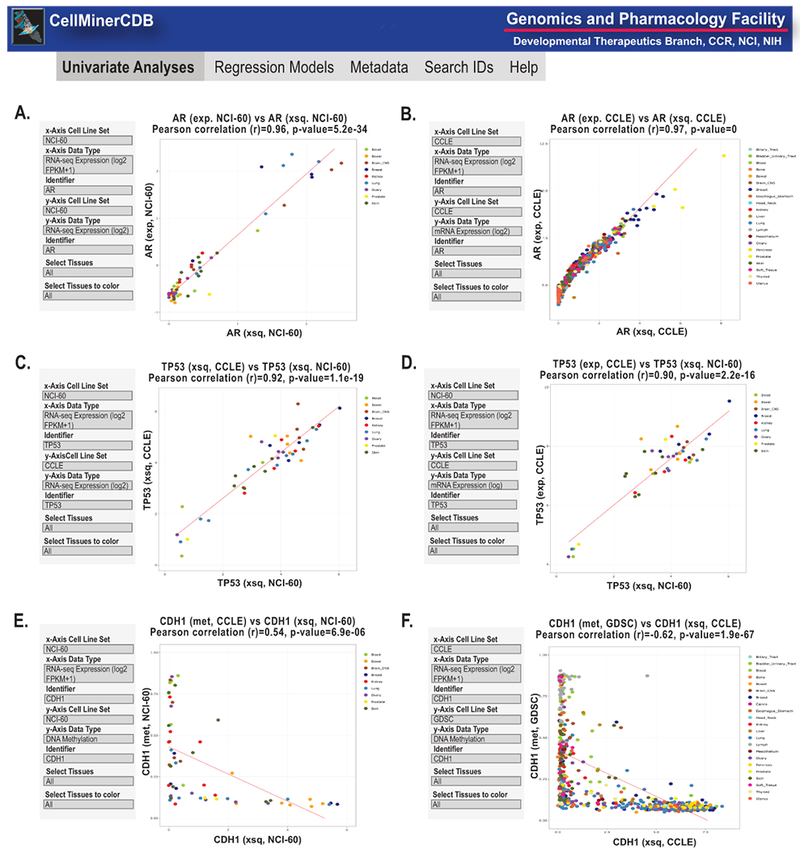
Snapshot of CellMinerCDB univariate analyses2. A-B. AR (androgen receptor) transcript expression comparisons of RNA-seq (log2 FPKM + 0.1) and microarrays for the NCI-60 and CCLE. C-D. TP53 transcript expression comparisons of RNA-seq (log2 FPKM +1) and microarrays for the NCI-60 and CCLE. E-F. CDH1 (E-cadherin) comparisons of RNA-seq (log2 FPKM +1) expression and DNA methylation levels. In all panels, the input parameters are shown on the left. Each dot is a cell line, with the color code defined by the legend on the right and in CellMinerCDB2. Regression lines are included. x- and y-axes, correlations and p values are as defined within each panel. Exp. is microarray expression using z score, xsq is RNA-seq expression using log2 FPKM +1 and met is DNA methylation.
Isoform expression, characterization, implications, and functional category enrichment
Figure 4A shows that the number of isoforms per gene (Supplemental Table 2) varies widely across genes; most genes expressing a single isoform while the histone H3K27me3 demethylase UTY (Ubiquitously Transcribed Tetratricopeptide Repeat Containing, Y-Linked) gene encoded on the Y-chromosome expressed 45 isoforms. The MUC1 gene has 18 isoforms, with its two most abundant forms having mean abundance ratios of 0.42 and 0.46 (Supplemental Table 2). Both are truncated from the reference full-length MUC1 gene coding for a 1255 polypeptide (tumor-associated epithelial membrane antigen expressed in epithelial cells) and code for polypeptides 475 and 484 amino acids in length.
Figure 4.
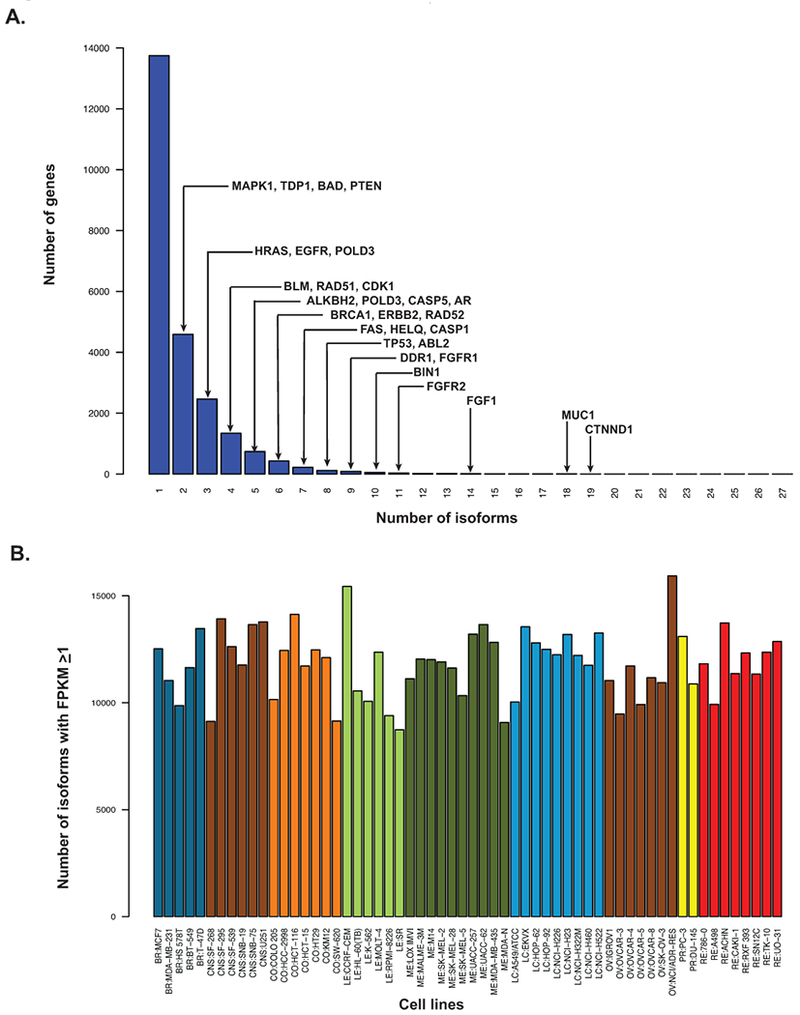
Isoform number features. A. Bar-graph distribution of the number of isoforms per gene. The x-axis is the number of isoforms. The y-axis is the number of genes with the indicated number of isoforms. Examples of genes with the indicated numbers of isoforms are indicated. B. Bar-graph of the total isoform variability per cell line. The x-axis is the NCI-60 cell lines. The y-axis is the number of isoforms present within that cell line with FPKM≥1.
The number of isoforms per cell line was also found to vary (Figure 4B). Variation in the number of bases sequenced per cell line had minimal contribution to this variation (~8%), suggesting biological source. As RNA processing genes affect isoforms, we checked for significant correlations between the composite transcript expression levels (Supplemental Table 1) of several core RNA processing genes and the number of isoforms pattern (Figure 4B) using our Pattern comparison tool (26). Significant (p < 0.002) correlations were found for core RNA processing genes including CDK12, DDX5, NONO, PPP2R1A, SF3B1 and ZRSR2. An additional comparison was done at the omic level using pre-ranked Gene Set Enrichment Analysis on the p-values from that same Pattern comparison analysis. RNA processing gene sets were found to be significantly enriched (Table 1). The Table 1 enriched gene sets all fall within the top 4% and have false discovery q-values < 3.5E-07. These results reveal the potential impact of RNA processing genes on the number of gene isoforms determined by RNA-seq of the NCI-60.
Table 1:
Enriched categories with significant correlation between the total isoform number pattern (Figure 3B) and their composite transcript expression levels (Supplemental Table 1) a
| RNA processing gene sets | Gene group size | Enrichment score | NES b |
|---|---|---|---|
| GO_MRNA_PROCESSING | 409 | 0.45 | 10.45 |
| GO_RNA_SPLICING | 348 | 0.46 | 9.84 |
| REACTOME_MRNA_PROCESSING | 153 | 0.55 | 7.77 |
| GO_SPLICEOSOMAL_COMPLEX | 162 | 0.50 | 7.27 |
| REACTOME_MRNA_SPLICING | 104 | 0.55 | 6.47 |
| KEGG_SPLICEOSOME | 123 | 0.49 | 6.43 |
Categories identified using Gene set enrichment analysis (GSEA) [23]. The input was p-values for correlations between gene composite expression levels (Supplemental Table 1), and the isoform numbers per cell line pattern (Figure 3B). The upper filter is set at 510. There are 11,173 gene sets with positive, and 3,048 with negative correlations. All Table 1 gene sets have false discovery rate q-values < 3.5E-07, fall within the top 4% of gene sets, and have positive correlations.
Normalized enrichment score after permutations.
As isoforms may affect cancer progression and/or pharmacological response, we checked for genes that affect these processes and have isoforms that lead to altered numbers of amino acids. Multiple examples were found. These include the oncogenes ABL1, ERBB2, HRAS, and PIK3CB, the tumor suppressors APC, BRCA1, E2F3, PTEN, TSC1 and VHL, the DNA-damage response genes PARP2, RAD51 and XRCC6, the chromatin affecting genes SIRT2, SMARCB1 and KDM5C, the tumor drivers ABI1, CCND3, MITF, MYH11, PPARG, PRDM1 and RALGDS, and the apoptosis affecting genes CASP1, 2, 5, 6, 7, 8, and 9, BIRC2, 5, and 7, BCL2, BCL2L1, DAP and DAP3.
Isoforms and pharmacological response
Significant correlation was found between the number of isoforms pattern (Figure 4B) and the drug responses for only two of 201 FDA-approved or clinical trial drugs, mithramycin and vorinostat (r = −0.332, p = 009 and r =0.336, p = 0.010, respectively). Mithramycin’s negative correlation indicates reduced activity in the presence of increased numbers of isoforms, and vorinostat’s positive correlation the opposite.
Table 2 exemplifies gene-drug comparisons for which the gene isoforms (Supplemental Table 2) provided improved predictability as compared to the composite expression (Supplemental Table 1). In each of these examples, we required: i) that the isoforms resulted in truncated proteins, ii) a minimum isoform abundance of 10% of the total, and iii) that the gene had known biological link to the drugs mechanisms of action. These links are direct for BCR-imatinib (the drug targets BCR-ABL tyrosine kinase), ALKBH2-benzaldhyde (a DNA alkylation repair protein and an alkylating agent), both CDK1 and CDK20-palbociclib (the drug targets CDKs) and FGFR3-pazopanib (the drug targets FGFRs). The link is through molecular pathway for AKT3-vandetanib and MAPK10-AZD-9291 (two genes in the EGFR pathway and two EGFR inhibitors). The links are functional for CASP1-imexon, CASP8-arsenic trioxide and API5-dasatinib (three apoptosis or survival genes and three apoptosis affecting drugs), both POLD3-ifosfamide and DDX11-bendamustine (two DNA-damage repair genes and two DNA-damaging drugs), and PDPK1-7hydroxystaurosporine (a protein kinase and a kinase affecting drug). The link is through prior literature for MAPK8-oxaliplatin from (29).
Table 2:
Isoform expression-drug activity pairs wth enhanced correlations to isoforms as compared to composite gene expression
| Gene |
Transcript |
Protein |
Drug |
Gene expression vs. drug activity P values i |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Names a | Type b | RefSeq c | Abundance d | Amino Acids e | Drug Name f | NSC g | Mech.(s) of action h | Isoform | Composite |
| BCR | GTPase-activating | NM_021574 | 0.13 | 1227 / 1271 | Imatinib | 743414 | BCR-ABL|PK:YK | 0.00001 | 0.2036 |
| CASP1 | Apoptotic | NM 001257119 | 0.216 | 383 / 404 | Imexon | 714597 | Apo | 0.00005 | 0.00201 |
| CASP8 | Apoptotic | NM_001080124 | 0.151 | 464 / 479 | Arsenic trioxide | 92859 | Apo | 0.00092 | 0.74096 |
| MAPK8 | Apoptotic | NM_001278548 | 0.277 | 308 / 427 | Oxaliplatin | 266046 | AlkAg | 0.00002 | 0.04352 |
| API5 | Survival | NM_006595 | 0.290 | 504 / 524 | Dasatinib | 759877 | PK:YK,PDGFR,KIT | 0.00010 | 0.34951 |
| ALKBH2 | DDR | NM_001001655 | 0.200 | 157 / 261 | Benzaldehyde (BEN) | 281612 | AlkAg | 0.00104 | 0.32629 |
| DDX11 | DDR | NM_004399 | 0.160 | 856 / 970 | Bendamustine | 138783 | AlkAg | 0.00313 | 0.41420 |
| POLD3 | DDR | NR_046409 | 0.122 | na | Ifosfamide | 109724 | AlkAg | 0.00002 | 0.04390 |
| CDK1 | Protein kinase | NM_033379 | 0.088 | 240 / 297 | Palbociclib | 758247 | PK:STK,CDK | 0.00010 | 0.01554 |
| CDK20 | Protein kinase | NM_001039803 | 0.229 | 346 / 346 | Palbociclib | 758247 | PK:STK,CDK | 0.00001 | 0.00206 |
| AKT3 | Protein kinase | NM_001206729 | 0.099 | 465 / 479 | Vandetanib | 760766 | PK:YK,EGFR | 0.00056 | 0.20447 |
| PDPK1 | Protein kinase | NM_031268 | 0.360 | 429 / 556 | 7-Hydroxystaur. | 638646 | PK:STK | 0.00225 | 0.39567 |
| MAPK10 | Protein kinase | NM_002753 | 0.49 | 422 / 464 | AZD-9291 | 779217 | PK:EGFR | 0.00005 | 0.01349 |
| FGFR3 | Signalling | NM_022965 | 0.253 | 694/806 | Pazopanib | 752782 | PK:YK,PDGFR,FGFR | 0.00003 | 0.00780 |
Gene name as defined by the UCSC Table Browser at https://genome.ucsc.edu/cgi-bin/hgTables.
DDR is DNA damage repair. Chromatin is chromatin modifying. MT is methylation transferase. Genes may fall into additional functional categories.
Transcript reference sequence as defined by NCBI at http://www.ncbi.nlm.nih.gov/nucleotide.
Abundance of isofrom as compared to other isoforms for the same gene.
Number of amino acids in protein.
All drugs are either Food and Drug Administration approved or in clinical trial. 7-Hydroxystaur. is 7-Hydroxystaurosporine.
Cancer Chemotherapy National Service Center (NSC) number.
Mech. is mechanism. Apo is apoptosis inducer. AlkAg is alkylating agent. PK is protein kinase inhibitor. YK is tyrosine kinase inhibitor. PDGFR is PDGFR inhibitor. KIT is KIT inhibitor. Db is DNA binder. T2 is topoisomerase 2 inhibitor. STK is Serine threonine kinase inhibitor. CDK is CDK inhibitor. EGFR is EGFR inhibitor. PDGFR is PDGFR inhibitor. BRAF is BRAF inhibitor. HDAC is histone deacetylase. FGFR is FGFR inhibitor.
Calculated from Pearson’s correlations. The FPKM values for both composite gene and isoform expression is were log2 transformed to make appropriate comparison to the log10 transformed drug activity data.
Discussion
Working with the NCI-60 data in CellMiner enables access to multiple forms of molecular data in easy-to-compare formats for casual and experienced users. Supplemental Figure 1 provides examples of this, comparing RNA-seq composite transcript expression data to four other relevant molecular data types. A brief description of data accession is given in association with Figure 2, with full documentation provided within CellMiner.
Supplemental Figure 1A compares RNA-seq and microarray transcript data, demonstrating concordance between the two. Identical genes exhibit high correlations and positive association with variance in both microarray and RNA-seq, with higher variance associated with higher correlation. Supplemental Figures 1B–D compare RNA-seq and transcript microarray data to aCGH, DNA methylation and protein expression and reveals largely concordant results for the two forms of transcript data with a more well separated bimodal distribution for the transcript microarray comparisons to the aCGH. Use of Spearman’s as opposed to Pearson’s correlations is largely concordant, highlighting data reliability and the fact that correlation are not driven by outliers. Existing variability between the RNA-seq and transcript microarray data is likely due to a combination of both the intrinsic difference in the two data forms (hybridization versus sequence counting), as well as their handling. The transcript microarray data is a combination of five microarrays that have undergone quality control that minimize the influence of variable isoforms (26).
Characterization of isoform expression levels in the NCI-60 in Supplemental Table 2 demonstrates multiple types of transcript variability, including: i) the number of isoforms per gene (Figure 4A), ii) individual isoform abundance levels by cell, iii) isoform abundance summed across the NCI-60 (the mean abundance ratio values from column 3 of Supplemental Table 2), iv) total number of isoforms per cell (Figure 4B), and v) number of amino acids present in isoforms of individual genes (column 7 of Supplemental Table 2). Thus, both the expression and functional landscapes become more complex when considering RNA-seq isoform levels. This also may have translational implication, as exemplified by the MUC1 isoform variability. Cancer cells expressing MUC1 are currently targeted in clinical trials in which its 20 amino acid variable number tandem repeat is used to elicit immune response. As the number of these repeats occurring in normal individuals is felt to vary from ~ 20 to 120 times and the forms expressed in all the NCI-60 cell lines have only one full length repeat (a reduction that could easily affect immune response), determination of repeat number of a patients expressed isoform would seem a desirable biomarker for patients enrolling in these trials.
The ability to test consistency across platforms, as exemplified using CellMiner in Supplemental Figure 1A–D, as well as across both platforms and institutes, as tested using CellMinerCDB in Figure 3A–F, provides quality control that gives the user confidence when results match. The ability to compare different cell line sets using CellMinerCDB allows users to easily make comparisons between data that only exists in one of the cell line sets, as in the Figure 3F comparison of CCLE RNA-seq and GDSC DNA methylation data for CDH1 (9).
The pattern of the total number of isoforms per cell line visualized in Figure 4B provides a functional measurement of overall RNA processing activity across cell lines. Comparison of the pattern of these isoforms to the composite transcript expressions (Supplemental Table 2) shows significant correlation to multiple core RNA processing genes that have previously been recognized in human malignancies leading to splicing alterations (15,30). This, in addition to the enrichment of RNA processing gene sets demonstrated in Table 1, provides a putative transcript regulatory explanation for the observed isoform variation across cell lines.
The genes we list in results with isoform variability that might affect cancer progression and/or pharmacological response highlight the potential widespread effects of isoform variations. From our NCI-60 RNA-seq analyses, we identified specific tumor drivers reported to have isoform variation resulting in altered protein products in cancer including ABI1, CCND3, MITF, MYH11, PPARG, PRDM1 and RALGDS (31). Chromatin factors and apoptosis genes are known to have altered transcripts in cancer (15,30,32–34). Splicing alterations in cancer have also previously been reported in oncogenes, and genes involved in proliferation, invasion, DNA repair, DNA damage and drug resistance (30,31,33,35).
The concept of RNA splicing dysregulation in refractory cancers has been proposed as an avenue for therapy (33). That just two FDA-approved drugs showed significant correlations to the number of isoforms per cell line pattern, mithramycin (a DNA alkylating agent and RNA synthesis inhibitor) and vorinostat (an HDAC inhibitor), is not surprising in the statistical context, as the employed p value of 0.01 would predict that number of drugs by chance. However, pharmacologically, it is plausible it is a reflection that the current FDA-approved and clinical trial drugs for which we have data were not designed to affect overall RNA processing and are unlikely to act selectively in that capacity. Potentially more valuable in this context, if they are screened, will be the anti-cancer splicing modulators (36,37).
When we considered individual genes with isoform alterations known to be associated to drug response, some notable correlations were found. Table 2 provides examples of isoforms with significant correlation to biologically-linked pharmacological responses. In these examples, the isoform provides a more predictive indicator of pharmacological response than the composite gene expression. MAPK8 is included as its isoforms have previously been proposed to affect oxaliplatin response (29). Taken into consideration with the previously reported interrelationships between RNA processing alterations, cancer and pharmacology, these results emphasize the need for consideration of isoforms in addition to composite expression when mining biomarkers for use in making pharmacological prediction. Additional study will be required to determine the influence of the multiple factors that may affect reliability of this sort of analysis, such as data pipeline employed, abundance of transcript, abundance of isoform, number of isoforms per gene, variability of pattern for these parameters, and depth of sequence reads.
The transcription factor androgen receptor (AR) has perhaps the most intensively studied and clinically-relevant cancer-related isoforms (38,39). Multiple forms of these splicing variants (AR-Vs) bind DNA, activating programs of gene expression distinguishable from that of full-length AR (AR-FL) (40). AR-V7, the most well characterized of these, has been shown to: i) bind DNA, ii) retain its N-terminal transactivation domain, iii) delete its C-terminal ligand-binding domain (LBD), iv) result in a constitutively active, ligand-independent transcription factor, v) be associated with prior AR-targeted therapy, vi) be predictive of resistance to abiraterone and enzalutamide in castration-resistant prostate cancer, and vii) be prognostic (38). AR-V4 dimerizes with both full length and V7 forms of AR and mitigates enzalutamide’s inhibition of full-length AR (41). The NCI-60 cells express AR-FL, AR-V7, and AR-V4 (NM000044, NM001348063, and NM001348063, respectively), along with two more severely truncated versions (Supplemental Table 2). AR-FL and AR-V7 are both expressed in eighteen cell lines, with AR-V7 expressed at lower levels, consistent with prior reports (39). All nine NCI-60 tissue of origin types are positive for expression of AR-V4, V7 and/or full-length AR. This broad-based expression of multiple forms of AR supports the current interest in AR as a drug target or prognostic indicator for non-prostatic indications (38,42,43). Clinically, AR variants are detectable either in circulating tumor cells or by digital mRNA analysis platforms usable in fresh, frozen or formalin-fixed paraffin-embedded tissue, and they have been shown to be predictive of resistance to AR-targeted therapy (38,42,44). While additional clarification is required, the AR experience in prostate cancer, demonstrates that analysis of variant isoforms can and should be done to facilitate a precision medicine approach to therapy (42,45). These same approaches may be applied to other genes known to have either cancer or pharmacological importance, and isoform variability.
Currently the attempt is being made to provide patient treatment with greater consideration of their specific cancer’s molecular alterations, primarily using DNA mutational changes and occasionally expression level as biomarkers. As demonstrated in our genome-wide study of the NCI-60, cancer cells contain multiple functionally relevant isoforms, including pharmacologically important genes. As many of these isoforms have substantial amino acid losses, it is probable that their functional effect is as important as DNA mutational changes. In addition, isoform alterations can be expected to have cumulative effects for those cancers with increased levels of RNA processing instability. Isoform variability should be considered going forward when analyzing and interpreting RNA-seq data in clinical samples, and potentially for looking for biomarkers of both cancer progression and pharmacological intervention.
Supplementary Material
Statement of significance.
The current study provides RNA sequencing data for the NCI-60 cell lines made accessible through both CellMiner and CellMinerCDB and is an important pharmacogenomics resource for the field.
Acknowledgements
Our studies are supported by the Intra-mural Program of the National Cancer Institute, Center for Cancer Research (Z01 BC 011497).
Footnotes
There are no conflicts of interest to report.
References
- 1.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther 2010;9:1451–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, et al. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst 1990;82:1113–8 [DOI] [PubMed] [Google Scholar]
- 3.Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet 2000;24:236–44 [DOI] [PubMed] [Google Scholar]
- 4.Chabner BA. NCI-60 Cell Line Screening: A Radical Departure in its Time. J Natl Cancer Inst 2016;108. [DOI] [PubMed] [Google Scholar]
- 5.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483:570–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 2016;533:333–7 [DOI] [PubMed] [Google Scholar]
- 8.Wilks C, Cline MS, Weiler E, Diehkans M, Craft B, Martin C, et al. The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database (Oxford) 2014;2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, et al. CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 2018;10:247–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 2012;109:15030–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohlhagen G, Paull K, Cushman M, Nagafufuji P, Pommier Y. Protein-linked DNA strand breaks induced by NSC 314622, a non-camptothecin topoisomerase I poison. Mol Pharmacol 1998;54:50–8 [DOI] [PubMed] [Google Scholar]
- 12.Reinhold WC, Varma S, Sunshine M, Rajapakse V, Luna A, Kohn KW, et al. The NCI-60 Methylome and its Integration into CellMiner. Cancer Res 2017;77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holbeck SL, Camalier R, Crowell JA, Govindharajulu JP, Hollingshead M, Anderson LW, et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res 2017;77:3564–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casamassimi A, Federico A, Rienzo M, Esposito S, Ciccodicola A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016;35:2413–27 [DOI] [PubMed] [Google Scholar]
- 16.Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 2016;17:257–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keam SP, Caramia F, Gamell C, Paul PJ, Arnau GM, Neeson PJ, et al. The Transcriptional Landscape of Radiation-Treated Human Prostate Cancer: Analysis of a Prospective Tissue Cohort. Int J Radiat Oncol Biol Phys 2018;100:188–98 [DOI] [PubMed] [Google Scholar]
- 18.Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 2015;33:306–12 [DOI] [PubMed] [Google Scholar]
- 19.Parasramka M, Serie DJ, Asmann YW, Eckel-Passow JE, Castle EP, Stanton ML, et al. Validation of Gene Expression Signatures to Identify Low-risk Clear-cell Renal Cell Carcinoma Patients at Higher Risk for Disease-related Death. Eur Urol Focus 2016;2:608–15 [DOI] [PubMed] [Google Scholar]
- 20.Safikhani Z, Smirnov P, Thu KL, Silvester J, El-Hachem N, Quevedo R, et al. Gene isoforms as expression-based biomarkers predictive of drug response in vitro. Nat Commun 2017;8:1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szalat R, Avet-Loiseau H, Munshi NC. Gene Expression Profiles in Myeloma: Ready for the Real World? Clin Cancer Res 2016;22:5434–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H, D’Andrade Petula, Fulmer-Smentek Stephanie, Lorenzi Philip, Kohn Kurt W., Weinstein John N., et al. mRNA and microRNA expression profiles of the NCI-60 integrated with drug activities. MCT 2010;9(5):1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 2012;28:1530–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo T, Luna A, Koh CC, Rajapakse V, Wu Z, Menden MP, et al. Rapid proteotyping reveals cancer biology and drug response determinants in the NCI-60 cells. 2018 [Google Scholar]
- 26.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, et al. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res 2012:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varma S, Pommier Y, Sunshine M, Weinstein JN, Reinhold WC. High resolution copy number variation data in the NCI-60 cancer cell lines from whole genome microarrays accessible through CellMiner. PloS one 2014;9:e92047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishizuka S, Charboneau L, Young L, Major S, Reinhold WC, Waltham M, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A 2003;100:14229–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasilevskaya IA, Selvakumaran M, Hierro LC, Goldstein SR, Winkler JD, O’Dwyer PJ. Inhibition of JNK Sensitizes Hypoxic Colon Cancer Cells to DNA-Damaging Agents. Clin Cancer Res 2015;21:4143–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA 2016;22:1285–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sebestyen E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res 2015;43:1345–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koschmann C, Nunez FJ, Mendez F, Brosnan-Cashman JA, Meeker AK, Lowenstein PR, et al. Mutated Chromatin Regulatory Factors as Tumor Drivers in Cancer. Cancer Res 2017;77:227–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meliso FM, Hubert CG, Galante PAF, Penalva LO. RNA processing as an alternative route to attack glioblastoma. Hum Genet 2017;136:1129–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dasgupta A, Nomura M, Shuck R, Yustein J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int J Mol Sci 2016;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010;24:2343–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bates DO, Morris JC, Oltean S, Donaldson LF. Pharmacology of Modulators of Alternative Splicing. Pharmacol Rev 2017;69:63–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonarakis ES. AR Signaling in Human Malignancies: Prostate Cancer and Beyond. Cancers (Basel) 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu C, Luo J. Decoding the androgen receptor splice variants. Transl Androl Urol 2013;2:178–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Lonergan PE, Nacusi LP, Wang L, Schmidt LJ, Sun Z, et al. The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J Urol 2015;193:690–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhan Y, Zhang G, Wang X, Qi Y, Bai S, Li D, et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol Cancer Res 2017;15:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christenson JL, Trepel JB, Ali HY, Lee S, Eisner JR, Baskin-Bey ES, et al. Harnessing a Different Dependency: How to Identify and Target Androgen Receptor-Positive Versus Quadruple-Negative Breast Cancer. Horm Cancer 2018;9:82–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitkov M, Joseph R, Copland J 3rd. Steroid hormone influence on melanomagenesis. Mol Cell Endocrinol 2015;417:94–102 [DOI] [PubMed] [Google Scholar]
- 44.Bastos DA, Antonarakis ES. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev Mol Diagn 2018;18:155–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun 2016;7:13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.